
Differentiating aspartic acid isomers and epimers with charge transfer dissociation mass spectrometry (CTD-MS)
Halle M.
Edwards
a,
Hoi-Ting
Wu
b,
Ryan R.
Julian
 b and
Glen P.
Jackson
*ac
b and
Glen P.
Jackson
*ac
aC. Eugene Bennett Department of Chemistry, West Virginia University, Morgantown, WV, USA. E-mail: glen.jackson@mail.wvu.edu
bDepartment of Chemistry, University of California, Riverside, CA, USA
cDepartment of Forensic and Investigative Science, West Virginia University, Morgantown, WV, USA
First published on 16th February 2022
Abstract
The ability to understand the function of a protein often relies on knowledge about its detailed structure. Sometimes, seemingly insignificant changes in the primary structure of a protein, like an amino acid substitution, can completely disrupt a protein's function. Long-lived proteins (LLPs), which can be found in critical areas of the human body, like the brain and eye, are especially susceptible to primary sequence alterations in the form of isomerization and epimerization. Because long-lived proteins do not have the corrective regeneration capabilities of most other proteins, points of isomerism and epimerization that accumulate within the proteins can severely hamper their functions and can lead to serious diseases like Alzheimer's disease, cancer and cataracts. Whereas tandem mass spectrometry (MS/MS) in the form of collision-induced dissociation (CID) generally excels at peptide characterization, MS/MS often struggles to pinpoint modifications within LLPs, especially when the differences are only isomeric or epimeric in nature. One of the most prevalent and difficult-to-identify modifications is that of aspartic acid between its four isomeric forms: L-Asp, L-isoAsp, D-Asp, and D-isoAsp. In this study, peptides containing isomers of Asp were analyzed by charge transfer dissociation (CTD) mass spectrometry to identify spectral features that could discriminate between the different isomers. For the four isomers of Asp in three model peptides, CTD produced diagnostic ions of the form cn+57 on the N-terminal side of iso-Asp residues, but not on the N-terminal side of Asp residues. Using CTD, the L- and D forms of Asp and isoAsp could also be differentiated based on the relative abundance of y- and z ions on the C-terminal side of Asp residues. Differentiation was accomplished through a chiral discrimination factor, R, which compares an ion ratio in a spectrum of one epimer or isomer to the same ion ratio in the spectrum of a different epimer or isomer. The R values obtained using CTD are as robust and statistically significant as other fragmentation techniques, like radical directed dissociation (RDD). In summary, the extent of backbone and side-chain fragments produced by CTD enabled the differentiation of isomers and epimers of Asp in a variety of peptides.
Introduction
Proteins in the human body perform various vital functions to maintain health and homeostasis. Most proteins have relatively short lifetimes, on the order of days or weeks, and are regenerated frequently. However, other proteins—like elastin, collagen, nuclear pores, and eye lens crystalline—have long lifetimes on the order of decades.1–3 Throughout a protein's lifetime, spontaneous modifications such as oxidation, isomerization and epimerization can occur, and these modifications can accumulate in long-lived proteins that are not regularly replaced.4 These accumulated modifications can lead to conformational changes in the protein structure, aggregation and loss of function, which can be a root cause of many debilitating diseases that are linked to degeneration.Alzheimer's is one such neurodegenerative disease that is marked by a loss of synaptic function in the brain and can be linked to aggregations of amyloid beta and tau proteins.5,6 Similarly, other degenerative diseases—like Parkinson's, Huntington's, cystic fibrosis and certain cancers—likely originate from protein misfolding and subsequent aggregation.7 Cataracts, which is the leading cause of blindness worldwide, develops due to the breakdown of eye lens crystalline over time and results in an altered protein structure that is less transparent than the properly folded form.8,9 In addition to the importance in studying degenerative diseases, knowledge of protein structure and post translational modifications is important to the development of therapeutic antibodies, especially because loss of function can decrease antigen binding, thereby limiting the effectiveness of treatments.10,11
All twenty amino acids that make up proteins within the human body can undergo racemization from the preferred L-form to the D-form, but the rates of racemization vary considerably. For example, aspartic acid racemizes at least four times more quickly than other amino acids.12 Due to its rapid racemization, L-Asp to D-Asp isomerization has been more widely observed in biological systems and has been more widely studied.13–15 It is well known that aspartic acid in a protein—whether from translation or from deamidation of asparagine—is prone to forming a stable succinimide ring intermediate following self-nucleophilic attack. Subsequent ring opening and/or stereo-inversion converts aspartate to one of four isomeric forms: L-Asp, D-Asp, L-isoAsp, and D-isoAsp.16 All four forms of Asp have been detected in the human brain, although L-Asp is the original form produced via translation.17,18 Accumulation of D-Asp is often observed in long-lived proteins, and D-Asp is linked to age related diseases like cataracts.19–21 Additionally, D-Asp is found at higher concentrations than L-Asp in the brains of Alzheimer's patients.22 In fact, the link between aging and racemization is so well formed that measurements of D-enantiomers can be used as a tool to estimate the age of biological material in forensic and archaeologic applications.14,23
Many different mass spectrometry methods have attempted to differentiate the four forms of Asp, with varying degrees of success.24,25 Although they are isomeric, the structural differences between Asp and isoAsp are distinguishable because they can produce some unique fragment ions or fragment ions with different relative abundances. Commonly observed bn+H2O and yn-46 ions have been reported in fast atom bombardment mass spectrometry (FAB-MS),26 low-energy collision induced dissociation (CID),27,28 high-energy CID,29 matrix-assisted laser desorption/ionization (MALDI) photodissociation (PD),25 and MALDI post-source decay (PSD).25 Electron transfer dissociation (ETD) and electron capture dissociation (ECD) produce reliable cn+57 and zn-57 ions that arise from cleavage between the Cα and the additional carbon incorporated into the backbone of isoAsp residues.30–36 Additionally, side chain cleavages in the form of w-, d-, and v ions are often only observed for Asp residues and not isoAsp residues.25,2918O-Labeling of deamidation products can provide mass distinction between Asp/isoAsp when paired with reversed phase liquid chromatography.37,38
Whereas unique ions characteristic of isoAsp are preferable for identification, differences in relative ion abundances can also provide insight into the identity of the questioned residue. FAB-MS39 and CID40 produce less abundant b- and a ions and more abundant y ions at isoAsp residues relative to the same ions observed for Asp, and Asp also tends to form a more intense immonium ions.41 MALDI free-radical initiated peptide sequencing (FRIPS) provides differences in the abundance of neutral losses—particularly H2O and CO2—between Asp and isoAsp, and it generally favors more intense peaks for Asp residues.25 Also, ETD has been shown to provide more intense z ions for isoAsp relative to Asp.33
Unique ions and differences in relative ion abundances can both distinguish Asp and isoAsp residues from one another in peptides; however, such diagnostics cannot differentiate L- and D epimers of Asp and isoAsp because the epimers only differ in their stereochemistry and cannot yield unique mass fragments. One method that has shown great potential for chiral differentiation is radical-directed dissociation (RDD).42–46 RDD is a radical based fragmentation technique that generates a radical through site-specific cleavage of a carbon-iodine bond by photodissociation.47 Peptides are first modified to include a C–I bond by attaching a chromophore, such as iodobenzoic acid to the N-terminus or by inserting an iodine into an existing tyrosine residue. The peptide is then ionized, isolated in a 2D or 3D ion trap and subjected to a 266 nm pulsed laser to induce photodissociation of the C–I bond and create a radical. The radical product is then subjected to collisional activation to create radical-induced cleavages.45
RDD spectra often show significantly different spectra for L- and D epimers of the same peptide sequence, with many peaks having different relative abundances between the two epimers. To quantitate this degree of differentiation, an R value can be calculated, which compares the intensity ratio of a pair of peaks in the spectrum of the L-Asp epimer to the same pair of peaks in the spectrum of the D-Asp epimer according to eqn (1).48RA and RB represent the pair of peaks with the largest difference in abundance between the two epimers.
