DOI:
10.1039/D0AN02305A
(Paper)
Analyst, 2021,
146, 1188-1197
A new tandem enrichment strategy for the simultaneous profiling of O-GlcNAcylation and phosphorylation in RNA-binding proteome†
Received
30th November 2020
, Accepted 29th December 2020
First published on 4th January 2021
Abstract
RNA-protein interactions play important roles in almost every step of the lifetime of RNAs, such as RNA splicing, transporting, localization, translation and degradation. Post-translational modifications, such as O-GlcNAcylation and phosphorylation, and their “cross-talk” (OPCT) are essential to the activity and function regulation of RNA-binding proteins (RBPs). However, due to the extremely low abundance of O-GlcNAcylation and the lack of RBP-targeted enrichment strategies, large-scale simultaneous profiling of O-GlcNAcylation and phosphorylation on RBPs is still a challenging task. In the present study, we developed a tandem enrichment strategy combining metabolic labeling-based RNA tagging for selective purification of RBPs and HILIC-based enrichment for simultaneous O-GlcNAcylation and phosphorylation profiling. Benefiting from the sequence-independent RNA tagging by ethynyluridine (EU) labeling, 1115 RBPs binding to different types of RNAs were successfully enriched and identified by quantitative mass spectrometry (MS) analysis. Further HILIC enrichment on the tryptic-digested RBPs and MS analysis led to the first large-scale identification of O-GlcNAcylation and phosphorylation in the RNA-binding proteome, with 461 O-GlcNAc peptides corresponding to 300 RBPs and 671 phosphopeptides corresponding to 389 RBPs. Interestingly, ∼25% RBPs modified by two PTMs were found to be related to multiple metabolism pathways. This strategy has the advantage of high compatibility with MS and provides peptide-level evidence for the identification of O-GlcNAcylated RBPs. We expect it will support simultaneous mapping of O-GlcNAcylation and phosphorylation on RBPs and facilitate further elucidation of the crucial roles of OPCT in the function regulation of RBPs.
Introduction
Post-translational modifications (PTMs) are the covalent modifications of a protein after its synthesis; they can broaden the functionality range of a protein by regulating its structure, stability, and spatial localization. Many PTMs exist on the same protein simultaneously. These co-existing PTMs or the so-called “PTM crosstalk” reciprocally or codependently regulate the protein activity1 and play an important role in the occurrence and development of chronic diseases, such as diabetes and cancer.2 One of the most biologically intriguing PTM crosstalk is that between O-GlcNAcylation and phosphorylation (O–P), which occurs reciprocally or sequentially on serine/threonine residues. It is becoming increasingly accepted that O-GlcNAcylation rivals phosphorylation and the interplay between two modifications acts as an on/off switch in various cellular pathways.3,4 For example, decreased O-GlcNAcylation of lipid droplet-associated perilipin 1 (PLIN1) leads to elevated PLIN1 phosphorylation and enhanced lipolysis,5 and enhanced O-GlcNAcylation of MYPT1 blocks its phosphorylation and affects diabetic wound healing.6 Therefore, the systematic identification and quantification of proteins, namely, O-GlcNAcylation and phosphorylation, may contribute greatly to the in-depth investigation of O–P crosstalk and therefore discovering its regulating mechanism in the key cellular process.
The gene oncology (GO) annotation analysis of known O-GlcNAc proteins reveals that a substantial portion of them are RNA-binding proteins (RBPs).7 Furthermore, many RBPs are also phosphoproteins,8,9 indicating the possibility of co-existence and the “crosstalk” of O-GlcNAcylation and phosphorylation modifications on the same RBP. Individual O–P-modified RBPs have been previously discovered;8–11 however, their large-scale investigation has not been reported due to methodological limitations. The past decade has witnessed rapid progress in comprehensively capturing and identifying RBPs, of which the most well-adopted is the polyadenylated [poly(A)] tail-dependent oligo(dT) enrichment strategy.12–14 However, the incapability of capturing RBPs bound on nonpoly(A) RNAs limits their application. Fortunately, emerging technologies such as click chemistry-assisted methods15,16 and organic–aqueous phase separation–based methods17–19 enable the capture of RBPs bound on various types of RNA, irrespective of their poly(A) status. Although these RBP-capturing strategies effectively facilitate the identification and function studies of RBPs, a large-scale investigation of RBPs regulated by O-GlcNAcylation, phosphorylation and O–P “crosstalk” (OPCT) has not been reported due to the lack of methods for simultaneous enrichment and omics-based profiling of O-GlcNAcylation and phosphorylation on RBPs.
Mass spectrometry is currently the most powerful tool for identification of large-scale proteins and PTMs; it shows high sensitivity, a wide dynamic range, and high-throughput capability. However, global investigation of O-GlcNAcylation and phosphorylation on RBPs simultaneously is a highly challenging task. On one hand, dynamic O-GlcNAc and phosphorylation often occur at low stoichiometry, accounting for less than 1% of the total protein, respectively;20,21 this creates severe signal suppression by the co-eluted non-modified peptides. On the other hand, the commonly adopted PTM enrichment strategies, including antibody immunoprecipitation,22,23 TiO2-based strategies,24 lectin enrichment,25 and metabolic labeling-based enrichment via “click chemistry”,26,27 focus primarily on a specific type of PTM, and each requires specialized enrichment conditions. The incompatibility of these PTM enrichment methods with each other and with the RBP enrichment approaches is currently the major obstacle that limits in-depth mapping of O-GlcNAcylation and phosphorylation on RBPs. Simply combining the different enrichment methods in a sequential way leads to severe sample loss in each enrichment step and may not be a feasible way to profile the low-abundant O-GlcNAcylation, phosphorylation and OPCT on RBPs.
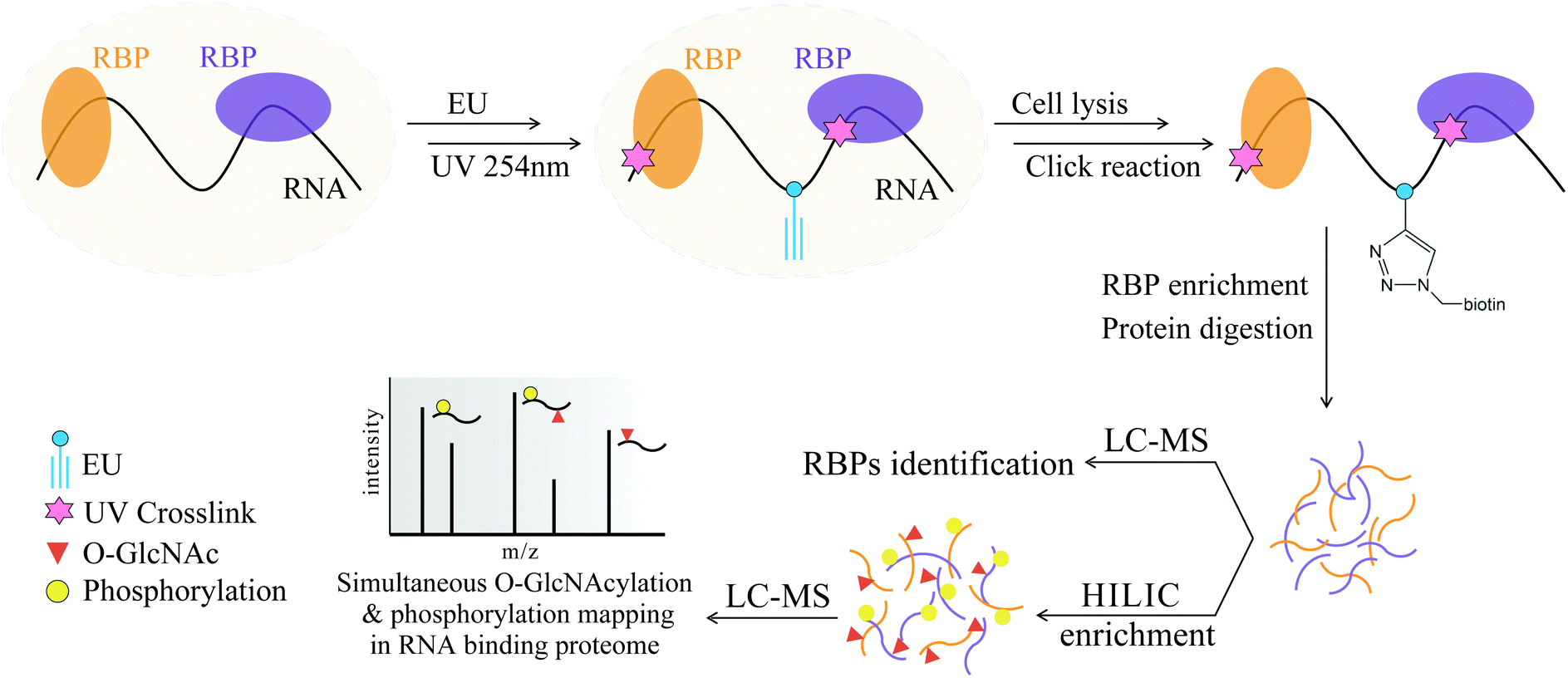
Hydrophilic interaction liquid chromatography (HILIC) is a powerful technique that efficiently separates polar/hydrophilic compounds from their counterparts.28,29 Considering the hydrophilicity of the GlcNAc and PO3 functional groups and the possibility of their co-existence on the same peptides in OPCT, we think that HILIC enrichment may be a solution for simultaneous enrichment of the two PTMs. Herein, we developed a tandem enrichment strategy for large-scale RBP-targeted enrichment and identification of O-GlcNAc peptides and phosphopeptides (Fig. 1). In this strategy, RNAs are first tagged by metabolic labeling of ethynyluridine (EU), which substitutes the natural uridine with the alkyne-modified analog. After 254 nm UV irradiation for RNA and RBP crosslinking, all kinds of RNAs and their binding RBPs can be enriched via alkyne–azide cycloaddition (CuAAC click chemistry).30 Next, the enriched RBPs are digested into peptides, and a small portion of the peptides are analyzed by LC-MS/MS for RBPs identification. The majority of the digested peptides were subjected to HILIC enrichment and subsequent LC-MS/MS analysis to identify the O-GlcNAc peptides and phosphopeptides of the RBPs. In this way, we successfully enriched and identified 1115 RBPs from HeLa cells by MS analysis, including 461 O-GlcNAc peptides corresponding to 300 O-GlcNAc RBPs and 671 phosphorylated peptides corresponding to 389 phosphorylated RBPs. Benefiting from the high selectivity of EU-based RNA labeling and “click chemistry”-based RBP enrichment, broad affinity of HILIC enrichment for the two hydrophilic PTMs, and high compatibility between the two enrichment methods, the first large-scale peptide level identification of O-GlcNAc and phosphorylation of RBPs was achieved.
