
Mechanism and structure–activity relationship of H2 and CO2 activation at the ZnO/Cu catalyst interface†

Abstract
Cu/ZnO/Al2O3 catalysts are the most well-known heterogeneous catalysts for the hydrogenation of CO and CO2 into methanol. Herein, density functional theory calculations were performed to investigate the mechanism of H2 activation and the effects of hydrogen spillover on CO2 adsorption and activation at the interfacial site of the ZnO/Cu model catalyst, which was simulated by loading ZnO ribbons of different sizes on the Cu(111), Cu(100), and Cu(211) surfaces. The ZnO/Cu interface is found to facilitate the formation of H adsorbates from the dissociation of H2 molecules, which promotes the facile formation of oxygen vacancy (VO) sites in the ZnO component due to its reducibility and the hydrogen spillover effect. The resulting interfacial structure of the ZnO/Cu model catalyst can contain perfect, hydroxylated, and oxygen-vacancy-present ZnO sites, which may act as the adsorption and activation sites for CO2. Further calculations show that molecular CO2 adsorbed at the VO site 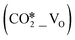 can be efficiently activated by direct dissociation or hydrogenation to the HCOO* species. In addition, the smaller ZnO structure and less exposure of the Cu(211) facet facilitate hydrogen spillover and the formation of the interfacial VO site. This study provides important insights into the structure–activity relationship for the active sites of the ZnO/Cu model catalyst and the mechanisms of CO2 activation and hydrogenation.
can be efficiently activated by direct dissociation or hydrogenation to the HCOO* species. In addition, the smaller ZnO structure and less exposure of the Cu(211) facet facilitate hydrogen spillover and the formation of the interfacial VO site. This study provides important insights into the structure–activity relationship for the active sites of the ZnO/Cu model catalyst and the mechanisms of CO2 activation and hydrogenation.

 Please wait while we load your content...
Please wait while we load your content...

