
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Analysis of illicit pills and drugs of abuse in urine samples using a 3D-printed open port probe hyphenated with differential mobility spectrometry-mass spectrometry†
Piotr
Sosnowski
 ,
Victor
Marin
,
Xiaobo
Tian
and
Gérard
Hopfgartner
*
,
Victor
Marin
,
Xiaobo
Tian
and
Gérard
Hopfgartner
*
Life Sciences Mass Spectrometry, Department of Inorganic and Analytical Chemistry, University of Geneva, Quai Ernest Ansermet 24, 1211, Geneva, Switzerland. E-mail: gerard.hopfgartner@unige.ch
First published on 9th August 2022
Abstract
The present work describes the application of an in-house developed 3D-printed open port probe (3DP-OPP) with differential ion mobility spectrometry (DMS) mass spectrometry. Targeted quantitative analysis in urine was performed with a triple quadrupole mass spectrometer in the selected reaction monitoring mode (OPP-DMS-SRM/MS) and illicit pill screening using data independent acquisition (OPP-DMS-SWATH/MS). The combination of compensation voltage (CoV) scanning in DMS using modifiers with SWATH/MS acquisition for MS/MS spectrum generation enabled the differentiation of isobaric signals with a large dynamic range and enhance the information contained in the screening of illicit ecstasy pills. As for any direct MS introduction technique where no chromatographic separation is applied DMS with acetonitrile as a modifier allows the separation of cocaine and tramadol, and their isomeric metabolites in urine samples. Quantitative application using OPP-DMS-SRM/MS is presented without the need for sample preparation with a lower limit of quantification at 10–25 ng mL−1 for the analytes and less than 40 seconds for sample to sample analysis.
Introduction
One of the methods to prepare prototypes of in-house developed tools is the use of the commonly accessible three-dimensional printing (3DP) method. Stereolithography (SLA) printing,1 where liquid photocurable resin is used to prepare a solid model through UV photopolymerization, provides excellent resolution for making high-quality objects with details down to 50–100 μm. Alternatively, fused deposition modelling (FDM),2 where the model is constructed with layers of melted material, can be used for models that require less high-resolution details. We previously applied 3DP to modify and further optimize the sampling3 of the open port probe (OPP) proposed by Van Berkel et al.4 This interface is based on coaxial, vertically aligned tubes and can be used with atmospheric pressure ionization sources for mass spectrometry experiments. The outer tube provides a self-cleaning sampling dome by pumping the mobile phase through it, while the inner tube aspirates from the dome into the API source by setting nebulization gas at a high rate that is slower than the mobile phase rate of the outer tube. This reduces the risk of contamination of the ion source as direct contact is omitted, and at the same time provides a Gaussian-like shape of the recorded signal.To thoroughly benefit from the potential of OPP, especially when focusing on quantitative aspects, an automatic and reproducible way of sample introduction is necessary. One of the efficient methods of sampling is acoustic droplet ejection (ADE).5 With this technique, small droplets of ∼2.5nL are being ejected through an ultrasonic pulse into the sampling dome of the OPP, which greatly reduces the ion suppression effect due to high dilution (1000×). The injected droplets generate very sharp peaks with a width of ∼1.2s. This provides high-throughput analysis and was recently applied to the in situ kinetics of dextromethorphan conversion to dextrorphan,6 quality control of dose–response and chemical synthesis experiments,7 and screening of diacylglycerol acyltransferase 2 inhibitors.8
An alternative way of sampling is the use of a robotic autosampler operating with single-use, low volume (10 μL) pipette tips.9 Here, the droplet falls on top of the OPP sampling dome through non-contact pneumatic ejection with a syringe plunger. With 3D printing, robotic samplers can be adapted and modified to work with different brands of labware and custom tools. This sampling solution seems to be more cost-effective, compared to ADE which requires a compatible custom microtiter plate. Additionally, even though the volume of the sample used is 5–15 μL, only a limited amount of sample (∼3%)9 is being aspirated due to the self-cleaning counterflow of the mobile phase. This volume is further diluted in the sampling dome of 3DP-OPP in the flow of the mobile phase, which in sum reduces the ionization suppression from the matrix.
Due to the lack of any separation prior to the MS, it may be an issue to quickly and reliably analyze multiple compounds using 3DP-OPP. Full scan and fixed precursor/product ion scan modes require either prior information about targeted compounds or multiple runs to obtain optimized data. Alternatively, data-dependent acquisition (DDA) could be considered, but this method is not optimal in the case of coelution of multiple species as it may lead to missed signals.10
Sequential window acquisition of all theoretical mass spectra (SWATH-MS) is an example of the data-independent acquisition (DIA)11,12 method which enables the collection of all precursors and all fragments. MS/MS spectra are being acquired for all molecules that are passing through the quadrupole filter, set in Q1 window isolation (e.g. m/z 100–150) mode. Multiple fixed or variable size windows can be set to cover the full mass range.13 This way of recording data enables the gathering of information about every ionized analyte in the introduced sample (MS/MSALL). It can provide structural information, with the specificity increasing by reducing the window size.14 Yet smaller isolation windows also increase the number of scans and total cycle time, which can be reduced by decreasing the scan period, on the other hand, may negatively affect data quality. Thus, either compromise needs to be made between specificity and sensitivity, or an additional level of separation needs to be provided by hyphenating an MS analyzer with e.g. liquid chromatography or ion mobility.
