
Native mass spectrometry for the design and selection of protein bioreceptors for perfluorinated compounds†
Elise
Daems‡
 abc,
Giulia
Moro‡
acd,
Herald
Berghmans
e,
Ligia M.
Moretto
d,
Silvia
Dewilde§
e,
Alessandro
Angelini
df,
Frank
Sobott
*bgh and
Karolien
De Wael
*ac
abc,
Giulia
Moro‡
acd,
Herald
Berghmans
e,
Ligia M.
Moretto
d,
Silvia
Dewilde§
e,
Alessandro
Angelini
df,
Frank
Sobott
*bgh and
Karolien
De Wael
*ac
aAXES Research Group, University of Antwerp, Groenenborgerlaan 171, 2020 Antwerp, Belgium. E-mail: karolien.dewael@uantwerpen.be
bBAMS Research Group, University of Antwerp, Groenenborgerlaan 171, 2020 Antwerp, Belgium. E-mail: F.Sobott@leeds.ac.uk
cNANOlab Centre of Excellence, University of Antwerp, Groenenborgerlaan 171, 2020 Antwerp, Belgium
dDepartment of Molecular Science and Nanosystems, Università Ca'Foscari, Via Torino 155, 30172 Mestre, Italy
ePPES Research Group, University of Antwerp, Universiteitsplein 1, 2610 Wilrijk, Belgium
fEuropean Centre for Living Technology (ECLT), Ca'Bottacin, Dorsoduro 3911, Calle Crosera, 30123 Venice, Italy
gAstbury Centre for Structural Molecular Biology, University of Leeds, Leeds LS2 9JT, UK
hSchool of Molecular and Cellular Biology, University of Leeds, Leeds LS2 9JT, UK
First published on 18th January 2021
Abstract
Biosensing platforms are answering the increasing demand for analytical tools for environmental monitoring of small molecules, such as per- and polyfluoroalkyl substances (PFAS). By transferring toxicological findings in bioreceptor design we can develop innovative pathways for biosensor design. Indeed, toxicological studies provide fundamental information about PFAS-biomolecule complexes that can help evaluate the applicability of the latter as bioreceptors. The toolbox of native mass spectrometry (MS) can support this evaluation, as shown by the two case studies reported in this work. The analysis of model proteins’ (i.e. albumin, haemoglobin, cytochrome c and neuroglobin) interactions with well-known PFAS, such as perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS), demonstrated the potential of this native MS screening approach. In the first case study, untreated albumin and delipidated albumin were compared in the presence and absence of PFOA confirming that the delipidation step increases albumin affinity for PFOA without affecting protein stability. In the second case study, the applicability of our methodology to identify potential bioreceptors for PFOS/PFOA was extended to other proteins. Structurally related haemoglobin and neuroglobin revealed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex, whereas no binding was observed for cytochrome c. These studies have value as a proof-of-concept for a general application of native MS to identify bioreceptors for toxic compounds.
:1 complex, whereas no binding was observed for cytochrome c. These studies have value as a proof-of-concept for a general application of native MS to identify bioreceptors for toxic compounds.
Introduction
Biosensing platforms are answering the increasing demand for analytical tools for environmental monitoring.1–3 A key step in the development of biosensors is the selection of the bioreceptor or biorecognition element, a biomolecule (e.g. protein or nucleic acid) which enables the specific recognition of the analyte and undergoes biochemical changes that can be further translated into a detectable signal.4 The possibility to combine a protein bioreceptor within a biosensing platform depends mainly on the affinity and specificity of the recognition, the compatibility with the matrix of interest (e.g. waste water, biological fluids, etc.), the production costs and the physicochemical stability under the required working conditions.5 These criteria should be carefully evaluated while screening new bioreceptors for small molecules, such as per- and polyfluorinated alkyl substances (PFAS).PFAS, a class of more than 3000 manmade chemicals, represent a global issue due to their persistent, bioaccumulative and toxic behaviour, which has adverse effects on the ecosystem and human health.6–8 In the last two decades, improved regulatory plans and phase-out initiatives supported by in-depth toxicological and environmental studies limited the usage of PFAS, such as perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS).9–12 However, Barceló and Ruan13 pointed out that “this is not the end of the story” since novel fluorinated compounds are continuously entering the market to replace the old ones, following the trend predicted by Wang and co-workers.14 The monitoring of long-chain and new generation PFAS through fluorescence-based, optical and electrochemical sensors is giving promising results.15–18 However, examples of biosensing platforms are still limited.19 Transposing toxicological studies, particularly the ones focused on PFAS–protein interactions, to bioreceptor design is of great importance to develop new sensing platforms. Indeed, toxicological studies provide fundamental information about PFAS–protein complexes by clarifying binding site distributions, affinity constants, stoichiometries, etc. Liu et al. offered a complete overview of the analytical techniques in use for the characterisation of these compounds giving emphasis to their limits and potential.20 For instance, PFOA and PFOS affinity towards serum proteins such as albumin and haemoglobin was extensively characterised by multi-analytical approaches based on fluorescence spectroscopy, mass spectrometry (MS), circular dichroism, isothermal titration calorimetry, X-ray crystallography, and molecular docking.21–24 Multi-analytical studies confirmed the strong affinity of PFOA for human serum albumin (hSA): PFOA's carboxylate head and fluorinated C8 tail mimic the structure of fatty acids promoting PFOA binding into hSA's hydrophobic pockets.25,26 Similarly to fatty acids, hSA–PFOA interactions are non-covalent, mainly hydrophobic, and influenced by protein conformation and environmental conditions (such as pH).27 These findings suggest the possibility to use hSA as a bioreceptor.19 Also other proteins were found to be good candidates: in 2016, Wang et al. described how PFOS influences the stability and conformation of haemoglobin, providing key information about its half-life in blood. PFOS was found to interact with the protein, leading to a significant conformational change and exposure of the haem group.21
In this context, native MS offers a rich toolbox to support bioreceptor screening studies. Native MS is widely applied to study biomolecules, such as proteins. Because a soft ionisation technique is used, even relatively weak non-covalent interactions are preserved upon transfer to the gas-phase, allowing the analysis of non-covalent protein–target complexes.28 Besides identifying and characterising individual species, native MS can also provide information on the stoichiometry and composition of complexes. Moreover, native MS allows analysis of heterogeneity within proteins and their complexes.29–31 Other advantages of native MS are that only low sample amounts are required (a few μL) at relatively low concentrations (low μM-range), no labelling is required and it is relatively fast (minutes)32 compared to other techniques often used for the characterisation of PFAS–protein complexes.20 Combining MS with ion-mobility spectrometry (IM-MS) adds the arrival time dimension, which corresponds to the global size and shape of the ions, to the analysis and provides information on the structure, conformational heterogeneity and topology of the complexes. When performing collision induced unfolding (CIU) experiments, the conformational stability of proteins and their complexes can be examined.33
To show the potential of native MS in bioreceptor screening, we describe two case studies dedicated to PFAS–protein complexes. PFOA/PFOS were used as model analytes and albumin, cytochrome c (cyt c) and neuroglobin (NGB) as possible bioreceptors. In the first case study we assessed the applicability of hSA isolated and purified from blood as a bioreceptor for PFOA (Fig. 1A). Considering the fatty acid-mimicking nature of PFOA, untreated hSA and delipidated hSA were first compared. Although the delipidation step improved PFOA affinity by removing residual fatty acids present in the hSA isolated from blood, it could also lead to a lower stability of the protein itself. Native MS was applied to characterise the hSA–PFOA complexes, while the complex stability was probed by CIU.