| Rchiral = RA/RB | (1) |
Charge transfer dissociation (CTD) also generates radical species in peptides through interactions of protonated or deprotonated precursors with a beam of kiloelectronvolt helium cations. The fast helium cations effectually abstract an electron from the precursor.50 CTD is effective at providing numerous backbone cleavages of peptides—including a-, b-, c-, x-, y-, z-, d-, w- and v ions—the last three of which are especially useful side chain losses.51 The radical-driven nature of fragmentation in CTD implies that it might perform similarly to RDD for the discrimination of L- and D epimers of Asp. Additionally, the numerous fragments produced by CTD could provide distinction between Asp and isoAsp. The current work therefore investigated synthetic versions of peptides derived from crystallin proteins containing isomers of Asp using both CTD and CID to identify discriminatory features of the spectra that can provide distinction between the different isomeric forms.
Methods
Instrumentation
A Bruker amaZon 3D ion trap mass spectrometer, modified to perform CTD, was used for all experiments. The instrument modifications are described elsewhere.51 Ultra-high purity (UHP) helium was used as the CTD reagent gas.Samples
Synthetic versions of crystallin peptides were synthesized following an accelerated FMOC-protected solid-phase peptide synthesis protocol52 and provided by the Julian Laboratory (University of California Riverside, Riverside, CA). Each peptide was reconstituted in a water/acetonitrile/formic acid mixture (49.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49.5:1, v/v/v) with final concentrations between 50–100 μM. The peptides included FVIFLDVK and HFSPEDLTVK, which are found within sheep42 and human53 αA crystallin, and GYQYLLEPGDFR, which is common to mouse βB1 crystallin.54 Each peptide was fabricated in four different versions, with either L-Asp, L-isoAsp, D-Asp, or D-isoAsp as the D residue.
:49.5:1, v/v/v) with final concentrations between 50–100 μM. The peptides included FVIFLDVK and HFSPEDLTVK, which are found within sheep42 and human53 αA crystallin, and GYQYLLEPGDFR, which is common to mouse βB1 crystallin.54 Each peptide was fabricated in four different versions, with either L-Asp, L-isoAsp, D-Asp, or D-isoAsp as the D residue.
Method
Peptide solutions were ionized by a static nanospray source with a voltage of 1500–1800 V. An isolation width of 4 Da was used during precursor isolation, and the low mass cutoff was set to m/z 250 during CTD. For comparison experiments, CID experiments were performed with a reaction amplitude between 0.5–2.0 V for 50 ms, with Smartfrag disabled. For CTD experiments, the pressure in the vacuum chamber was maintained at ∼1.2 × 10−5 mbar, and the ion beam was pulsed on for 100 ms with a voltage of 5–7 kV. The ions gain ∼80% of the anode potential so have between 4–5.6 keV of kinetic energy. Product ion spectra were collected for 1–2 minutes in enhanced-resolution mode. After CTD, unreacted precursor ions were removed using resonance ejection at the MS3 level to minimize space-charge effects and improve the mass accuracy and signal-to-noise ratio.Data analysis
Spectra were converted to mzML format using MSConvert (http://proteowizard.sourceforge.net/download.html) and worked up in mmass.55–57 The averaged spectra were normalized to the base peak and automated peak picking was performed using a signal-to-noise threshold of 5.0 and an absolute intensity threshold of 0.3. Fragmentor (https://sites.google.com/ucr.edu/jlab/software/fragmentor?authuser=0) was used to predict the masses of peptide fragments and aid in annotation. Peaks were only labeled if they exceeded the S/N thresholds, were within ±0.2 Da of the theoretical mass, and if the 13C isotope peak met or exceeded the expected abundance relative to the 12C isomer. R values of epimers were calculated using RIsomer (https://sites.google.com/ucr.edu/jlab/software/r-isomer?authuser=0). Single-factor analysis of variance (ANOVA) was performed with SPSS v28 to identify significantly different peaks between isomers and epimers of aspartic acid.Results & discussion
CTD fragmentation produced numerous types of backbone and side chain cleavages for the peptides studied and provided 100% sequence coverage, in most cases. In addition to the b- and y ions commonly observed with CID, CTD produced a series of a-, x- and z ions and some c ions (Fig. 1). Aligned with previous observations, many of the backbone cleavages were radical species, like the a+1, x+1, and z+1 ions that are typically found in other high energy fragmentation techniques.50,58 As a generalization, increasing the charge state of the precursor ion from 1+ to 2+ increased the number of observed fragments, and CID produced primarily b- and y ions, as well as neutral losses and a few a ions.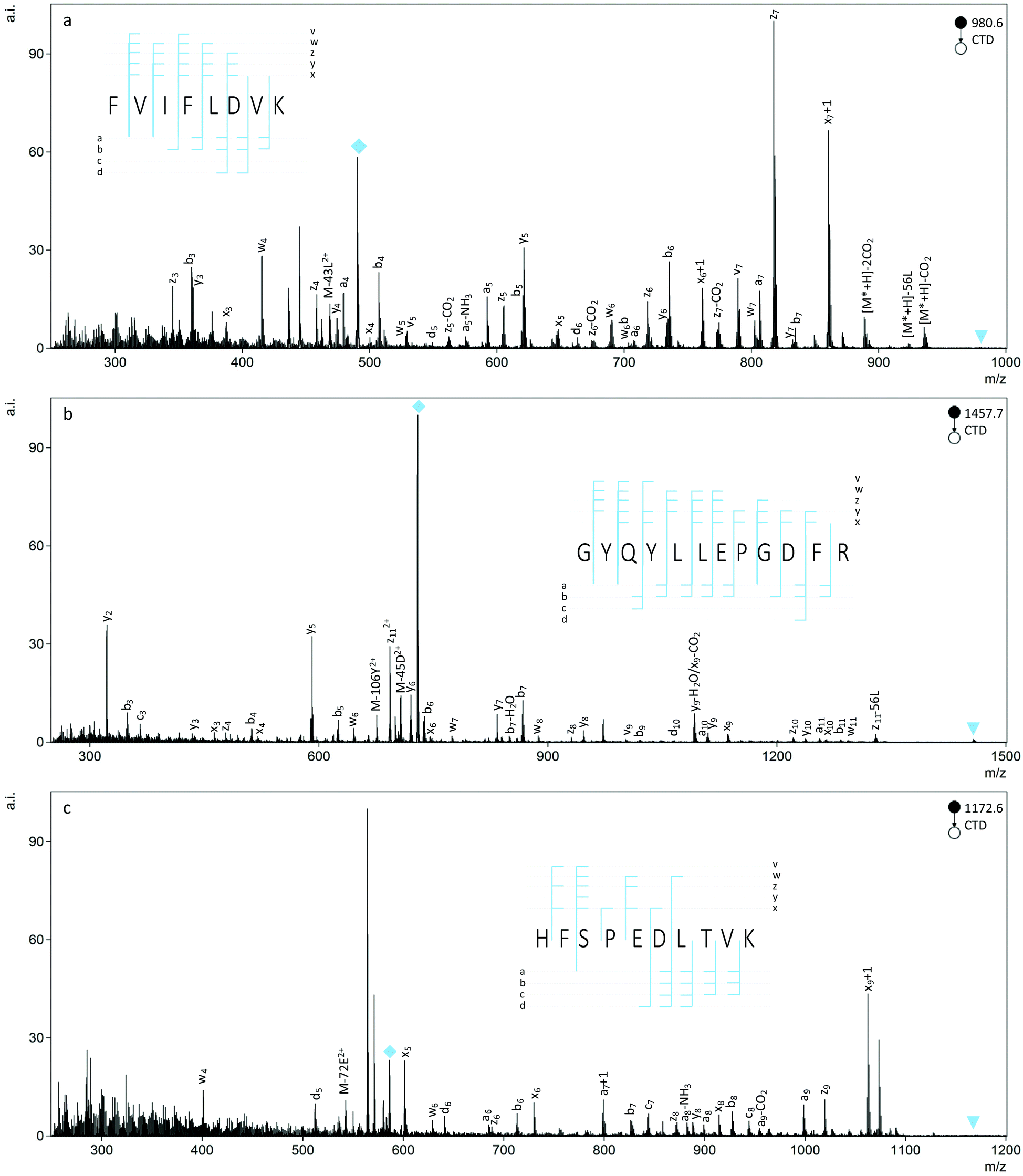 | ||
