
Photocatalytic conversion of CO2 to CO by Ru(ii) and Os(ii) octahedral complexes: a DFT/TDDFT study†
 *a
and
*a
and
Abstract
The reaction mechanisms of the photocatalytic reduction of CO2 to CO catalyzed by [(en)M(CO)3Cl] complexes (M = Ru, Os, en = ethylenediamine) in the presence of triethanolamine (TEOA), R3N (R = –CH2CH2OH), in DCM and DMF solvents, were studied by means of DFT/TDDFT electronic structure calculations. The geometric and free energy reaction profiles for two possible reaction pathways were calculated. Both reaction pathways studied, start with the 17e−, catalytically active intermediate, [(en)M(CO)3]˙+ generated from the first triplet excited state, T1 upon reductive quenching by TEOA which acts as a sacrificial electron donor. In the first possible pathway, TEOA− anion binds to the metal center of the catalytically active intermediate, [(en)M(CO)3]˙+ followed by CO2 insertion into the M-OCH2CH2NR2 bond. The latter upon successive protonations releases a metal ‘free’ [R2NCH2CH2OC(O)(OH)] intermediate which starts a new and final catalytic cycle, leading to the formation of CO and H2O while regenarating TEOA. In the second possible pathway, the 17e−, catalytically active intermediate, [(en)M(CO)3]˙+ captures CO2 molecule, forming an η1-CO2 complex. Upon 2H+/2e− successive protonations and reductions, CO product is obtained along with regenarating the catalytically active intermediate [(en)M(CO)3]˙+. The nature of the proton donor affects the reaction profiles of both mechanisms. The nature of the solvent does not affect significantly the reaction mechanisms under study. Finally, since photoexcitation and T1 reductive quenching are common to both pathways, we have srutinized the photophysical properties of the [(en)M(CO)3Cl] complexes along with their T1 excited states reduction potentials, 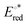 . The [(en)M(CO)3Cl] complexes absorb mainly in the UV region while the absolute
. The [(en)M(CO)3Cl] complexes absorb mainly in the UV region while the absolute 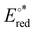 are in the range 6.4–0.9 eV.
are in the range 6.4–0.9 eV.

 Please wait while we load your content...
Please wait while we load your content...

