
Influence of protein ion charge state on 213 nm top-down UVPD†
Simon
Becher
a,
Huixin
Wang
b,
Michael G.
Leeming
c,
William A.
Donald
 d and
Sven
Heiles
*a
d and
Sven
Heiles
*a
aInstitute of Inorganic and Analytical Chemistry, Justus Liebig University Giessen, 35392 Giessen, Germany. E-mail: sven.heiles@anorg.chemie.uni-giessen.de
bMark Wainwright Analytical Centre, University of New South Wales, New South Wales, Australia
cBio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Victoria, Australia
dSchool of Chemistry, University of New South Wales, Sydney, New South Wales, Australia
First published on 14th May 2021
Abstract
Ultraviolet photodissociation (UVPD) is a powerful and rapidly developing method in top-down proteomics. Sequence coverages can exceed those obtained with collision- and electron-induced fragmentation methods. Because of the recent interest in UVPD, factors that influence protein fragmentation and sequence coverage are actively debated in the literature. Here, we performed top-down 213 nm UVPD experiments on a 7 T Fourier-transform ion cyclotron resonance mass spectrometer (FT-ICR MS) for the model proteins ubiquitin, myoglobin and cytochrome c that were electrosprayed from native, denaturing and supercharging solutions in order to investigate the effect of protein charge states on UVPD fragments. By performing UVPD in ultrahigh vacuum, factors associated with collisional cooling and any ion activation during transfer between mass analyzers can be largely eliminated. Sequence coverage increased from <10% for low charge states to >60% for high charge states for all three proteins. This trend is influenced by the overall charge state, i.e., charges per number of amino acid residues, and to a lesser degree by associated structural changes of protein ions of different charge states based on comparisons to published collision-cross section measurements. To rationalize this finding, and correlate sequence ion formation and identity with the number and location of protons, UVPD results were compared to protonation sites predicted based on electrostatic modelling. Assuming confined protonation sites, these results indicate the presence of two general fragmentation types; i.e., charge remote and charge directed. For moderately high protein charge states, fragment ions mostly originate in regions between likely protonation sites (charge remote), whereas sequence ions of highly charge protein ions occur either near backbone amide protonation sites at low-basicity residues (charge directed) or at charge remote sites (i.e., high-basicity residues). Overall, our results suggest that top-down 213 UVPD performance in the zero-pressure limit depends strongly on protein charge states and protonation sites can influence the location of backbone cleavages.
Introduction
Sequencing peptides generated by enzymatic digestion of proteins, so called bottom-up proteomics, is the method of choice to investigate complex protein mixtures via mass spectrometry (MS) and tandem MS (MSn) approaches, whereas dissociation of intact protein ions, i.e. top-down proteomics, is increasingly employed to reveal proteoform complexity. The increasing prevalence of top-down investigations is driven by a growing appreciation that single amino acid alterations, isomerism or post-translational modifications (PTMs) can impact the functional context of proteins.1 For example, Fournier et al. showed that acetylation of lysine can influence protein activity and function.2 Using high-energy collisional dissociation the authors demonstrated that human lysine acetyltransferases KAT2A and KAT2B are capable of activating and deactivating polo-like kinase 4 by acetylation of PLK4 kinase residues K45 and K46.On the other hand, the improvement of mass spectrometric performance, such as sensitivity, mass resolution, mass range, as well as development of novel MSn workflows give access to proteins/protein complexes formed by electrospray ionization from native-like solutions and reveals proteoform complexity that is hard to capture with bottom-up methodologies.3–5 However, the success of proteoform identification as well as sequence coverages depends on the method of ion activation.
Collision-induced dissociation (CID) is the most widely used MSn method in top-down proteomics.6,7 Upon CID, protein ions are heated by collisions with the gas atoms/molecules resulting in protein ion fragmentation. Due to the low energy per collision of most CID methods (typically 10 kJ mol−1 per collision is transferred to protein ions in low-energy CID), the introduced energy after every collision equilibrates throughout the protein before bond cleavage occurs. Therefore, most prominent fragment ion signals are often associated with the lowest bond dissociation enthalpies. For example, Donald and co-workers demonstrated that CID of intact highly charged ubiquitin, cytochrome c, lysozyme and β-lactoglobulin ions results in preferential cleavage sites near the first low-basicity amino acid residue that is predicted to be protonated with increasing charge state yielding highly selective ion fragmention.8 The highest sequence coverages are obtained for charge states without backbone protonation, because nonspecific cleavage of the amide backbone occurs. By comparison to predictions of charge carrier positions benchmarked against gas-phase basicity measurements, hybrid quantum mechanics and molecular dynamics simulations of dissociation pathways and energy-resolved CID measurements, the dominance of the observed selective fragment ions can be attributed to the lowered energetic demand of backbone cleavage due to protonation.9 However, slow heating during CID triggers preferential loss of labile groups complicating the ability to identify the sites of some PTMs and causing hydrogen/deuterium (H/D) scrambling.10,11 Infrared multiphoton dissociation (IRMPD) of protein ions utilizes the absorption of multiple IR photons, resulting in fragmentation patterns that are often similar to CID results.12,13
An alternative means of fragmentation in top-down proteomics involves generating hydrogen-rich radical protein ions that subsequently fragment through intramolecular bond homolysis.14 Electrons can be introduced directly (electron-capture dissociation ECD) or via ion–ion reactions involving electron transfer (electron-transfer dissociation ETD). Molina et al. and Zenaidee et al. showed that cleavage of protein bonds in ETD and ECD tend to occur near the likely locations of charge carriers and depends strongly on charge state.15,16 That is, fragmentation in ECD and ETD experiments is influenced by the number and position of charge carriers, which impacts the sequence coverage. Resulting sequence coverage values for top-down proteomics via ETD and ECD are typically comparable to that for CID or higher, and importantly PTM fragmentation can be minimized in ETD/ECD.17
Another method for ion activation that recently has received considerable attention is ultraviolet photodissociation (UVPD). The method employs high energy UV photons for the electronic excitation of analyte ions to trigger fragmentation. The energy of a single UV photon can be used to form fragment ions and access high-energy dissociation pathways.18–20 The wavelengths mostly employed for UVPD of protein and peptide ions are 266 nm, 213 nm, 193 nm, and 157 nm.
UVPD has been employed in numerous case studies. For example, zwitterionic salt bridges in gaseous ions were identified by Julian and co-workers by irradiating protein ions with 266 nm laser light creating photoelectrons in close proximity to salt bridges as indicated by the detected c-fragments.21 Wavelengths below 266 nm allow excitation of amide and ester bonds. In a top-down proteomics approach Shaw et al. reported 99%, 93% and 87% sequence coverages using 193 nm photons on ubiquitin (9 kDa; 7+ to 13+), myoglobin (17 kDa; 16+, 18+, 20+, 22+ and 24+) and carbonic anhydrase II (29 kDa; 34+), respectively.17 Studies on apo- and holo-myoglobin showed fragment suppression for sites that bind to the heme unit as well as for the helical core of the molecule. This indicates the sensitivity of UVPD for selected charge states to probe primary, secondary, tertiary and quaternary protein structures.22
Application of 157 nm photons from the vacuum ultraviolet (VUV) range increases possible fragmentation sites due to absorption by C–C and carbon–heteroatom bonds leading to homolytic bond cleavage in peptides.23,24 However, in addition to complications arising from containing the VUV laser beam, fragmentation can also result in extensive cleavage of side chain residues resulting in very complex tandem mass spectra.