| Fig. 1 CTD spectra of all-L peptides: (a) FVIFLDVK (b) GYQYLLEPGDFR and (c) HFSPEDLTVK with inset fragment maps. Triangles represent the resonantly ejected precursor and diamonds represent the CTnoD product ion. | ||
L-Asp vs.L-isoAsp
Before comparing the CTD spectra of Asp and isoAsp, we first identified commonly observed isoAsp peaks from other methods of tandem mass spectrometry, as outlined in Table 1. After annotating the CTD spectra of peptides containing either Asp or isoAsp, we compared the fragments obtained through CTD to those observed using other methods. For instance, the b6 ion in FVIFLDVK was about 80% less intense for the isoAsp version relative to the Asp version. However, there was no meaningful difference in ion abundance of the b10 ions for the Asp and isoAsp versions of GYQYLLEPGDFR, and the b6 ion in HFSPEDLTVK was actually more intense for isoAsp than for Asp, which is in contrast to the trend observed using CID40 and FAB-MS.39 As a generalization, CTD produced a ions that are enhanced for Asp residues and y- and z ions that are enhanced for isoAsp residues. For example, the a ions for Asp in FVIFLDVK1+ and GYQYLLEPGDFR2+ are significantly more intense (p < 0.05) than the same ions for isoAsp residues. The a10 ion for GYQYLLEPGDFR1+ was also observed to be slightly more intense for Asp than isoAsp, but the difference was less significant (p = 0.114). These results are similar to those obtained with FAB-MS.39| isoAsp identifiers | Observed with | CTD observations |
|---|---|---|
| Decreased b ions | FAB-MS39 | Inconsistent |
| CID40 | ||
| Decreased a ions | FAB-MS39 | Consistent |
| Increased y ions | CID40 | Inconsistent |
| MALDI-PSD25 | ||
| Increased z ions | MALDI-FRIPS25 | Consistent |
| ETD33 | ||
| b n+H2O, yn-46 | CID27–29 | Not observed |
| FAB-MS26 | ||
| MALDI-PSD25 | ||
| MALDI-PD25 | ||
| c n+57, zn-57 | ECD30–32 | Inconsistent |
| ETD35,36,59 | ||
| MALDI-FRIPS25 | ||
| Decreased -CO2 | MALDI-FRIPS25 | Not observed |
Whereas enhanced a ions in a CTD spectrum can help confirm the presence of Asp residues in a peptide, enhanced z- and y ions are helpful in identifying isoAsp residues. Regardless of the precursor charge state, the z3 ion for FVIFLDVK was significantly more intense (p < 0.05) for the sequence containing isoAsp rather than Asp in the sixth position. Likewise, the z5 ion was significantly more intense (p < 0.05) for isoAsp relative to Asp in HFSPEDLTVK2+. These observations are consistent with those of MALDI-FRIPS25 and ETD.33 The z3 ion for GYQYLLEPGDFR was too low in abundance for confident assignment. When observed, y ions were also significantly more intense (p < 0.05) for isoAsp relative to Asp in all but one case. In that exception, the y3 ion was isobaric with the 13C peak of the b3 ion of FVIFLDVK at m/z 360.2. The general trends observed for Asp and isoAsp in the various CTD spectra are consistent with those reported for CID40 and MALDI-PSD.25
Table 1 contains a summary of CTD observations and the common isoAsp identifiers reported in the literature and only apply to L-forms of Asp/isoAsp; D-forms of Asp/isoAsp provide a more complicated problem that will be addressed in a different section.
The helpful bn+H2O and yn-46 ions observed in collisionally activated peptides of isoAsp residues were not observed with CTD, but a single c5+57 ion was observed at m/z 694.4 for FVIFLDVK2+ (Fig. 2). This unique cn+57 ion for iso-Asp was first observed with ECD, and it has become a reliable diagnostic ion in ECD and ETD to differentiate Asp and isoAsp residues.32 The cleavage between the Cα and the extra carbon inserted into the peptide backbone of isoAsp results in a fragment at cn+57 that cannot be produced when an Asp residue is present. Notably, we observe this unique ion present in both the L form and D form of isoAsp in FVIFLDVK, which suggests that the mechanism is not sensitive to chiral differences. The c5+57 fragment was only observed for this particular peptide, though, so although we demonstrated that CTD is capable of producing this diagnostic ion in one example, the formation of this type of product ion in CTD is not reliably present in all peptides that contain Asp/isoAsp.