Interfacing ion mobility spectrometry (IMS) and mass spectrometry (MS) can provide multiple benefits in the cases of targeted and untargeted analyses. Selectivity tuning on an example of sulphonamide isomers in human plasma15 or orthogonality to HPLC-MS methods for metabolomic studies on patients with chronic kidney disease16 was presented in the past. The application of IMS has been described for the analysis of amino acids, peptides, proteins, lipids, oligonucleotides, carbohydrates, and pharmaceutical and recreational drugs.17–21 Differential ion mobility spectrometry (DMS),22 alternatively called high-field asymmetric waveform ion mobility spectrometry (FAIMS),23 utilizes separation voltage (SV) as an alternating electric field to force trajectory on the ion based on its collisional cross section. An additional DC voltage named compensation voltage (CoV) is also applied to correct the trajectory and enable the ions to pass through the DMS cell. This provides an additional level of separation and confirmation based on the compound structure, which can be applied for molecules that may not be distinguishable by mass spectrometry only.
In this report, we present the hyphenation of a 3D printed open port probe with SWATH-MS, DMS-SWATH-MS, and DMS-SRM/MS. The design, optimization, and application of 3DP-OPP were already described in our previous work.3 In the first part, the hyphenation of a 3DP-OPP with high-resolution mass spectrometry (HRMS), by utilizing SWATH-MS and DMS-SWATH-MS methods is presented. Applied methods can be used for the detection of illegal drugs in unknown origin solid samples, where DMS provides an additional dimension of separation and confirmation for isobaric compounds. This can also reduce false-positive results that could be generated by interferences. In the second part, we present quantification of cocaine and tramadol, and their metabolites including isomeric compounds (benzoylecgonine/norcocaine, O-desmethyltramadol/N-desmethyltramadol), separated in a DMS cell prior to entering the mass analyzer set in the selected reaction monitoring mode. The application of the method for spiked urine and samples obtained during traffic control is described.
Experimental
Chemicals
LC-MS grade water was purchased from Huberlab (Aesch, Switzerland); methanol was purchased from VWR (Darmstadt, Germany); formic acid was purchased from Sigma-Aldrich (Buchs, Switzerland). Benzoylecgonine, norcocaine, cocaine, cocaine-d3, methylecgonine, cocaethanol, tramadol, O-desmethyltramadol, and N-desmethyltramadol were purchased from Lipomed AG (Arlesheim, Switzerland). A pool of human urine was made using nine anonymous donors and was stored at −20 °C prior to analysis.Computer-aided design and 3D printing
3DP-OPP and other models were designed in Autodesk Fusion360 software (San Rafael, USA). Source files in .stl format containing various parts are available in the ESI.† A FormLabs Form 2 SLA printer (Artsupport GmbH, RümLang, Switzerland) was used for printing a 3DP-OPP with High Temp resin due to its compatibility with methanol and a printing liquid handler tool with Clear resin because it is more flexible and resistant to impact. The model was oriented at 45° towards the table and sliced at 0.05 mm resolution using the PreForm software (FormLabs, Somerville, USA). The density of the supports was set to 0.60 and the point size was set to 0.50 mm. An X400CE FDM printer and PLA filament (German RepRap GmbH, Feldkirchen, Germany) were used for printing other parts.Ecstasy pills and urine samples
Illicit Ecstasy tablets and urine samples from traffic control were from the Institute of Forensic Medicine, University of Bern, Switzerland. Urine samples were provided anonymously from the Institute of Forensic Medicine for analytical method development and collected with the informed consent of the subjects.3DP-OPP-MS general conditions