Another UVPD wavelength that employs solid state lasers is 213 nm that is absorbed by aromatic groups, amides as well as sulfides, and is now available as part of a commercial mass spectrometer.25 213 nm UVPD systems have been used by multiple researchers to investigate peptides and protein ions. For example, cleavage of C–S and S–S bonds of cysteine moieties by 213 nm UVPD can result in the formation of unique thioether, disulfide and trisulfide fragments, which facilitate identification of cysteine linkages in antibodies.26 The major factor that determines the performance of 213 nm UVPD top-down and bottom-up proteomics experiments is the absorption of UV light by the amide n–π* transition.27 Typically amide groups exhibit a local absorption maximum around 210 nm.28 In a study by Fornelli et al., the authors found that proline residues increase fragmentation efficiencies compared to other amino acids in 213 nm UVPD.29 Unique fragments of proline were also found by Dugourd and co-workers corresponding to b + 2, a + 2 and y − 2 ions formed in 213 nm UVPD.30 The authors explained the formation of these 213 nm UVPD specific ions by homolytic bond cleavage after excitation of n–π* transitions in proline moieties. The dependence of protein charge states on 213 nm performance has been investigated by Fornelli et al. for ubiquitin (8+ to 13+), myoglobin (14+ to 25+) and carbonic anhydrase (29+ to 43+). This study was performed on an orbital trapping mass spectrometer but did not reveal a pronounced charge state dependence.29
Here, we present top-down 213 nm UVPD results for a wide range of charge states for a set of model proteins in the zero-pressure limit in order to probe the intrinsic response of proteins to 213 nm light irradiation and rationalize fragmentation patterns in terms of charge state dependence. We examined ubiquitin, cytochrome c and myoglobin with charge states ranging from 5+ to 17+, 7+ to 24+, and 9+ to 34+ with top-down 213 nm UVPD, respectively. Cyclic alkyl carbonate solution additives were used to form protein ion charge states in very high charge states.9,31 Experimental UVPD cleavage sites and ion abundances for all protein charge states are compared to predicted protonation sites in order to rationalize the impact of charge location and overall protein charge on 213 nm UVPD performance.
Experimental section
Safety considerations
Methanol (MeOH), 4-vinyl-1,3-dioxolan-2-one (vinyl ethylene carbonate, VEC), propylene carbonate (PC) (≥99.7%) and formic acid (FA) were used for protein solutions. Heated electrospray ionization (HESI) uses high voltage and high temperatures. The high voltage area and hot surfaces were shielded by the source. Lasers in the UV range are dangerous for the human eye. To avoid danger, laser safety goggles were worn all the time and a laser safety officer examined the instrument.Materials
Ubiquitin from bovine erythrocytes (≥98%), cytochrome c from bovine heart (≥95%), myoglobin from equine skeletal muscle (≥95%), propylene carbonate (PC) (≥99.7%), 4-vinyl-1,3-dioxolan-2-one (vinyl ethylene carbonate, VEC) (≥99%) were purchased from Sigma-Aldrich (St Louis, MO, USA). Ammonium acetate (NH4Ac) (≥97%) was purchased from Alfa Aesar (Kandel, Germany). Methanol (HPLC grade) was purchased from Merck (Darmstadt, Germany). Water (HPLC grade) was purchased from VWR (Darmstadt, Germany). Formic acid (FA) (∼98%) was purchased from Honeywell Fluka (Seelze, Germany).Sample preparation
Proteins were diluted to final concentrations of 10−4 to 10−8 M. For native MS, 0.2% NH4Ac and 1% FA in water were used as solvent.32 For denaturing solutions, proteins were prepared in 44.5% MeOH, 54.5% water and 1% FA. Protein supercharging was achieved by adding PC (1% to 20%) or VEC (5% to 10%) to the denaturing solutions (Fig. S1†).33,34 The heme associated signal at m/z 616.2 found in the supercharged cytochrome c spectrum is most likely a result of in-source activation of the highly charged ions.Mass spectrometry
All experiments were performed on a 7 T LTQ FT Ultra (Thermo Fisher Scientific, Bremen, Germany), operated with 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 resolution at m/z 400. HESI was used for the ionization and typical parameters were: 3.5 to 5.5 kV spray voltage, 310 °C cone temperature, 2 to 20 μL min−1 flow rate, 4 to 25 (arbitrary units) sheath gas rate, 0 to 5 (arbitrary units) aux gas rate, 0 to 20 (arbitrary units) sweep gas rate. All reported uncertainties are standard deviations of triplicate measurements. For the data evaluation, 120 UVPD spectra were averaged per charge state and protein (Fig. S2†). The influence on the signal-to-noise (S/N) ratio was evaluated by measuring 800 spectra using 100000 resolution at m/z 400 and 300 spectra using 750000 resolution at m/z 400.
000 resolution at m/z 400. HESI was used for the ionization and typical parameters were: 3.5 to 5.5 kV spray voltage, 310 °C cone temperature, 2 to 20 μL min−1 flow rate, 4 to 25 (arbitrary units) sheath gas rate, 0 to 5 (arbitrary units) aux gas rate, 0 to 20 (arbitrary units) sweep gas rate. All reported uncertainties are standard deviations of triplicate measurements. For the data evaluation, 120 UVPD spectra were averaged per charge state and protein (Fig. S2†). The influence on the signal-to-noise (S/N) ratio was evaluated by measuring 800 spectra using 100000 resolution at m/z 400 and 300 spectra using 750000 resolution at m/z 400.
For UVPD experiments a 213 nm laser was installed as described previously (Fig. S3a†).35 Briefly, the fifth harmonic of a solid state Nd:YAG (Tempest, New Wave Research, Portland, USA) was guided in the ICR-cell through a UV-grade fused silica viewport (Hositrad, Hoevelaken, Netherlands). The laser was operated at 20 Hz providing 0.7 mJ per pulse with a 3.5 mm beam diameter. To limit the influence of secondary fragmentation, the dependence of the sequence coverage and the PY on pulses per spectra was investigated (Fig. S4–6†). For all proteins the sequence coverage increases with the number of pulses per spectrum, reaches a maximum and decreases for the highest pulse numbers. The decrease of sequence coverage most likely stems from extensive secondary fragmentation. To limit the impact of secondary fragments but also to obtain reasonably high sequence coverage, the pulse number at which the sequence coverage reaches a maximum (ubiquitin = 40 pulses per spectrum Fig. S4,† cytochrome c = 10 pulses per spectrum Fig. S5,† myoglobin = 20 pulses per spectrum Fig. S6†) was used. Precursor ions were isolated using Δm/z = ± 10 to isolate the complete isotopic distribution of every charge state. Holo-myoglobin was fragmented in the native charge state 9+, all other experiments were carried out on apo-myoglobin.
To minimize the influence of the precursor signal intensity on UVPD results, the number of ions in the ICR-cell was controlled by adjusting the automatic gain control target, protein concentrations and HESI parameters. Stability of the spray conditions were checked by monitoring precursor signal intensity over 40 spectra (20 before and 20 after the UVPD measurement). The normalized level (NL) of the trapped ions was adjusted from 8 × 103 to 7 × 104 (Fig. S7†).
Data analysis
Data evaluation of UVPD-MS2 data was performed using a custom in-house MATLAB algorithm. The Roepstorff nomenclature is used to describe peptide fragment ions.36 Details of the algorithm are described in the ESI (Fig. S8†). The signal-to-noise (S/N) level was evaluated manually for each measurement. The S/N ratio used for data analysis was set to 3. All isotopes with a theoretical abundance of 65% relative to the most abundant isotope need to be present for signal annotation. The maximum absolute deviation was set to 10 ppm. PTMs, H2O or NH3 loss and multiply cleaved fragments were not taken into account. Only fragments meeting all these requirements were used for the sequence coverage and PY calculations. Fragment ion intensities were normalized such that the sum of all fragment intensities is 1. The efficiency of dissociation is represented by the product yield (PY), which is defined as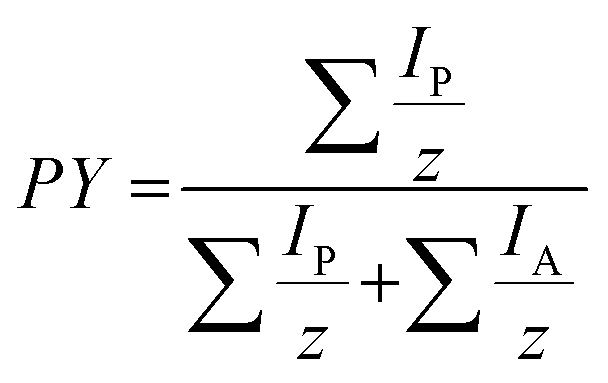 | (1) |
Protonation of the proteins was investigated theoretically by calculating the frequency of protonation at different amino acid residues using the freeware PredictPrPlus,8 which is based on the method reported by Williams and co-workers.37 Specifically, the energies of proton configurations were calculated by assuming the proteins are in highly elongated 1D strings, treating each amino acid residue as a node with an intrinsic gas-phase basicity and accounting for coulombic repulsion between charge sites. Our model does not adequately describe the distributions of protons for native-like protein ion structures. Therefore, linking fragmentation patterns to native-like protein ion structures should be avoided. The energies are then optimized using a Monte Carlo-type ‘pseudo-random walk’ method.37 This approach can be used to predict the configuration of ionizing protons on an elongated protein, thereby estimate the measured basicity values16 and collision-induced cleavage sites of intact protein ions.8 Here, PredictPrPlus was modified to also calculate and output the electrostatic potential experienced by a ‘probe’ charge placed at neutral backbone amide moieties as a function of the residue number, and the electrostatic repulsion between fragment ions at a given cleavage site prior to fragmentation as a function of amino acid residue number. Input parameters used for the calculations are given in Table S1.†
Results and discussion
Effects of MS parameters on UVPD results
To probe the intrinsic propensity of proteins to fragment upon 213 nm UVPD, the effects of experimental MS parameters on the number, intensity and identity of fragment ions were investigated. Firstly, the background pressure can impact UVPD results.38 Here, we assume that gas-protein ion collisions during the trapping time of at most 2 s are negligible at a background pressure of ∼2 × 10−10 mbar in the vacuum chamber surrounding the FT-ICR cell.39Four further factors can influence the PY and S/N of UVPD results. These are: (a) number of trapped ions, (b) the mass spectrometric performance settings (e.g. resolution and scan number), (c) the number of averaged spectra and (d) the number of laser pulses per spectrum.