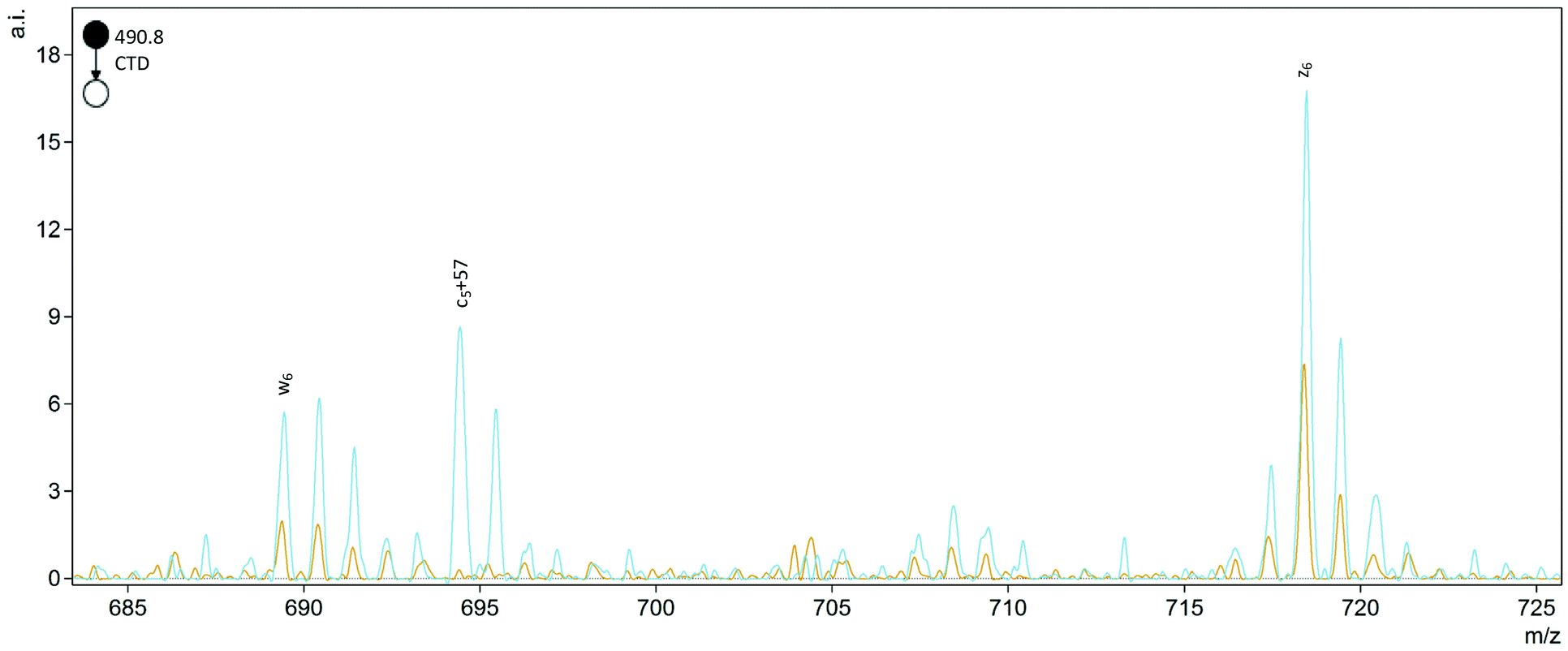 | ||
Fig. 2 Comparison of the D epimers of FVIFL![[D with combining low line]](https://www.rsc.org/images/entities/b_char_0044_0332.gif) ![[i with combining low line]](https://www.rsc.org/images/entities/b_char_0069_0332.gif) VK (blue) and FVIFLVK (orange). The peak at m/z 694.4 for c5+57 is unique to the isoAsp residue. VK (blue) and FVIFLVK (orange). The peak at m/z 694.4 for c5+57 is unique to the isoAsp residue. | ||
L/D Epimers of Asp
To differentiate L- and D epimers of Asp using CTD, we first identified peaks that were significantly different in abundance between the epimers. We also used CID spectra of the same peptides as a benchmark. Significant differences in ion abundances were determined using one-way analysis of variance (ANOVA) using Asp epimerization as the fixed factor. Each test included three replicate measurements of each peptide. To be considered for one-way ANOVA, peaks had to be present with a signal-to-noise (S/N) ratio greater than 5 in at least one spectrum of the two epimers. The number of significant differences and the significance of the differences—as assessed by the significance, p, of the F values—were both considered as metrics for the reliability of epimer discrimination.Fragmentation of singly charged peptide precursors produced a similar number of peaks that contained significant (p < 0.05) abundance differences in both CTD and CID spectra. However, for the doubly charged peptides, CTD produced a greater number of significantly different peaks than CID (Table 2). The identities of the significant peaks indicate a few trends in the types of ions that may be useful for discriminating between L/D forms of Asp. The most promising trend is a potential side chain loss (bn-45D) from aspartic acid (where the postscript D indicates the neutral loss is most likely form the aspartic acid residue). When observed, the bn-45D peak is more intense for the D-epimer (Fig. 3). Unfortunately, without higher mass resolution, we cannot distinguish bn-45D ions from an-NH3 ions because they are nominal isobars. Whether the ion is the loss of the Asp/isoAsp side chain or a neutral loss of ammonia is less important than the observation that the peak occurs at a greater abundance for one epimer over the other; either way, the peak can still be used to positively identify the D epimer. If the ion is in fact the loss of the aspartic acid side chain, this observation suggests the side chain is more readily lost from the D form relative to the L-form and presumably relates to the re-arrangements that are made possible by the three-dimensional configuration of the peptide.
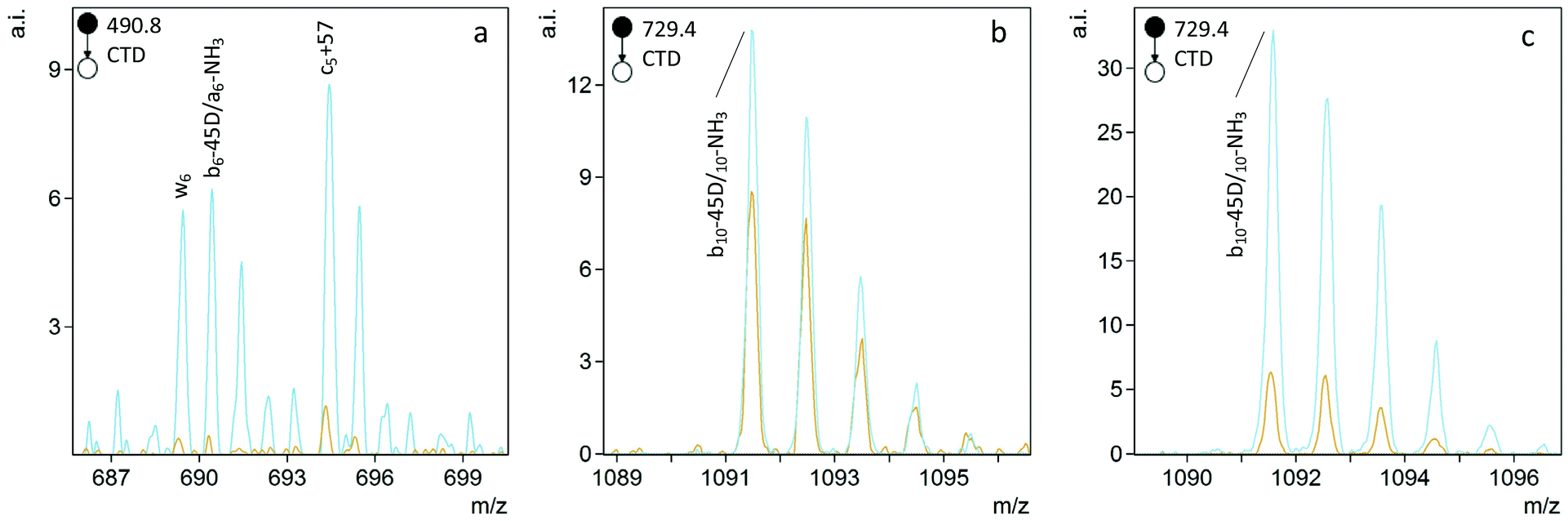 | ||
| Fig. 3 CTD of L- and D epimers of (a) FVIFLVK, (b) GYQYLLEPGFR, and (c) GYQYLLEPGFR showing more intense ions corresponding to bn-45D/an-NH3 for the D epimer of Asp (blue) relative to the L epimer (orange). | ||
| Sequence | CTD | CID | CTD | CID |
|---|---|---|---|---|
| 1 + precursor | 1 + precursor | 2 + precursor | 2 + precursor | |
| Significance assessed using one-way ANOVA using D epimer as the fixed factor and p < 0.05.a The sequences of the peptides are identified by single letter codes with Di representing isoaspartic acid. Bold, underlined residues correspond to the site of epimerization.b CID data not collected. | ||||
| FVIFLVK |
30 | 25 | 51 | 12 |
| FVIFLVK |
24 | 45 | 51 | 13 |
| GYQYLLEPGFR |
9 | —b | 40 | 18 |
| GYQYLLEPGFR |
48 | —b | 50 | 25 |
| HFSPELTVK |
19 | 20 | 83 | 15 |
| HFSPELTVK |
19 | 11 | 55 | 20 |
Other neutral losses from backbone cleavages, like -H2O and -CO2, are more abundant for the D epimer than the L epimer in several cases. For example, b10-CO2 is more abundant for D epimers of GYQYLLEPG![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif) FR and GYQYLLEPG
FR and GYQYLLEPG![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) FR, whereas an x5-CO2 is more abundant for the D epimer of HFSPELTVK. Additionally, the z3-H2O and z5-H2O ions are more abundant for the D epimers of FVIFLVK and HFSPELTVK, respectively. Although not observed in every case, these neutral losses seem to be preferred for the D epimers and thus could be helpful in identifying the chirality of Asp residues.