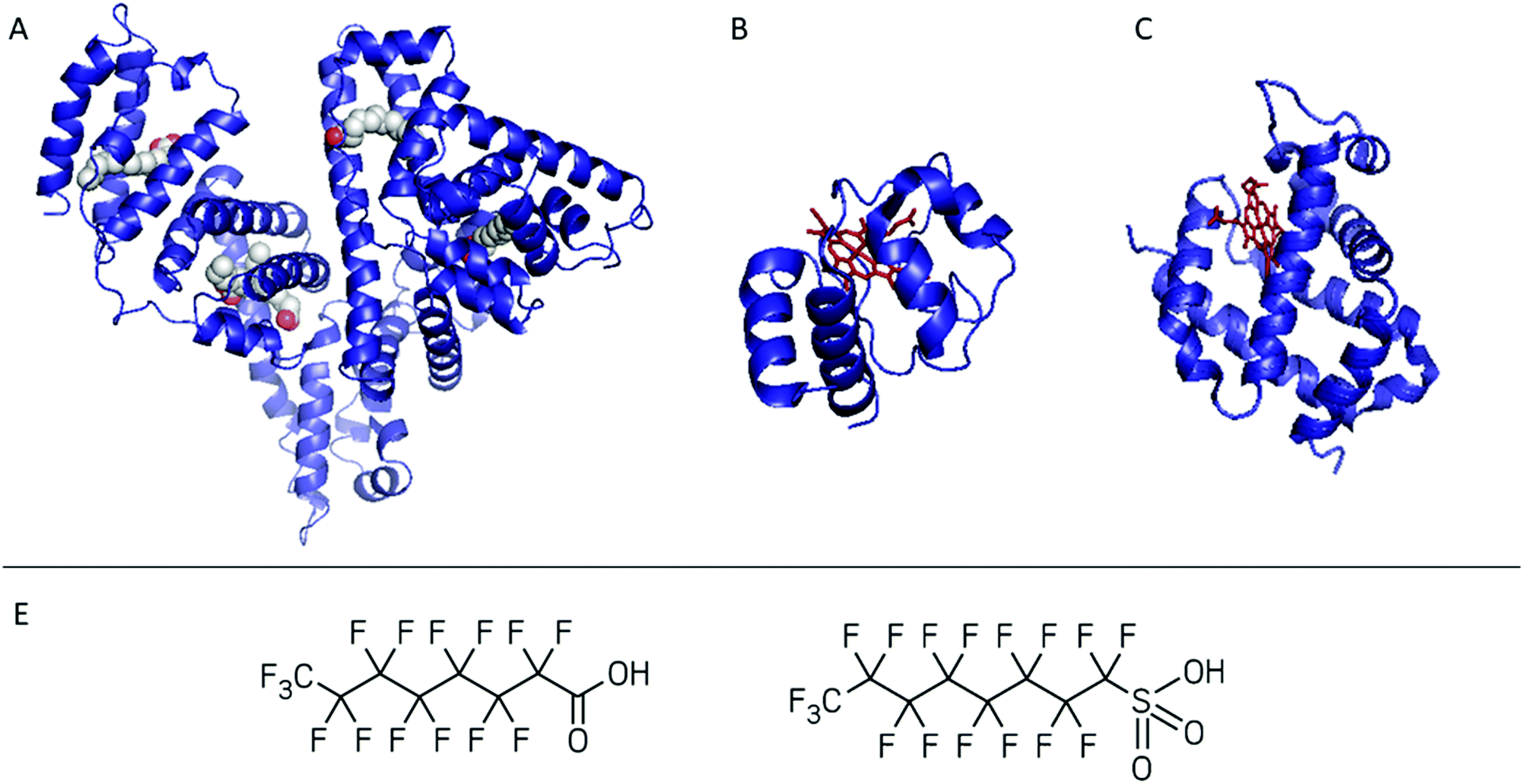 | ||
| Fig. 1 Summary of the proteins tested as bioreceptors: (A) human serum albumin in complex with myristic acid, (B) cytochrome c, and (C) neuroglobin. The haem group is shown in red. The proteins were screened against PFOA and/or PFOS (D from the left to the right). PDB databank ID is reported in Table S1.† | ||
Since the interactions of haemoglobin (Hb) with PFAS have been previously studied, we probe the use of other haemoproteins such as NGB and cyt c (Fig. 1B and C) in the second case study. Cyt c is a 12 kDa, water-soluble protein and can be considered as a model protein, easy to combine with sensing platforms such as electrochemical ones.34–36 However, its interaction with PFAS is unclear and its applicability in PFAS biosensing was never tested. NGB is a 17 kDa globin with a 3/3 fold expressed in the central and peripheral nervous system, cerebrospinal fluid, retina and endocrine tissues.37,38 In contrast to cyt c, NGB has not been applied in biosensing so far. Therefore, the possibility to include cyt c and NGB in a PFAS bioreceptor library was screened by native MS. Through these case studies we aim to demonstrate the use of native MS as a complementary method for the design and characterisation of bioreceptors.