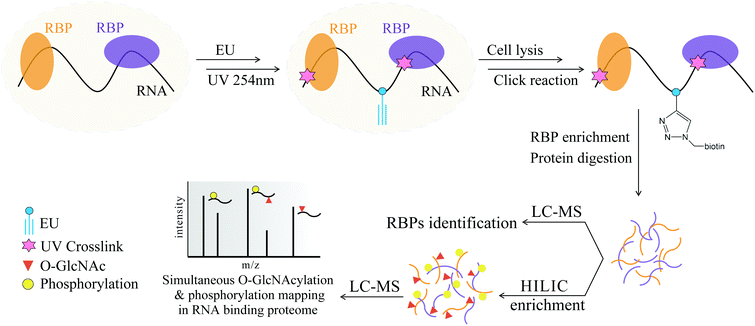 |
| | Fig. 1 Schematic overview of the tandem enrichment strategy for simultaneous profiling of O-GlcNAcylation and phosphorylation in the RNA-binding proteome. | |
Experimental section
The materials and reagents used are described in the ESI.†
Cell culture and evaluation of concentration and duration for EU incorporation
The conditions for metabolic-based EU labeling followed those reported in the literature, with modification and optimization.15,16,31,32 Briefly, HeLa cells were cultured in DMEM (Corning, USA) supplemented with 10% (v/v) FBS (Gibco, USA), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin (Gibco, USA) at 37 °C in a 5% CO2 humidified incubator. Once 80%-90% confluency was achieved, the HeLa cells were cultured in DMEM with EU at varied concentrations (0.01 mM, 0.1 mM, 0.5 mM and 2 mM) for 3 h, 6 h, 9 h and 12 h, respectively. Next, the cells were washed twice with ice-cold PBS, fixed with 3.7% formaldehyde (in PBS) for 30 min at room temperature (RT), washed twice with ice-cold PBS and permeabilized with 0.5% Triton X-100 (in PBS) for 30 min at RT. After being washed three times with ice-cold PBS, the permeabilized cells were incubated with 50 μL click reaction buffer (50 mM Tris-HCl, pH 7.4; 0.5 mM CuSO4, 10 mM sodium L-ascorbate (freshly prepared), 0.5 mM THPTA and 10 μM Cy5-azide for 30 min at RT. The cells were washed with ice-cold PBS and treated with 50 μL Hoechst (10 μg mL−1, in PBS) for 5 min at RT. Next, the cells were washed with ice-cold PBS three times and maintained in PBS in the dark at 4 °C. Images were taken using a confocal laser scanning microscope (CLSM, Nikon, A1R Si+ STORM).
EU labeling efficiency determination by LC-MS/MS
HeLa cells were cultured with 0.5 mM EU for 9 h. The total RNAs were extracted from the cells with TRIzol reagent, digested with S1 nuclease and alkaline phosphatase, and subjected to LC-MS/MS analysis to quantify the uridine (U) and EU. The LC/ESI-MS/MS was conducted on a Shimadzu LC-20AD HPLC (Tokyo, Japan) with two LC-20AD pumps and an AB 3200 QTRAP mass spectrometer (Applied Biosystems, Foster City, CA). The MS data were processed using AB Sciex Analyst 1.5 software (Applied Biosystems, Foster City, CA). The LC separation was performed on an Inertsil ODS-3 column (250 mm × 2.0 mm i.d., 5 μm, Tokyo, Japan). Formic acid in water (0.1%, v/v, solvent A) and MeOH (solvent B) were employed as the mobile phase. A gradient of 5% B for 5 min, 5%–60% B over 15 min, 60% B for 10 min, and 5% B for 14 min was applied. The flow rate of the mobile phase was set at 0.2 mL min−1. The nucleosides and derivatives were monitored in multiple reaction monitoring (MRM) mode (precursor ions → product ions) of EU (269.1 → 137.1) and U (245.2 → 113.3).
Click reaction and streptavidin bead enrichment
200 mM CuSO4–THPTA mixture (premixed at the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and 25 mM biotin-N3 in DMSO were added to the cell lysate (the final concentrations were 0.5 mM for Cu2+ and 0.1 mM for biotin-N3, respectively). Next, 1 M freshly prepared sodium L-ascorbate was added (final concentration 5 mM) to initiate the reaction. The samples were rotated for 1 h at RT in the dark. After the click reaction, the solution was passed through an Amicon Ultra 10 kDa cutoff device (0.5 mL device) to remove excess biotin-azide by repeated PBS washing. Then, 50 μL streptavidin beads and 200 μL biotinylated proteins were mixed and incubated for 1 h under gentle rotation. The beads were washed twice with 200 μL PBS containing 0.2% SDS (pH 7.4), twice with 200 μL PBS containing 6 M urea, and twice with 200 μL PBS. The resulting sample was then used for analysis on SDS/PAGE with silver staining and MS-based proteomic analysis.
:1) and 25 mM biotin-N3 in DMSO were added to the cell lysate (the final concentrations were 0.5 mM for Cu2+ and 0.1 mM for biotin-N3, respectively). Next, 1 M freshly prepared sodium L-ascorbate was added (final concentration 5 mM) to initiate the reaction. The samples were rotated for 1 h at RT in the dark. After the click reaction, the solution was passed through an Amicon Ultra 10 kDa cutoff device (0.5 mL device) to remove excess biotin-azide by repeated PBS washing. Then, 50 μL streptavidin beads and 200 μL biotinylated proteins were mixed and incubated for 1 h under gentle rotation. The beads were washed twice with 200 μL PBS containing 0.2% SDS (pH 7.4), twice with 200 μL PBS containing 6 M urea, and twice with 200 μL PBS. The resulting sample was then used for analysis on SDS/PAGE with silver staining and MS-based proteomic analysis.
RBPs enrichment
HeLa cells cultured in 15 cm dishes (Corning) were grown to 80%-90% confluence and were treated with or without 0.5 mM EU for 9 h. The plates were washed three times with ice-cold PBS, then irradiated with 0.25 J cm−2 UV light at 254 nm on ice by an ultraviolet crosslinker (CL-1000, UVP Company, USA). For the UV-omitted control group, the cells were treated with 0.5 mM EU and kept in the dark for the following process. The cells were harvested by scraping and stored at −80 °C until use. Next, the obtained HeLa cells were lysed by 0.5% SDS (PBS, pH 7.4, containing protease inhibitor cocktail and 10 mM RNase inhibitor) and passed through a syringe with a narrow needle to homogenize them, followed by incubating on ice for 20 min. Next, four times volume of 0.2% Triton X-100 (PBS, pH 7.4, containing protease inhibitor cocktail and 10 mM RNase inhibitor) were added to dilute the SDS to less than 0.1%, then incubated on ice for another 20 min. The lysates were centrifuged (16000g for 15 min at 4 °C) and the supernatants were collected, followed by a click reaction and bead enrichment, as described in the ESI (Click reaction and streptavidin-beads enrichment). Next, the beads-enriched proteins were isolated on a magnetic stand and washed twice with 200 μL 0.2% SDS, 6 M urea and PBS, respectively, to remove the nonspecifically bound proteins. Then, the washed beads were dispersed in PBS and treated with 10 μg mL−1 RNase A for 1 h at 37 °C to release the RBPs. For the RNase pre-digested control groups, the bead-enriched proteins were treated with 10 μg mL−1 RNase A before being washed with the reagents described above. Then, all the experimental and control samples were subjected to SDS-PAGE (silver stained) and LC-MS/MS analysis.
Protein digestion and desalting
The samples were dispersed in 50 mM NH4HCO3, reduced with 10 mM DTT at 56 °C for 1 h and alkylated by 20 mM IAA at RT in the dark for 1 h. Next, the proteins were digested into peptides by trypsin at a mass ratio of 1:50 overnight at 37 °C. The tryptic peptides were desalted using a 100 μL pipette tip containing three pieces of 3 M Empore C18 discs. The pipette tips were pretreated by sequential addition of 80 μL ACN, 80 μL 50% H2O/50% ACN (v/v) and 80 μL H2O, which were all pre-cooled on ice. The tryptic-digested peptides were loaded into the pipette tips, washed with 80 μL H2O twice, and eluted with 80 μL 50% H2O/50% ACN (v/v) twice. Finally, the samples were vacuum-dried and redissolved with 0.1% formic acid prior to LC-MS/MS analysis. The RBPs were identified using label-free quantitative mass spectrometry with consistent quality control.
HILIC enrichment of O-GlcNAc and phosphorylated peptides
Nucleocytoplasmic proteins extracted by NE-PER Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher, USA) were used to optimize the HILIC enrichment conditions. Tryptic-digested nucleocytoplasmic proteins or RBPs obtained using the above method were treated with 100 units of PNGase F in 25 mM ammonium bicarbonate (pH 7.8) at 37 °C overnight; next, they were treated with 10 units of galactose oxidase and 15 units HRP in 25 mM sodium phosphate buffer (pH 7.0) containing 5% DMSO for 1 h at 35 °C using the FASP protocol33 to remove the N-glycans and destroy the O-galactosamine modifications on the residual oligosaccharide modified N/O-glycoproteins while sparing the O-GlcNAc modifications on the peptides. Next, the peptides were dissolved in 200 μL 80% ACN/1% TFA and incubated for 30 min with 5 mg ZIC-HILIC materials. Then, the ZIC-HILIC materials were washed with 100 μL 80% ACN/1% TFA three times to remove nonspecific peptides. Finally, the enriched O-GlcNAc and phosphorylated peptides were eluted three times with 100 μL 0.5% FA and vacuum-dried. The obtained O-GlcNAc and phosphorylated peptides were then subjected to LC-MS/MS analysis.
NanoLC-MS/MS analysis
The resulting peptide mixture was analyzed with an Orbitrap Fusion mass spectrometer coupled with an EASY-nLC 1000 nano-LC system (Thermo Fisher Scientific). Peptide separation was performed on an in-house-made 15 cm length reverse phase C18 column (150 nm id, 1.9 μm, 100 Å) using A and B buffers (buffer A: 0.1% FA in water; buffer B: 0.1% FA in ACN) at a constant flow rate of 600 nL min−1. The gradient was set as follows: 6%–9% B for 2 min, 9%–13% B for 8 min, 13%–26% B for 40 min, 26%–38% B for 20 min, 38%–100% for 1 min, and 100% B for 7 min. The dynamic exclusion duration of data-dependent MS2 acquisition (DDA) was 18 s. For the MS1 scan, mass spectra were acquired in the positive-ion mode over the range of 300–1400 m/z with a resolution of 120000 and a maximum ion injection time of 100 ms. The MS2 spectra was acquired with an automatic gain control target value of 5.0e3 and a maximum injection time of 35 ms with higher-energy collision (HCD) with a normalized collision energy of 32%.
Database search and data processing
The MS raw data were searched using MaxQuant software (version 1.6.17.0) against the UniPort Human database (updated on July 21, 2015). The digestion mode was set as trypsin with two missed cleavages allowed. Acetyl N-terminal and oxidation (methionine) modifications were set as variable modifications, and carbamidomethyl was set as a fixed modification. The mass tolerances were set as 20 ppm and 4.5 ppm for the first search and main search, respectively. The false discovery rate (FDR) was set as ≤0.01 at the spectra, protein and modification levels. The minimum score and delta score for the modified peptides were set as ≥40 and ≥6, respectively. For RBP identification, the process for both the experimental and the control groups was conducted three times. The search results were further processed by Perseus software (version 1.6.10.43). Significance was assessed by Student's t-test using p-values to identify potential RBPs. Proteins with P < 0.01, at least two identified unique peptides, and a fold change of greater than four (experimental versus control) were considered as RBP candidates. Only candidates identified in all three control sets are considered to be RBPs. For O-GlcNAc and phosphorylated RBPs identification, HexNAc (ST) and Phospho (ST) were added to the variable modifications, and a localization probability filter of ≥0.75 was applied. GO annotation analysis was performed in DAVID 6.8, and the proteins annotated in multiple terms were counted multiple times.
Results and discussion
Optimization of the RBPs-enrichment procedures
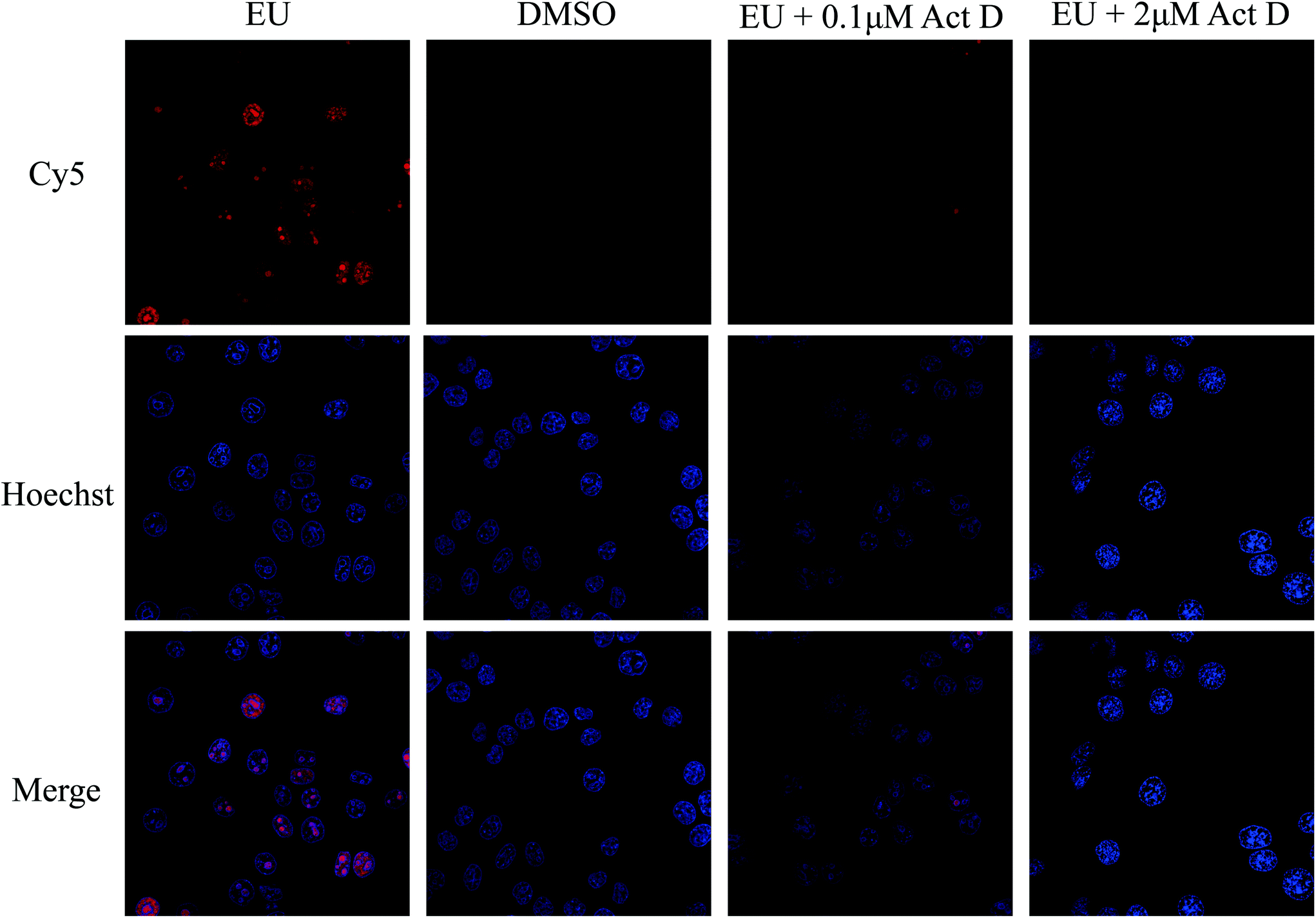
To achieve the optimized EU labeling efficiency, the EU concentration and incubation times used in the cell culture were evaluated. A fluorescent dye containing an azide group, Cy5-azide, was used to validate whether the labeling was successful. As shown in Fig. 2, the EU-treated cells showed very strong fluorescence signals compared to the cells in the DMSO-treated group without EU treatment. Next, actinomycin D, an RNA polymerase I inhibitor,34 was added with EU to incubate with HeLa cells. A dose-dependent reduction of the fluorescence signal was discovered, indicating that EU was specifically labeled to the RNA in the living cells (Fig. 2). The labeling efficiency of EU in RNAs is a key factor that influences the enrichment of RBPs. To achieve higher labeling efficiency, the concentrations of EU (0.01 to 2 mM) and the incubation time (3 to 12 h) were optimized (Fig. S1†). The labeling efficiency N (N = N1/N2; N1, number of Cy5-labeled cells, namely EU-labeled cells; N2, number of Hoechst-labeled cells, namely total cells) was used to evaluate the labeling efficiency. As shown in Fig. S1A,† with increasing concentration of EU, the fluorescence signal was enhanced significantly and the N increased accordingly, indicating improvement of the labeling efficiency. When the concentration of EU reaches 2 mM, many globular structures appeared in the cells, presumably due to the abnormal physiological state of the cells induced by the overdose of EU. Thus, 0.5 mM EU was chosen as the optimized condition. As shown in Fig. S1B,† N increased with increasing incubation time from 3 to 9 h, and it decreased when the incubation time reached 12 h. This phenomenon may be due to cell death caused by overlong incubation with EU. Thus, 9 h was chosen as the optimized incubation time.
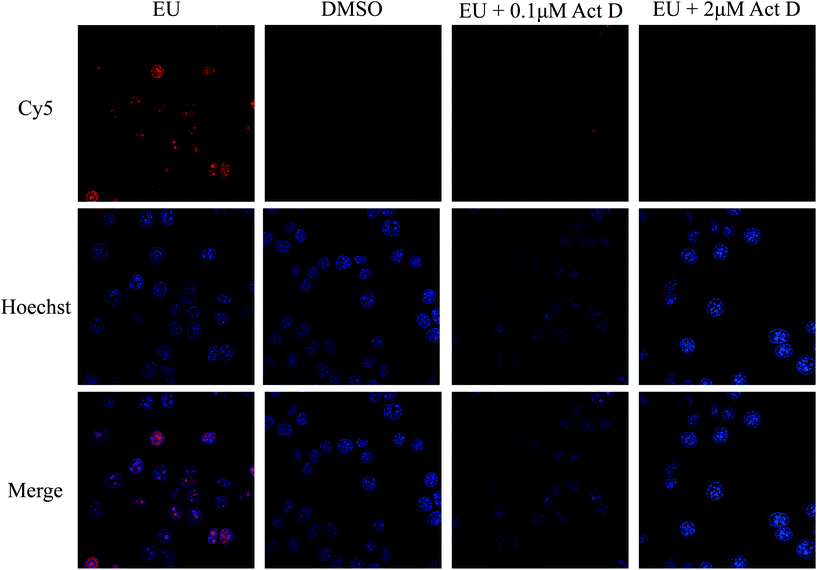 |
| | Fig. 2 Confocal laser scanning microscopy (CLSM) images of HeLa cells treated with EU, DMSO, EU + 0.1 μM Act D, and EU + 2 μM Act D. Act D, Actinomycin D. | |
To determine the actual EU labeling efficiency, the HeLa cells were incubated with EU at a concentration of 0.5 mM for 9 h, and the total RNAs were extracted by TRIzol reagent. The total RNAs were digested and subjected to LC-MS for the quantification of EU and uridine. The quantification results showed that EU accounted for 4.48% of the total uridines, corresponding to approximately 1.1% of the total nucleosides. Considering that almost all RNA species contain more than 100 nucleotides and the majority of them have more than 1000 nucleotides,35 we think that >1% labeling efficiency is enough for the global enrichment of RBPs.
Evaluation and validation of the “click chemistry”-based enrichment method
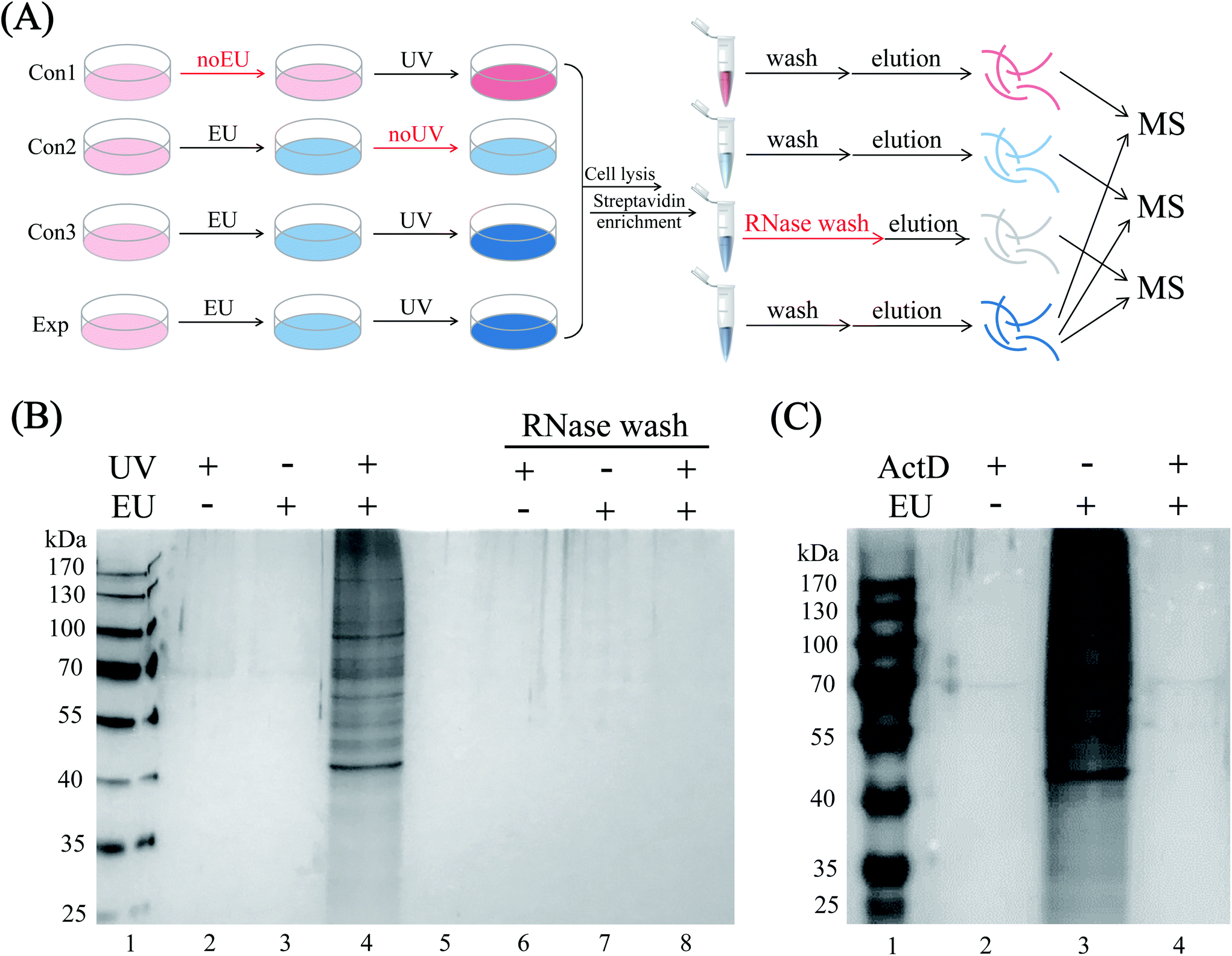
HeLa cells were EU-labeled and photo-crosslinked under the optimized conditions, and the cell lysates were click-tagged with biotin-N3. The biotinylated RNA-protein complexes were enriched by streptavidin beads, and the RBPs were released by RNase A digestion. To increase the confidence of RBP identification, we performed three control sets, namely EU-omitted (con1), UV-omitted (con2) and RNase pre-digested (con3) samples, respectively (Fig. 3A). As shown in the SDS-PAGE gel image (Fig. 3B), the enriched sample exhibited protein bands with a diverse mass range of RBPs (lane 4), while the three controls yielded minimal background signals with almost no nonspecific bands (lanes 2, 3 and 8). The actinomycin D-treated group also yielded almost no protein bands (Fig. 3C, lane 4) because of the inhibition of RNA synthesis by actinomycin D, which limited the incorporation of EU into RNA. The above results proved the highly selective and RNA-dependent enrichment by the EU-labeling and “click chemistry” method with only marginal non-specific adsorption.
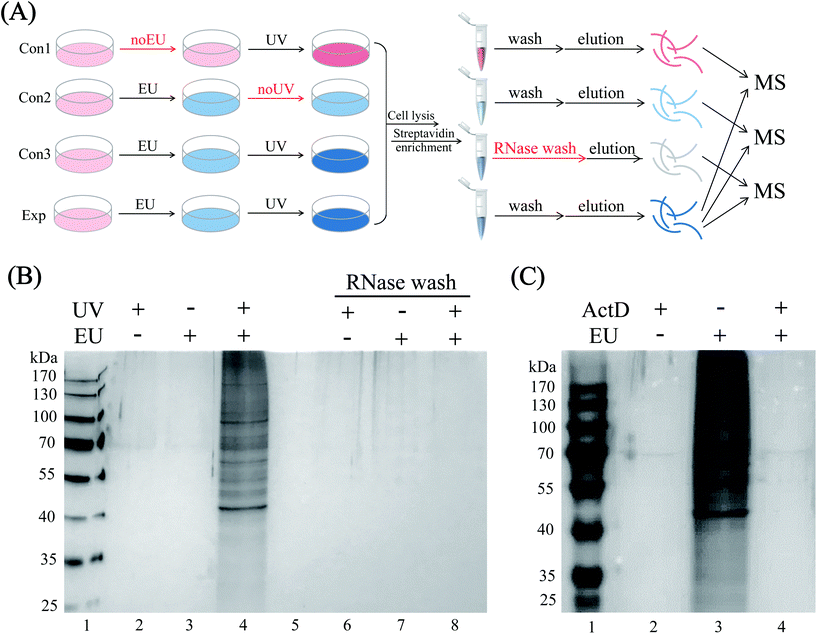 |
| | Fig. 3 (A) Sample preparation workflow for RBP enrichment and identification. Three sets of negative control experiments were performed, including the EU-omitted, UV-omitted, and RNase pre-digested samples, respectively. (B) In-gel analysis of the RBPs enrichment strategy. HeLa cells were treated with EU alone, UV alone, and EU and UV, respectively. All lysates were reacted with biotin-N3 and enriched by streptavidin beads. After treatment with or without RNase A, the samples were resolved by SDS-PAGE and silver staining. (C) HeLa cells were treated with EU and actinomycin D and enriched by the methods described above. ActD: actinomycin D. | |
Proteomic identification of RBPs
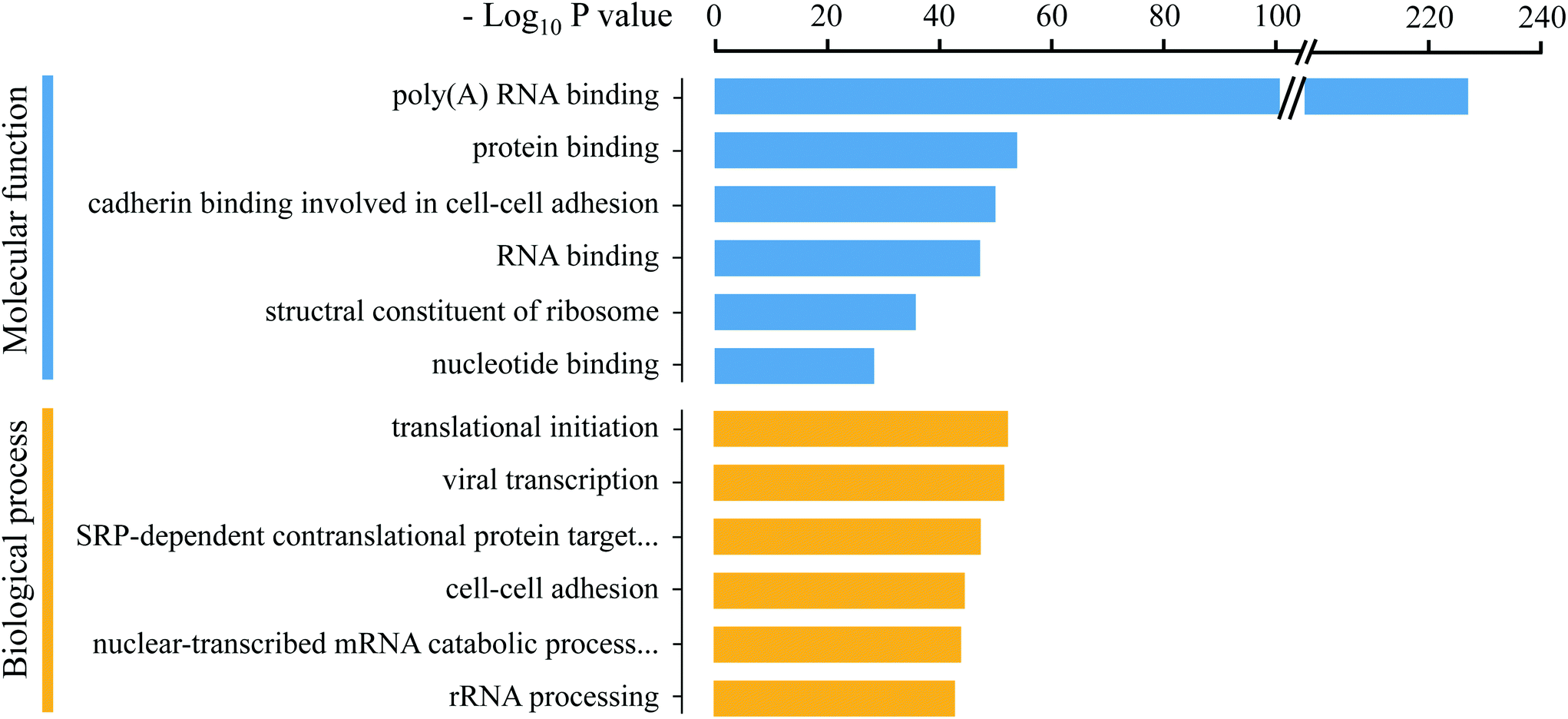
Previous methods for RBP enrichment mainly rely on the poly (A) tail of the mRNAs to specifically purify their binding proteins. However, many types of RNAs do not have the poly (A) tail, especially non-coding RNAs, which have attracted much attention in recent years due to their important roles in gene expression and epigenetic regulation. Therefore, the Eu-tagging and “click chemistry”-based RBP enrichment method is advantageous for achieving a more comprehensive profiling of the RNA-binding proteome. To remove the residual unspecifically adsorbed non-RNA-binding proteins from the MS identification results, quantitative comparisons between the enrichment groups and three control groups (noEU, noUV, and RNase pre-digest before washing) were conducted, respectively (Fig. 3A). In total, 1115 RBPs were obtained after stringent filtering (Dataset S1†), which is 54.86% more than the numbers reported in previous studies.15,16 The improved RBP enrichment may be attributed to the optimized EU labeling conditions and efficiency (the 1.1% EU labeling in the total nucleosides ensured that majority of the RNA molecules can be tagged with the alkyne handle), which is crucial for achieving deeper mapping of PTMs on RBPs. GO annotation analysis using DAVID 6.836 revealed that the identified RBPs were enriched in the molecular function terms “poly(A) RNA binding” and “RNA binding”, and most were related to various RNA processes, such as translational initiation, mRNA splicing and regulation of mRNA stability (Fig. 4); this further proved the reliability of this method for RBP enrichment.
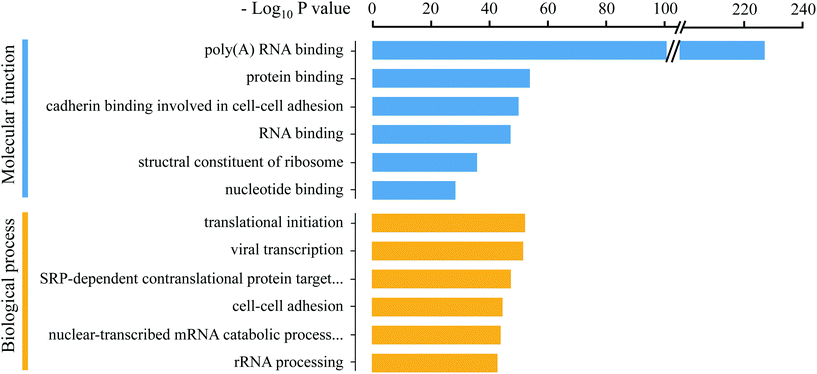 |
| | Fig. 4 GO enrichment analysis of the RBPs. | |
Establishment of the HILIC enrichment strategy for simultaneous profiling of O-GlcNAcylation and phosphorylation
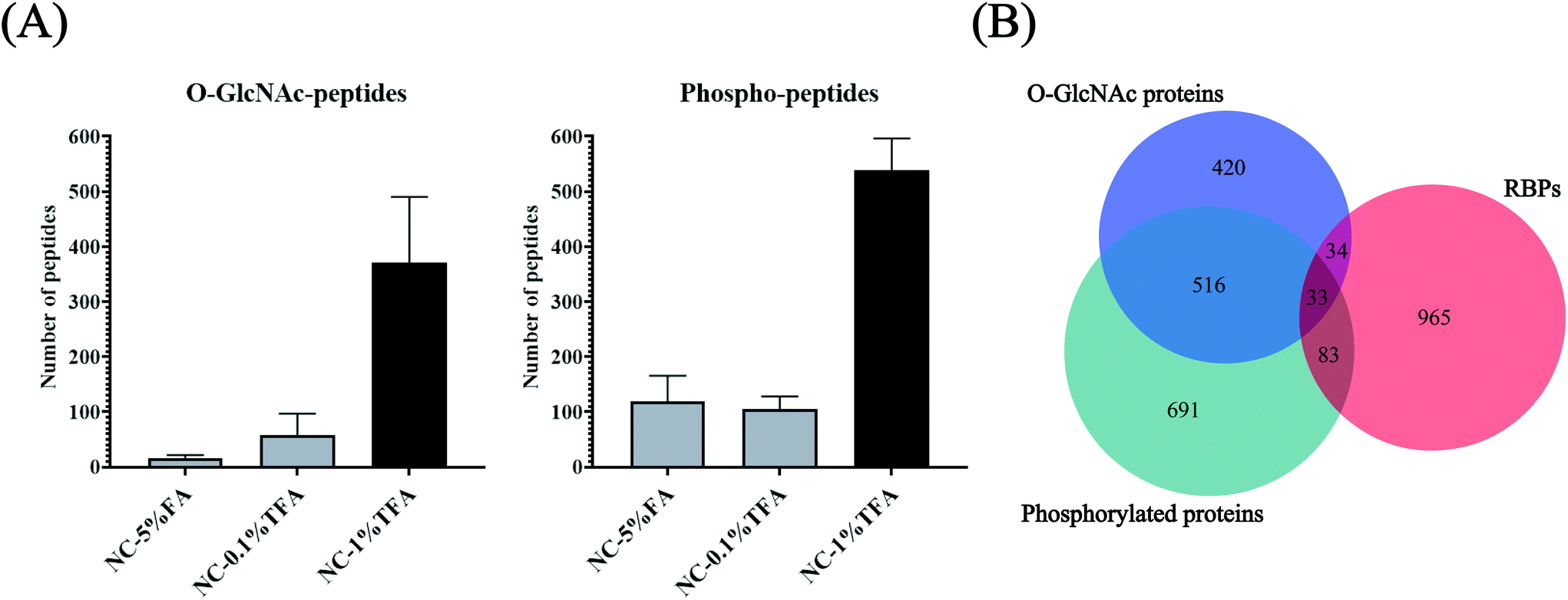
O-GlcNAcylation is notoriously difficult to enrich due to its small size and uncharged characteristics. The enrichment efficiency using lectin (WGA) or antibodies is low.37 Although metabolic labeling may tag O-GlcNAcylation with an enrichment handle,38 the potential conflict of azide/alkyne labeling and enrichment for both glycan and RNA makes this approach unsuitable for O-GlcNAcylation profiling for RBPs. Furthermore, considering the limited amount of RBPs can be obtained (∼10 μg from 108 cells), enriching O-GlcNAcylation and phosphorylation separately using different methods may not be feasible and may lead to severe sample loss. Inspired by the hydrophilicity of the O-GlcNAc and PO3 groups and the possibility of their co-existence in OPTC, we proposed a HILIC method for simultaneous enrichment of both O-GlcNAcylation and phosphorylation-modified peptides. Considering that O-GlcNAc modification only exists in cytoplasmic, nuclear and mitochondrial proteins, nucleocytoplasmic proteins were chosen as the model sample to optimize the HILIC enrichment conditions to reduce the co-eluting non-glycopeptides/non-phosphopeptides before actual application for RBPs. Among the three different binding/washing buffer conditions, 1% TFA resulted in the most identified O-GlcNAc and phosphorylated peptides, which was at least 2.7 times more than that obtained using 5% FA and 0.1% TFA (Fig. 5A and Table S1†). We attributed the improved enrichment for both O-GlcNAc and phosphorylated peptides to the application of TFA, a hydrophobic ion-pairing reagent, in the mobile phase, which may increase the selectivity of HILIC to hydrophilic components.39 TFA forms hydrophobic neutral ion pairs with positively charged peptides under acidic conditions, while O-GlcNAc and phosphorylated peptides are not affected due to their stronger hydrophilicity. However, due to the insufficient TFA concentration, the selectivity in the NC-0.1% TFA group was not improved significantly. Taken together, 80% ACN in 1% TFA was chosen as the optimized HILIC condition for enrichment of RBP-O-GlcNAc/phosphorylation modifications. For clearance, because the low pH condition (1% TFA in 80% ACN, pH 0.84) was only adopted for a relatively short time (about 30 min) and the HILIC materials were only used once, the influence of the pH-related degradation was negligible. Due to the limited abundance of RBPs, only 13.5% of them can be covered by HILIC enrichment from the tryptic-digested nucleocytoplasmic proteins (Fig. 5B), indicating the necessity of using RBP and HILIC tandem enrichment for efficient profiling of the two PTMs in the RNA-binding proteome.
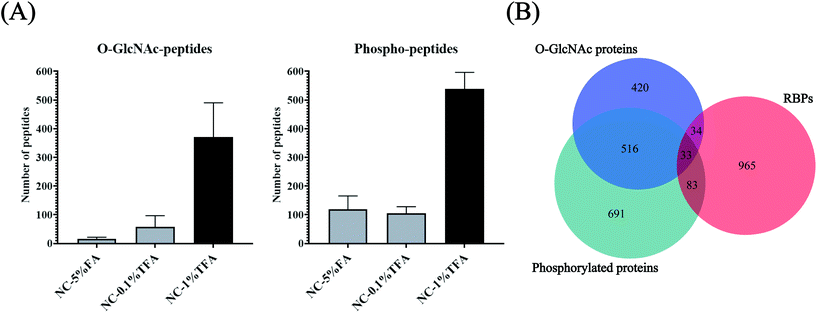 |
| | Fig. 5 (A) Number of O-GlcNAc-peptides and phospho-peptides enriched by HILIC under different conditions. NC, nucleocytoplasmic proteins. (B) Overlap of the O-GlcNAc proteins and phosphorylated proteins obtained by HILIC enrichment of the tryptic-digested nucleocytoplasmic proteins with the RBPs identified by metabolic labeling and “click chemistry” enrichment. | |
Simultaneous enrichment and identification of O-GlcNAcylation and phosphorylation in the RNA-binding proteome
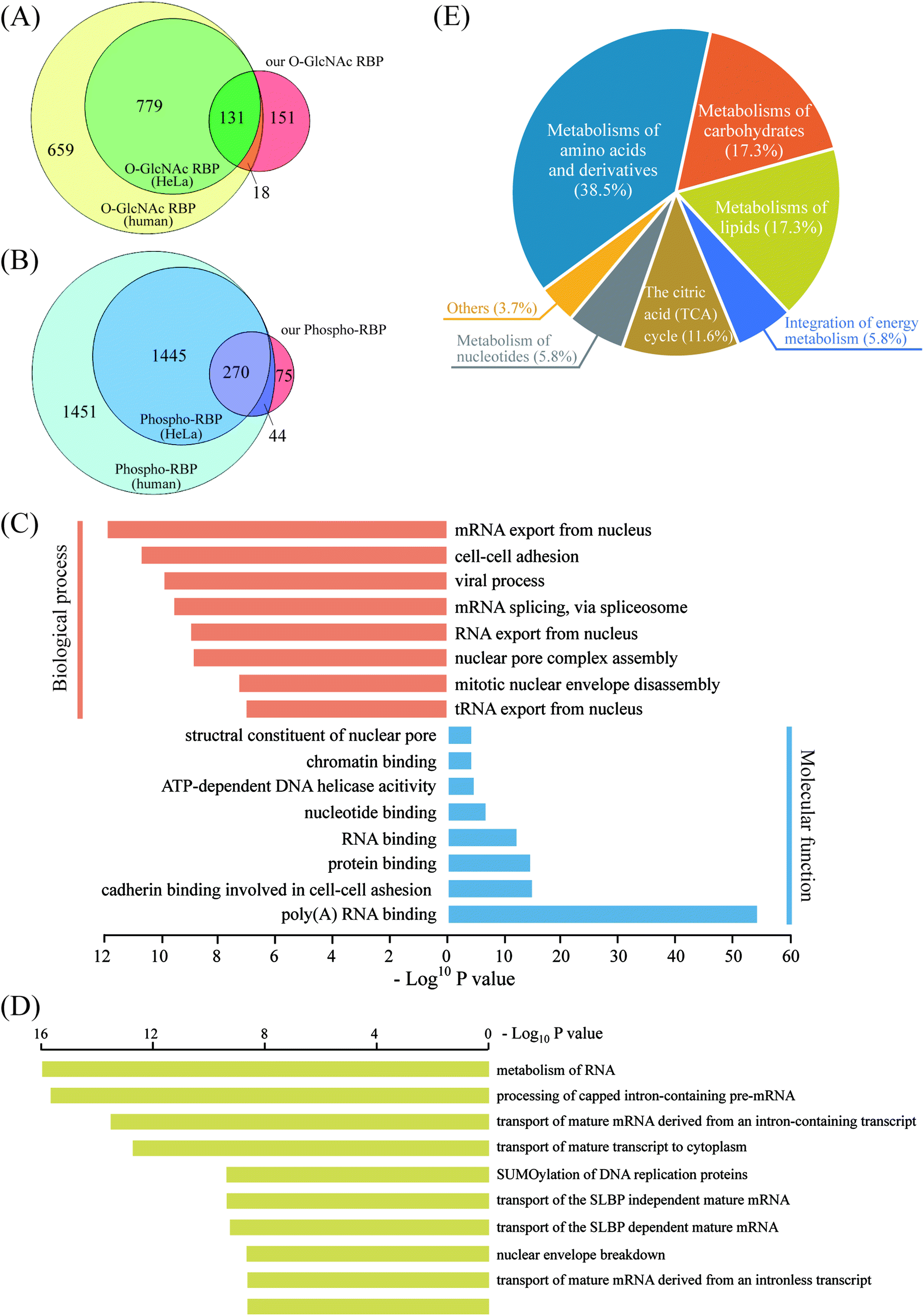
As revealed in the above experiments, direct enrichment of O-glcNAc/phosphorylated peptides from tryptic-digested nucleocytoplasmic proteins resulted in poor mapping of the two PTMs in the RNA-binding proteome. Therefore, we proposed an RBP-targeted PTM profiling approach via HILIC enrichment of the O-GlcNAc peptides and phosphopeptides from the metabolic-labeled and “click chemistry”-enriched RBPs. In addition to the reduced sample loss by the simultaneous enrichment of the two PTMs, improved PTM targeting on RBPs can be expected by this tandem enrichment strategy via removal of the majority of the non-RBPs before HILIC enrichment. This strategy led to the first large-scale profiling of O-GlcNAcylation and phosphorylation in the RNA-binding proteome. 461 O-GlcNAc peptides corresponding to 300 RBPs and 671 phosphopeptides corresponding to 389 RBPs were obtained by overlapping the MS-identified RBPs with the PTM proteins/peptides obtained by HILIC enrichment (Dataset S2, Tables S2 and S3†). At least three times more O-GlcNAc/phosphorylated RBPs were identified than that obtained by direct HILIC enrichment from tryptic-digested nucleocytoplasmic proteins, which demonstrated the advantage of using our tandem enrichment strategy. In order to further analyze our O-GlcNAc/phosphorylated RBPs identified in HeLa cells, we compiled a list of 1587 human O-GlcNAc RBPs, including RBPs in HeLa cells (910) as well as in other human cell lines, by overlapping the O-GlcNAc proteins and the RBPs that have been identified to date.15–19,37,40–42 Additionally, a list of 3210 human phospho-RBPs (1715 in HeLa) was obtained in the same way. Of the 300 O-GlcNAc RBPs obtained by our tandem enrichment strategy, 149 (49.67%) were overlapped with known human O-GlcNAc RBPs, most of which were also identified in known HeLa O-GlcNAc RBPs (Fig. 6A). Similarly, of the 389 phospho-RBPs, 314 (80.72%) were overlapped with known human phospho-RBPs (Fig. 6B). As a result, our tandem enrichment strategy identified 151 O-GlcNAc RBPs and 75 phospho-RBPs that were not previously identified. Furthermore, we discovered 213 RBPs that were modified by both PTMs, indicating potentially extensive OPTC regulations that deserve future investigation. GO annotation analysis using the bioinformatics tool DAVID 6.8 was conducted to better understand the O–P-modified RBPs (Fig. 6C). The molecular function results were consistent with those of all RBPs, mainly related to poly(A) RNA binding and RNA binding. Interestingly, biological process analysis indicated that the O–P-modified RBPs were more enriched in mRNA exported from the nucleus. Pathway analysis of the O–P-modified RBPs by Reactome43 revealed strong signatures of RNA metabolism and transport processes, in line with the GO biological process results. Another interesting finding was that ∼25% of the O–P-modified RBPs (46 RBPs) were involved in metabolism pathways not directly related to RNA, such as metabolism of carbohydrates, lipids, amino acids and derivatives (Fig. 6D and E). Considering the complex correlation among RNA, RBPs, PTM and metabolism,44 the above discovery provides a valuable dataset for further study of the PTM-regulated translational machinery and protein (enzyme)-RNA interactions.
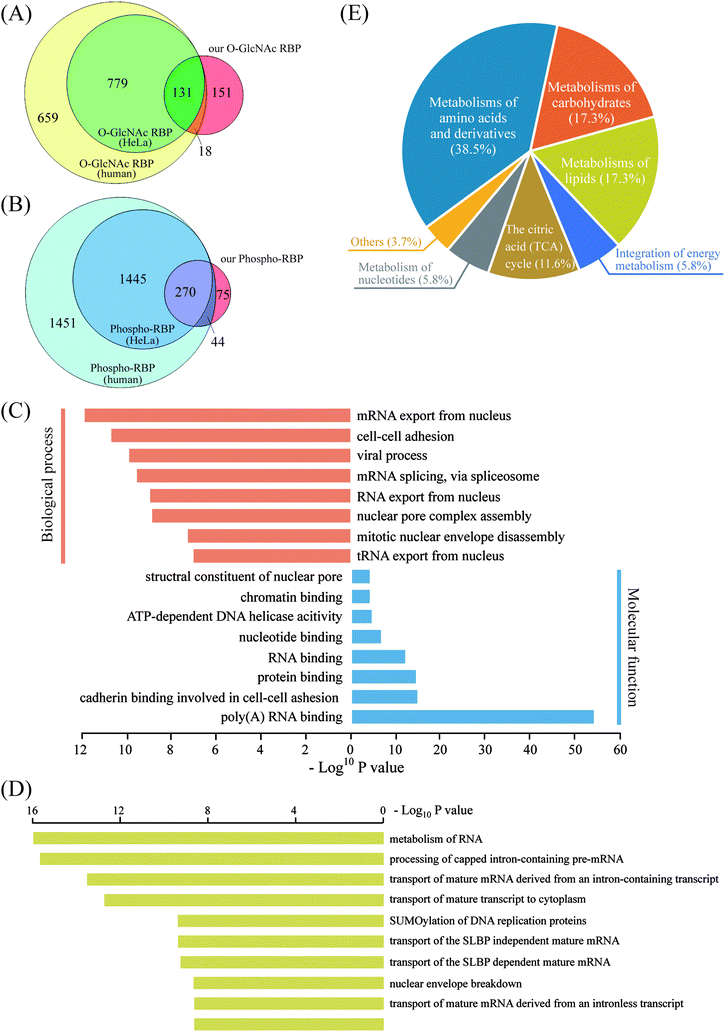 |
| | Fig. 6 (A) Overlap of the known O-GlcNAc RBPs and O-GlcNAc RBPs obtained by our tandem enrichment method. (B) Overlap of the known phospho-RBPs with the phospho-RBPs obtained by our tandem enrichment method. (C) GO analysis of the 213 O–P-modified RBPs. (D) Reactome pathway analysis of 213 O–P-modified RBPs. (E) Distribution of the 46 metabolic enzymes from the 213 O–P-modified RBPs in specific metabolic pathways. | |
Conclusion
In the present study, we developed a tandem method for simultaneous enrichment and identification of O-GlcNAcylation and phosphorylation in the RNA-binding proteome. By combined application of metabolic labeling and the click reaction for purification of RBPs and HILIC enrichment for the two types of PTMs, we identified 1115 RBPs in HeLa cells and 461 O-GlcNAc peptides and 671 phosphopeptides from these RBPs. Approximately 20% of these RBPs are modified by both PTMs, indicating the potentially wide existence of OPTC in RBPs and the advantage of our strategy for further study of the OPTC-regulated function of RBPs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Key R&D Program of China [grant numbers 2016YFA0501403, 2018YFC0910302, 2017YFA0505002]; and National Natural Science Foundation of China [no. 21675172].
References
- A. S. Venne, L. Kollipara and R. P. Zahedi, Proteomics, 2014, 14, 513–524 CrossRef CAS.
- J. D. Rotty, G. W. Hart and P. A. Coulombe, Nat. Cell Biol., 2010, 12, 847–849 CrossRef CAS.
- H. B. Ruan, J. P. Singh, M. D. Li, J. Wu and X. Yang, Trends Endocrinol. Metab., 2013, 24, 301–309 CrossRef CAS.
- H. Nie, H. Ju, J. Fan, X. Shi, Y. Cheng, X. Cang, Z. Zheng, X. Duan and W. Yi, Nat. Commun., 2020, 11, 36 CrossRef CAS.
- Y. Yang, M. Fu, M.-D. Li, K. Zhang, B. Zhang, S. Wang, Y. Liu, W. Ni, Q. Ong, J. Mi and X. Yang, Nat. Commun., 2020, 11, 181 CrossRef CAS.
- N. J. Pedowitz, A. R. Batt, N. Darabedian and M. R. Pratt, Nat. Chem. Biol., 2020 DOI:10.1038/s41589-020-0640-8.
- S.-L. Xu, R. J. Chalkley, J. C. Maynard, W. Wang, W. Ni, X. Jiang, K. Shin, L. Cheng, D. Savage, A. F. R. Hühmer, A. L. Burlingame and Z.-Y. Wang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E1536–E1543 CrossRef CAS.
- Y. Watanabe, S. Shinozaki-Yabana, Y. Chikashige, Y. Hiraoka and M. Yamamoto, Nature, 1997, 386, 187–190 CrossRef CAS.
- I. Lisitsky and G. Schuster, Nucleic Acids Res., 1995, 23, 2506–2511 CrossRef CAS.
- T. Ohn, N. Kedersha, T. Hickman, S. Tisdale and P. Anderson, Nat. Cell Biol., 2008, 10, 1224–1231 CrossRef CAS.
- J. Xiao, S. Xu, C. Li, Y. Xu, L. Xing, Y. Niu, Q. Huan, Y. Tang, C. Zhao, D. Wagner, C. Gao and K. Chong, Nat. Commun., 2014, 5, 4572 CrossRef CAS.
- A. Castello, B. Fischer, K. Eichelbaum, R. Horos, B. M. Beckmann, C. Strein, N. E. Davey, D. T. Humphreys, T. Preiss, L. M. Steinmetz, J. Krijgsveld and M. W. Hentze, Cell, 2012, 149, 1393–1406 CrossRef CAS.
- A. Castello, R. Horos, C. Strein, B. Fischer, K. Eichelbaum, L. M. Steinmetz, J. Krijgsveld and M. W. Hentze, Nat. Protoc., 2013, 8, 491–500 CrossRef CAS.
- A. G. Baltz, M. Munschauer, B. Schwanhausser, A. Vasile, Y. Murakawa, M. Schueler, N. Youngs, D. Penfold-Brown, K. Drew, M. Milek, E. Wyler, R. Bonneau, M. Selbach, C. Dieterich and M. Landthaler, Mol. Cell, 2012, 46, 674–690 CrossRef CAS.
- R. Huang, M. Han, L. Meng and X. Chen, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E3879–E3887 CrossRef CAS.
- X. Bao, X. Guo, M. Yin, M. Tariq, Y. Lai, S. Kanwal, J. Zhou, N. Li, Y. Lv, C. Pulido-Quetglas, X. Wang, L. Ji, M. J. Khan, X. Zhu, Z. Luo, C. Shao, D. H. Lim, X. Liu, N. Li, W. Wang, M. He, Y. L. Liu, C. Ward, T. Wang, G. Zhang, D. Wang, J. Yang, Y. Chen, C. Zhang, R. Jauch, Y. G. Yang, Y. Wang, B. Qin, M. L. Anko, A. P. Hutchins, H. Sun, H. Wang, X. D. Fu, B. Zhang and M. A. Esteban, Nat. Methods, 2018, 15, 213–220 CrossRef CAS.
- R. M. L. Queiroz, T. Smith, E. Villanueva, M. Marti-Solano, M. Monti, M. Pizzinga, D. M. Mirea, M. Ramakrishna, R. F. Harvey, V. Dezi, G. H. Thomas, A. E. Willis and K. S. Lilley, Nat. Biotechnol., 2019, 37, 169–178 CrossRef CAS.
- J. Trendel, T. Schwarzl, R. Horos, A. Prakash, A. Bateman, M. W. Hentze and J. Krijgsveld, Cell, 2019, 176, 391–403 CrossRef CAS.
- E. C. Urdaneta, C. H. Vieira-Vieira, T. Hick, H. H. Wessels, D. Figini, R. Moschall, J. Medenbach, U. Ohler, S. Granneman, M. Selbach and B. M. Beckmann, Nat. Commun., 2019, 10, 990 CrossRef.
- J. E. Rexach, C. J. Rogers, S. H. Yu, J. Tao, Y. E. Sun and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2010, 6, 645–651 CrossRef CAS.
- J. Villén and S. P. Gygi, Nat. Protoc., 2008, 3, 1630–1638 CrossRef.
- C. F. Teo, S. Ingale, M. A. Wolfert, G. A. Elsayed, L. G. Nöt, J. C. Chatham, L. Wells and G.-J. Boons, Nat. Chem. Biol., 2010, 6, 338–343 CrossRef CAS.
- S. R. Fuhs, J. Meisenhelder, A. Aslanian, L. Ma, A. Zagorska, M. Stankova, A. Binnie, F. Al-Obeidi, J. Mauger, G. Lemke, J. R. Yates 3rd and T. Hunter, Cell, 2015, 162, 198–210 CrossRef CAS.
- M. W. H. Pinkse, S. Mohammed, J. W. Gouw, B. van Breukelen, H. R. Vos and A. J. R. Heck, J. Proteome Res., 2008, 7, 687–697 CrossRef CAS.
- K. Vosseller, J. C. Trinidad, R. J. Chalkley, C. G. Specht, A. Thalhammer, A. J. Lynn, J. O. Snedecor, S. Guan, K. F. Medzihradszky, D. A. Maltby, R. Schoepfer and A. L. Burlingame, Mol. Cell. Proteomics, 2006, 5, 923 CrossRef CAS.
- Y. Zhu, L. I. Willems, D. Salas, S. Cecioni, W. B. Wu, L. J. Foster and D. J. Vocadlo, J. Am. Chem. Soc., 2020 DOI:10.1021/jacs.0c04121.
- J. F. Alfaro, C.-X. Gong, M. E. Monroe, J. T. Aldrich, T. R. W. Clauss, S. O. Purvine, Z. Wang, D. G. Camp, J. Shabanowitz, P. Stanley, G. W. Hart, D. F. Hunt, F. Yang and R. D. Smith, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7280 CrossRef CAS.
- B. Buszewski and S. Noga, Anal. Bioanal. Chem., 2012, 402, 231–247 CrossRef CAS.
- N. Sun, H. Wu, H. Chen, X. Shen and C. Deng, Chem. Commun., 2019, 55, 10359–10375 RSC.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS.
- D. Schulz and A. Rentmeister, ChemBioChem, 2014, 15, 2342–2347 CrossRef CAS.
- C. Y. Jao and A. Salic, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15779 CrossRef CAS.
- J. R. Wiśniewski and M. Mann, Anal. Chem., 2012, 84, 2631–2637 CrossRef.
- O. Bensaude, Transcription, 2011, 2, 103–108 CrossRef.
- E. Jankowsky and M. E. Harris, Nat. Rev. Mol. Cell Biol., 2015, 16, 533–544 CrossRef CAS.
- D. W. Huang, B. T. Sherman and R. A. Lempicki, Nat. Protoc., 2009, 4, 44–57 CrossRef CAS.
- B. Huo, W. Zhang, X. Zhao, H. Dong, Y. Yu, J. Wang, X. Qian and W. Qin, Chem. Commun., 2018, 54, 13790–13793 RSC.
- K. Qin, Y. Zhu, W. Qin, J. Gao, X. Shao, Y. L. Wang, W. Zhou, C. Wang and X. Chen, ACS Chem. Biol., 2018, 13, 1983–1989 CrossRef CAS.
- S. Mysling, G. Palmisano, P. Højrup and M. Thaysen-Andersen, Anal. Chem., 2010, 82, 5598–5609 CrossRef CAS.
- H. Chu, H. Zheng, J. Yao, N. Sun, G. Yan and C. Deng, Chem. Commun., 2020, 56, 11299–11302 RSC.
- J. Li, Z. Li, X. Duan, K. Qin, L. Dang, S. Sun, L. Cai, L. C. Hsieh-Wilson, L. Wu and W. Yi, ACS Chem. Biol., 2019, 14, 4–10 CrossRef CAS.
- W. Qin, K. Qin, X. Fan, L. Peng, W. Hong, Y. Zhu, P. Lv, Y. Du, R. Huang, M. Han, B. Cheng, Y. Liu, W. Zhou, C. Wang and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 1817–1820 CrossRef CAS.
- B. Jassal, L. Matthews, G. Viteri, C. Gong, P. Lorente, A. Fabregat, K. Sidiropoulos, J. Cook, M. Gillespie, R. Haw, F. Loney, B. May, M. Milacic, K. Rothfels, C. Sevilla, V. Shamovsky, S. Shorser, T. Varusai, J. Weiser, G. Wu, L. Stein, H. Hermjakob and P. D'Eustachio, Nucleic Acids Res., 2019, 48, D498–D503 Search PubMed.
- A. Castello, M. W. Hentze and T. Preiss, Trends Endocrinol. Metab., 2015, 26, 746–757 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0an02305a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Jian
Li
a,
Tong
Liu
a,
Zheng
Zhang
*ab,
Weijie
Qin
*a and
Xiaohong
Qian
*a
a,
Jian
Li
a,
Tong
Liu
a,
Zheng
Zhang
*ab,
Weijie
Qin
*a and
Xiaohong
Qian
*a