FR, whereas an x5-CO2 is more abundant for the D epimer of HFSPELTVK. Additionally, the z3-H2O and z5-H2O ions are more abundant for the D epimers of FVIFLVK and HFSPELTVK, respectively. Although not observed in every case, these neutral losses seem to be preferred for the D epimers and thus could be helpful in identifying the chirality of Asp residues.
We also observed significant differences in ion abundances for peaks that are not as obviously related to the proximity of the D residue. For example, the z4 fragment for FVIFLDVK is significantly more intense for the version containing D-Asp relative to L-Asp, but this cleavage site is one amino acid residue removed from the site of epimerization. Since the epimers differ only in their stereochemistry, these differences in ion abundances using CTD indicate that fragmentation behavior is sufficiently sensitive to conformational changes as to be readily observable at cleavage sites not directly related to the site of epimerization.
To quantify the degree of chiral discrimination possible with CTD, R values were calculated for pairs of epimers with paired peak lists that were already determined to be significantly different between the spectra of the two epimers, as identified through one-way ANOVA. The R values shown in Tables 3 and 4 were calculated from the mean of three replicate peak abundances for each peak for each epimer. In some cases, CTD produced one or more unique peaks for one epimer, such as the b6-45D/a6-NH3 peak for FVIFLVK in Fig. 3, and the a9-H2O peak for HFSPEDLTVK in Fig. 4. In these cases, CTD provides unequivocal differentiation between the two epimers. In contrast, CID did not provide any unique peaks for any of the peptides relative to their epimers.
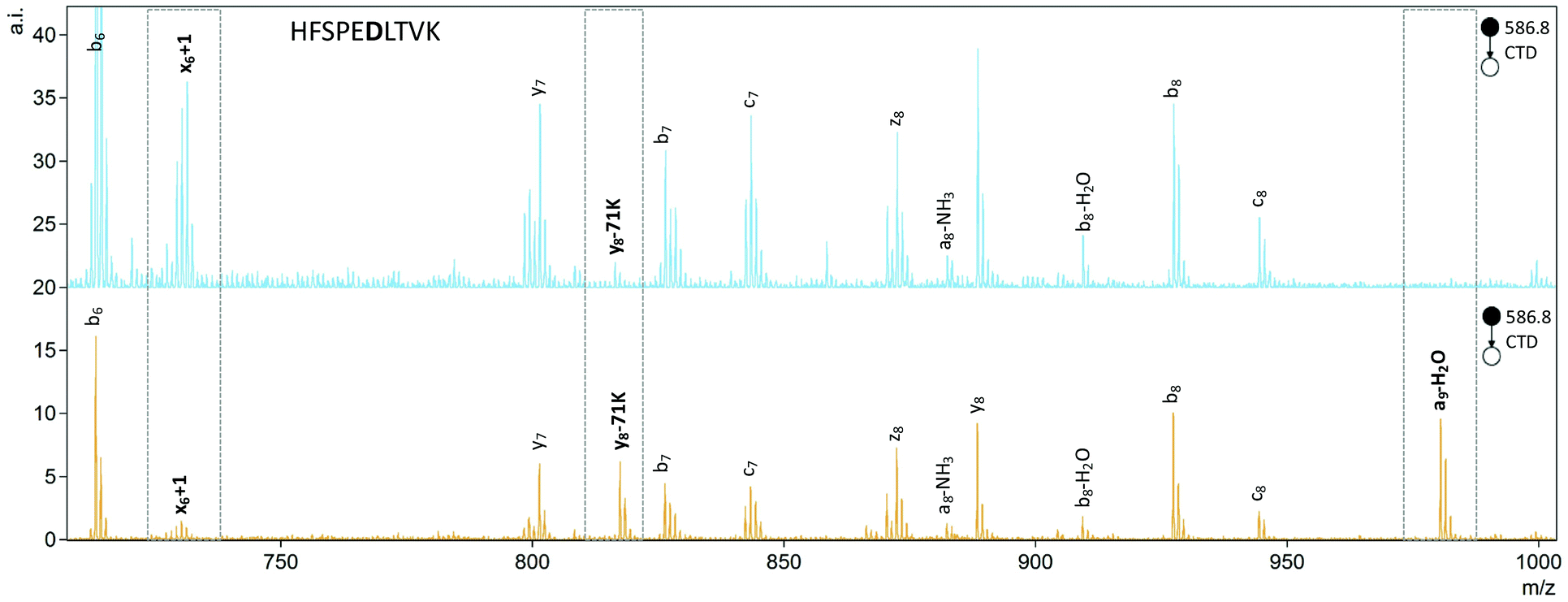 | ||
| Fig. 4 CTD of L- (orange) and D- (blue) epimers of HFSPEDLTVK2+ showing the differences in relative peak abundances between spectra. The peaks that vary the most between the two spectra are the y8-71K ion and the x6+1 ion, which together give an R value = 69.8. Additionally, the a9+H2O ion is unique to L-Asp. The peaks of interest are indicated by boxes. | ||
| Sequence | 1+ precursor | 2+ precursor | ||
|---|---|---|---|---|
| CTD | CID | CTD | CID | |
| a The sequences of the peptides are identified by single letter codes with Di representing isoaspartic acid. Bold, underlined residues correspond to the site of epimerization. b The unexpectedly large R values present with CID may be attributed to unintentional differences in the accumulation times between two CID spectra during data collection. c CID data not collected. | ||||
| FVIFLVK |
5.8 | 18.5b | 9.3 | 1.0 |
| FVIFLVK |
10.2 | 11.0b | 16.8 | 1.0 |
| GYQYLLEPGFR |
1.6 | —c | 2.8 | 4.1 |
| GYQYLLEPGFR |
19.9 | —c | 48.2 | 5.3 |
| HFSPELTVK |
26.3 | 1.2 | 69.8 | 5.0 |
| HFSPELTVK |
41.0 | 7.9 | 37.5 | 2.8 |
| Sequence | CTD | CID | ||||||
|---|---|---|---|---|---|---|---|---|
| 1+ precursor | 2+ precursor | 1+ precursor | 2+ precursor | |||||
| R value | Related peaks | R value | Related peaks | R value | Related peaks | R value | Related peaks | |
| a The sequences of the peptides are identified by single letter codes with Di representing isoaspartic acid. Bold, underlined residues correspond to the site of epimerization. b The unexpectedly large R values present with CID may be attributed to unintentional differences in the accumulation times between two CID spectra during data collection. c CID data not collected. | ||||||||
| FVIFLVK |
5.8 | x 6 -H2O+ | 9.3 | a 7 + | 18.5b | Unassigned | 1.0 | |
| b 5 + | z 5-58K2+ | y 4 + | ||||||
| FVIFLVK |
10.2 | a 5 + | 16.8 | b 6 + | 11.0b | M-H2O+ | 1.0 | |
| c 4-15V+ | b 3 + | Unassigned | ||||||
| GYQYLLEPGFR |
1.6 | M*-45D2+ | 2.8 | M-CO23+ | —c | —c | 4.1 | y 8 + |
| x 2 + | b 3 + | Unassigned | ||||||
| GYQYLLEPGFR |
19.9 | M-H2O2+ | 48.2 | y 9-H2O | —c | —c | 5.3 | y 8 + |
| y 6-CO2+ | y 5 2+ | b 7 + | ||||||
| HFSPELTVK |
26.3 | M*-59E-56L+ | 69.8 | y 8-71K | 1.2 | b 9 + | 5.0 | b 6 + |
| x 5 + | x 6 + 1+ | y 8 + | Unassigned | |||||
| HFSPELTVK |