The 3D printed open port probe model used was of co-axial tube design and included an outer tube and was mounted directly on an ion source. The inner tube was a PEEK capillary (0.794 mm OD, 250 μm ID, 20 cm length). Methanol with a 0.1% formic acid mixture was used as a solvent and delivered using an LC-30AD pump (Shimadzu, Reinach, Schweiz) with a flow rate of 600 μL min−1. About 500 μL min−1 was aspirated down the PEEK capillary to the Turbo V ion source attached to either QTRAP 6500+ or QTOF 6600+ (Sciex, Concord, Canada). The configuration of OPP to ESI is shown in Fig. S1.† Generally, 10 μL of samples were delivered onto OPP by the liquid handler that works with dispensable pipette tips. In every experiment, the OPP was used in this “spill over mode”, where the aspiration rate is slightly lower than the solvent pump rate. The standard ESI nebulizer and emitter capillary (375 and 100 μm ID, respectively) were replaced by a larger internal diameter set (635 and 150 μm ID).3DP-OPP-DMS-SRM/MS conditions
The mass spectrometer was operated in selected reaction monitoring (SRM) mode. Positive ion mode ESI was used with ion source nitrogen gas settings GS1 = 90; GS2 = 50; curtain gas = 20; TEM = 500 °C and ISVF = 2500 V. Acetonitrile pumped at 170 μL min−1 was used as a modifier for DMS and with following settings: SV = 2600, DR = Off, DT = Low (150 °C). Analyst software version 1.7.1 was used for data acquisition, and Peak View 2.2 and MultiQuant 2.1 were used for data processing. Optimized SRM transitions and CoV values are presented in Table S3.†3DP-OPP-SWATH/MS conditions
The mass spectrometer was operated in mixed HRMS/SWATH/MS acquisition mode with a total cycle time of 1131 ms. The MS1 experiment was performed in TOFMS mode in the mass range m/z 90–1000 with 100 ms accumulation time. The MS2 experiment involved 32 independent 6 Da Q1 SWATH windows covering mass ranges of m/z 90–250 (1st run) and m/z 240–400 (2nd run) with 30 ms accumulation time for each Q1 window. To induce fragmentation, the collision energy voltage (CE) was set to 30 eV and the collision energy spread voltage (CES) was set to 15 eV. Positive ion mode ESI was used with ion source nitrogen gas settings GS1 = 80; GS2 = 40; curtain gas = 25; TEM = 500 °C and ISVF = 4500 V. Peak View Research 1.2.20 with Library Search utility and commercial forensic MS/MS spectral library from Sciex (version 1.1; 1700 entries; obtained with Sciex TripleTOF instruments at a collision energy of 35 eV and a collision energy spread of 15 eV) was used for compound identification.3DP-OPP-DMS-SWATH/MS conditions
The mass spectrometer was operated in mixed HRMS/SWATH/MS acquisition mode with a total cycle time of 1131 ms. The MS1 experiment was performed in TOFMS mode in the mass range 90–1000 with 100 ms accumulation time. The MS2 experiment involved 32 SWATH windows set to a fixed m/z value of 90–250. Each window had an individually set CoV value to cover the range from −35 V to −4 V with a step size of 1 V and with 30 ms accumulation time for each window. To induce fragmentation, the collision energy voltage (CE) was set to 30 V and the collision energy spread voltage (CES) was set to 15 V. Positive ion mode ESI was used with ion source nitrogen gas settings GS1 = 80; GS2 = 40; curtain gas = 25; TEM = 500 °C and ISVF = 4500 V. Acetonitrile pumped at 170 μL min−1 was used as a modifier for DMS and with following settings SV = 3600, DR = Off, DT = Low (150 °C). Peak View Research 1.2.20 with Library Search utility and commercial forensic MS/MS spectral library from Sciex (version 1.1; 1700 entries; obtained with Sciex TripleTOF instruments at a collision energy of 35 V and a collision energy spread of 15 V) was used for compound identification.Method performance