(a) The influence of the number of trapped ions on the UVPD results was investigated by monitoring the sequence coverage as a function of precursor ion abundance. A representative example of a plot of sequence coverage vs. precursor ion abundance for [ubiquitin + 13H]13+ is shown in Fig. S9.† The sequence coverage increases with increasing precursor abundance. However, ion abundances cannot be necessarily readily tuned for each charge state as the detected intensity of protein charge states is determined by the spray solution composition as well as the ESI process.40–42 To reduce the influence of the precursor ion abundance between different protein charge states, precursor abundances were adjusted to 8 × 103–7 × 104 (Fig. S7†) by tuning the concentration and ion collection time, which was the values typically encountered for the lowest intensity protein charge states in all experiments.
(b) Because fragment ions can potentially overlap in m/z in top-down tandem MS, increasing the mass resolution will ultimately yield higher sequence coverage values compared to lower mass resolution settings. This is also true for our UVPD results as demonstrated in Fig. S10.† Compromising between measurement time and resolution all experiments were performed at mass resolution settings of 100000 at m/z 400.
(c) The number of averaged spectra also influences the sequence coverage by affecting S/N levels. As expected, the noise decreases when increasing the number of averaged spectra (Fig. S2†), which increases the experiment time. To compromise between increased sequence coverage and measurement time, 120 spectra were averaged in all experiments.
(d) Another experimental parameter that influences fragment ion S/N levels is the number of laser pulses per spectrum. The influence of the number of laser pulses on the sequence coverage was studied for [ubiquitin + 13H]13+, [cytochrome c + 19H]19+ and [myoglobin c + 21H]21+, and the results are shown in Fig. S4–6.† Initially the sequence coverage increases when more laser pulses are used, hits a maximum and starts decreasing for the largest laser pulse numbers. But PY values stay mostly within the error margin of the experiments for laser pulse numbers above 10 (or 500 ms of irradiation time). The decrease of sequence coverage with increased number of laser pulses is likely caused by extensive secondary fragmentation of fragment ions. Laser pulse numbers with the maximum sequence coverage were selected and used throughout this study (40, 10 and 20 shots per spectrum for ubiquitin, cytochrome c and myoglobin, respectively). The constant PY for high laser pulse numbers also indicates that the influence of a kinetic shift on the PY and sequence coverage values is negligible under our experimental conditions. This is because increasing the laser pulse number goes along with an increased time for irradiated proteins to fragment until ion detection. If a kinetic shift would prevent fragmentation prior to ion detection, PY values should continuously increase with increasing laser pulse number.
Overall, the selected settings enabled the influence of experimental and instrumental parameters on the UVPD results to be minimized as the aim was to exclusively probe the intrinsic UVPD response of protein ions as a function of charge state. However, these settings are not intended to maximize the sequence coverage. If sequence coverage is near 100%, then any effects of charge state on the UVPD of protein ions will be more challenging to determine.
Influence of charge state on UVPD photoproduct yield
The PY of proteins subjected to tandem MS methods, such as CID or ETD, is known to be affected by many factors including primary, secondary, tertiary and quaternary protein structures as well as protein charge state.8,43,44 In order to investigate the dependence of the PY on these factors in 213 nm UVPD, ubiquitin, myoglobin and cytochrome c were sprayed fragmented and analyzed from native, denaturing and supercharging solutions.The resulting PYs as a function of charge state are shown in Fig. 1. Charge states analyzed from native, denaturing and unfolded solutions are color-coded yellow, blue and red, respectively.45 The PY of all proteins increases with increasing charge states. A linear fit of PY values as a function of precursor ion charge state results in moderate to good coefficients of determination. In particular, R2 values are 0.70 for ubiquitin (circles), 0.90 for cytochrome c (triangles) and 0.84 for myoglobin (crosses). Absolute deviations to the linear regression are at most ± 0.25 for all three proteins.
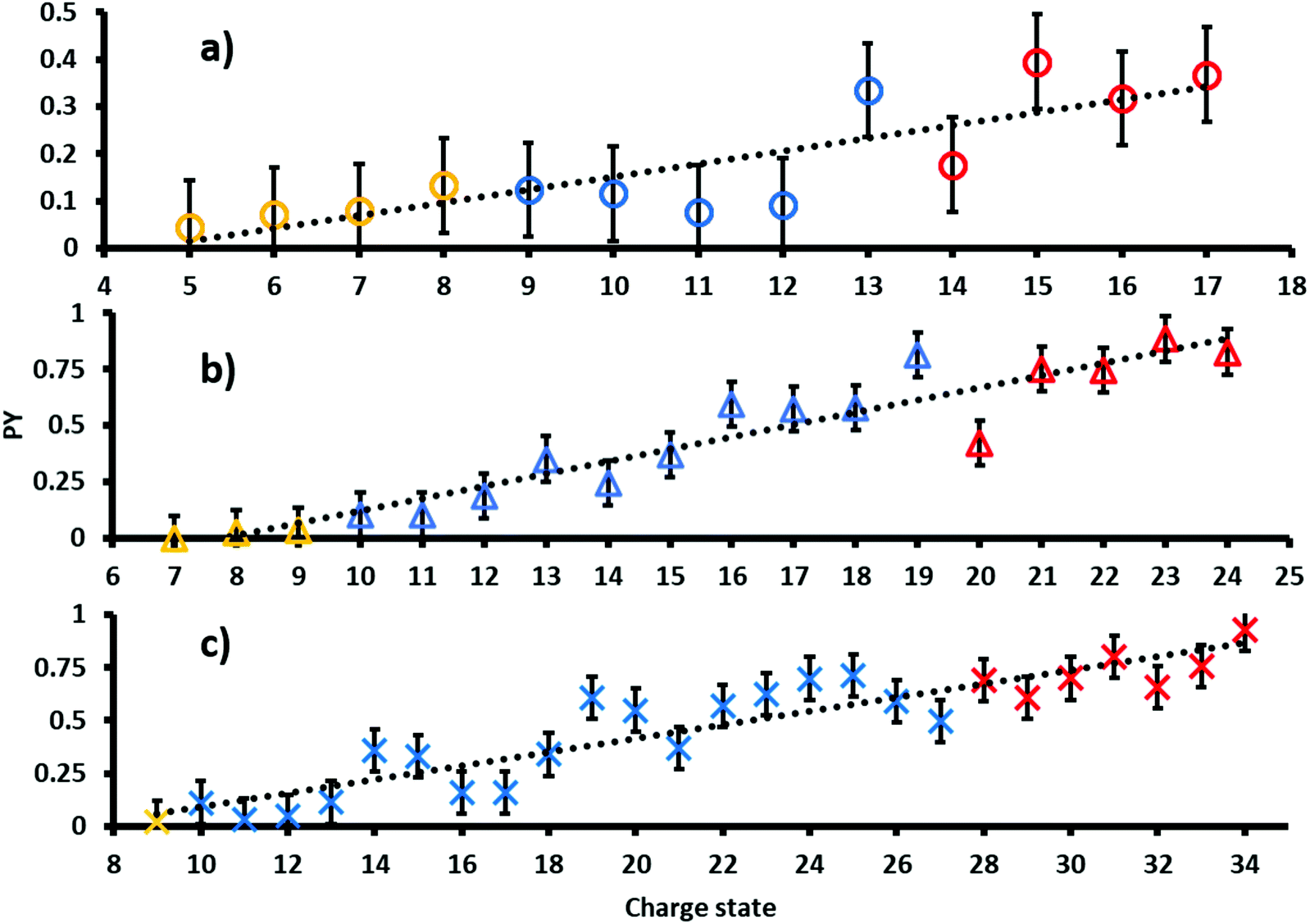 | ||
| Fig. 1 Photoproduct yields for (a) ubiquitin, (b) cytochrome c and (c) myoglobin ions as a function of precursors charge state. Proteins formed from native, denaturing and supercharging solutions are color-coded yellow, blue and red, respectively. As a guide to the eye linear trend lines are shown in black. | ||
The collision cross sections (CCS) increase with protein charge state. However, the extent of the increase in CCS per charge state depends on ESI spray solution conditions and typically exhibits characteristic “jumps” in the slope of CCS vs. charge between charge states obtained from different solution conditions. For example, the CCS values in He for cytochrome c increases by ∼200 Å2 per charge for ions generated from native solutions, whereas that for denaturing and supercharging solutions increases by only ∼100 Å2 and ∼50 Å2 per charge respectively.46 Because the PY values follow a largely linear trend for all precursor charge states, including charge states formed via different solution conditions, including presence of holo- (native MS) and apo-myoglobin (denaturing and supercharging), these results suggest that protein structure and associated structural changes do not influence the overall UVPD PY as strongly as CCS values under these conditions. Therefore, our data implies that the most important factor affecting the magnitude of PYs is the overall charge state. Potentially, coulombic repulsion and the location of protons can more strongly affect the efficiency of fragmentation in 213 nm protein UVPD. These data are consistent with results reported by Kolbowski et al. who used 213 nm UVPD to identify the peptide charge density as driving force for peptide backbone cleavage.43
Sequence coverage as a function of charge state
In contrast to the PY, the sequence coverage does not necessarily increase when fragment ion intensities increase. For example, multiple fragment ions originating from cleavage between the same adjacent residues (e.g., detecting complementary a/x ions only vs. a/x, b/y, and c/z) do not increase the sequence coverage, whereas the PY is increased. The increase in sequence coverage for ubiquitin, cytochrome c and myoglobin is plotted vs. the protein charge state in Fig. 2. For all three proteins, the UVPD sequence coverage under our experimental conditions for the lower charge states is below 10%, increases with approximately 5% per charge state as the charge state increases, and then plateaus at a maximum of 50–60%. This non-linear increase in the sequence coverage with an increase in the charge state directly contrasts with previous top-down CID results.8 In CID, the sequence coverage first increases as protein charge states increase, and then drops significantly for higher protein charge states. Therefore, the charge state of protein ions influences the sequence coverage in 213 nm UVPD in a substantially different manner than in CID experiments for the same proteins and charge states.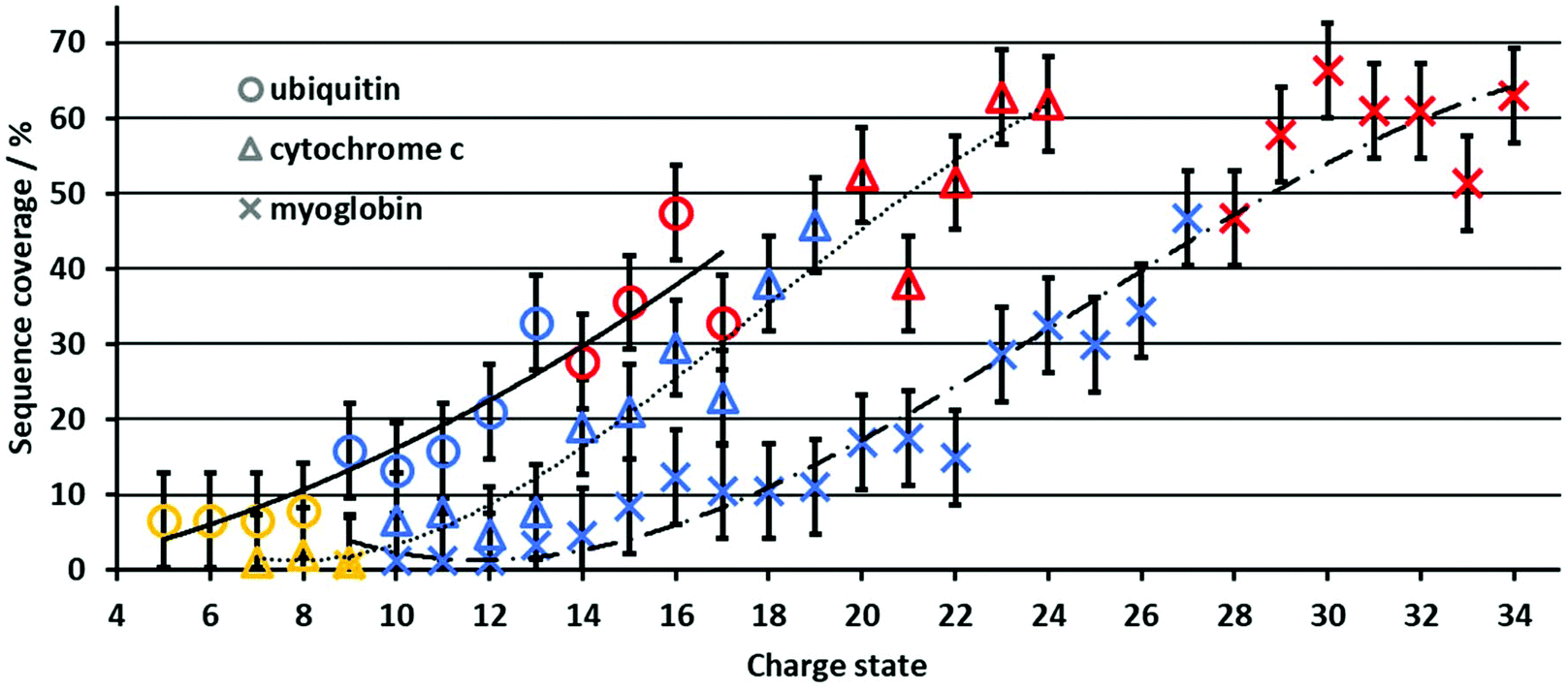 | ||
| Fig. 2 Sequence coverage for ubiquitin, cytochrome c and myoglobin ions as a function of precursor charge state. Ubiquitin, cytochrome c and myoglobin are represented by circles, triangles and crosses, respectively. Proteins formed from native, denaturing and supercharging solutions are colored yellow, blue and red, respectively. The black lines serve as a guide to the eye. | ||
Although the size and overall charge state of ubiquitin, cytochrome c and myoglobin are different, the increase in the sequence coverage with an increase in charge state follows a similar trend for all three proteins (Fig. 2). This implies that the increase of the sequence coverage in 213 nm UVPD with charge state is not substantially affected by the protein identity, size or overall charge but by the protein ion charge with respect to the protein size, i.e. the number of charges per amino acid. Therefore, Fig. 3 shows the sequence coverage as a function of charges per amino acids. The maximum charge states of the three proteins are 17+, 24+, 34+ for ubiquitin, cytochrome c and myoglobin, respectively. The charges per amino acid for all three proteins are in the range of 0.06 to 0.22.
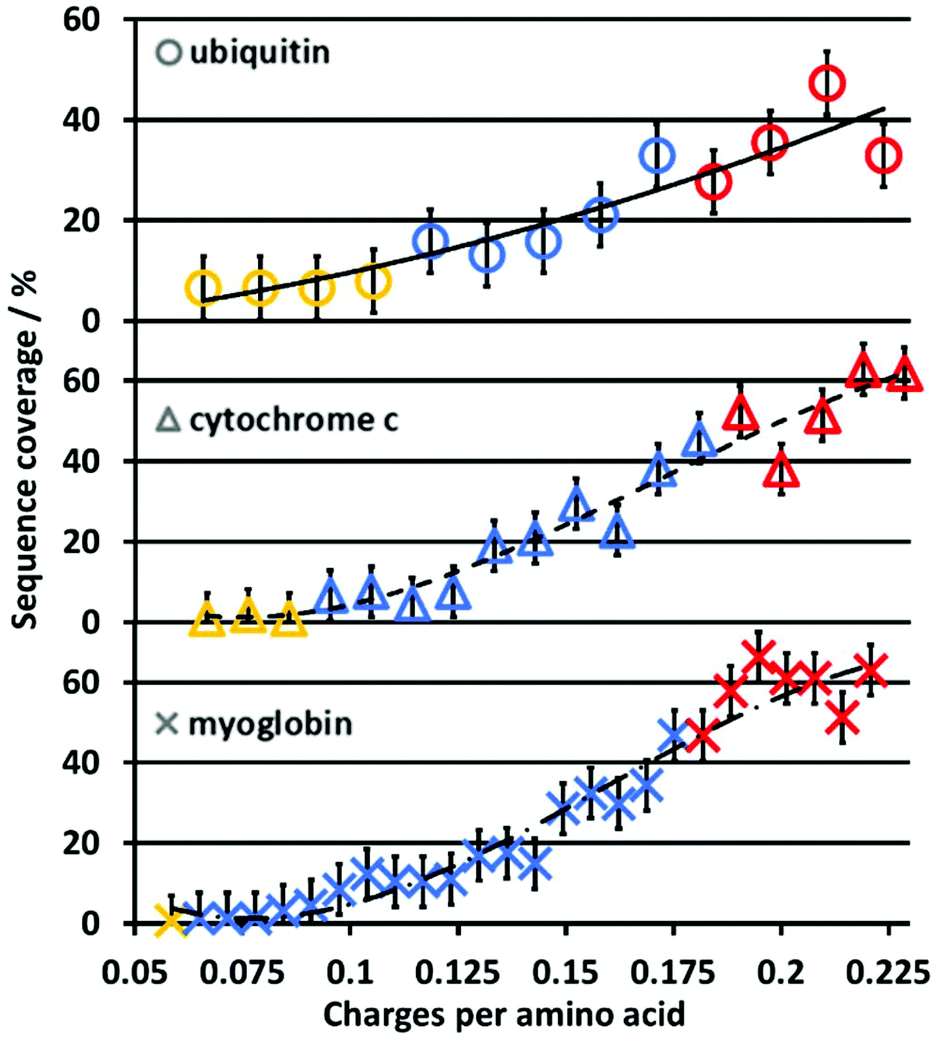 | ||
| Fig. 3 Sequence coverage for ubiquitin, cytochrome c and myoglobin ions as a function of charges per amino acid residues. Ubiquitin, cytochrome c and myoglobin are represented by circles, triangles and crosses, respectively. Proteins formed from native, denaturing and supercharging solutions are colored yellow, blue and red, respectively. The black lines serve as a guide to the eye. | ||
The slope corresponding to the change in sequence coverage vs. an increase in charge density for all three proteins differs in the ranges of 0.06 to 0.11, 0.11 to 0.18 and 0.18 to 0.22 charges per residue. In the range of 0.06 to 0.11 there is essentially no increase in sequence coverage. From 0.11 to 0.18 the sequence coverage increases dramatically by 200–600% for ubiquitin, cytochromc c and myoglobin, respectively. In contrast, from 0.18 to 0.22 charges per residue, there is virtually no change in sequence coverage. The general trend is very similar for all three proteins. This suggests that the most important descriptor for the change of sequence coverage with charge state is the number of charges per amino acid.
Increasing the number of charges per amino acid of the protein has multiple effects. The distance between charges decreases on average and the repulsive Coulomb force between charge sites increases substantially. Moreover, at moderate to high charge states, nearly all basic residues should become protonated resulting in the protonation of the amide backbone at higher charge states.8,47
Influence of the charge state on fragment ion identities
We next investigated the type of fragment ions formed in 213 nm UVPD as a function of charge state. In Fig. 4, the normalized number of each fragment ion type formed upon UVPD of ubiquitin 5+ to 17+ is shown. Fragments with masses −1 Da and +1 Da were included for each ion type, corresponding to hydrogen-transfer reactions that can occur as fragment ions separate. Data for myoglobin and cytochrome c are provided in the ESI (Fig. S11 and 12†). Due to the limited sequence coverage obtained under our experimental conditions for low ion charge states, only a few fragments are present for charge states <9+. For charge states >9+, y-fragments are most abundant and for high charge states (>15+), all fragment types are formed in very similar abundances. The dominance of y ions for medium charge states is in line with studies by Dugourd and co-workers, who found y-fragments were the most prominent in 213 nm top-down UVPD of ubiquitin in an orbitrap instrument.48 The dominance of y-type fragments is even more abundant in our data. This could be an effect of the trap pressure. While formation of b- and y-fragments is suppressed by collisional cooling in high pressure regimes, the b- and y-ion intensities increase at lower pressures.38 These data are consistent with enhanced intramolecular vibrational relaxation from electronically excited states in the ultrahigh vacuum of the FT-ICR cell, resulting in the formation of relatively higher abundant y-ions, which are usually a distinctive sequence ion in top-down CID experiments.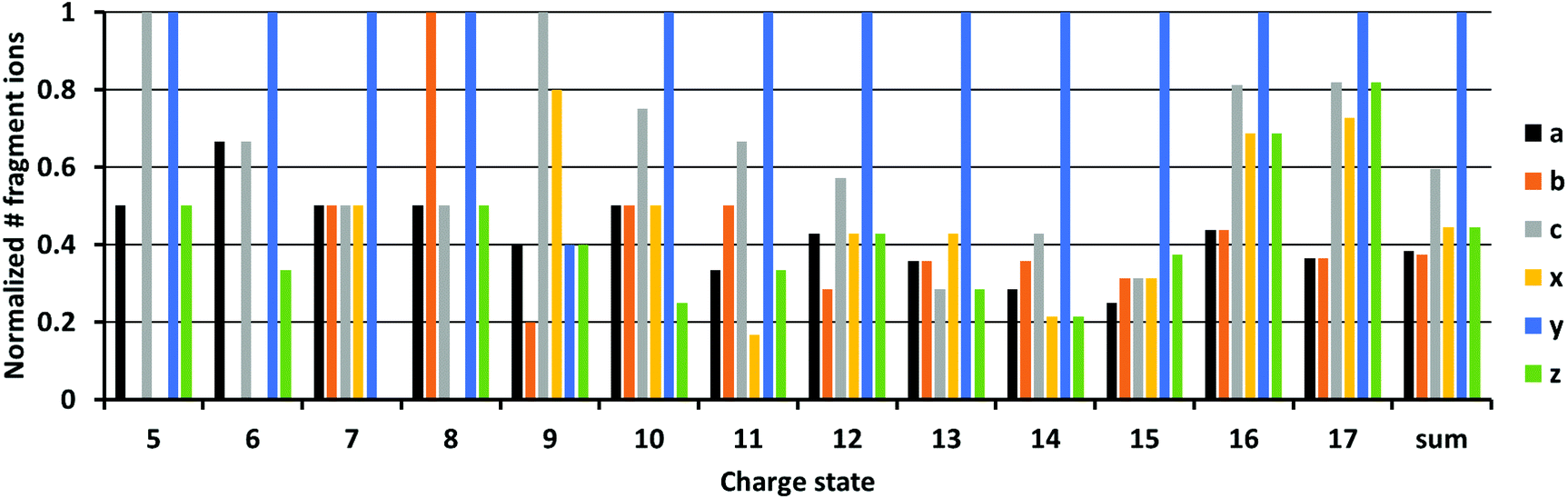 | ||
| Fig. 4 Normalized number of fragment ions of ubiquitin charge states 5+ to 17+. Fragment ions of a, b, c, x, y, z are shown in black, orange, grey, yellow, blue and green, respectively. Fragments with masses of −1 and +1 are included. | ||
The relative abundances of a, b, c, x, y and z fragments did not change substantially from the 8+ to 13+ charge states. Relative fragment type abundances were also reasonably constant up to charge state 17+. However, the difference in abundance between y-ions and all other ion types decreased for the 16+ and 17+ charge states. Results obtained for myoglobin and cytochrome c differ to ubiquitin (Fig. S11 and 12†). However, no trend for the fragment types is found for increasing charge states. Overall, these results indicate that the charge state of protein ions does not influence the relative abundance of 213 nm UVPD derived fragment ion types to a large extent, unlike all other parameters, such as PY and sequence coverage that depended strongly on the protein ion charge state.
Correlating fragmentation sites to charge carrier positions
In order to rationalize the impact of charge on top-down UVPD results, charge carrier positions and electrostatic potentials were predicted using PredictPrPlus and compared to experimental results. Corresponding mass spectrometric fragment ion abundances (black bars), electrostatic potentials (white squares connected by a green line), protonation frequencies of side chains (blue bars) and the amide backbone (orange bars) are shown in Fig. 5 for [ubiquitin + 12H]12+ and [ubiquitin + 17H]17+. Results for all charge states of ubiquitin, cytochrome c and myoglobin are included in the SI (Fig. S13–15†). As the proton position of native-like protein ions are not accurately predicted, only fragment ions of unfolded charge states are quantitively compared to model predictions.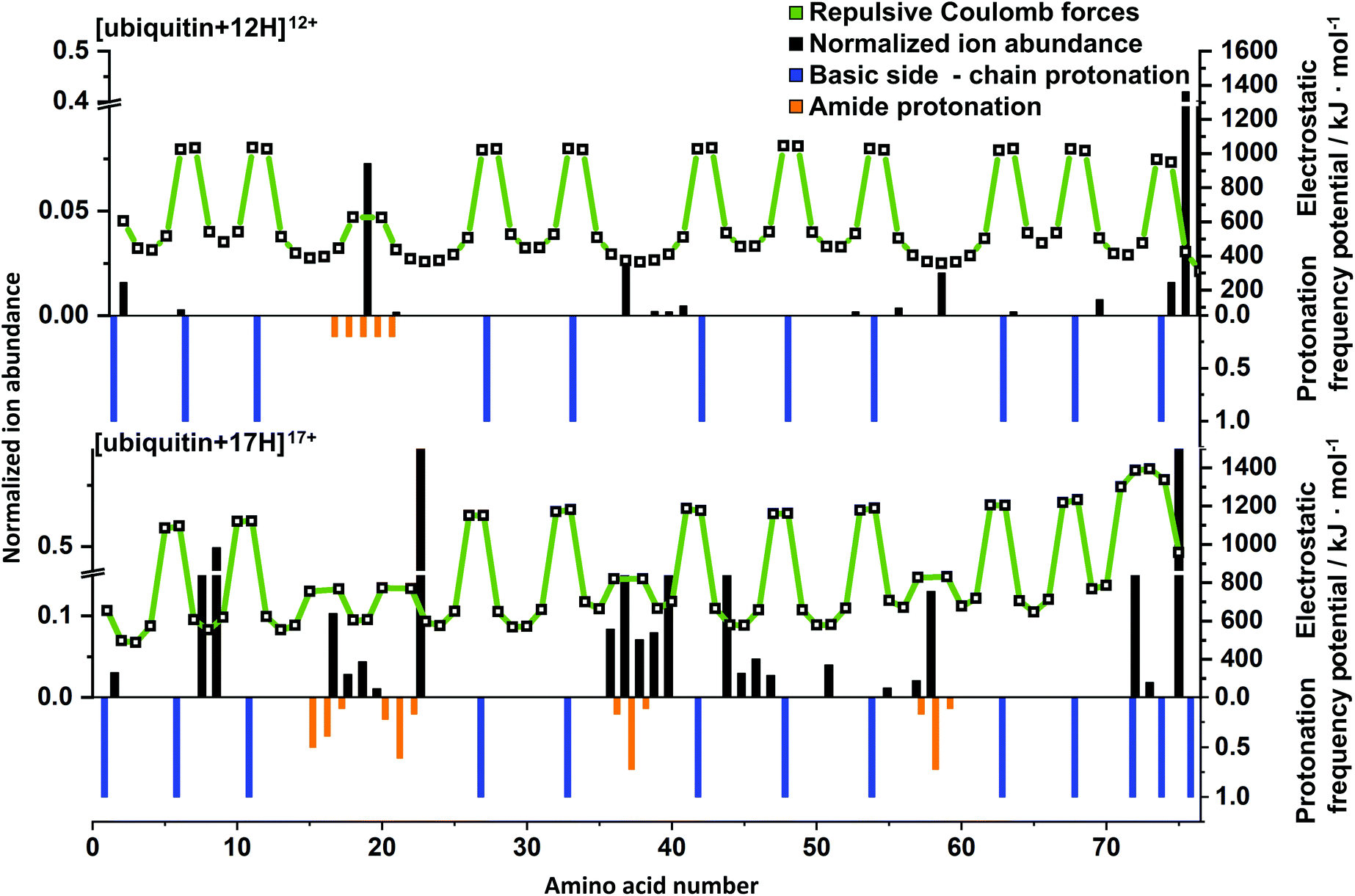 | ||
| Fig. 5 Normalized experimental fragment ion abundances are shown as black bars. The calculated electrostatic potential at each residue is shown with white squares, which are connected with green lines to guide to the eye. Predicted protonation frequencies at basic side-chain amino acid residue (or N/C-terminus) and the amide backbone are shown in blue and orange bars, respectively. | ||
[ubiquitin + 12H]12+ is the first charge state of ubiquitin for which the number of protons is predicted to exceed the number of high-basicity amino acid moieties. For the UVPD of this ion, the experimental fragment ions are dominated by bond cleavage at Glu18 (Fig. 5, top). Comparison to the predicted protonation sites reveals that the excess proton that is predicted to be located at a low basicity amide backbone site is likely to be located between Glu16 and Ser20. This suggests that amide bond cleavage correlates with amide backbone protonation. This is also in line with results for [ubiquitin + 17H]17+ which contains four protons that are predicted to protonate the amide backbone (Fig. 5, bottom). These protonation sites (i.e. two protons between Leu15 and Thr22, one between Ile36 and Pro38 and one between Ser57 and Tyr59) correlate to experimental cleavage sites that result in high fragment ion intensities. This suggests that the presence of a charge on the amide backbone can facilitate and direct protein ion fragmentation, consistent with a charge-directed fragmentation event.30,49,50
In contrast, some fragment ions for [ubiquitin + 12H]12+, [ubiquitin + 17H]17+ and also other ubiquitin charge states (Fig. S13†) are not associated with protonation of the amide backbone. For example, fragments close to Ile36 – Asp39, Asp52, Thr55 in [ubiquitin + 12H]12+ as well as Leu8, Thr9 and Ile44 to Gly47 in [ubiquitin + 17H]17+ do not coincide with the presence of amide bond protonation. However, the location of these fragment ions in the amino acid sequence correlates with local or global minima of the Coulomb potential. In [ubiquitin + 17H]17+ fragmentation between Ile44 to Gly47 would lead to most equally charged 9+ and 8+ fragments, resulting in a maximal decrease of intermolecular electrostatic potentials, (Fig. S16†) which is represented by high fragmentation yields between Ile36 to Asp39 and Ile44 to Gly47 for [ubiquitin + 17H]17+. The correlation of these fragment ions with a decrease of electrostatic potentials suggests that formation of these fragmentation ions is facilitated by maximizing the Coulomb energy decrease due to fragment ion formation. As formation of these fragment ions are not directly affected by the presence of charges but indirectly by remote influence of the Coulomb potential, the corresponding fragment ions can be classified as charge-remote fragment ions. Similar trends were observed for charge dependent fragment ions of myoglobin and cytochrome c (Fig. S14 and S15†). For example, [cytochrome c + 21H]22+ has intense signals at Thr29, Gly35 – Phe37, Lys56 and Ser48, Met66 – Tyr68, Leu95 which coincide with minima of the electrostatic potential and protonation of the amide backbone, respectively. On the other hand, fragmentation at Lys14 and Ile76 does not correspond to charge-directed or charge-remote fragmentation. Therefore, all investigated protein ions contain between 10–15% of fragment ions not in line with the simplistic dissociation scenarios discussed above. However, the large majority of the fragments formed upon 213 nm are in agreement with the charge remote or charge directed fragmentation pathways. To explain the remaining 213 nm fragment ions, more future research is needed.
In CID experiments fragmentation at amino acid Glu18 is highly selective for the 12+.8 Donald and co-workers attributed this selectivity to protonation of the amide bond of the first low basicity amino acid residue that is protonated with increasing charge (Glu18) based on mixed QM/MM modelling, energy resolved CID experiments, and electrostatic calculations. Protonation of the amide bond was calculated to reduce the dissociation barrier by 50 kJ mol−1 in charge-induced vs. charge-remote fragmentation mechanisms. In contrast to CID, 213 nm UVPD experiments of the 12+ ubiquitin yields additional fragments at Ile36 to Gln40 and Asp52 reducing the selectivity of the formation of Glu18 associated fragment ions in 213 nm UVPD compared to CID.
Differences in the selectivity of Glu18 associated ions between CID and UVPD most likely arise due to excitation and energy redistribution events characteristic to the excitation methods. During CID, molecules are heated by less than 10 kJ mol−1 per collision, followed by redistribution of the excess energy to the vibrational heat bath of the protein ion.51 This ultimately leads to dynamical sampling of accessible protein conformers by the CID-heated ions, preferentially accessing fragmentation pathways with the lowest dissociation barriers. Hence, fragmentation preferentially occurs for the energetically weakest bond.
In contrast, 213 nm UVPD introduces 562 kJ mol−1 per photon to activate ions. While some degree of energy redistribution will still occur, the energy introduced by UV photons can be sufficient to directly cleave amide bonds of approximately 335 kJ mol−1 or less (depending on Coulomb repulsion) prior to or after only partial energy redistribution or photon emission in gas-phase fluoresence.52,53 Therefore, 213 nm UVPD mass spectra should contain fragment ions also observed in CID experiments (particularly in ultrahigh vacuum with limited collisional cooling) but also additional fragments from direct bond cleavage. This is in line with our observation for ubiquitin, cytochrome c and myoglobin hinting to fragment ions arising due to charge-remote (side chain protonation) and charge-induced dissociation (amide protonation) in 213 nm UVPD.
Conclusions
In this work we investigated the effect of the overall protein ion charge state on the 213 UV photodissociation of native-like, unfolded and supercharged charge states of ubiquitin, cytochrome c and myoglobin in terms of the extent of fragmentation, the type of fragment ions that are formed, and fragmentation sites. The goal was not to explore the limits of sequence coverage in top-down UVPD measurements but to control the experimental conditions in order to systematically probe the intrinsic UVPD fragmentation of protein ions as a function of protein charge state. Our data strongly indicates that the photoproduct yields and sequence coverages increase as the charge state increases. The major factor that impacts top-down 213 nm UVPD fragmentation of protein ions is the overall protein charge state, and the effects of secondary/tertiary structural changes of these protein ions are less pronounced under these conditions. The extent of protein ion charging correlates with the sequence coverage and the PY value but the relative number of different fragment ion types that are formed is largely independent of protein charge state. In particular, the relative sequence coverage increase depends more strongly on the number of charges per amino acid residues than on the masses or identity of the proteins. For unfolded and supercharged charge states these results are consistent with a major driving force in the dissociation of intact protein ions by 213 nm top-down UVPD being the Coulomb repulsion between individual localized charges in these multiply charged protein ions.Comparison of the protein cleavage sites and abundances to predicted charge locations and the resulting calculated electrostatic potentials reveal that the majority of cleavage sites are consistent with two mechanisms. For protein ions in which the proton number does not exceed the number of high-basicity residues in which protons are located primarily on high-basicity amino acid residue side chains, the majority of fragment ions stem from sites of local coulombic minima between adjacent charges. Unlike, fragmentation in ECD and ETD in which fragmentation sites are directed by the location of protons, the majority of fragments in our UVPD results arise at sites with a relatively large distance between predicted protonation sites thereby minimizing coulombic repulsion upon dissociation.34 For some supercharged protein ions the number of protons exceed the number of basic sites and protonation of the amide backbone is predicted. For these highly charged protein ions, cleavage sites are not only determined by the Coulomb repulsion but also by amide protonation as high sequence ion abundance is observed in the same region in which amide backbone protonation is predicted. This, however, is in marked contrast to top-down CID results for the same protein ion, which is dominated by highly selective fragment ions formed typically at the first amide backbone site that is predicted to be protonated with increasing charge state.8
In contrast to our results, another study has investigated the protein charge state dependence in 213 nm top-down UVPD experiments on an orbitrap instrument and found that sequence coverage does not depend on the charge density of highly charged protein ions.29,48 A potential explanation for these differences is the pressure regimes in which UVPD is performed. In our FT-ICR MS, pressures of 2 × 10−10 mbar minimizes ion-neutral collisions during UVPD experiments, whereas frequent deactivating collision at about 10−5 mbar could minimize fragmentation arising from vibrational excitation owing to intramolecular vibrational relaxation from electronically excited states. Such an effect of pressure has been studied by Hao et al. who demonstrated that the intensities of UVPD-like fragments can decrease at lower pressures.38
Thus, these results can be considered a first step towards rationalizing fragmentation sites in top-down 213 nm UVPD as a function of charge state but more work is required to understand the differences between different UVPD wavelength, the impact of the wavelengths on sequence coverage results, influences of locally different UV absorption cross sections, and additional effects of experimental conditions on the UVPD charge-state dependence.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
S.H. acknowledges the Fonds der chemischen Industrie for financial support through a Liebig fellowship. The authors thank Alexander S. Berninger for initial support during this project and Marion Girod for providing their UVPD data. W.A.D. thanks the Australian Research Council for funding (FT200100798).References
- C. Xia, Y. Tao, M. Li, T. Che and J. Qu, Exp. Ther. Med., 2020, 20, 2923–2940 CrossRef CAS PubMed.
- M. Fournier, M. Orpinell, C. Grauffel, E. Scheer, J.-M. Garnier, T. Ye, V. Chavant, M. Joint, F. Esashi, A. Dejaegere, P. Gönczy and L. Tora, Nat. Commun., 2016, 7, 13227 CrossRef CAS PubMed.
- S. K. Wierenga, M. J. Zocher, M. M. Mirus, T. P. Conrads, M. B. Goshe and T. D. Veenstra, Rapid. Commun. Mass Spectrom., 2002, 16, 1404–1408 CrossRef CAS PubMed.
- S. Hildonen, T. G. Halvorsen and L. Reubsaet, Proteomics, 2014, 14, 2031–2041 CrossRef CAS PubMed.
- S. Doll and A. L. Burlingame, ACS Chem. Biol., 2015, 10, 63–71 CrossRef CAS PubMed.
- S. C. Beu, M. W. Senko, J. P. Quinn, F. M. Wampler III and F. W. McLafferty, J. Am. Soc. Mass Spectrom., 1993, 4, 557–565 CrossRef CAS PubMed.
- W. Cai, H. Guner, Z. R. Gregorich, A. J. Chen, S. Ayaz-Guner, Y. Peng, S. G. Valeja, X. Liu and Y. Ge, MCP, 2016, 15, 703–714 CAS.
- H. Wang, M. G. Leeming, J. Ho and W. A. Donald, Chem. – Eur. J., 2019, 25, 823–834 CrossRef CAS PubMed.
- H. Wang, G. Yong, S. L. Brown, H. E. Lee, M. A. Zenaidee, C. T. Supuran and W. A. Donald, Anal. Chim. Acta, 2018, 1003, 1–9 CrossRef CAS PubMed.
- J. F. Nemeth-Cawley, S. Karnik and J. C. Rouse, J. Mass Spectrom., 2001, 36, 1301–1311 CrossRef CAS PubMed.
- N. I. Brodie, R. Huguet, T. Zhang, R. Viner, V. Zabrouskov, J. Pan, E. V. Petrotchenko and C. H. Borchers, Anal. Chem., 2018, 90, 3079–3082 CrossRef CAS PubMed.
- N. C. Polfer, Chem. Soc. Rev., 2011, 40, 2211–2221 RSC.
- D. P. Little, J. P. Speir, M. W. Senko, P. B. O'Connor and F. W. McLafferty, Anal. Chem., 1994, 66, 2809–2815 CrossRef CAS PubMed.
- J. J. Coon, J. Shabanowitz, D. F. Hunt and J. E. P. Syka, J. Am. Soc. Mass Spectrom., 2005, 16, 880–882 CrossRef CAS PubMed.
- H. Molina, R. Matthiesen, K. Kandasamy and A. Pandey, Anal. Chem., 2008, 80, 4825–4835 CrossRef CAS PubMed.
- M. A. Zenaidee, M. G. Leeming, F. Zhang, T. T. Funston and W. A. Donald, Angew. Chem., Int. Ed., 2017, 56, 8522–8526 CrossRef CAS PubMed.
- J. B. Shaw, W. Li, D. D. Holden, Y. Zhang, J. Griep-Raming, R. T. Fellers, B. P. Early, P. M. Thomas, N. L. Kelleher and J. S. Brodbelt, J. Am. Chem. Soc., 2013, 135, 12646–12651 CrossRef CAS PubMed.
- J. A. Madsen, D. R. Boutz and J. S. Brodbelt, J. Proteome Res., 2010, 9, 4205–4214 CrossRef CAS PubMed.
- T. Ly and R. R. Julian, Angew. Chem., Int. Ed., 2009, 48, 7130–7137 CrossRef CAS PubMed.
- J. S. Brodbelt, L. J. Morrison and I. Santos, Chem. Rev., 2020, 120, 3328–3380 CrossRef CAS PubMed.
- J. Bonner, Y. A. Lyon, C. Nellessen and R. R. Julian, J. Am. Soc. Mass Spectrom., 2017, 139, 10286–10293 CAS.
- M. B. Cammarata and J. S. Brodbelt, Chem. Sci., 2015, 6, 1324–1333 RSC.
- W. Cui, M. S. Thompson and J. P. Reilly, J. Am. Soc. Mass Spectrom., 2005, 16, 1384–1398 CrossRef CAS PubMed.
- M. S. Thompson, W. Cui and J. P. Reilly, Angew. Chem., Int. Ed., 2004, 43, 4791–4794 CAS.
- S. Becher, P. Esch and S. Heiles, Anal. Chem., 2018, 90, 11486–11494 CrossRef CAS PubMed.
- J. Bonner, L. E. Talbert, N. Akkawi and R. R. Julian, Analyst, 2018, 143, 5176–5184 RSC.
- A. Y. Pereverzev, Z. Koczor-Benda, E. Saparbaev, V. N. Kopysov, E. Rosta and O. V. Boyarkin, J. Phys. Chem. Lett., 2020, 11, 206–209 CrossRef CAS PubMed.
- A. Chakir, G. Solignac, A. Mellouki and D. Daumont, Chem. Phys. Lett., 2005, 404, 74–78 CrossRef CAS.
- L. Fornelli, K. Srzentić, T. K. Toby, P. F. Doubleday, R. Huguet, C. Mullen, R. D. Melani, H. Dos Santos Seckler, C. J. DeHart, C. R. Weisbrod, K. R. Durbin, J. B. Greer, B. P. Early, R. T. Fellers, V. Zabrouskov, P. M. Thomas, P. D. Compton and N. L. Kelleher, Mol. Cell. Proteomics, 2020, 19, 405–420 CrossRef CAS PubMed.
- M. Girod, Z. Sanader, M. Vojkovic, R. Antoine, L. MacAleese, J. Lemoine, V. Bonacic-Koutecky and P. Dugourd, J. Am. Soc. Mass Spectrom., 2015, 26, 432–443 CrossRef CAS PubMed.
- E. D. B. Foley, M. A. Zenaidee, R. F. Tabor, J. Ho, J. E. Beves and W. A. Donald, Anal. Chim. Acta, 2019, 1, 100004 CrossRef CAS PubMed.
- A. Theisen, R. Black, D. Corinti, J. M. Brown, B. Bellina and P. E. Barran, J. Am. Soc. Mass Spectrom., 2019, 30, 24–33 CrossRef CAS PubMed.
- C. A. Teo and W. A. Donald, Anal. Chem., 2014, 86, 4455–4462 CrossRef CAS PubMed.
- M. A. Zenaidee and W. A. Donald, Anal. Methods, 2015, 7, 7132–7139 RSC.
- S. Becher, B. Spengler and S. Heiles, Eur. J. Mass. Spectrom., 2018, 24, 54–65 CrossRef CAS PubMed.
- P. Roepstorff and J. Fohlman, Biomed. Mass Spectrom., 1984, 11, 601 CrossRef CAS PubMed.
- P. D. Schnier, D. S. Gross and E. R. Williams, J. Am. Soc. Mass Spectrom., 1995, 6, 1086–1097 CrossRef CAS PubMed.
- C. Hao, J. C. Y. Le Blanc, U. H. Verkerk, K. W. M. Siu and A. V. Loboda, Rapid Commun. Mass Spectrom., 2010, 24, 2262–2268 CrossRef CAS PubMed.
- A. V. Tolmachev, E. W. Robinson, S. Wu, L. Paša-Tolić and R. D. Smith, Int. J. Mass Spectrom., 2009, 281, 32–38 CrossRef PubMed.
- S. H. Lomeli, S. Yin, R. R. Ogorzalek Loo and J. A. Loo, J. Am. Soc. Mass Spectrom., 2009, 20, 593–596 CrossRef CAS PubMed.
- A. T. Iavarone, J. C. Jurchen and E. R. Williams, J. Am. Soc. Mass Spectrom., 2000, 11, 976–985 CrossRef CAS PubMed.
- C. A. Cassou, H. J. Sterling, A. C. Susa and E. R. Williams, Anal. Chem., 2013, 85, 138–146 CrossRef CAS PubMed.
- L. Kolbowski, A. Belsom and J. Rappsilber, J. Am. Soc. Mass Spectrom., 2020, 31, 1282–1290 CrossRef CAS PubMed.
- J. P. O‘Brien, W. Li, Y. Zhang and J. S. Brodbelt, J. Am. Chem. Soc., 2014, 136, 12920–12928 CrossRef PubMed.
- S. Vahidi, B. B. Stocks and L. Konermann, Anal. Chem., 2013, 85, 10471–10478 CrossRef CAS PubMed.
- J. C. May, E. Jurneczko, S. M. Stow, I. Kratochvil, S. Kalkhof and J. A. McLean, Int. J. Mass Spectrom., 2018, 427, 79–90 CrossRef CAS PubMed.
- D. S. Gross and E. R. Williams, J. Am. Chem. Soc., 1995, 117, 883–890 CrossRef CAS.
- M. A. Halim, M. Girod, L. MacAleese, J. Lemoine, R. Antoine and P. Dugourd, J. Am. Soc. Mass Spectrom., 2016, 27, 1435–1442 CrossRef CAS PubMed.
- M. J. Nold, C. Wesdemiotis, T. Yalcin and A. G. Harrison, Int. J. Mass Spectrom., 1997, 164, 137–153 CrossRef CAS.
- A. Somogyi, V. H. Wysocki and I. Mayer, J. Am. Soc. Mass Spectrom., 1994, 5, 704–717 CrossRef CAS PubMed.
- A. P. Horton, S. A. Robotham, J. R. Cannon, D. D. Holden, E. M. Marcotte and J. S. Brodbelt, Anal. Chem., 2017, 89, 3747–3753 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- M. F. Czar, F. Zosel, I. König, D. Nettels, B. Wunderlich, B. Schuler, A. Zarrine-Afsar and R. A. Jockusch, Anal. Chem., 2015, 87, 7559–7565 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1an00571e |
| This journal is © The Royal Society of Chemistry 2021 |