41.0 | c 6 + | 37.5 | z 6 + | 7.9b | M-H2O–NH3+ | 2.8 | M-H2O+ |
| M*-45D-43L+ | a 7 2+ | Unassigned | y 4 + | |||||
Only ions present in both spectra with S/N greater than 5 were considered for R value calculations. In almost every case, CTD produced R values that were notably larger than CID. In fact, the R values obtained for CTD match or exceed those of RDD, which is the current gold standard and typically delivers R values in the range of 2.0–30.0.45,49
The identities of the pairs of peaks used to calculate R values were also investigated to see if they were obviously related to the altered Asp or isoAsp residues. Epimerization can disturb the three-dimensional structure of the entire peptide, so differences in fragment ion abundances may not always be obviously related to the Asp residue. In fact, with CID, most of the peaks are backbone cleavages or involve neutral losses unrelated to the Asp/isoAsp residue, so there is little, or no, information one can gain about what types of cleavages can be enhanced or hindered with L/D epimers or how differences in fragment ion abundances can be used to provide predictions for new Asp-containing peptides. For CID, there are only two cases in which at least one of the peaks are adjacent to the Asp/isoAsp residues. For the L- and D epimers of HFSPELTVK, a b6 ion on the C-terminal side of Asp is significantly different, and for HFSPELTVK, a y4 ion on the C-terminal side of isoAsp is significantly different. For CTD, many of the significant peaks are adjacent to, or one residue removed from, the Asp/isoAsp residue (Table 4).
Herein, CTD demonstrates a high degree of chiral discrimination that is similar to, or greater than, RDD and ETD. Furthermore, D epimers produced more abundant bn-45D/an-NH3 ions that can provide confidence in assigning the chirality of an Asp residue in an unknown peptide. Though this specific ion is not always observed, the high R values obtained with CTD can be used to identify peptides with epimerization present in the sequence. Comparisons with standards of known chirality could then provide additional clarity in identifying the location and type of epimerization, which could be useful in the analysis of peptide mixtures when coupled to LC.
a-/x-Ion formation
The abundance of an+1 radical ions in the CTD spectra are reminiscent of those produced by UVPD,50 which indicates that the fragmentation mechanism in CTD could follow similar homolytic cleavage of the Cα–C bond to form the an+1 ions.60 In CTD, the homolytic cleavage of the Cα–C bond may be instigated by ionization of the nearby lone pair on the carbonyl oxygen atom, in a similar mechanism to that proposed for metastable atom-activated dissociation (MAD).61 Among the two isoforms of the three peptides studied, numerous an+1 ions were observed in every case. More frequently than the an+1 ions, several xn+1 ions were also observed for each peptide. These findings are consistent with CTD of other low-charge state peptides using CTD.50Among the fragmentation methods capable of producing a/x ions and their radical counterparts, the an+1 ions are most commonly observed. For example, in UVPD, absorption of a 157 nm photon leads to homolytic cleavage to produce an+1 and xn+1 ions. These primary fragments then undergo hydrogen elimination to form the even electron a/x species.60,62,63 In addition to the mechanism described above, where CTD fragmentation begins with the radical cation localized on the carbonyl oxygen, the radical could instead be localized on the amide nitrogen, as described by Kjeldsen and coworkers for EDD.64 As proposed in Scheme 1, α-cleavage of the amide backbone would create an even-electron a ion and an xn+1 ion. Since both an+1 and xn+1 species are observed, it is possible that fragmentation could proceed via various competing pathways in CTD. However, given that xn+1 ions are generally more abundant than an+1 ions, excitation of, or radical location on, the amide nitrogen may be preferred in this case. In principle, the xn+1 ions with a radical on the carbonyl carbon in Scheme 1 could readily form z-type ions through the loss of a neutral molecule of isocyanic acid, or CONH.