Experimental
Materials
Perfluorooctanoic acid (PFOA, ≥96%), perfluorooctanesulfonic acid potassium salt (PFOS, ≥98%), human haemoglobin and cytochrome c from human heart were purchased from Sigma-Aldrich (Belgium). Human serum albumin was isolated from blood and purified as previously described by Chen et al.39 Recombinant human neuroglobin was expressed and purified as reported previously.40 The 100 mmol L−1 ammonium acetate solution at pH 6.8 was prepared using ammonium acetate (>98%) purchased from Sigma-Aldrich (Belgium). All other reagents were of analytical grade and solutions were prepared using double distilled deionised water.Sample preparation
Prior to use, untreated hSA, delipidated hSA and Hb were dialysed overnight in 100 mmol L−1 ammonium acetate pH 6.8 using Slide-a-Lyzer Mini dialysis units with a molecular weight cut-off of 3.5 kDa (Thermo Fisher Scientific, Belgium). The denatured samples were directly prepared in 50/50 acetonitrile/MilliQ water with 0.1% formic acid. The concentrations of the protein solutions were verified using a Nanodrop2000 (Thermo Fisher Scientific) using the parameters in Table S1.† The average of three triplicate measurements was considered. NGB and cyt c were buffer exchanged to 100 mmol L−1 ammonium acetate pH 6.8 using Micro Bio-spin columns (Bio-gelP6, Bio-rad). Samples of 10 μmol L−1 protein with the desired concentration of PFOA/PFOS were prepared in 100 mmol L−1 ammonium acetate pH 6.8. Once prepared the protein:PFOA/PFOS solutions were kept for max 5 h in a fridge at 4 °C. No incubation time was needed before running the measurements.
Native MS
A sample volume of approximately 2–4 μL protein solution was required for each measurement and introduced into the mass spectrometer using nano-electrospray ionisation (nano-ESI) with in-house made gold-coated borosilicate capillaries. Two different instruments were used in this work. The hSA measurements were performed on a Q-TOF2 instrument (MSVision) which is modified for the transmission of high-mass macromolecular assemblies.41 The spray capillary voltage ranged between 1.2 and 1.8 kV, the source temperature was set to 30 °C and the sample and extractor cone were set to 25 V and 10 V, respectively. The collision energy was fixed to 50 V. Gas pressures were 10 mbar and 1 × 10−2 mbar for the backing and collision gas, respectively. The measurements of the other proteins and the CIU experiments of hSA were performed on a Synapt G2 HDMS instrument (Waters) with N2 gas (purity 99.9999%, Messer) in the IM cell and the following instrument settings were used: spray capillary voltage 1.2–1.9 kV, source temperature 30 °C, sampling cone 25 V, extraction cone 1 V, trap collision energy 5 V (for the CIU experiments this was varied from 10 to 150 V), transfer collision energy 0 V, trap DC bias 45 V, IMS wave height 35 V and IMS wave velocity 700 m s−1. The backing pressure was set to 3.9 mbar, the source pressure to 2.8 × 10−3 mbar, the trap pressure to 2.4 × 10−2 mbar, the IMS pressure to 3.0 mbar, and the transfer pressure to 2.5 × 10−2 mbar.Data elaboration
All data were analysed using MassLynx v4.2 (Waters) and Driftscope v2.3 (Waters). The Gaussian peak deconvolution algorithm within OriginPro 2018 was used to separate main and shoulder peaks and bound and unbound states. Afterwards, the intensity of these peaks was used to estimate the abundance of each species. The Kd of the untreated hSA–PFOA complex was estimated from a single point measurement, as described by Göth et al., for all charge states and the mean Kd was reported.42 Arrival times were extracted at the full width at half maximum from the whole peak, including salt adducts and complexes (where appropriate). This corresponds to an m/z-window from 4435 to 4455 m/z and 4435 to 4520 m/z for the unbound and bound hSA, respectively. Experimental TWCCSN243 values were calculated using a previously reported method relying on a calibration with other native proteins under the same experimental conditions.44 During the calibration procedure, a linear fitting was applied according to the study by Thalassinos et al.45 The CIUSuite 2 software (v2.2) developed by Polasky et al. was used to generate the CIU plots and analyse these data.46Results and discussion
Testing hSA's applicability as a PFOA-bioreceptor
In this first case study, the possibility of improving hSA–PFOA binding affinity and stoichiometry was tested by comparing untreated and delipidated hSA. To assess the stability of the delipidated hSA–PFOA complex, the CIU threshold and pattern were compared with those of the untreated hSA–PFOA.Toxicological studies showed high affinity of PFOA for hSA fatty acid binding sites.25–27 To facilitate access to hSA cavities, a delipidation pretreatment for hSA was included, as previously reported.39 The removal of endogenous fatty acids is expected to lead to better reproducibility of the interaction and a higher sensitivity. To verify these hypotheses, we determined the number of binding sites and compared the affinity of delipidated hSA to that of untreated hSA towards PFOA. In Fig. 2A, the mass spectrum of untreated hSA shows the presence of the protein monomer (charge states 17+ to 13+), non-specific dimer (charge states 23+ to 20+) and non-specific trimer (charge states 28+ to 26+). The experimentally determined mass of hSA was found to be around 66.7 ± 0.3 kDa, which corresponds well with the mass of mature albumin which is subjected to cleavage of the signal peptide and the propeptide resulting in 585 amino acids (66.5 kDa).47 Moreover, a small amount of the complete hSA consisting of 609 amino acids (including propeptide and signal peptide) can be observed as the minor charge state distribution in between the main peaks, with an experimentally determined mass of 68.9 ± 0.9 kDa. Analysis of the monomer revealed the presence of a shoulder to the main peak with an intensity of 52% compared to the main peak (Fig. 2B). This is not observed upon denaturing of the protein (Fig. S1†) and the intensity of the shoulder decreases to 31% upon applying higher collision energies (Fig. S2†). Therefore, this feature is noncovalent in nature and can possibly be ascribed to the presence of long- or short-chain fatty acids.48,49 Upon addition of PFOA (10-fold excess), a 1:1 stoichiometry complex was formed as indicated by the appearance of the second peak in Fig. 2C. Over all charge states present, ca. 49% of the untreated hSA is present in its unbound state and 51% has one PFOA bound. The Kd was estimated to be 90 ± 5 μmol L−1. This value is in agreement with the previously reported Kd of 100 ± 90 μmol L−1 by Beesoon et al. for the hSA–PFOA complex.50 The observed stoichiometry is lower compared to other values reported in the literature in which bovine serum albumin and rat serum albumin were used.25,27 The exact concentration, type and state of the protein used can influence the stoichiometry.