Calibration curves were generated by spiking urine with analytes and their deuterated derivatives. For method validation linearity, accuracy, precision, sensitivity, dilution and carry-over were tested. Data for accuracy and precision validation were obtained from 3 runs made on 2 separate days. Each run consisted of 1 repetition for each calibrator and 5 repetitions for each QC sample.8 calibrators and 4 quality control samples were prepared by spiking pooled urine. 50 μL of urine standards were diluted with 450 μL (10 ng mL−1) of internal standard ice-cold methanolic solution. Samples were next vortexed and centrifuged. 100 μL of each solution was transferred into a 96-well plate, which was delivered using a liquid handler mounted on an autosampler (Fig. S2†).
Calibration curves were constructed by plotting the ratio of integrated SRM signals of drugs and their corresponding internal standards (Aanalyte/AIS) as a function of concentration (canalyte). 1/x2 weighting was applied for all compounds, except methylecgonine (1/x).
Results and discussion
Design of 3D printed tools
A schematic of the introduction of a liquid sample and the acquisition workflow is summarized in Fig. 1.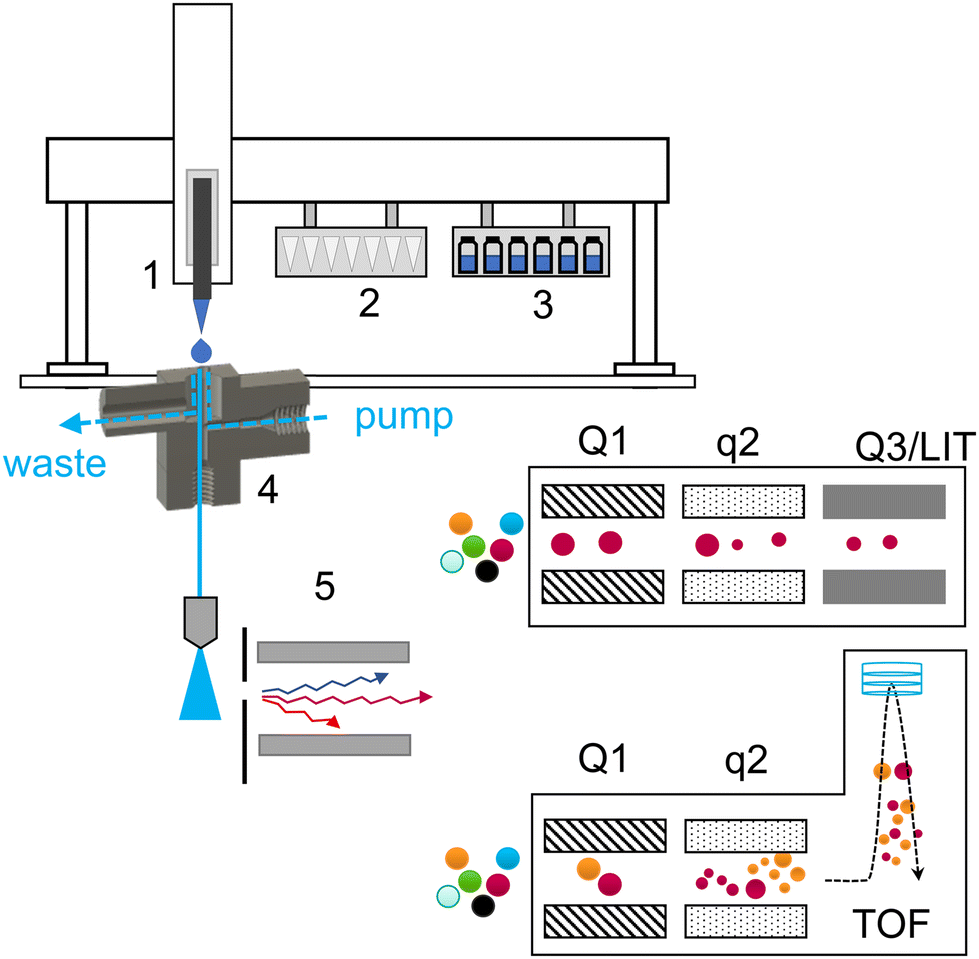 | ||
| Fig. 1 General setup for the introduction of liquid samples and mass spectrometric acquisition: (1) modified CTC syringe holder to accommodate standard Eppendorf pipette tips, (2) pipette tip rack, (3) refrigerated sample rack, (4) 3D printed OPP and (5) differential ion mobility cell. Two different types of MS were used: triple quadrupole in the selected ion monitoring mode and (ii) quadrupole time of flight with data independent acquisition. | ||
The majority of used tools were already described in detail in our previous work (Table S1†).3 A 3DP-OPP holder was redesigned to improve the robustness of the setup. When mounted on the PAL system, we observed that rapid movement of the robotic arm could generate strong vibrations and influence the position of the pipette tip above the sampling dome during injection. Therefore, the holder is now always directly installed on the turbo ion source and 3DP-OPP is inserted into the port with spherical insets, locking it in place (Fig. S3†). On the other hand, the waste box and tip removal tool are now assembled together with a single metal screw, to ensure that the position of the box will not change during analysis or during emptying the box, as this may result in collision and damage to pipette tool (Fig. S4†). An injection cycle was also updated. The pipette tip position over the OPP sampling dome is calibrated for position 1 of the tip rack. When taking the new tip, it is gently pressed onto the rack to attach it. We observed that for other positions, the pipette tip may be attached but shifted slightly to the left or right, which resulted in an incorrect position during droplet fall. To prevent that, after taking a new tip the pipette tool is returned to position 1 of the tip holder and presses the tip again. It does not affect the analysis time as it occurs during previous sample acquisition, and at the same time it ensures a reproducible tip attachment.
3DP-OPP-SWATH/MS and 3DP-OPP-DMS-SWATH/MS for untargeted qualitative analysis
Seized illegal ecstasy pills are interesting samples due to the lack of appropriate quality control. Additionally, the formulation ingredients of the pill may give insights into the origin of the sample. The individual components may be present either by “enriching” the powder used for pressing the pills or due to differences in the synthesis and purification steps. We previously analyzed a single pill using a targeted approach with the SRM-IDA-EPI method and a QqQ analyzer. In the current experiment, 6 pills were analyzed using the untargeted approach with the SWATH/MS method and a QTOF mass analyzer, with library screening for identification. To prepare the sample, the surface of ecstasy pills was gently scratched using a single-use sharp toothpick, where a small amount of powder was attached to a porous wooden material. The toothpick was next manually transferred directly to the sampling dome of the OPP for approximately 10 seconds, dissolving the sample in the OPP liquid dome. After that, the toothpick was discarded. This resulted in the generation of a peak-shaped signal (Fig. 2A). We found this method superior to directly inserting the whole pill into the sampling dome, as there is no risk of cross-contamination between the samples and the liquid is not being absorbed into the pill. Compounds were tentatively identified by accurate mass considering an error of 10 ppm (Table S2†). Product ion spectra were used for identity confirmation by applying the Library Search (in-house SWATH library) utility incorporated into PeakView software.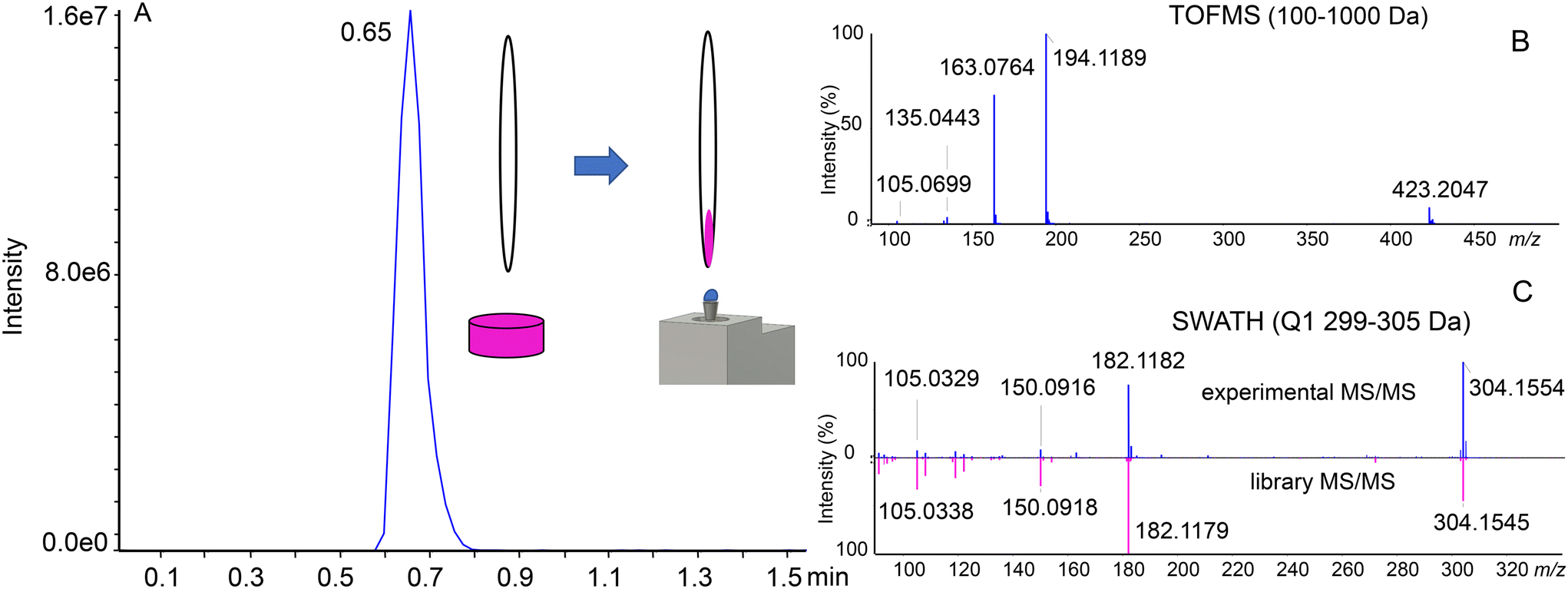 | ||
| Fig. 2 (A) Total ion chromatogram obtained during sampling of pill 2 using a toothpick. (B) TOFMS scan. Mass spectrum showing intense MDMA-related signals ([M + H]+m/z 194.1189) MDMA in-source decay fragments (m/z 105.0699, 135.0443, 163.0764) and a 2MDMA + HCl cluster (m/z 423.2047). (C) Product ion spectrum of the precursor at m/z 304.1554, Q1 window 299–305 Da, CE = 35 eV and CES = 15 eV (top) and the library extracted MS/MS spectrum of cocaine (bottom). | ||
The advantages of SWATH/MS (DIA) over DDA are visible when considering Fig. 2B and C. For DDA, without setting an exclusion list, precursor selection will be done according to the signal intensity. In this situation, the first 5 precursors for the MS/MS experiment selected would be MDMA (m/z 194.1), MDMA in-source decay fragments (m/z 105.1, 135.0, 163.1) and a 2MDMA + HCl cluster (m/z 423.2). SWATH/MS, on the other hand, has a predetermined precursor selection window, which enables the identification of cocaine (Fig. 2C). However, in DDA the precursor ion selection is based on the intensity and the peak of cocaine or the even lower intensity peaks have a risk to be missed which is a general limitation of DDA. All of the results were next manually reviewed to ensure similarity in the fragmentation pattern (Fig. S5†).Table 1 presents the compounds identified in the set of 6 analyzed pills. The main ingredient of each sample is MDMA, but in all of them also MDEA is present, an MDMA analog extended by a single –CH2– group in the alkyl chain on the amine group. Cocaine was detected in half of the analyzed set. Caffeine, amphetamine, and ketamine were present in single samples. Less common compounds were also found. MDHOET is an MDMA analog with an extended alkyl chain by –C2H4– and terminated with a hydroxyl group.24 DPIA, an impurity of amphetamine synthesis, which through biotransformation may form amphetamine,25 was present only in the pill in which amphetamine itself was detected. The presence of 2DPMP, a long-lasting stimulant that can cause dangerous agitation and psychosis, is particularly interesting.26 Unfortunately, the used library did not include MDHOET and DPIA reference mass spectra and we were not able to obtain analytical standards for these compounds. Tentative identification of these molecules was based on accurate mass and proposed fragments (Fig. S6†).
One of the challenges that are present when using flow injection methods, which OPP-MS is an example of, is that all of the compounds are eluting at the same time. While HRMS is already separating the compounds based on their m/z ratios, in the case of isobaric or isomeric compounds it may not be possible to distinguish them without prior chromatographic separation. We observed for 3DP-OPP-SWATH/MS that the signal corresponding to intact MDMA and its in-source decay fragments was multiple times more abundant, compared to other analytes. This is not surprising as MDMA is supposed to be the main ingredient of these pills. In the case of pill 1, in the m/z 194–200 Q1 window, we observed extra fragment ions not related to MDMA, but the ones that could indicate the presence of caffeine. Yet we did not observe the caffeine precursor ion (m/z 195.0877). At the same time, the 2nd isotopic peak of the MDMA [M + H]+ signal was shifted (theoretical mass m/z 195.1209, found mass m/z 195.0936, 140 ppm error), which could be related to the limited resolving power (Fig. 3A).
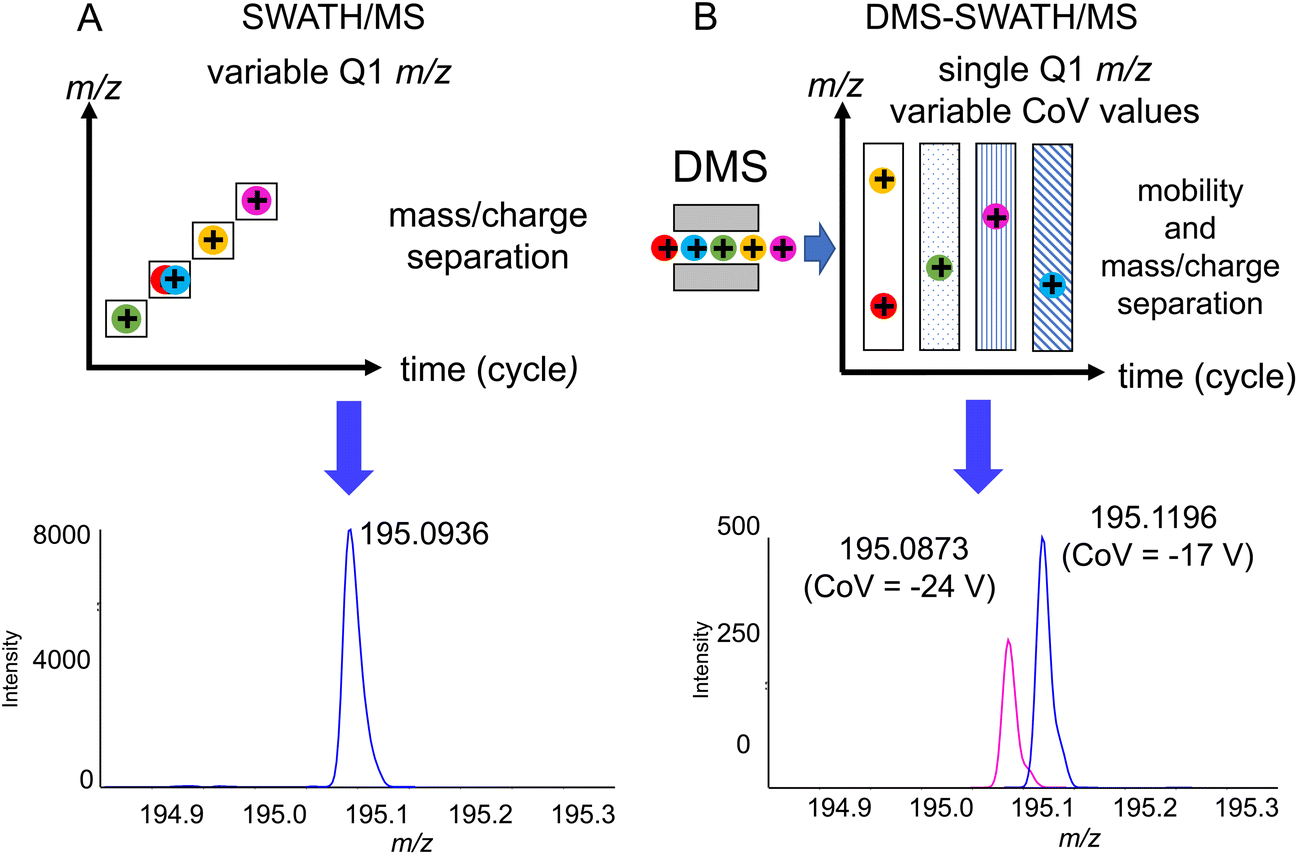 | ||
| Fig. 3 Schematic illustration of the method and representative zoom of mass spectra (range m/z 194.8–195.3) obtained from ecstasy pill 1 with (A) 3DP-OPP-SWATH/MS with multiple m/z Q1 windows and (B) 3DP-OPP-DMS-SWATH/MS with a single m/z window and multiple CoV values (13C isotope of MDMA m/z 195.1196 and [M + H]+ of caffeine at m/z 195.0873). | ||
To confirm the presence of caffeine, an additional level of separation using DMS was applied, where instead of recording multiple different m/z windows, a single m/z 90–250 window was set for all experiments, while CoV values were increased incrementally (1 V) for each experiment in the range of −34 V to −4 V. Acetonitrile was selected as the DMS modifier, providing substantial separation in our previous results.21 This provided separation of MDMA, which was transmitted through the cell at −17 CoV, from caffeine transmitted at −24 CoV (Fig. 3B). While 5x–10x loss of sensitivity was observed, ion mobility separation filtering still provided good quality MS/MS spectrathat could be used for the identification of caffeine. If necessary, the method itself could be further optimized by multiple runs with smaller m/z windows, or as a single experiment with a longer cycle time.
3DP-OPP-DMS-MS for quantitative analysis of drugs in urine
Quantitation of the compounds that are present in complex matrices may require appropriate separation before obtaining signals with a mass analyzer. The main reason for that is the possibility of false-positive signals produced by the molecules already present in the sample. While the most common for liquid samples is the use of liquid chromatography and separation on a column, this method may require a lengthy analysis time, depending on the nature of the analyte. A way to substitute chromatographic separation and drastically decrease the analysis time is the combination of ion mobility and mass spectrometry. Applying the next fast and reliable way of sample introduction using a 3DP-OPP and robotic sampler, which will generate a Gaussian-like signal, may in sum provide a quick way to quantify compounds of interest below approved cut-off levels.To test the application of our new setup for rapid analysis of drugs in urine, with DMS providing an additional level of separation, we prepared a series of dilutions of cocaine and tramadol, and their metabolites in pooled urine. Internal standards (cocaine-d3 and tramadol-d3) at a concentration of 8 ng mL−1 were used to minimize quantitation error. DMS parameters were optimized to ensure that isomeric compounds will be resolved at the baseline level (Fig. 4 and Fig. S7†) to minimize multiple different ions passing through Q1 and Q3 in the same scan. This separation is important as e.g. benzoylecgonine and norcocaine share SRM transition (m/z 290 > m/z 168) which may lead to incorrect estimation of the concentration of the compounds. It also gives an additional level of confirmation for the cocaine/tramadol intake and the presence of their metabolites.
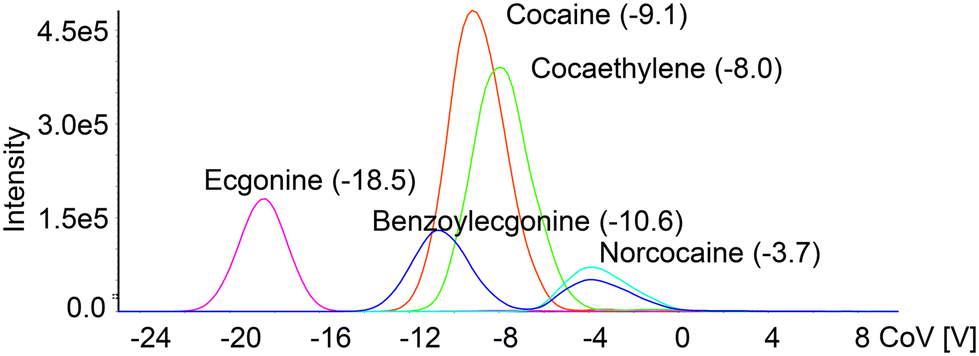 | ||
| Fig. 4 Ionograms obtained during the CoV value ramp for cocaine and its metabolites infused at 100 ng mL−1. The separation voltage was 2600 V and the modifier was acetonitrile. | ||
Selected validation results are presented in Table 2 and full validation can be found in the ESI (Tables S4–S10 and Fig. S8†). The total run time for each sample was 36s with peak widths of 6–10s. The method preserved linearity over the applied range of concentrations (0–1000 and 0–2500 ng mL−1) with 1/x2 weighting applied, except methylecgonine where 1/x weighting was used. Correlation coefficient (r) values were always above 0.99. The lower limit of quantitation (LLOQ) was the lowest non-zero concentration of the calibration curve, the values of which are 25 ng mL−1 for benzoylecgonine, norcocaine, and methylecgonine, and 10 ng mL−1 for other tested compounds. The precision of all of the quality control samples was within ≤11% difference and the accuracy was within ±13% (16% for QCLLOQ). The back calculated accuracy for at least 6 out of 7 non-zero calibrators (>75%) was within ±15% (±20% for LLOQ; Table S6†). The typical confirmatory cut-off applied for all the compounds in urine is 100 ng mL−1. When 3 blank samples were analyzed directly after the highest calibration point, the carryover did not exceed 20% of LLOQ.
| QC sample | Nominal concentration | Within-run accuracy (%; 3 runs) | Between runs accuracy (%) | Within-run precision (%CV; 3 runs) | Between runs precision (%CV) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Benzoylecgonine | QCLLOQ | 25 | 94.8 | 90.7 | 102 | 96.0 | 8.0 | 7.0 | 3.3 | 6.2 |
| QCL | 75 | 105 | 92.5 | 103 | 100 | 3.1 | 3.1 | 3.2 | 6.6 | |
| QCM | 400 | 100 | 98.2 | 106 | 101 | 1.2 | 3.2 | 4.4 | 3.7 | |
| QCH | 2000 | 101 | 101 | 108 | 103 | 2.6 | 5.0 | 1.8 | 4.1 | |
| Norcocaine | QCLLOQ | 25 | 98.2 | 89.7 | 88.2 | 92.0 | 4.0 | 10.0 | 5.4 | 5.8 |
| QCL | 75 | 112 | 92.0 | 101 | 102 | 2.0 | 3.6 | 1.1 | 9.8 | |
| QCM | 400 | 102 | 99.8 | 98.7 | 100 | 4.1 | 3.9 | 2.1 | 1.6 | |
| QCH | 2000 | 106 | 103 | 107 | 105 | 4.8 | 5.4 | 7.0 | 2.3 | |
| O-Desmethyltramadol | QCLLOQ | 10 | 106 | 116 | 108 | 110 | 6.0 | 7.4 | 4.8 | 6.7 |
| QCL | 30 | 110 | 110 | 110 | 110 | 6.8 | 2.8 | 0.1 | 2.5 | |
| QCM | 200 | 104 | 100 | 99.3 | 101 | 3.4 | 1.3 | 2.4 | 3.2 | |
| QCH | 800 | 110 | 105 | 106 | 107 | 5.6 | 3.0 | 2.6 | 1.9 | |
| N-Desmethyltramadol | QCLLOQ | 10 | 107 | 101 | 97.3 | 99.2 | 4.4 | 8.2 | 2.5 | 2.7 |
| QCL | 30 | 107 | 102 | 113 | 107 | 5.4 | 5.9 | 2.9 | 6.9 | |
| QCM | 200 | 100 | 102 | 100 | 101 | 3.0 | 1.2 | 2.9 | 0.9 | |
| QCH | 800 | 104 | 107 | 106 | 107 | 2.5 | 2.7 | 3.7 | 0.9 | |
The method was next applied to measure the concentrations of cocaine and tramadol, and their metabolites in urine samples taken during traffic control. The cut-off applied for all the compounds was 100 ng mL−1, based on SAMSHA requirements.27 If necessary, the samples were diluted 100× using pooled urine to fit in the calibration curve. Out of 49 measured samples, 8 were positive for cocaine or its metabolites and 3 were positive for tramadol or its metabolites. Benzoylecgonine (8), norcocaine (3), methylecgonine (7), cocaine (4), cocaethylene (3), tramadol (3), O-desmethyltramadol (1) and N-desmethyltramadol (3) were all detected. Cocaethylene is especially interesting, as it is a metabolite that is present when both cocaine and ethanol are being taken. Table 3 shows the median concentration of each compound for positive samples.
Conclusions
In this report, we have demonstrated an application of in-house designed 3DP tools to analyze forensic samples using high and low-resolution mass spectrometry with DIA and ion mobility applied. Updated 3D printed tools and analysis workflow removed minor issues observed during previous experiments. SWATH/MS and DMS-SWATH/MS are applied as methods for qualitative analysis of illicit ecstasy pills, providing data that show the individual components of the samples. The SWATH Q1 approach enables the improvement of the detection dynamic range of analytes and to identify a low amount of components present in the sample. DMS with a single SWATH Q1 window further enhanced this capability in addition to the separation of isobars and isomers.The DMS-SRM/MS method is applied for validated fast and robust quantification of cocaine and tramadol, and their metabolites in urine, with the validated method applied for samples obtained during traffic control with improved selectivity. Overall, both SWATH-MS and DMS-MS can be successfully applied to supplement direct analysis with a 3D printed open port probe.
Author contributions
Piotr Sosnowski: conceptualization, methodology, investigation, and writing – original draft. Victor Marin: methodology and investigation. Xiaobo Tian: investigation, review & editing. Gérard Hopfgartner: conceptualization, methodology, writing – review & editing, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Stefan König (University of Bern) for providing traffic control urine samples.References
-
C. W. Hull, U.S. Pat, US4575330A, 1986 Search PubMed
.
-
S. S. Crump, U.S. Pat, US5121329A, 1992 Search PubMed
- P. Sosnowski and G. Hopfgartner, Talanta, 2020, 215, 120894 CrossRef CAS PubMed
- G. J. Van Berkel and V. Kertesz, Rapid Commun. Mass Spectrom., 2015, 29, 1749–1756 CrossRef CAS PubMed
- I. Sinclair, R. Stearns, S. Pringle, J. Wingfield, S. Datwani, E. Hall, L. Ghislain, L. Majlof and M. Bachman, J. Lab. Autom., 2015, 21, 19–26 CrossRef PubMed
- H. Zhang, C. Liu, W. Hua, L. P. Ghislain, J. Liu, L. Aschenbrenner, S. Noell, K. J. Dirico, L. F. Lanyon, C. M. Steppan, M. West, D. W. Arnold, T. R. Covey, S. S. Datwani and M. D. Troutman, Anal. Chem., 2021, 93, 10850–10861 CrossRef CAS PubMed
- J. Zhang, Y. Zhang, C. Liu, T. Covey, J. Nielsen, S. Li, H. Weller and W. Shou, SLAS Technol., 2021, 26, 178–188 CrossRef CAS PubMed
- X. Wen, C. Liu, L. Ghislain, K. Tovar, V. Shah, S. J. Stout, S. Cifelli, S. Satapati, G. O'Donnell, P. R. Sheth, M. J. Wildey, S. S. Datwani, T. R. Covey, K. P. Bateman and D. G. McLaren, Anal. Chem., 2021, 93, 6071–6079 CrossRef CAS PubMed
- G. J. Van Berkel, V. Kertesz, M. Orcutt, A. Bentley, J. Glick and J. Flarakos, Anal. Chem., 2017, 89, 12578–12586 CrossRef CAS PubMed
- T. Sajic, Y. Liu and R. Aebersold, Proteom. – Clin. Appl., 2015, 9, 307–321 CrossRef CAS PubMed
- G. Hopfgartner, D. Tonoli and E. Varesio, Anal. Bioanal. Chem., 2012, 402, 2587–2596 CrossRef CAS PubMed
- R. Bonner and G. Hopfgartner, TrAC, Trends Anal. Chem., 2019, 120, 115278 CrossRef
- Y. Zhang, A. Bilbao, T. Bruderer, J. Luban, C. Strambio-De-Castillia, F. Lisacek, G. Hopfgartner and E. Varesio, J. Proteome Res., 2015, 14, 4359–4371 CrossRef CAS PubMed
- M. Raetz, E. Duchoslav, R. Bonner and G. Hopfgartner, Anal. Bioanal. Chem., 2019, 411, 5681–5690 CrossRef CAS PubMed
- D. Ruskic and G. Hopfgartner, Anal. Chem., 2019, 91, 11670–11677 CrossRef CAS PubMed
- S. Wernisch and S. Pennathur, Anal. Bioanal. Chem., 2019, 411, 6297–6308 CrossRef CAS PubMed
- K. L. Davidson and M. F. Bush, Anal. Chem., 2017, 89, 2017–2023 CrossRef CAS PubMed
- R. A. Harris, J. C. May, C. A. Stinson, Y. Xia and J. A. McLean, Anal. Chem., 2018, 90, 1915–1924 CrossRef CAS PubMed
- F. Mashayekhy Rad, C. Leck, L. L. Ilag and U. Nilsson, Rapid Commun. Mass Spectrom., 2018, 32, 942–950 CrossRef CAS PubMed
- C. Wu, W. F. Siems, J. Klasmeier and H. H. Hill, Anal. Chem., 2000, 72, 391–395 CrossRef CAS PubMed
- T. Porta, E. Varesio and G. Hopfgartner, Anal. Chem., 2013, 85, 11771–11779 CrossRef CAS PubMed
- I. A. Buryakov, E. V. Krylov, E. G. Nazarov and U. K. Rasulev, Int. J. Mass Spectrom. Ion Processes, 1993, 128, 143–148 CrossRef CAS
- R. W. Purves, R. Guevremont, S. Day, C. W. Pipich and M. S. Matyjaszczyk, Rev. Sci. Instrum., 1998, 69, 4094–4105 CrossRef CAS
- C. Koper, E. Ali-Tolppa, J. S. Bozenko Jr., V. Dufey, M. Pütz, C. Weyermann and F. Zrcek, Microgram J., 2005, 3, 166–174 CAS
- V. Ottaviano, C. Furnari and F. Rosati, Ann. Ist. Super. Sanita, 2002, 38, 331–335 CAS
- O. Lapatto-Reiniluoto, U. Tacke and K. Hoppu, Suom. Lääkäril., 2011, 66, 1398–1401 Search PubMed
- https://www.samhsa.gov, July 28, 2022.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2an00925k |
| This journal is © The Royal Society of Chemistry 2022 |