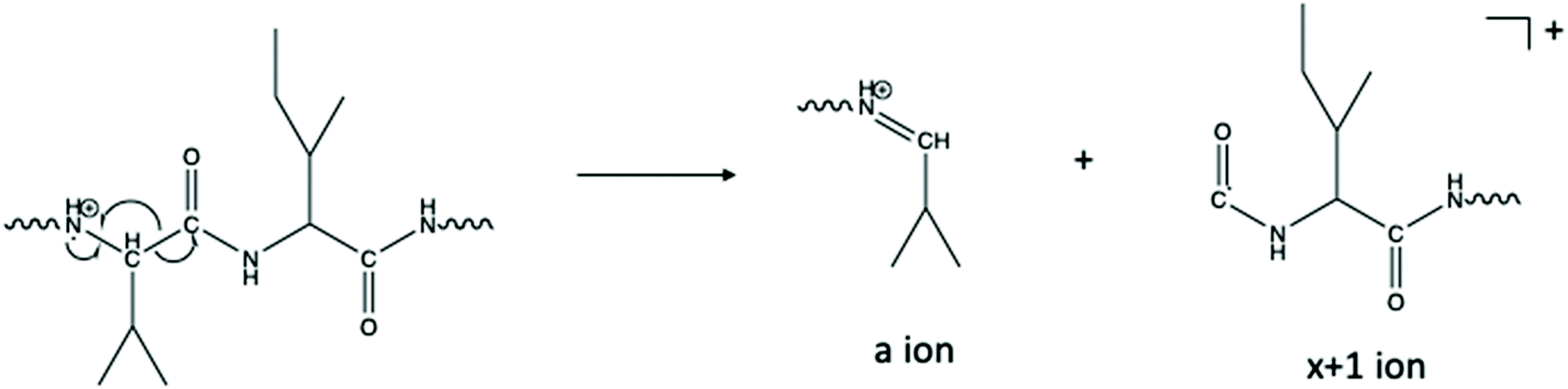 | ||
| Scheme 1 One proposed pathway for the formation of a- and x+1 ions.64 Alternative pathways are also possible.60,62,63 | ||
Conclusions
CTD demonstrates an ability to distinguish isomeric forms of Asp and isoAsp in various peptides on a benchtop instrument without chemical modification of the peptide. For CTD of peptides containing Asp and isoAsp, the increased abundance of y- and z ions in IsoAsp peptides relative to Asp peptides can be useful in identifying isoAsp residues. In addition, a ions tend to be more abundant in Asp-containing peptides relative to IsoAsp. CTD can generate unique cn+57 ions for isoAsp residues, in a similar fashion to ECD and ETD, and for the L- and D epimers of Asp and isoAsp, CTD demonstrates a degree of chiral discrimination that is similar to, or better than, RDD. Comparison of relative peak abundances in epimer pairs of three Asp-containing peptides and three isoAsp-containing peptides provided R values ranging from 2.6–70. Furthermore, a bn-45D/an-NH3 ion was found to be a reliable indicator for the D isomers of Asp/isoAsp relative to the L isomers. Improved mass resolution would clarify the specific identity of this beneficial fragment, which could then establish its relevance to L/D discrimination of Asp within peptides containing all-L amino acids. These findings show that CTD can provide reliable and structurally meaningful fragments that are sensitive to conformational differences of peptides in the gas phase.Conflicts of interest
No conflicts of interest to declare.Acknowledgements
The authors would like to thank Yana A. Lyon for synthesizing the FVIFLDVK peptide used in this study. This work was supported by the National Science Foundation (NSF) (CHE-1710376). The opinions, findings and conclusions or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the views of NSF.References
- N. H. Thayer, C. K. Leverich, M. P. Fitzgibbon, Z. W. Nelson, K. A. Henderson, P. R. Gafken, J. J. Hsu and D. E. Gottschling, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14019–14026 CrossRef CAS PubMed.
- J. N. Savas, B. H. Toyama, T. Xu, J. R. Yates and M. W. Hetzer, Science, 2012, 335, 1–4 CrossRef PubMed.
- E. F. Fornasiero, S. Mandad, H. Wildhagen, M. Alevra, B. Rammner, S. Keihani, F. Opazo, I. Urban, M. S. Sakib, M. K. Fard, K. Kirli, T. P. Centeno, R. O. Vidal, R.-U. Rahman, E. Benito, A. Fischer, S. Dennerlein, P. Rehling, I. Feussner, S. Bonn, M. Simons, H. Urlaub and S. O. Rizzoli, Nat. Commun., 2018, 9, 1–17 CrossRef CAS PubMed.
- P. A. C. Cloos and S. Christgau, Matrix Biol., 2002, 21, 39–52 CrossRef CAS PubMed.
- T. L. Spires-Jones and B. T. Hyman, Neuron, 2014, 82, 756–771 CrossRef CAS PubMed.
- S. Forner, D. Baglietto-Vargas, A. C. Martini, L. Trujillo-Estrada and F. M. LaFerla, Trends Neurosci., 2017, 40, 347–357 CrossRef CAS PubMed.
- T. K. Chaudhuri and S. Paul, FEBS J., 2006, 273, 1331–1349 CrossRef CAS PubMed.
- W. G. Hodge, J. P. Whitcher and W. Satariano, Epidemiol. Rev., 1995, 17, 336–346 CrossRef CAS PubMed.
- D. Lam, S. K. Rao, V. Ratra, Y. Liu, P. Mitchell, J. King, M.-J. Tassignon, J. Jonas, C. P. Pang and D. F. Chang, Nat. Rev. Dis. Primers, 2015, 1, 1–15 Search PubMed.
- Y. Tian and B. T. Ruotolo, Analyst, 2018, 143, 2459–2468 RSC.
- Z. Zhang, H. Pan and X. Chen, Mass Spectrom. Rev., 2009, 28, 147–176 CrossRef CAS PubMed.
- C. R. McCudden and V. B. Kraus, Clin. Biochem., 2006, 39, 1112–1130 CrossRef CAS PubMed.
- J. L. Bada, Methods Enzymol., 1984, 106, 98–115 CAS.
- S. Ohtani, Y. Yamada, T. Yamamoto, S. Arany, K. Gonmori and N. Yoshioka, J. Forensic Sci., 2004, 49, 1–5 CrossRef.
- N. K. Shah, B. Brodsky, A. Kirkpatrick and J. A. M. Ramshaw, Biopolymers, 1999, 49, 297–302 CrossRef CAS PubMed.
- T. Geiger and S. Clarke, J. Biol. Chem., 1987, 262, 785–794 CrossRef CAS.
- X. Zheng, L. Deng, E. S. Baker, Y. M. Ibrahim, V. A. Petyuk and R. D. Smith, Chem. Commun., 2017, 53, 7913–7916 RSC.
- H. Fukuda, T. Shimizu, M. Nakajima, H. Mori and T. Shirasawa, Bioorg. Med. Chem. Lett., 1999, 9, 953–956 CrossRef CAS PubMed.
- P. M. Helfman, J. L. Bada and M. Y. Shou, Gerontology, 1977, 23, 419–425 CrossRef CAS PubMed.
- P. M. Masters, J. L. Bada and J. S. Zigler, Jr., Nature, 1977, 268, 71–73 CrossRef CAS PubMed.
- P. M. Masters, J. L. Bada and J. S. Zigler, Jr., Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 1204–1208 CrossRef CAS PubMed.
- G. H. Fisher, A. D'Aniello, A. Vetere, G. P. Cusano, M. Chávez and L. Petrucelli, Neurosci. Lett., 1992, 143, 215–218 CrossRef CAS PubMed.
- E. R. Waite, M. J. Collins, S. Ritz-Timme, H. W. Schutz, C. Cattaneo and H. I. M. Borrman, Forensic Sci. Int., 1999, 103, 113–124 CrossRef CAS PubMed.