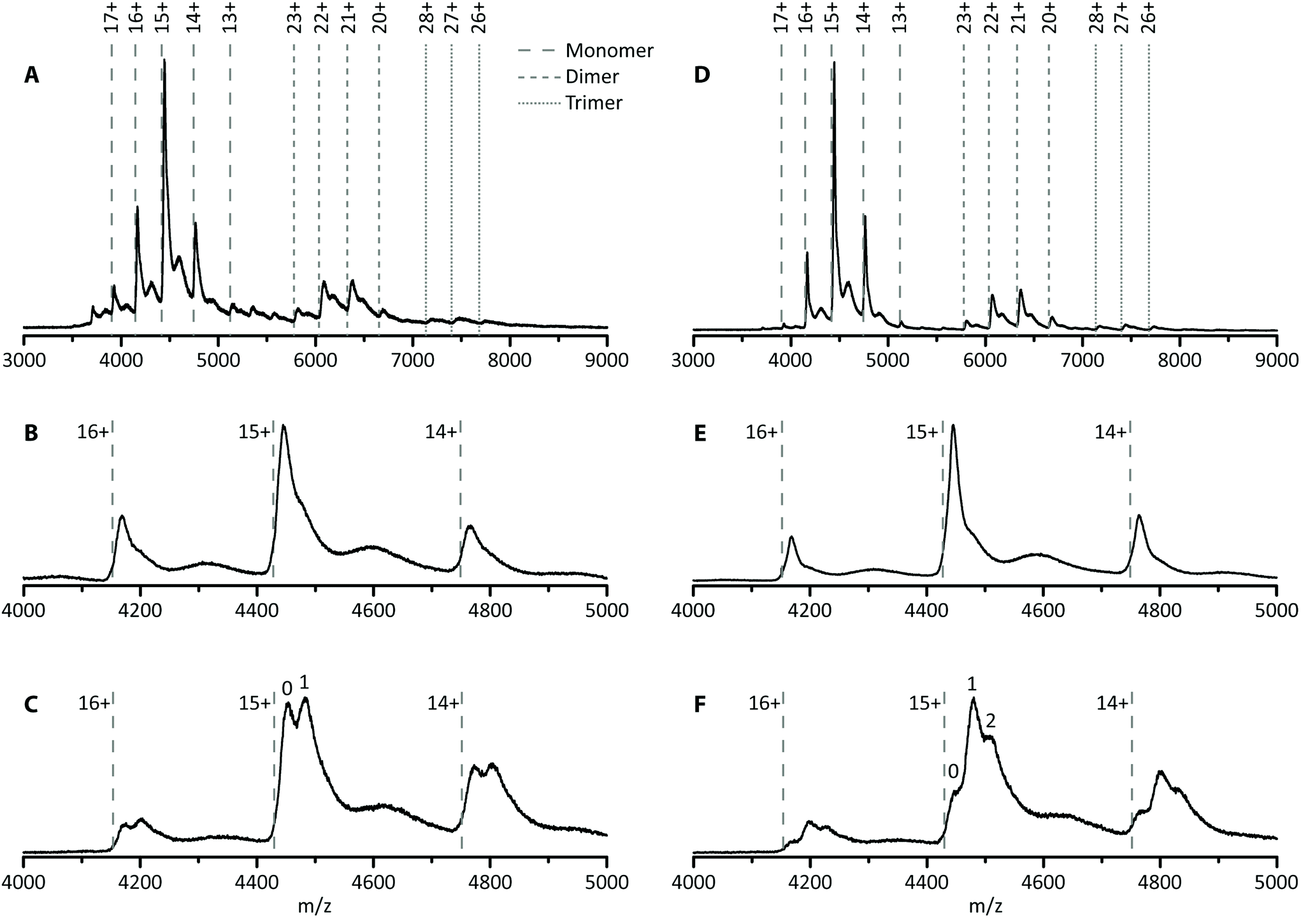 | ||
| Fig. 2 Native MS spectra of untreated (A–C) and delipidated (D–F) hSA. The full MS spectra of hSA are given (A and D), together with a zoom of the monomer region (B and E) and spectra of hSA in complex with PFOA at a 1:10 protein:ligand ratio (C and F). The dashed lines indicate the theoretical m/z-values of the monomer, dimer and trimer of hSA. | ||
The same experiments were performed with the delipidated hSA and an experimental mass of 66.6 ± 0.2 kDa was obtained. Again, a shoulder is observed on the main peak, but with a lower intensity of only 31% compared to 52% for the untreated hSA, suggesting that the delipidation process removed the noncovalent feature (most likely fatty acids) at least partially (Fig. 2E). Addition of a 10-fold excess of PFOA to delipidated hSA yielded a complex with different stoichiometry (1:2, Fig. 2F). Moreover, the intensity of the hSA peak without any PFOA bound is significantly lower for the delipidated hSA (20%) than for the untreated hSA (49%), suggesting a shift towards more complex formation. Approximately 45% of the hSA has one PFOA bound, while 35% has two PFOA molecules bound. The increase of the binding stoichiometry can be attributed to the delipidation step. As such, native MS confirmed that hSA delipidation is important to attain higher binding stoichiometries of the proposed bioreceptor.
From the plots in Fig. 3A and C and the corresponding feature detection in Fig. S3A and C,† it is possible to observe that both proteins showed a similar TWCCSN2 (4469 ± 22 Å2 and 4458 ± 22 Å2 for the native untreated and delipidated hSA, respectively) until a collision energy of 825 eV, which corresponds with an applied potential difference of 55 V; afterwards a multi-step unfolding process started.52,53 The first transition occurs between 60 and 70 V (Fig. S4†) and results in a state with a TWCCSN2 of 5284 ± 22 Å2 and 5262 ± 22 Å2 for the untreated and delipidated hSA, respectively (Fig. S3†). Eschweiler et al. assigned this second conformation to the unfolding of domain II.52 The second transition takes place just after 100 V and generates a state with a TWCCSN2 of 5563 ± 22 Å2 and 5551 ± 22 Å2 for the untreated and delipidated hSA, respectively, which corresponds to the partial unfolding of domain III. A final unfolding step occurs between 120 and 130 V, which results in a state with a TWCCSN2 of 5686 ± 22 Å2 and 5641 ± 22 Å2 for the untreated and delipidated hSA, respectively. This final transition can be ascribed to the unfolding of domain I and the remaining domain III.52 The consistency of the unfolding patterns, with a relatively low root-mean-square deviation (RMSD) of 9.26, suggested that the delipidation process did not affect the stability of hSA. No significant differences in the unfolding products or changes in the threshold collision energies were observed. These preliminary results proved that the delipidation treatment did not affect the protein stability.
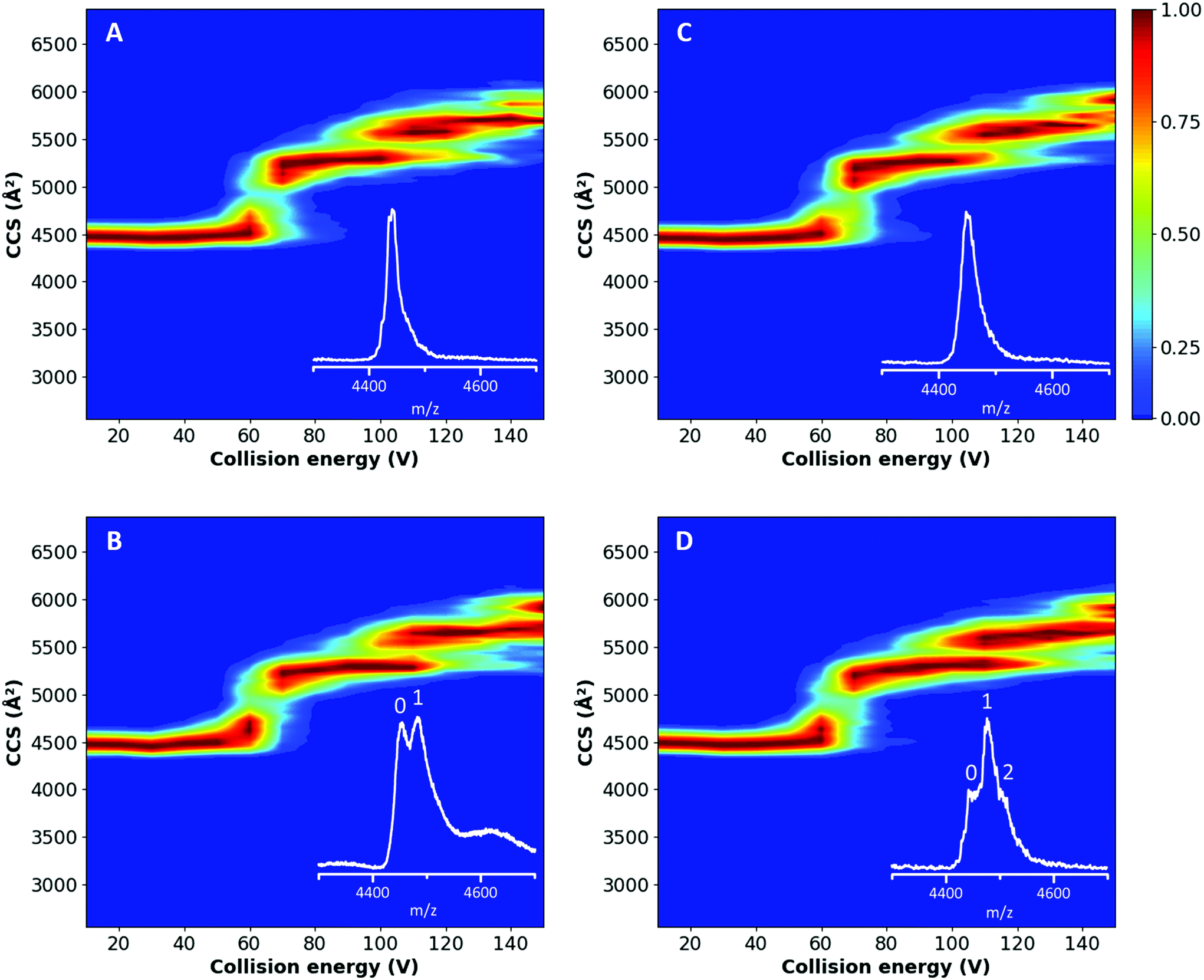 | ||
| Fig. 3 CIU plots of untreated (A) and delipidated (C) hSA and of the untreated (B) and delipidated (D) hSA in complex with PFOA (1:10 protein:PFOA ratio). The TWCCSN2 is plotted as a function of the trap collision energy and the intensity is shown using a colour scale (red is 100% and blue 0%). The insets show the peaks selected (15+ charge state) for extraction of the TWCCSN2 at a trap collision energy of 60 V. Arrival times were extracted from the whole peak, including salt adducts and complexes (where appropriate). | ||
The CIU experiments were also extended to the hSA–PFOA complexes, to test whether the presence of ligands influenced the stability of the proteins, particularly the delipidated form. A 10-fold excess of PFOA to hSA was tested and the unfolding pathways of treated and untreated hSA are shown in Fig. 3B and D. The initial conformation has a TWCCSN2 of 4480 ± 22 Å2 (Fig. S3†) and unfolds between 60 and 70 V (Fig. S4†) to generate a state with a TWCCSN2 of 5284 ± 22 Å2 and 5306 ± 22 Å2 for untreated and delipidated hSA, respectively. The final state has a TWCCSN2 of 5641 ± 22 Å2. This compares well with the fourth and final state of both proteins without PFOA indicating that the third feature, corresponding to the partial unfolding of domain III, disappears in the unfolding process of the complex. In general, the unfolding pathways of the complexes were found to be comparable between the two proteins (RMSD of 7.90).
Throughout all conformational transitions and until the highest energy applied (Fig. S6†), PFOA is still bound to hSA (both untreated and delipidated) which illustrates that the strength of ligand binding is comparable to the protein's intramolecular interactions, i.e. conformational stability. This indicates that the binding site for PFOA is not significantly affected by the initial unfolding, and implies that the PFOA is most likely buried in the structure of domain I or III,52 rather than binding peripherally to the surface. Since our experimental data show that the transition corresponding to the partial unfolding of domain III is absent for the hSA–PFOA complexes (Fig. 3B and D and Fig. S3B and D†), it is clear that the binding of PFOA influences the stability of domain III. This further implies that PFOA is the most likely bound in domain III rather than in domain I for both the untreated and delipidated hSA.
This method provides a useful preliminary control to assess the applicability of a protein as the bioreceptor under different working conditions. Since no differences in the unfolding paths and stabilities have been observed, our study supports the possibility to use delipidated hSA as a bioreceptor in different sensing platforms. This is also supported by previous work concerning the design of a proof-of-concept impedimetric sensor for PFOA, where delipidated hSA was immobilised on portable screen-printed electrodes modified with pyrrole carboxylic acid.2 Then, PFOA–hSA complex formation was followed at a confined surface. Despite the limited sensitivity of the platform, this sensing strategy is a first proof of the applicability of delipidated hSA as a bioreceptor for PFAS monitoring in water.
:1 stoichiometry is observed, proving the coherence of the native MS data with the literature (Fig. S7C†).21 After verifying the formation of the Hb–PFOS complex, we studied the interaction of PFOA with Hb. Again, a 1:1 Hb–PFOA complex was observed after addition of a 10-fold excess of the target. The intensity of the peak corresponding to this complex (93%) is higher than that of the Hb–PFOS complex (51%) (Fig. S7D†). In both cases, the haem groups are not removed from the Hb upon binding of PFOA/PFOS, indicating that there is no competition between the haem groups and PFOS or PFOA.
These findings are in agreement with the Hb–PFOS complex structure suggested by molecular modelling by Wang et al.21 in which the target molecule was suggested to bind the domain proximal to the haem without any direct interactions with the prosthetic group. Therefore, Hb–PFOA/PFOS screening confirmed the formation of complexes and their stability (i.e. no loss of the haem group) as well as the consistency of MS data with the literature. Once we confirmed the capability of Hb to bind PFAS, it was possible to proceed with the screening of other haemoproteins, such as cyt c and NGB.
Cyt c was chosen because it is well-known to bind to lipid bilayers. To check whether cyt c can bind PFOA, native MS experiments were performed (Fig. 4A). Cyt c shows two charge states (7+ and 6+) which are heavily adducted with sodium ions (a common contamination in native MS). Upon adding a 10-fold excess of PFOA, no new peaks appeared indicating the lack of a cyt c-PFOA complex (Fig. 4B). To verify this observation, the negatively charged PFOS was used as well, but again no complex was observed (Fig. S8†). This is unexpected since cyt c is well-known to interact with anionic phospholipids, such as cardiolipin.54 Over the years, multiple binding sites for these anionic lipids were discovered and it was found that electrostatic forces play a key role in the cyt c-lipid interaction.55–58 Since PFOA and PFOS mimic fatty acids and are negatively charged at neutral pH,59,60 it was plausible that cyt c could interact with these PFAS and could work as a bioreceptor. One possible explanation for the absence of complex is that cyt c specifically binds cardiolipins and that the PFAS used here are structurally too different to interact with cyt c.
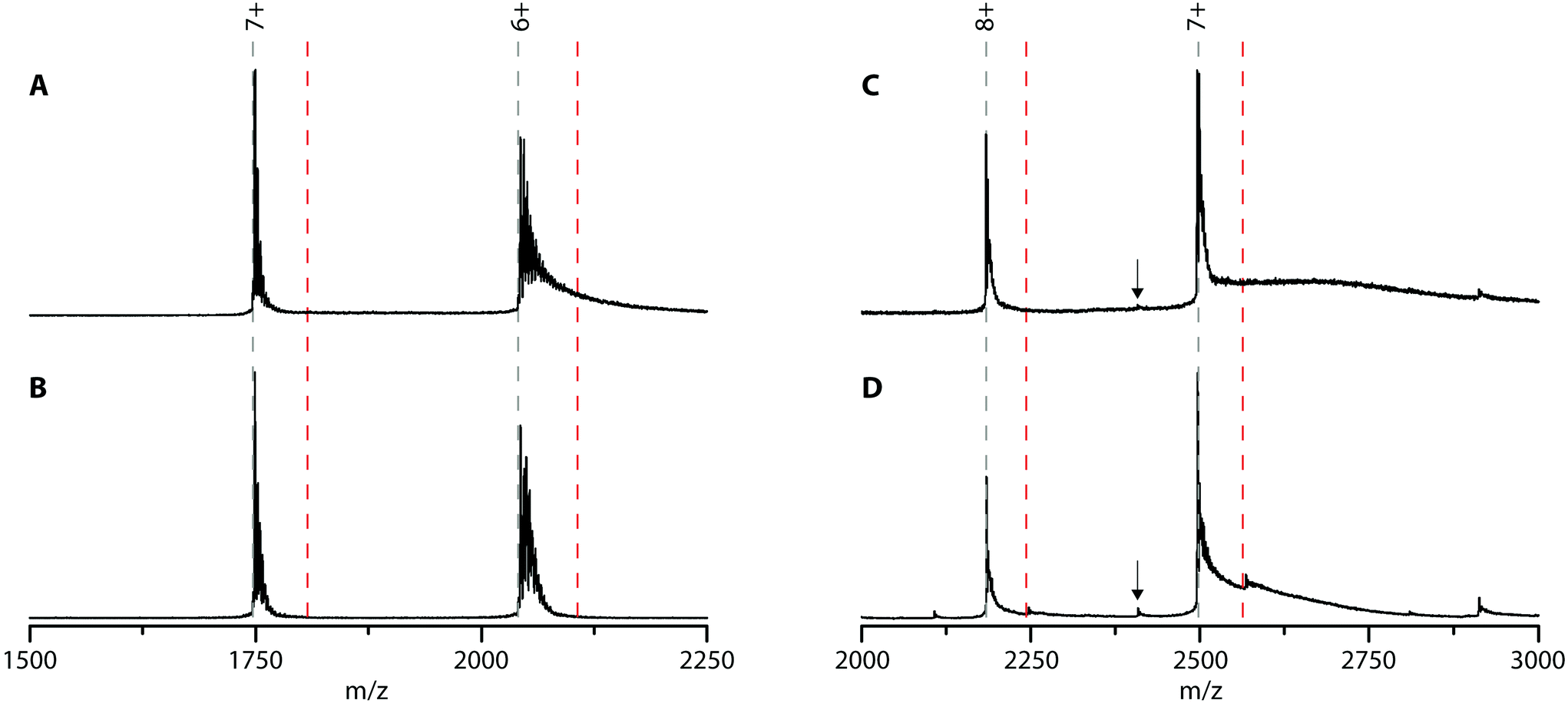 | ||
| Fig. 4 Native MS spectra of cyt c (A and B) and NGB (C and D) without (A and C) and in the presence of PFOA in a 1:10 protein:target ratio (B and D, respectively). The dashed lines indicate the theoretical m/z-values of the protein and 1:1 protein-PFOA complex in grey and red, respectively. The arrows indicate the theoretical m/z-value of NGB without the haem group. | ||
Next, possible NGB–PFOA interactions were investigated. Since the ability of Hb to bind PFOA was confirmed using native MS, it is possible that another globin, NGB, could also bind to PFOA and work as a bioreceptor. To our knowledge, NGB–PFOA interactions have not been investigated so far. The mass spectrum of NGB shown in Fig. 4C is characterised by two prominent charge states (8+ and 7+). Upon adding a 10-fold excess of PFOA only small intensity peaks corresponding to the 1:1 complex were observed (Fig. 4D). Overall, 87% of the NGB is present in its unbound state and only 13% has one PFOA bound. This indicates that NGB is only able to bind PFOA weakly and with a lower affinity than Hb. The most intense peaks correspond to NGB with haem group and the very low intensity peaks around 2110 and 2410 m/z that are present after addition of PFOA can be assigned to NGB without the haem group. This could suggest that PFOA is affecting the stability of NGB. Furthermore, there is no evidence of NGB without a haem group binding to PFOA.
Finally, the NGB-PFOS complex formation was investigated (Fig. S9†). The spectra showed that the amount of the NGB-PFOS complex formed (7%) is even lower than for NGB-PFOA and is therefore not investigated in more detail. From this screening, NGB is found to be a less suitable bioreceptor for PFAS compared to Hb.
Conclusions
The two case studies reported here underline the potential of applying native MS in the screening of biorecognition elements and implementation of bioreceptor libraries for environmental contaminants such as PFAS.Native MS revealed the higher PFOA binding capacity of delipidated hSA with respect to the untreated hSA. Removal of fatty acids from the hydrophobic pockets facilitates binding of PFOA to the cavities. CIU experiments showed that fatty acid removal did not affect the conformational stability of the protein or the complex itself. PFOA appears to bind tightly to hSA–PFOA as the complex is retained even at high collision energies. This evidence correlates well with protein stability and implies that PFOA is buried in the protein structure.
Furthermore, it was shown that native MS can be used to screen other possible bioreceptors for PFAS. Hb, a protein known to bind PFOS, can also bind PFOA. Conversely, the structurally related NGB displays a lower binding affinity toward both compounds. Finally, cyt c, a well-known lipid-binding protein, does not show any binding.
These examples illustrate how native MS can contribute to the screening and characterisation of possible bioreceptors. Although many challenges still remain, our approach has the potential to play an important role in the transfer of toxicological findings to the design of bioreceptors for innovative biosensing strategies. One of the major challenges will be the screening of short-chain PFAS which will possibly require a more extensive sample preparation and multiple desalting steps to enhance the peak separation in native MS. Future research efforts should be oriented toward testing novel proteins and assessing binding affinity and stoichiometry using different molar ratios.
Author contributions
Elise Daems: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review and editing. Giulia Moro: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review & editing. Herald Berghmans: resources, validation. Ligia M. Moretto: conceptualization, funding acquisition, supervision, validation, writing – review and editing. Silvia Dewilde: resources, supervision. Alessandro Angelini: conceptualization, resources, supervision, validation, writing – review & editing. Frank Sobott: funding acquisition, methodology, resources, supervision, validation, writing – review & editing. Karolien De Wael: conceptualization, funding acquisition, project administration, resources, supervision, validation, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Fund for Scientific Research (FWO) Flanders [grant numbers G054819N and 1S65717N]. Thanks to Dr Stanislav Trashin for the many fruitful discussions on haemoproteins, particularly neuroglobin. We dedicate this paper to our colleague, Prof. Sylvia De Wilde, who passed away much too early on October 3rd, 2020.Notes and references
- C. Bosch-Orea, M. Farré and D. Barceló, Compr. Anal. Chem., 2017, 77, 337–383 CrossRef.
- G. Moro, F. Bottari, S. Liberi, S. Covaceuszach, A. Cassetta, A. Angelini, K. De Wael and L. M. Moretto, Bioelectrochemistry, 2020, 134, 107540 CrossRef CAS.
- B. Liu, J. Zhuang and G. Wei, Environ. Sci.: Nano, 2020, 7, 2195–2213 RSC.
- H. Liu, J. Ge, E. Ma and L. Yang, in Biomaterials in Translational Medicine: A Biomaterials Approach, 2019, pp. 213–255 Search PubMed.
- S. B. Patil, V. F. Annese and D. R. S. Cumming, in Advances in Nanosensors for Biological and Environmental Analysis, Elsevier Inc., 2019, pp. 133–142 Search PubMed.
- M. Ateia, A. Maroli, N. Tharayil and T. Karanfil, Chemosphere, 2019, 220, 866–882 CrossRef CAS.
- H. Feng, Y. Ruan, K. Zhang and P. K. S. Lam, TrAC, Trends Anal. Chem., 2019, 121, 115372 CrossRef CAS.
- Y. Wang, W. Chang, L. Wang, Y. Zhang, Y. Zhang, M. Wang, Y. Wang and P. Li, Ecotoxicol. Environ. Saf., 2019, 182, 109402 CrossRef CAS.
- S. F. Nakayama, M. Yoshikane, Y. Onoda, Y. Nishihama, M. Iwai-Shimada, M. Takagi, Y. Kobayashi and T. Isobe, TrAC, Trends Anal. Chem., 2019, 121, 115410 CrossRef CAS.
- S. Liu, R. Yang, N. Yin, Y. L. Wang and F. Faiola, Ecotoxicol. Environ. Saf., 2019, 169, 564–572 CrossRef CAS.
- J. Rovira, M. Á. Martínez, R. P. Sharma, T. Espuis, M. Nadal, V. Kumar, D. Costopoulou, I. Vassiliadou, L. Leondiadis, J. L. Domingo and M. Schuhmacher, Environ. Res., 2019, 175, 384–392 CrossRef CAS.
- F. Sánchez-Soberón, R. Sutton, M. Sedlak, D. Yee, M. Schuhmacher and J. S. Park, Chemosphere, 2020, 252, 126454 CrossRef.
- D. Barceló and T. Ruan, TrAC, Trends Anal. Chem., 2019, 121, 2–3 Search PubMed.
- Z. Wang, I. T. Cousins, M. Scheringer, R. C. Buck and K. Hungerbühler, Environ. Int., 2014, 70, 62–75 CrossRef CAS.
- M. W. Glasscott, K. J. Vannoy, R. Kazemi, M. D. Verber and J. E. Dick, Environ. Sci. Technol. Lett., 2020, 7, 489–495 CrossRef CAS.
- N. Karimian, A. M. Stortini, L. M. Moretto, C. Costantino, S. Bogialli and P. Ugo, ACS Sens., 2018, 3, 1291–1298 CrossRef CAS.
- Z. Cheng, L. Du, P. Zhu, Q. Chen and K. Tan, Spectrochim. Acta, Part A, 2018, 201, 281–287 CrossRef CAS.
- X. Li, X. Wang, T. Fang, L. Zhang and J. Gong, Talanta, 2018, 181, 147–153 CrossRef CAS.
- N. Cennamo, L. Zeni, P. Tortora, M. E. Regonesi, A. Giusti, M. Staiano, S. D’Auria and A. Varriale, Talanta, 2018, 178, 955–961 CrossRef CAS.
- X. Liu, M. Fang, F. Xu and D. Chen, TrAC, Trends Anal. Chem., 2019, 116, 177–185 CrossRef CAS.
- Y. Wang, H. Zhang, Y. Kang, Z. Fei and J. Cao, Chem.-Biol. Interact., 2016, 254, 1–10 CrossRef CAS.
- Y. Liu, Z. Cao, W. Zong and R. Liu, RSC Adv., 2017, 7, 24781–24788 RSC.
- Q. Chi, Z. Li, J. Huang, J. Ma and X. Wang, Chemosphere, 2018, 198, 442–449 CrossRef CAS.
- M. Forsthuber, A. M. Kaiser, S. Granitzer, I. Hassl, M. Hengstschläger, H. Stangl and C. Gundacker, Environ. Int., 2020, 137, 105324 CrossRef CAS.
- X. Han, T. A. Snow, R. A. Kemper and G. W. Jepson, Chem. Res. Toxicol., 2003, 16, 775–781 Search PubMed.
- L. L. Wu, H. W. Gao, N. Y. Gao, F. F. Chen and L. Chen, BMC Struct. Biol., 2009, 9, 5–11 CrossRef.
- H. N. Bischel, L. A. MacManus-Spencer and R. G. Luthy, Environ. Sci. Technol., 2010, 44, 5263–5269 CrossRef CAS.
- F. Sobott, M. G. Mccammon, H. Hernández and C. V. Robinson, Philos. Trans. R. Soc., A, 2005, 363, 379–391 CrossRef CAS.
- A. C. Leney and A. J. R. Heck, J. Am. Soc. Mass Spectrom., 2017, 28, 5–13 CrossRef CAS.
- K. Ishii, M. Zhou and S. Uchiyama, Biochim. Biophys. Acta, Gen. Subj., 2018, 1862, 275–286 CrossRef CAS.
- J. R. Bolla, M. T. Agasid, S. Mehmood and C. V. Robinson, Annu. Rev. Biochem., 2019, 88, 85–111 CrossRef CAS.
- E. B. Erba and C. Petosa, Protein Sci., 2015, 24, 1176–1192 CrossRef.
- S. J. Hyung, C. V. Robinson and B. T. Ruotolo, Chem. Biol., 2009, 16, 382–390 CrossRef CAS.
- Y. Haldorai, S. K. Hwang, A. I. Gopalan, Y. S. Huh, Y. K. Han, W. Voit, G. Sai-Anand and K. P. Lee, Biosens. Bioelectron., 2016, 79, 543–552 CrossRef CAS.
- Z. S. Aghamiri, M. Mohsennia and H. A. Rafiee-Pour, Talanta, 2018, 176, 195–207 CrossRef CAS.
- M. Murphy, K. Theyagarajan, P. Ganesan, S. Senthilkumar and K. Thenmozhi, Appl. Surf. Sci., 2019, 492, 718–725 CrossRef CAS.
- T. Burmester, B. Weich, S. Reinhardt and T. Hankeln, Nature, 2000, 407, 520–523 CrossRef CAS.
- P. Ascenzi, A. di Masi, L. Leboffe, M. Fiocchetti, M. T. Nuzzo, M. Brunori and M. Marino, Mol. Aspects Med., 2016, 52, 1–48 CrossRef CAS.
- R. F. Chen, J. Biol. Chem., 1967, 242, 173–181 CrossRef CAS.
- S. Dewilde, K. Mees, L. Kiger, C. Lechauve, M. C. Marden, A. Pesce, M. Bolognesi and L. Moens, Methods Enzymol., 2008, 436, 341–357 CAS.
- F. Sobott, H. Hernández, M. G. McCammon, M. A. Tito and C. V. Robinson, Anal. Chem., 2002, 74, 1402–1407 CrossRef CAS.
- M. Göth, V. Badock, J. Weiske, K. Pagel and B. Kuropka, ChemMedChem, 2017, 12, 1201–1211 CrossRef.
- V. Gabelica, A. A. Shvartsburg, C. Afonso, P. Barran, J. L. P. Benesch, C. Bleiholder, M. T. Bowers, A. Bilbao, M. F. Bush, J. L. Campbell, I. D. G. Campuzano, T. Causon, B. H. Clowers, C. S. Creaser, E. De Pauw, J. Far, F. Fernandez-Lima, J. C. Fjeldsted, K. Giles, M. Groessl, C. J. Hogan, S. Hann, H. I. Kim, R. T. Kurulugama, J. C. May, J. A. McLean, K. Pagel, K. Richardson, M. E. Ridgeway, F. Rosu, F. Sobott, K. Thalassinos, S. J. Valentine and T. Wyttenbach, Mass Spectrom. Rev., 2019, 38, 291–320 CrossRef CAS.
- M. F. Bush, Z. Hall, K. Giles, J. Hoyes, C. V. Robinson and B. T. Ruotolo, Anal. Chem., 2010, 82, 9557–9565 CrossRef CAS.
- K. Thalassinos, M. Grabenauer, S. E. Slade, G. R. Hilton, M. T. Bowers and J. H. Scrivens, Anal. Chem., 2009, 81, 248–254 CrossRef CAS.
- D. A. Polasky, S. M. Dixit, S. M. Fantin and B. T. Ruotolo, Anal. Chem., 2019, 91, 3147–3155 CrossRef CAS.
- A. Dugaiczyk, S. W. Law and H. B. Brewer, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 71–75 CrossRef CAS.
- S. Curry, P. Brick and N. P. Franks, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 1999, 1441, 131–140 CrossRef CAS.
- S. Curry, H. Mandelkow, P. Brick and N. Franks, Nat. Struct. Biol., 1998, 5, 827–835 CrossRef CAS.
- S. Beesoon and J. W. Martin, Environ. Sci. Technol., 2015, 49, 5722–5731 CrossRef CAS.
- S. M. Dixit, D. A. Polasky and B. T. Ruotolo, Curr. Opin. Chem. Biol., 2018, 42, 93–100 CrossRef CAS.
- J. D. Eschweiler, R. M. Martini and B. T. Ruotolo, J. Am. Chem. Soc., 2017, 139, 534–540 CrossRef CAS.
- G. Li, K. DeLaney and L. Li, Nat. Commun., 2019, 10, 5038 CrossRef.
- V. E. Kagan, H. A. Bayir, N. A. Belikova, O. Kapralov, Y. Y. Tyurina, V. A. Tyurin, J. Jiang, D. A. Stoyanovsky, P. Wipf, P. M. Kochanek, J. S. Greenberger, B. Pitt, A. A. Shvedova and G. Borisenko, Free Radicals Biol. Med., 2009, 46, 1439–1453 CrossRef CAS.
- M. Rytömaa and P. K. J. Kinnunen, J. Biol. Chem., 1994, 269, 1770–1774 CrossRef.
- M. Rytömaa and P. K. J. Kinnunen, J. Biol. Chem., 1995, 270, 3197–3202 CrossRef.
- T. J. T. Pinheiro, G. A. Elöve, A. Watts and H. Roder, Biochemistry, 1997, 36, 13122–13132 CrossRef CAS.
- C. Kawai, F. M. Prado, G. L. C. Nunes, P. Di Mascio, A. M. Carmona-Ribeiro and I. L. Nantes, J. Biol. Chem., 2005, 280, 34709–34717 CrossRef CAS.
- D. C. Burns, D. A. Ellis, H. Li, C. J. Mcmurdo and E. Webster, Environ. Sci. Technol., 2008, 42, 9283–9288 CrossRef CAS.
- J. Cheng, E. Psillakis, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. A, 2009, 113, 8152–8156 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0an02005b |
| ‡ These authors contributed equally to this work. |
| § Deceased. |
| This journal is © The Royal Society of Chemistry 2021 |