- P. P. Hurtado and P. B. O'Connor, Mass Spectrom. Rev., 2012, 31, 609–625 CrossRef CAS PubMed.
- N. Degraan-Weber, J. Zhang and J. P. Reilly, J. Am. Soc. Mass Spectrom., 2016, 27, 2041–2053 CrossRef CAS PubMed.
- I. A. Papayannopoulos and K. Biemann, Pept. Res., 1992, 5, 83–90 CAS.
- P. Schindler, D. Muller, W. Marki, H. Grossenbacher and W. J. Richter, J. Mass Spectrom., 1996, 24, 967–974 CrossRef.
- L. J. González, T. Shimizu, Y. Satomi, L. Betancourt, V. Besada, G. Padrón, R. Orlando, T. Shirasawa, Y. Shimonishi and T. Takao, Rapid Commun. Mass Spectrom., 2000, 14, 2092–2102 CrossRef.
- S. A. Carr, M. E. Hemling, M. F. Bean and G. D. Roberts, Anal. Chem., 1991, 63, 2802–2824 CrossRef CAS PubMed.
- J. J. Cournoyer, C. Lin and P. B. O'Connor, Anal. Chem., 2006, 78, 1264–1271 CrossRef CAS PubMed.
- J. J. Cournoyer, C. Lin, M. J. Bowman and P. B. O'Connor, J. Am. Soc. Mass Spectrom., 2007, 18, 48–56 CrossRef CAS PubMed.
- J. J. Cournoyer, J. L. Pittman, V. B. Ivleva, E. Fallows, L. Waskell, C. E. Costello and P. B. O'Connor, Protein Sci., 2005, 14, 452–463 CrossRef CAS PubMed.
- P. B. O'Connor, J. J. Cournoyer, S. J. Pitteri, P. A. Chrisman and S. A. McLuckey, J. Am. Soc. Mass Spectrom., 2006, 17, 15–19 CrossRef PubMed.
- N. P. Sargaeva, C. Lin and P. B. O'Connor, J. Am. Soc. Mass Spectrom., 2011, 22, 480–491 CrossRef CAS PubMed.
- N. P. Sargaeva, C. Lin and P. B. O'Connor, Anal. Chem., 2011, 83, 6675–6682 CrossRef CAS PubMed.
- W. Y. K. Chan, T. W. D. Chan and P. B. O'Connor, J. Am. Soc. Mass Spectrom., 2010, 21, 1012–1015 CrossRef CAS PubMed.
- S. Wang and I. A. Kaltashov, Anal. Chem., 2013, 85, 6446–6452 CrossRef CAS PubMed.
- M. Liu, J. Cheetham, N. Cauchon, J. Ostovic, W. Ni, D. Ren and Z. S. Zhou, Anal. Chem., 2012, 84, 1056–1062 CrossRef CAS PubMed.
- J. R. Lloyd, M. L. Cotter, D. Ohori and D. L. Doyle, Biomed. Environ. Mass Spectrom., 1988, 15, 399–402 Search PubMed.
- W. D. Lehmann, A. Schlosser, G. Erben, R. Pipkorn, D. Bossemeyer and V. Kinzel, Protein Sci., 2000, 9, 2260–2268 CrossRef CAS PubMed.
- A. Schlosser and W. D. Lehmann, J. Mass Spectrom., 2000, 35, 1382–1390 CrossRef CAS.
- Y. A. Lyon, G. M. Sabbah and R. R. Julian, J. Proteome Res., 2017, 16, 1797–1805 CrossRef CAS PubMed.
- Y. Tao and R. R. Julian, Anal. Chem., 2014, 86, 9733–9741 CrossRef CAS PubMed.
- T. Ly and R. R. Julian, J. Am. Chem. Soc., 2008, 130, 351–358 CrossRef CAS PubMed.
- Y. Tao, N. R. Quebbemann and R. R. Julian, Anal. Chem., 2012, 84, 6814–6820 CrossRef CAS PubMed.
- L. Zhang and J. P. Reilly, J. Am. Soc. Mass Spectrom., 2009, 20, 1378–1390 CrossRef CAS PubMed.
- T. Ly, X. Zhang, Q. Sun, B. Moore, Y. Tao and R. R. Julian, Chem. Commun., 2011, 47, 2835–2837 RSC.
- W. A. Tao, D. Zhang, E. N. Nikolaev and R. G. Cooks, J. Am. Chem. Soc., 2000, 122, 10598–10609 CrossRef CAS.
- Y. A. Lyon, G. Beran and R. R. Julian, J. Am. Soc. Mass Spectrom., 2017, 28, 1365–1373 CrossRef CAS PubMed.
- W. D. Hoffmann and G. P. Jackson, J. Am. Soc. Mass Spectrom., 2014, 25, 1939–1943 CrossRef CAS PubMed.
- P. Li and G. P. Jackson, J. Am. Soc. Mass Spectrom., 2017, 28, 1271–1281 CrossRef CAS PubMed.
- C. Hood, G. Fuentes, H. Patel, K. Page, M. Menakuru and J. H. Park, J. Pept. Sci., 2008, 14, 97–101 CrossRef CAS PubMed.
- Y. A. Lyon, G. M. Sabbah and R. R. Julian, Exp. Eye Res., 2018, 171, 131–141 CrossRef CAS PubMed.
- X. Liu, M. Zhang, Y. Liu, P. Challa, P. Gonzalez and Y. Liu, Mol. Vision, 2008, 14, 2404–2412 CAS.
- M. Strohalm, M. Hassman, B. Košata and M. Kodíček, Rapid Commun. Mass Spectrom., 2008, 22, 905–908 CrossRef PubMed.
- M. Strohalm, D. Kavan, P. Novák, M. Volný and V. Havlíček, Anal. Chem., 2010, 82, 4648–4651 CrossRef CAS PubMed.
- T. H. J. Niedermeyer and M. Strohalm, PLoS One, 2012, 7, e44913 CrossRef CAS PubMed.
- P. Li, I. Kreft and G. P. Jackson, J. Am. Soc. Mass Spectrom., 2017, 29, 284–296 CrossRef PubMed.
- N. P. Sargaeva, C. Lin and P. B. O'Connor, Anal. Chem., 2009, 81, 9778–9786 CrossRef CAS PubMed.
- W. Cui, M. S. Thompson and J. P. Reilly, J. Am. Soc. Mass Spectrom., 2005, 16, 1384–1398 CrossRef CAS PubMed.
- S. L. Cook, O. L. Collin and G. P. Jackson, J. Mass Spectrom., 2009, 44, 1211–1223 CrossRef CAS PubMed.
- L. Zhang and J. P. Reilly, Anal. Chem., 2009, 81, 7829–7838 CrossRef CAS PubMed.
- L. Zhang, W. Cui, M. S. Thompson and J. P. Reilly, J. Am. Soc. Mass Spectrom., 2006, 17, 1315–1321 CrossRef CAS PubMed.
- F. Kjeldsen, O. A. Silivra, I. A. Ivonin, K. F. Haselmann, M. Gorshkov and R. A. Zubarev, Chem. – Eur. J., 2005, 11, 1803–1812 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |