
Identification of bacteria in mixed infection from urinary tract of patient's samples using Raman analysis of dried droplets†
Kateřina
Aubrechtová Dragounová‡
 ab,
Oleg
Ryabchykov
bc,
Daniel
Steinbach
d,
Vincent
Recla
e,
Nora
Lindig
e,
María José
González Vázquez
ab,
Susan
Foller
d,
Michael
Bauer
a,
Thomas W.
Bocklitz
bfg,
Jürgen
Popp
bf,
Jürgen
Rödel
e and
Ute
Neugebauer
*abf
ab,
Oleg
Ryabchykov
bc,
Daniel
Steinbach
d,
Vincent
Recla
e,
Nora
Lindig
e,
María José
González Vázquez
ab,
Susan
Foller
d,
Michael
Bauer
a,
Thomas W.
Bocklitz
bfg,
Jürgen
Popp
bf,
Jürgen
Rödel
e and
Ute
Neugebauer
*abf
aDepartment of Anaesthesiology and Intensive Care Medicine and Center for Sepsis Control and Care (CSCC), Jena University Hospital, Am Klinikum 1, 07747 Jena, Germany. E-mail: ute.neugebauer@leibniz-ipht.de
bLeibniz Institute of Photonic Technology (Leibniz-IPHT), a member of the Leibniz Centre for Photonics in Infection Research (LPI), Albert-Einstein-Straße 9, 07745 Jena, Germany
cBiophotonics Diagnostics GmbH, Am Wiesenbach 30, 07751 Jena, Germany
dDepartment of Urology, Jena University Hospital, Am Klinikum 1, 07747 Jena, Germany
eInstitute of Medical Microbiology, Jena University Hospital, Am Klinikum 1, 07747 Jena, Germany
fInstitute of Physical Chemistry and Abbe School of Photonics, Friedrich Schiller University Jena, Helmholtzweg 4, 07743 Jena, Germany
gInstitute of Computer Science, Faculty of Mathematics, Physics & Computer Science, University Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany
First published on 17th July 2023
Abstract
Urinary tract infections (UTI) are among the most frequent nosocomial infections. A fast identification of the pathogen and assignment of Gram type could help to prescribe most suitable treatments. Raman spectroscopy holds high potential for fast and reliable bacterial pathogens identification. While most studies so far have focused on individual pathogens or artificial mixtures, this contribution aims to translate the analysis to primary urine samples from patients with suspected UTIs. For this, we have included 59 primary urine samples out of which 29 were diagnosed as mixed infections. For Raman analysis, we first trained two classification models based on principal component analysis – linear discriminant analysis (PCA-LDA) with more than 3500 Raman spectra of 85 clinical isolates from 23 species in order to (1) identify the Gram type of the bacteria and (2) assign family membership to one of the six most abundant bacterial families in urinary tract infections (Enterobacteriaceae, Morganellaceae, Pseudomonadaceae, Enterococcaceae, Staphylococcaceae and Streptococcaceae). The classification models were applied to artificial mixtures of Gram positive and Gram negative bacteria to correctly predict mixed infections with an accuracy of 75%. Raman scans of dried droplets did not yet yield optimal classification results on family level. When translating the method to primary urine samples, we observed a strong bias towards Gram negative bacteria, on family level towards Morganellaceae, which reduced prediction accuracy. Spectral differences were observed between isolates grown on standard growth medium and bacteria of the same strain when characterized directly from the patient. Thus, improvement of the classification accuracy is expected with a larger data base containing also bacteria measured directly from the urine sample.
Introduction
Urinary tract infections (UTIs) are among the most frequent nosocomial infections, affecting annually the life of more than 150 million people worldwide, resulting in high health care costs (approximately 6 billion USD per year).1,2 UTIs have a high clinical significance in urology. Here they represent up to 40% of all nosocomial infections,3 where 80% of these infections are catheter-associated. Catheter-associated UTIs are connected with increased morbidity and mortality, and are the most common cause of secondary bloodstream infections.2,4 In addition, the risk of infection increases with prolonged catheterization.2,4 Another risk factor for increased prevalence of UTI is age.5 UTIs are mostly caused by Gram negative bacteria, such as Escherichia coli, Proteus spp., Enterobacter spp., Klebsiella spp., or Pseudomonas aeruginosa; but also Gram-positive bacteria like Enterococcus spp. and Staphylococcus spp. can cause infections in the urinary tract.2 UTIs can be caused by pathogens of one strain, but also by pathogens of different species causing a mixed (polymicrobial) infection. Almost all families and species interact with each other in polymicrobial infections. However, Morganellaceae are found almost exclusively in polymicrobial infections, presumably because they depend on other species. Streptococcaceae and Enterobacteriaceae promote the colonization of other species by influencing the host immune system. Pseudomonadaceae often lead to chronic infections through biofilm formation. Enterococcaceae, Enterobacteriaceae and Pseudomonadaceae show often multi-drug resistance.6 It is estimated that 3–24% of UTIs are mixed infections with probably slightly higher numbers in case of complicated UTIs and high prevalence of 30–86% in catheter associated UTIs. Age is also a risk factor for polymicrobial infections with 33% of mixed cultures originating from elderly patients.6–8 Furthermore, women are at increased risk. However, mixed UTIs in outpatients occur often due to preanalytic contaminations.9 Another risk factor for mixed infections is the presence of a catheter: samples obtained from catheterized patients show a higher probability for the occurrence of mixed infections,8,10–12 especially in long-term catheterization.13 Furthermore, polymicrobial infections are observed more frequently in complicated UTIs that require treatment, than in uncomplicated UTIs. Both, polymicrobial catheter-associated or complicated urinary tract infections are associated with increased mortality.6,14 It is assumed, that polymicrobial interactions can promote biomineralization, tissue damage, dissemination and modulation of the host immune system, which can promote diseases like urolithiasis or inflammation of several parts of the genitourinary tract.6 Finally, treatment failure or development of resistance of a mixed UTIs can lead to serious and life-threatening complications like renal failure or urosepsis, especially in high-risk patients.Despite the increasing interest on the clinical relevance of mixed UTIs in the last years,8,10–12 question how to deal with them was not yet addressed. In routine diagnostics, the presence of further pathogen in the culture is connected with complications. In one hand, if there is insufficient second pathogen concentration, such a culture is considered as contamination or colonization, and usually it may not be reported by laboratories. So, the piece of information relevant for long-term catheterization, elderly and high-risk patients is lost.12,15 On the other hand, when second pathogen counts are enough, there is no established routine how to identify responsible pathogen for particular UTI episode11 resulting in time-consuming, expensive, and labour-intensive identification and susceptibility testing of most present organism. Finally, both mentioned situations lead to broad-spectrum antibiotic treatment and the risk of bacterial resistance increase11 and may lead to potential life-threatening complications described above. Current conventional diagnostic methods, which are used as gold standard, require at least one cultivation step, so that the identity of the bacteria is available after 24 hours or more. In addition, the routine, based on monobacterial growth with significant count,8 is inherently designed for single pathogen UTIs. To obtain particular information about sample until then, some urine screen tests are applied before culturing, like Gram staining followed by microscopy, or tests on leukocyte esterase or nitrite via dipsticks indirectly indicating infection.5,7,16 Nevertheless, this approach suffers relatively low sensitivity and high false positive rate.5 On the other hand, other precise techniques able to shorten time of diagnosis, like PCR-based techniques, electrochemical DNA biosensors, mass spectrometry (MALDI-TOF) are expensive or have special requirements on preparation or personnel. On research level, Raman spectroscopy in combination with multivariate data analysis proved to be a powerful alternative to identify bacteria at the species level1,15,17 and their resistance pattern18–21 in real time with high accuracy, in a cultivation-independent manner and no special requirements on sample preparation.
However, to the best of our knowledge, most work on this topic focused on laboratory bacterial strains under optimal growth conditions (medium broth, agar plates) or selected clinical isolates. When translating the technique to primary urine samples, additional complexity is introduced as the exact chemical composition of the patient's urine is not known and might even change with disease state, but also with nutrition. Bacteria are likely to adapt their metabolic state and thus might be in different states than after cultivation under defined laboratory conditions.17,22 Furthermore, in case of real patients' samples studies, UTIs caused by single pathogens were included23 or if the mixture, the attention was focused only on one pathogen on purpose.17,20,24 Only few works15,25 were devoted to the Raman spectroscopic analysis of mixed infections in the form of artificial mixtures, i.e. again under optimal growth conditions.
The aim of this study is to present the potential of Raman spectroscopy for cultivation-independent bacteria identification directly from patient's urine samples based on a large Raman database covering the clinical spectrum of pathogens and also to shed more light onto spectroscopy-based diagnostic of polymicrobial infections. For this, bacteria are first classified according to Gram type as this information is valuable for selecting preliminary patient treatment. A second classification model evaluates the potential to also differentiate bacterial families to provide a more detailed picture on the infection.
Experimental
Patient's urine samples
The research involving human urine samples has been complied with all relevant national regulations, institutional policies and in accordance to the tenets of the Helsinki Declaration. The study was approved by the local ethics committee (approval number: 2021-2186-Material, ethic commission of Jena University Hospital). Informed written consent was given. All samples were fully anonymized. Two monovettes, each containing 10 ml of urine, were collected from the urine cup (midstream urine) or the urine bag (catheter urine) after informed consent by the Department of Urology of the Jena University Hospital. Sixty-four samples were collected from a total of 61 patients. Five samples were later excluded (see below, ESI Table S1†). The remaining 59 urine samples were collected from 19 catheterized patients and from further 38 patients with UTI suspicion (midstream urine) and from two patients with navel pouch or ileum-conduit.One monovette was subjected to routine microbiological analysis (gold standard), while the other monovette was subjected to Raman measurements at the same time. The maximal delay between sample collection and Raman sample preparation was 2 days, meanwhile urine was stored in the fridge at 4 °C.
Microbiological analysis of patient's urine samples
Within routine diagnostics, urine samples were streaked onto Columbia sheep blood agar and Drigalski lactose agar (Oxoid, Thermo Fisher Scientific, Wesel, Germany) using an BD Kiestra™ InoqulA automated sample processor (BD, Heidelberg, Germany). Four-quadrant streaking was applied with a volume of 20 μl. After overnight cultivation at 37 °C, plates were examined and grown bacterial colonies were identified using MALDI-TOF (Vitek MS, bioMérieux, Nürtingen, Germany). In the case, that no bacterial growth was detected from a patient's urine sample after three days, this patient was excluded from further comparison with Raman prediction (true for 4 out of 62 patients).Isolated bacterial cultures on blood or Drigalski agar plates were included in this study as clinical isolates, see ESI Table S2.†
Urine sample preparation for Raman measurements
At first, eukaryotic cells were removed from the urine samples using a 5 μm syringe filter (Filtropur). Bacteria in the remaining suspension were washed twice in 5000 μl of sterile deionized water (Hettich centrifuge with relative centrifugation force (rcf) 4190g, 150 rad mm−1 for 10 minutes), the supernatant was carefully removed with a pipette and the pellet was resuspend again in 1000 μl of sterile deionized water.Clinical isolates
Clinical isolates of bacteria from urinary tract infections originated from the strain collection of the Medical Microbiology at the Jena University Hospital as well as were directly isolated from the urine samples in this study. Bacteria were used from cryostocks or directly from isolation plates and re-cultivated on Müller Hinton 2 agar plates (Millipure®, Sigma-Aldrich) at 37 °C and 5% CO2. After overnight culture, bacteria were harvested and washed 3x times by resuspending them in 1000 μl of sterile deionized water after centrifugation (rcf 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500g for 1.5 minutes, Eppendorf microcentrifuge 5418).
500g for 1.5 minutes, Eppendorf microcentrifuge 5418).
It total, 85 bacterial strains belonging to 23 species were included in this study: 11 Escherichia coli strains, 14 Klebsiella spp. strains, 13 Pseudomonas spp. strains, 13 Enterococcus spp. strains, 6 Enterobacter cloacae strains, 7 Proteus mirabilis strains, 5 Streptococcus spp. strains, 9 Staphylococcus spp. strains, 2 Providencia rettgeri strains, 2 Citrobacter koseri strains, 1 Morganella morganii strain, 1 Acinetobacter ursingii strain, and 1 Corynebacterium amycolatum strain (ESI Table S2†).
Artificial mixtures
In total, four artificial mixtures were prepared to test the Raman analysis approach with defined samples under identical measurement conditions. Bacterial strains were chosen to fulfil following criteria: 1. To have one Gram negative and one Gram positive representatives in a mixed sample and 2. Should be a pathogen encountered in our clinical urine samples. As Gram negative strain we focussed on E. coli as it is the most common UTI-causing pathogen and selected four different isolates to include biological variation. As Gram-positive pathogen we selected two different E. faecalis strains as being the most common Gram positive species causing UTI. Furthermore, two different Staphylococcus species were chosen to represent both, coagulase-negative and -positive species. Artificial Mixture 1 was chosen to contain bacteria from the same patient isolate. The composition of the artificial mixtures is given in Table 1. Each mixture was measured twice in at least two independent batches.| Mixture name | Strain 1 | Strain 2 | Volume ratio strain 1:strain 2 |
|---|---|---|---|
| Mix_1 | E. coli urRP41 | E. faecalis urRP41 | 1:3 |
| Mix_2 | E. coli urRP59 | E. faecalis urRP56 | 1:3 |
| Mix_3 | E. coli urRP65 | S. warneri urRP20 | 1:1 |
| Mix_4 | E. coli urRP18 | S. aureus urRP022 | 1:3 |
Bacteria were cultivated overnight in 20 ml of AT2 medium in separate flasks. The optical density (OD) at 600 nm of the overnight culture was adjusted to yield 20 ml suspension with OD between 0.08–0.1 (cell Density Meter, Fisher Scientific, Fisherbrand), corresponding to McFarland standard 0.5. After that, each suspension was centrifugated at 4190 rcf for 10 min, supernatants were removed, and both pellets were resuspended into 1000 μl of diluted Raman medium (AT2 + 0.5 PBS). A total volume of 1000 μl of different artificial mixtures were created as outlined in Table 1. Different volume ratios were necessary to ensure that both bacterial strains were present in sufficient quantities. To verify the presence of strains in the mixture, 100 μl of the suspension was plated on MH2 agar plate. Afterwards, cells were washed twice in sterile deionized water (for 1.5 minutes at rcf 13500g Eppendorf centrifuge 5418), resuspend in 1000 μl of sterile deionized water.
Raman spectroscopic analysis of dried bacterial films
The sample volume of 3–5 μl (slightly varying cell concentration depending on sample and patient) were drop casted onto a CaF2 slide (Crystal GmbH, Germany) and allowed to dry in a heating chamber for 30 minutes at 50 °C. So totally, a rough time to prepare samples for Raman measurements ranges from 50 minutes (clinical isolates) to 90 minutes (patient samples). In case of patients’ samples, the delay between sample preparation and Raman measurement itself was up to 30 days, meanwhile dried droplets were stored in the fridge at 4 °C in isolated sterile Petri dish. Typical droplets are shown in ESI Fig. S1.†Raman measurement were performed using an upright CRM 300 WiTec micro-Raman system, equipped with UHTS spectrometer with 600 lines per mm grating, and air-cooled back-illuminated CCD camera (DV401 BV, ANDOR, 1024 × 127 pixels, cooled to −60 °C). Raman scattering was excited with 532 nm line of Nd:YAG laser with the power of 15 mW before passing the objective. The laser light was focused onto the sample using a 63× objective (Zeiss LD Plan-Neofluar Korr M27, NA 0.75), allowing a maximum spatial resolution of 355 nm under optimal conditions when using Abbe's formula (d = λ/2NA). Back-scattered Raman signal was collected and forwarded to the spectrograph by a multimode optical fibre with 50 μm core diameter. Performance and alignment check of the device was performed using silicon and 4-acetamidophenol on each measurement day.
Dried droplets of clinical isolates were measured in at least 3 independent batches per strain resulting in more than 3400 Raman spectra from the 23 different species. For each batch, at least 10 single spectra from different locations on the sample were recorded with acquisition time of 10 s.
Dried droplets of artificial mixtures and patient samples were measured as image scans in automated scanning mode covering an area of at least 20 μm × 20 μm, with a step size of 0.333 μm in XY directions with 5 s acquisition time per spectrum. Thus, at least 3600 spectra per sample were collected.
Data processing and statistical analysis
All computations were performed in programming language R 4.0.2.26 At first, spectra were despiked and wavenumber calibrated with available standard data (4-acetamidophenol).27 In next step, interpolation onto 610–3050 cm−1 range was performed with 2.5 cm−1 wavenumber step. Spectra were further baseline corrected with statistics-sensitive non-linear Iterative peak-clipping (SNIP)28,29 algorithm with 40 iterations and smoothing at 1st iteration, then the silent region (1750–2750 cm−1) was excluded. Further, vector normalization and quality check were done. As parameters for quality check filters, integrated background-to-signal ratio less than 50 and signal-to-noise ratio (SNR) above 1 were considered. The spectra with SNR above 50 were also removed because those were saturated spectra.After preprocessing and quality check using all spectral data, classification models were built using only spectra of clinical isolates. Principal component analysis (PCA) was carried out to reduce the dimensionality of the dataset, then, linear discriminant analysis (LDA) was utilized as a binary (Gram positive vs. Gram negative) classification model and a 6-class (bacterial families) model. In both cases, balanced model weights were used to get the optimal trade-off between the sensitivity and specificity. The number of principal components used in the LDA was optimized in the leave-one-replicate-out cross-validation.30 In such validation scheme for the total of N replicates, the model trained on N − 1 replicates is applied to the replicate excluded from the training. The procedure is repeated N times to obtain predictions for all replicates but avoids the situation when the spectra from the predicted replicate are included in training. Nine PCs were used in the Gram-model and 39 PCs were used in the Family-model. These two models were then used to predict bacteria identity in the artificial mixtures as well as the clinical urine samples.
Spectra of artificial mixtures and the patients’ samples were not utilized in training and were only used for result evaluation. In the case that after quality check, less than 10 spectra were kept from a patient, this patient was excluded from final evaluation (true for 1 patient in our set). Within each spectral scan, the number of spectra assigned by the model to each class were investigated. The predictions within each scan are normalized to the maximal value, thus limiting the normalized predictions between 0 and 1. All classes with more than 0.1 normalized predictions were considered present in the sample. Thus, each scan could be predicted as a member of a single class or multiple classes. Prediction accuracy was calculated by comparing with microbiological findings.
Results and discussion
The general idea of proposed approach is that Raman spectra of mixtures are composed of Raman spectra of each contained pathogen. Thus, in a first step, a Raman data base of clinical isolates from urinary tract infection is created and a classification model is built to assign Gram type and classify bacteria on family level based on their Raman spectra. After evaluation with cross-validation on clinical isolates, the model is applied to artificial mixtures created from the clinical isolates and finally translated to fresh patient's urine samples which are directly analysed without any additional cultivation step.Raman analysis of clinical isolates
In total, 3532 Raman spectra of 85 bacterial strains belonging to 23 species were recorded from clinical isolates: 1173 spectra of Gram positive bacteria (28 bacterial strains of 11 species) and 2359 spectra of Gram negative bacteria (57 bacterial strains of 12 species) (Table 2, ESI Table S2†). This data base provides a good representative selection of the most common UTI pathogens.2| a Two more families (Moraxellaceae and Corynebacteriaceae) were included in training the Gram type model, however, they were left out for training the family model. |
|---|

|
Raman mean spectra together with their standard deviation of Gram negative and positive bacteria included in the study are depicted in Fig. 1a. Typical spectral features of bacteria can be identified, e.g., 783 cm−1 (cytosine, thymine ring breathing31,32), 1005 cm−1 (phenylalanine ring breathing33), 1080 cm−1 (C–N stretch of proteins31), 1097 cm−1 (PO2− stretching in DNA33), 1250 cm−1 (amide III32,33), 1340 cm−1 (adenine, guanine, CN-stretching in purine nucleobases33), 1450 cm−1 (deformations of CH2 scissoring31,34), 1578 cm−1 (ring stretching of guanine, adenine24), 1670 cm−1 (amide I, lipids31,34), and overlapping bands at 2850 cm−1 and 2935 cm−1 (CH3 and CH2 stretching35). A detailed assignment of the Raman bands is given in ESI Table S4 and Fig. S2.† Clear spectral differences are visible between the Raman spectra of Gram positive and Gram negative bacteria. Fig. 2 shows the computed difference spectrum of the Raman mean spectra. The most visible difference is found around 748 cm−1 (position 7 in Fig. S2, Table S4†), range 900–1000 cm−1 (bands 14–18, Fig. S2, Table S4†), 1312 cm−1 (position 34) and 1578 cm−1 (position 45, Fig. S2, Table S4†). These differences can be mainly explained with the different cell wall compositions of Gram positive and negative bacteria. Similar results have been reported in previous studies.23
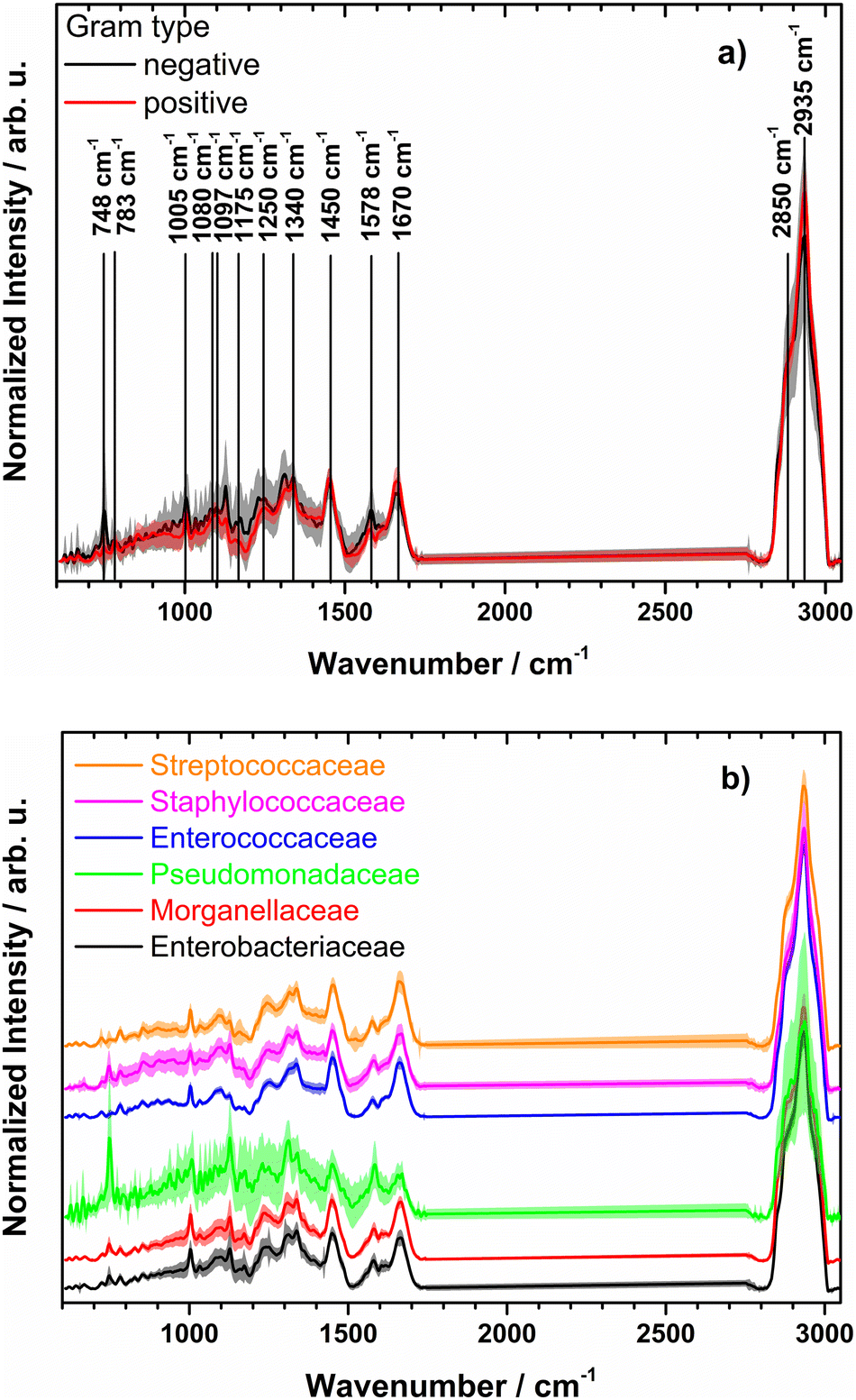 | ||
| Fig. 1 (a) Normalized mean Raman spectra of Gram negative (black) and Gram positive bacteria (red) together with standard deviation (shown as shadow). Raman band assignment is given in ESI Table S2.† (b) Mean preprocessed Raman spectra with standard deviations for families used for training the model (spectra are shifted on y axis for clarity). Genus and strains included per family are found in Table 2 and ESI Table S2,† respectively. | ||
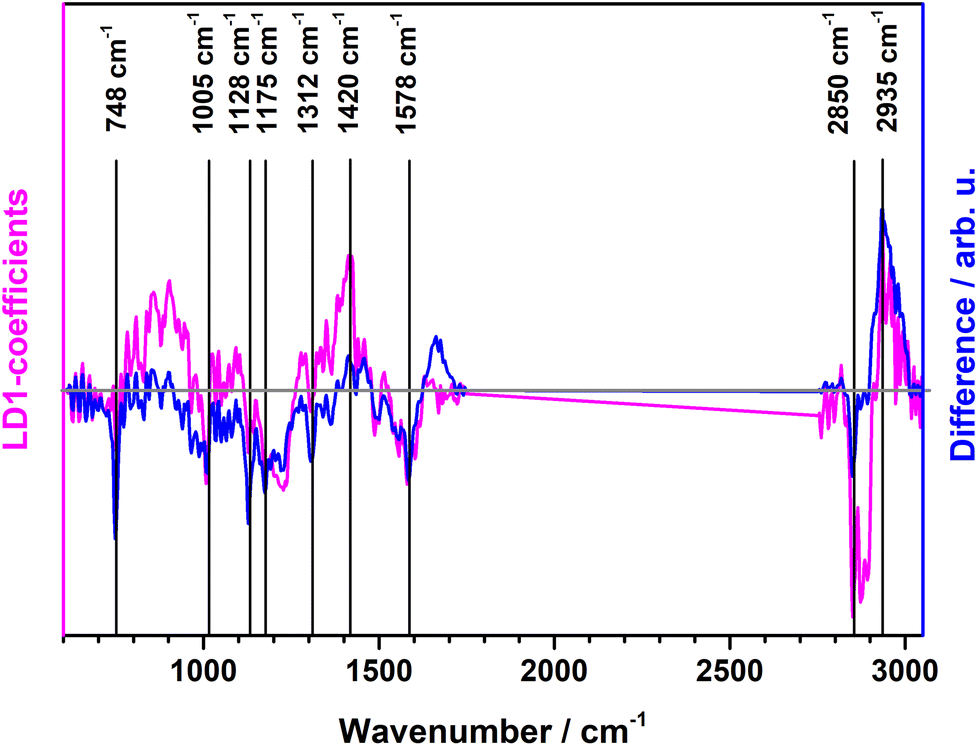 | ||
| Fig. 2 PCA-LDA loading plot of LD1 (pink) along with computed difference spectrum (Gram positive minus Gram negative) of the Raman mean spectra (blue). | ||
It has to be noted, that the standard deviation of Raman spectra from Gram negative species is higher than the standard deviation of Gram positive bacteria. This can be explained with the large variety of Gram negative bacteria included in this study (9 different bacterial genera compared to 4 Gram positive genera, see Table 2 and ESI Table S2†).
PCA-LDA classification model to differentiate Gram type of clinical isolates
A binary PCA-LDA classification model was trained with the clinical isolates to differentiate the Gram type of bacteria. A good differentiation was achieved as can be seen in the LD1 scatter plot (Fig. 3), where each point represents an individual bacterial Raman spectrum. The PCA-LDA loading of LD1 shows the same spectral features as the computed differences as can be seen in Fig. 2. The most prominent contribution to LD1 corresponds to the bands at 2850 cm−1, given by its intensity variance within Gram-type (more pronounced and less variance for Gram positive strains). Further prominent positive contribution to LD1 are found around 1420 cm−1 and indicated a higher abundance of this Raman band in Gram positive bacteria. It has been assigned to peptidoglycan.23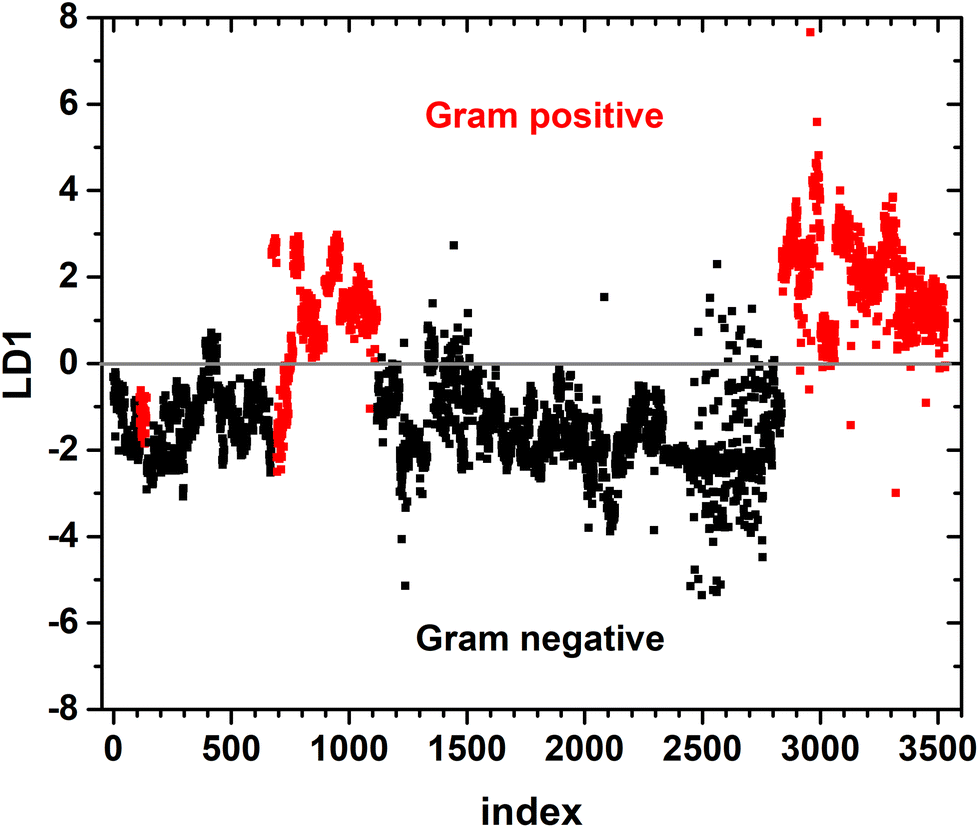 | ||
| Fig. 3 LDA score plot showing prediction of Gram type. The index number labels individual bacterial Raman spectra; colour codes the true Gram type (black: Gram negative, red: Gram positive). The grey line divides the graph into positive and negative parts of LD1 axis. | ||
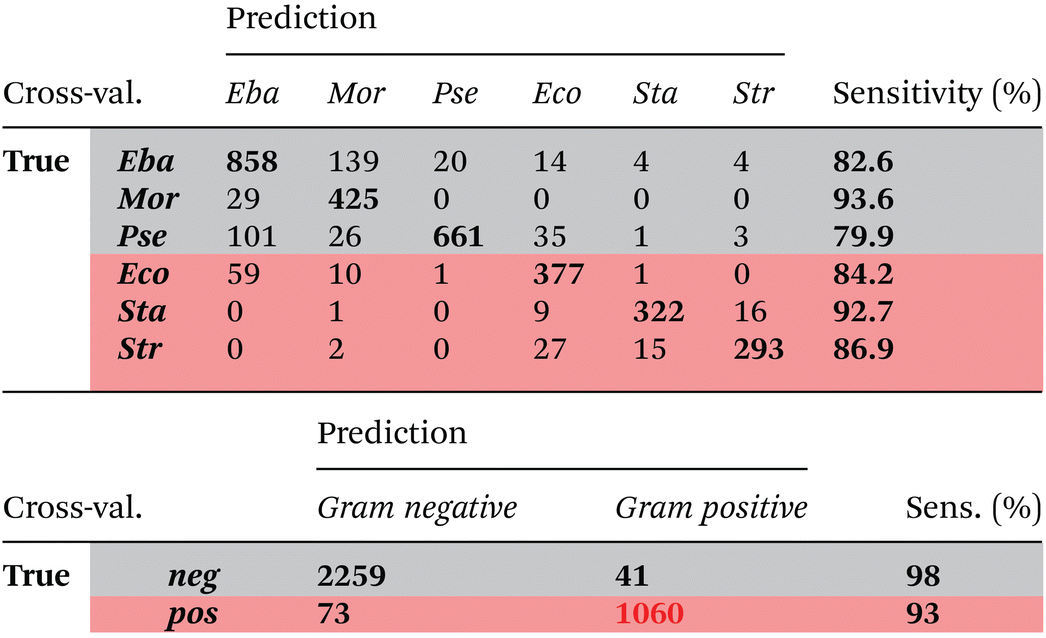
Quantitative results leave-one-out prediction are summarized in the confusion matrix in Table 3. High sensitivity and specificity for both classification levels are reached and exceed 90%, giving a balanced accuracy of 93.6%.
| Cross-validation | Prediction | Sensitivity (%) | ||
|---|---|---|---|---|
| neg. | pos. | |||
| True | neg. | 2256 | 103 | 95.6 |
| pos. | 99 | 1074 | 91.6 | |
PCA-LDA classification model to differentiate clinical isolates on family level
Fig. 1b depicts the mean preprocessed Raman spectra with standard deviations of the six different bacterial families. Some of the families, like Pseudomonadaceae, Staphylococcaceae and Enterobacteriaceae, demonstrate large intra-family variance. The former corresponds to observations of Rebrošová et al.31 Variations were explained with varying fluorescence background and production of exopolysaccharides as well as by different abundance levels of pigments, especially pyoverdine and fluorescein. Mean spectra per isolate of Pseudomonadaceae are presented in ESI Fig. S3.† Spectral variations have also been reported to occur within individual Staphylococcus species.36 Thus, a higher standard deviation within the Staphylococcaceae family is not surprising. The family of Enterobacteriaceae covers manifold bacterial species in comparison with another families, as can be clearly seen in Table 2 and Table S1,† which might introduce spectral variations due to different chemical compositions of the family members.Overall less spectra are included here as spectra of Moraxellaceae and Corynebacteriaceae were not used.
A six-class PCA-LDA model was trained to predict membership to respective bacterial family. Results of leave-one-replicate cross-validation are summarized in the confusion matrix (Table 4). An overall balanced accuracy of around 87% was reached. Best sensitivities are reached for Morganellaceae (>93%) and Staphylococcaceae (>92%). Lowest sensitivities were observed for Pseudomonadaceae (>79%) and Enterobacteriaceae (>82%) families. However, it has to be noted, that most mispredictions occurred within the same Gram type, namely between Pseudomonadaceae and Enterobacteriaceae as well as between Enterobacteriaceae and Morganellaceae. High similarity between Enterobacteriaceae and Morganellaceae is also seen in Fig. 1b. Both families belong to the order of Enterobacterales. High similarity of Raman spectra of bacteria from Enterobacteriaceae and Morganellaceae have been also reported previously,15 where it was not possible to separate spectra of E. coli and P. mirabilis using simple PCA clustering. The low sensitivity for predicting Pseudomonadaceae family might be due to the fact that within this family, a high standard deviation among individual strains is observed.
|
Application of the classification model to artificial mixtures
In the next step, artificial mixtures, containing a representative of Gram negative as well as Gram positive bacteria, were prepared and between 6600 and 21500 spectra were measured per mixture. The two-class Gram model as well as the 6-class bacterial family model were applied to predict the class membership for each individual spectrum. Predictions in the most abundant class were normalized to one and a relative prediction of less than 0.1 was not considered. Prediction results for the Gram model are shown in Table 5. An overall prediction accuracy of 75% is reached. Three out of four artificial mixtures are correctly predicted to be mixtures of Gram positive and Gram negative bacteria in all biological independent batches. For one mixture (Mix_1) very heterogeneous prediction results are observed for different biologically independent batches. Plating results of the bacterial suspensions indicate a relatively stable relative ratio of Gram positive and Gram negative bacteria in the mixture (ESI Table S3†), and therefore, cannot be the reason for the wrong predictions. Differences in the predicted ratio between batches (biological replicates) might originate from an inhomogeneous distribution of the bacteria within the dried droplet, which would result in a bias depending on the selected region for measurement.
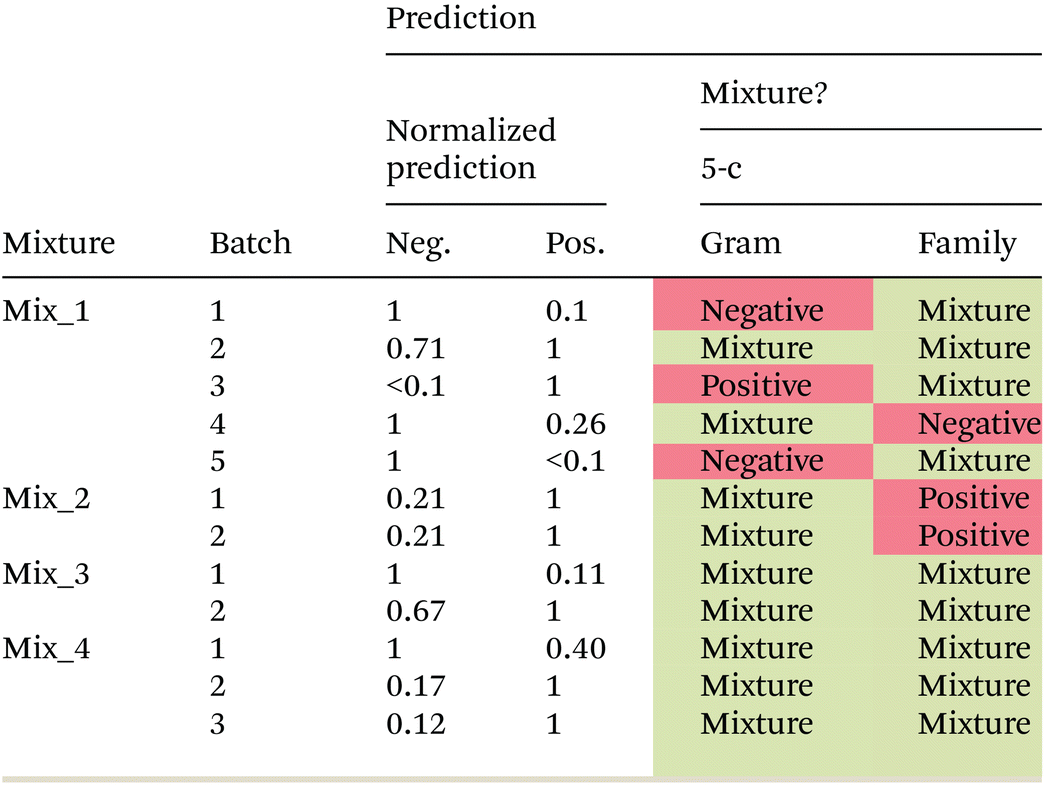
|
Prediction results for the artificial mixtures using the family model trained with the bacterial isolates are listed in Table 6. Except for artificial mixture 2 (Mix_2) and two batches of artificial mixture 1 (Mix_1), the truly present bacteria were always correctly predicted to be present in the mixture. However, a fully correct prediction was only achieved for batch 5 in artificial mixture 1. In all other cases (except batch 2 of Mix_1), also other families were predicted to be present in the mixture. In most cases, these other families contributed only to a minor proportion. However, in a very few cases they made up the majority (e.g., batch 2 in Mix_3 and batch 4 in Mix_4). Thus, it can be concluded that the current model did not proof yet powerful enough to predict correct family membership in mixed samples. One reason could be that image scans were recorded of dried droplets. We have chosen the step size (i.e. the pixel size of one point) with 0.333 μm rather small and also smaller than the average size of a bacterium (0.5–1 μm in diameter). However, it cannot be excluded that in a dried sample more than one bacterium was contributing to the spectrum and therefore making precise family assignment difficult. Nevertheless, as in most cases truly present bacteria were correctly predicted to be present, we aggregated the family predictions (Table 6) according to the Gram type and used this prediction to identify mixtures. The results are shown in the last column of Table 5. The same overall accuracy of 75% is reached as for the Gram type model. However, different batches were wrongly predicted to contain no mixtures.
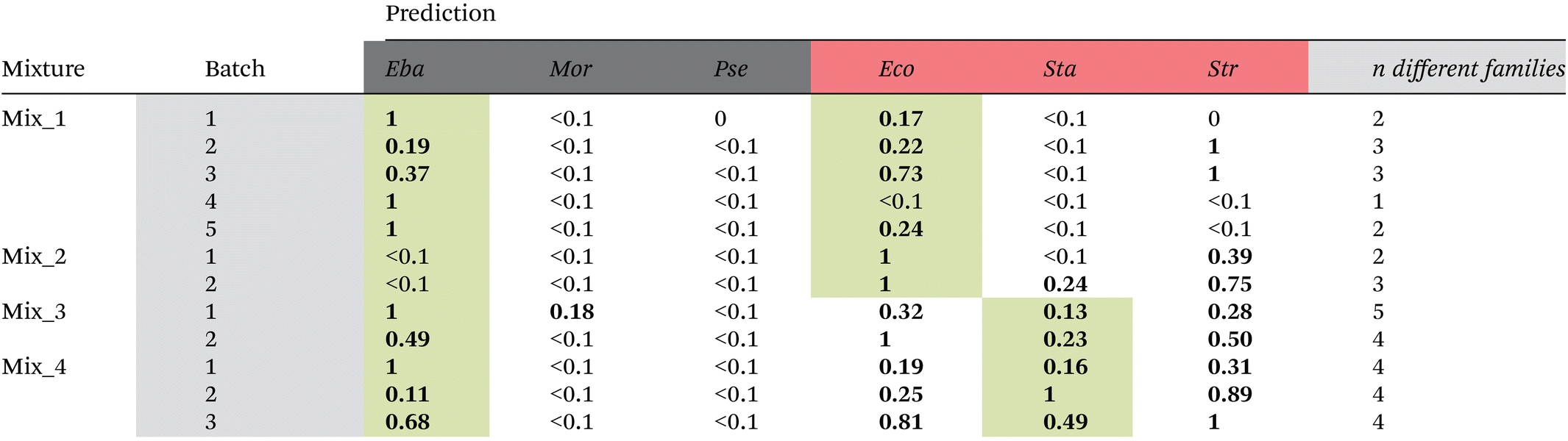
|
The achieved accuracies for mixture predictions with our PCA-LDA models are comparable to previously reported results of mixture analysis where prediction accuracies of up to 73 and 89% were achieved with PLS-DA and SVM, respectively.15
Translating the classification models to fresh patients’ samples
For the evaluation of our classification model with real world primary urine samples, a total of 64 patients’ urine samples were collected. Five samples had to be excluded, due to removal of all but one Raman spectrum during quality control (one patient) and no bacterial growth during microbiological cultivation (4 patients) (ESI Table S1†). Thus, for the following analysis, 59 patients’ urine samples were used. Among those, 21 samples were collected from catheter (incl. pouch) and 38 samples were midstream urine samples. Among the 56 patients, 20 were female (35.7%) and 36 were male (64.3%). Average age was 69 ± 15.3 years (female: 65.5 ± 17.8 years; male: 71 ± 13.3 years). One aliquot of the urine was analysed by the presented Raman algorithm without prior knowledge of its content. A second aliquot of the same urine sample was analysed by routine microbiological analysis.Results of Raman and microbiological analysis are provided for each of the 59 samples in ESI Table S5.† Microbiological analysis revealed that ∼50% of samples (29 out of 59 samples) showed mixed (polymicrobial) infections. In 18 samples with mixed infections, two pathogens were identified, in 11 samples three or more pathogens were identified (ESI Table S5†). Mixed infections were slightly more likely from catheter samples (11 out of 17 samples (∼65%) were mixed infections) than from midstream urine (18 out of 42 samples (∼43%) were mixed infections).
These findings are in line with results from other studies, where 30–86% of UTIs were reported to be true mixed infections.7,8 With an average age of 69 ± 15.2 years our cohort includes also a significant portion of elderly patients, which are more susceptible for mixed infections.12 Higher prevalence of mixed infections in samples originating from catheterized patients has also been reported.8,10–12
In very few cases, bacterial species were identified (always as part of mixed infections) which were not included in the training data set, e.g., Serratia marcescens in patient 21. Corynebacterium amycolatum (from patient 27) and Acinetobacter ursingii (from patient 29) were included in the Gram model, but not in the family model. In one patient, also fungi were identified as pathogen in mixed infections. However, with the currently applied sample preparation protocol, fungi are not expected in the Raman sample due to their size (>5 μm) and the filtration step at the beginning.
Table 7 summarized the results of the PCA-LDA classification analysis of the Raman data. At first, we applied the 2-class Gram type model to identify patients’ samples with mixed infections of Gram positive and Gram negative bacteria from infections with only Gram positive or Gram negative bacteria (Table 7, top). The model achieved an overall balanced accuracy of 49%, when considering correct predicting to one of the three options. The achieved accuracy is significantly lower than for the artificial mixtures where the three options could be predicted with an accuracy of 75%. When analysing the wrong predictions, a strong bias towards Gram negative bacteria is observed which is reflected in the high sensitivity and low specificity for this class in Table 7. This means, almost all Gram negative bacteria were correctly predicted to be Gram negative, while also many mixtures and Gram positive bacteria were wrongly predicted to be Gram negative bacteria. High specificity and low sensitivity were reached for Gram positive bacteria. This means, that no Gram negative bacteria or mixtures were predicted to be Gram positive bacteria.
| Bal. acc. 49% | Pred. Gram model | Sensitivity (%) | Specificity (%) | |||
|---|---|---|---|---|---|---|
| Neg. | mix. | Pos. | ||||
| True | Neg. | 26 | 5 | 0 | 83.9 | 28.6 |
| Mixture | 12 | 3 | 0 | 20 | 77.3 | |
| Pos. | 8 | 5 | 0 | 0.0 | 100.0 | |
| Bal. acc. 44% | Pred. family model | Sensitivity (%) | Specificity (%) | |||
|---|---|---|---|---|---|---|
| Neg. | mix. | Pos. | ||||
| True | Neg. | 21 | 10 | 0 | 67.7 | 39.3 |
| Mixture | 11 | 4 | 0 | 26.7 | 63.6 | |
| Pos. | 6 | 6 | 1 | 7.7 | 100.0 | |
A similar trend is observed when predicting the family membership with the 6-class family model and aggregating the family predictions according to the Gram type (Table 7, top). Here, an overall balanced accuracy of 44% is achieved. A similar bias towards predicting the presence of Gram negative bacteria in the sample as for the Gram model is observed. Upon closer investigation of the prediction on family level (ESI Table S5†), it can be seen, that in all, but two patients’ samples (patient 13 and patient 24), Gram negative Morganellaceae are predicted to be present in the sample. However, this is correct only for 8 patients’ samples and wrong for 47 patients’ samples.
For seven patients’ samples, two independent dried droplets were prepared and measured. In the analysis above, the samples with the most remaining spectra after automated Raman quality filtration were included. ESI Table S6† shows the prediction results of both batches for each of the seven urine samples. In most cases, a good overall agreement is found between the replicate samples.
It has to be noted that the classification models were trained with clinical isolates that were cultivated under ideal microbiological conditions, i.e. on Müller-Hinton 2 agar plates, while patients’ urine samples were directly analysed without any cultivation step. An influence of growth medium on Raman spectra has been reported in earlier studies22 and explained with a slightly changed overall chemical composition due to different nutrients in the medium. Storage of the samples prepared for Raman measurement might also play a role, as was shown in ref. 37.
In order to explore, if the growth medium effect could be relevant for our samples, we performed unsupervised principal component analysis with the Raman spectra of the pathogens when measured directly from the urine sample and after isolation and cultivation on Müller-Hinton 2 agar plates. For this, all 14 pairs of pathogens which were measured directly from urine and after isolation and cultivated were included in the analysis. Selected individual scores plots and the combined scores plot of all samples are presented in ESI Fig. S4.† Exemplarily, the PCA scores plot is shown for patient's sample number 16 in Fig. 4a. Raman spectra of the very same strain of Klebsiella pneumoniae measured directly from the urine sample (black dots) and after isolation and cultivation on Müller-Hinton 2 agar plates (red dots) show prominent differences already in the first principal component which describes 27% of the variation in the data set. Different measurement parameters of the isolates, such as single spectra (green dots) vs. image scan (red dots) do not have an effect on spectral variation as is clearly visible from coinciding green and red clusters in Fig. 4a. Differences in growth condition (in urine vs. Mueller-Hinton 2 agar plates) seem to be larger than differences between species and families as significant separation of growth conditions is visible already in PC1 in an unsupervised PCA model containing strains from different families (Fig. 4b and ESI Fig. S4†). The loading plot of PC1 (Fig. 4b) shows large contributions from the spectral region of CH-stretching. In addition, changes in bands corresponding to phenylalanine (1005 cm−1, 1590 cm−1), amide III (1250 cm−1), guanine (1310 cm−1), CH modes of glucosamines and proteins (1440 cm−1), and peptidoglycan (1590 cm−1) are visible in the fingerprint region. For all three selected bacterial strains, PC1 captures most differences.
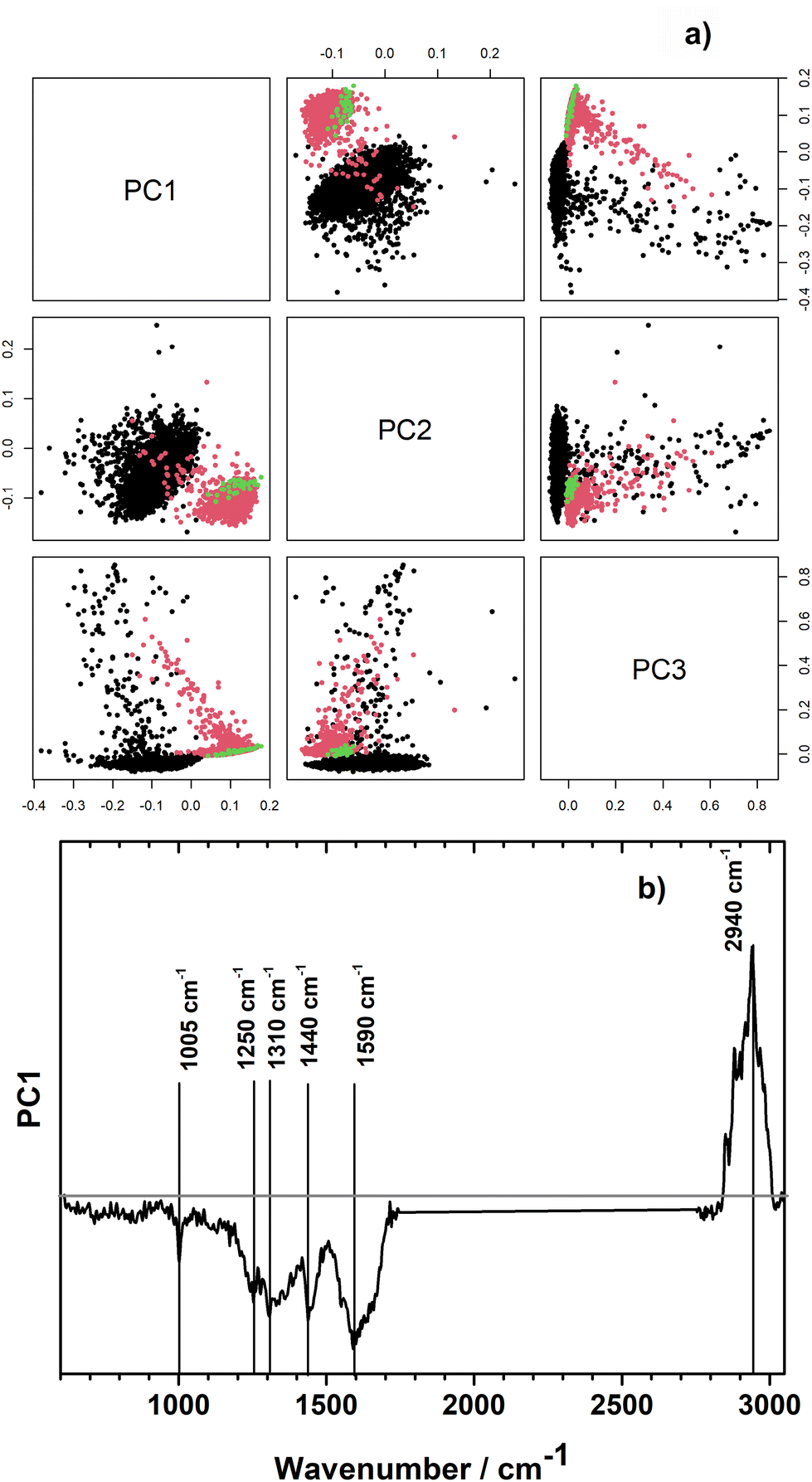 | ||
| Fig. 4 (a) PCA score plot showing the differentiation of one-pathogen patient sample no. 16 (black) and the same strain isolated for microbiological identification (red). Both measured under same measurement conditions (scans). Raman spectra of identical strain, reflecting different measurement mode (green, individual spectrum at 10 different position), is also added to discuss effect of different measurement conditions. (b) PC1 loadings plot. | ||
In order to avoid medium-induced effects on the classification model, it could be recommended to train the classification model with Raman spectra of bacteria directly from urine samples. In our small study we had only 30 urine samples with single pathogen infections (20 Gram negative and 10 Gram positive strains). For reliable classification models it is suggested to expand the data set. This will be within the scope of future work.
There are further factors that could affect the accuracy of the classification model to predict mixed infections. One would be the presence of bacterial species and families that were not included in the training model. In our case, we found Lactobacillus jensenii (Gram positive bacilli of family Lactobacillaceae) or Serratia marcescens (Gram negative rods of family Yersiniaceae). We assume that if the spectral data base is sufficiently large, also unknown species are assigned to the right Gram type or a closely related family.23
Furthermore, in literature, the application of different classification models has been discussed. In our study, we have chosen PCA-LDA, which is one of the most common approaches for classification tasks of isolated bacterial strains.24,33 However, other studies could demonstrate higher performance of other models, such as partial least squares-discriminant analysis (PLS-DA)15,38, k-Nearest Neighbours methods,23,31,36 or support vector machines (SVM),23 or deep learning methods39 to just name a few. A detailed comparison of different methods is beyond the scope of the manuscript and will be subject of further studies.
Excluded patients’ samples
During data analysis, a total of five patients had to be excluded from the direct comparison of Raman prediction and routine microbiological findings discussed above. Prediction results of Raman analysis of the urine from those patients are presented in ESI Table S7.† The reasons for excluding samples from the study were (1) less than 10 spectra were kept from a patient's sample after automated quality check of the Raman data (true for patient no. 60); (2) no bacteria were identified during routine microbiological analysis (patients no.: 61–64), which served as reference method in this study. As can be seen in the further descriptions on observations in the footnote of ESI Table S7,† in most of the latter cases, no pellet was seen by eye after sample preparation of patient's urine sample, being a first indication of low bacterial count in the sample.It has to be noted, that routine microbiological diagnostics relies on viable bacteria in the urine sample as it is a cultivation-based method that yields number of bacteria as colony forming units (CFU) per milliliter. Thus, bacteria that are already killed by a successful antibiotic treatment cannot be cultivated anymore, but might be still present (if not fully lysed yet) in the urine sample.
Furthermore, the current sample preparation workflow excludes fungal pathogens. If they should be included in future work, the filtration step needs to be modified.
Conclusions
Raman spectroscopy was demonstrated to be a powerful method to characterize bacteria. With a large data set of clinical isolates comprising 85 bacterial strains from 23 species a robust model could be trained to predict the Gram type of unknown bacterial samples with >93% accuracy. For predictions on family level, an overall balanced accuracy of around 87% was reached. Most misclassifications were observed within the same Gram type. When applied to artificial mixtures, the model could correctly predict mixtures of Gram positive and Gram negative bacteria with an accuracy of 75%, encouraging to translate this algorithm for the direct analysis of urine samples for which mixed infections are diagnosed in 30–86% of urinary tract infections. We have established a procedure for the fast preparation of bacterial samples for Raman spectroscopic analysis directly from 10 ml of patients’ urine samples. In our study 59 patients were included out of which 29 were found to have a mixed infection. When translating our classification model trained with clinical isolates cultivated under standard conditions to primary patients’ samples, we encountered a strong bias towards the prediction of Gram negative bacteria. PCA analysis revealed characteristic differences between bacteria from patients’ urine samples and after isolation and cultivation. It is therefore recommended to include Raman spectra of primary urine samples (e.g. with just single pathogen infections) into the training model to improve classification accuracies.Author contributions
KAD – Conceptualization, data curation, sample preparation, Raman measurement, supervision, validation, visualisation, writing-original draft, writing-review and editing. OR – Formal analysis, data curation, methodology, resources, software, validation, visualisation, writing-original draft, writing-review and editing. DS – Patient recruitment, data curation, investigation, methodology, project administration, resources, validation, writing-review and editing. VR & NL – Microbiological analysis, data curation, investigation, resources, writing-review and editing. MJGV – Sample preparation, Raman measurement, writing-review and editing. SF – Patient recruitment, writing-review and editing. MB – Resources, editing of manuscript. TWB – Software, supervision, writing-review and editing. JR – Microbiological analysis, supervision, writing-review and editing. JP – Supervision, writing-review and editing. UN – Conceptualization, funding acquisition, project administration, supervision, validation, writing-original draft, writing-review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the BMBF via ReHwIN (FKZ 13GW0432F) and CSCC (FKZ 01EO1502) is acknowledged. We acknowledge support by Photonics Research Germany (FKZ: Leibniz-IPHT LPI-BT4 13N15708), the European Union (Grant No. 861122: “ImageIN”), the Leibniz Society via the Leibniz ScienceCampus InfectoOptics (SAS-2015-HKI-LWC), the Jena Biophotonic and Imaging Laboratory (JBIL) and the Free State of Thuringia via ThIMEDOP (FKZ IZN 2018 0002).References
- U. Neugebauer, P. Rösch and J. Popp, Int. J. Antimicrob. Agents, 2015, 46, S35–S39 CrossRef CAS PubMed.
- A. Flores-Mireles, J. Walker, M. Caparon and S. J. Hultgren, Nat. Rev. Microbiol. Vol., 2015, 13, 269–284 CrossRef CAS PubMed.
- H. Rüden, P. Gastmeier, F. D. Daschner and M. Schumacher, Infection, 1997, 25, 199–202 CrossRef PubMed.
- R.-P. Vonberg, M. Behnke, C. Geffers, D. Sohr, H. Rüden, M. Dettenkofer and P. Gastmeier, Infect. Control Hosp. Epidemiol., 2006, 27, 357–361 CrossRef PubMed.
- E. Avci, N. S. Kaya, G. Ucankus and M. Culha, Anal. Bioanal. Chem., 2015, 407, 8233–8241 CrossRef CAS PubMed.
- J. R. Gaston, A. O. Johnson, K. L. Bair, A. N. White and C. E. Armbruster, Infect. Immun., 2021, 89, e00652–e00620 CrossRef CAS PubMed.
- B. S. Learman, A. L. Brauer, K. A. Eaton and C. E. Armbruster, Infect. Immun., 2020, 88, e00691–19 CAS.
- N. Nityadarshini, S. Mohapatra, H. Gautam, V. Jain, R. Chaudhry and A. Kapil, Trop. Doct., 2022, 52, 335–336 CrossRef PubMed.
- P. S. Whelan, A. Nelson, C. J. Kim, C. Tabib, G. M. Preminger, N. A. Turner, M. Lipkin and S. D. Advani, Antimicrob. Steward. Healthc. Epidemiol., 2022, 2, e29 CrossRef PubMed.
- Y. Siegman-Igra, T. Kulka, D. Schwartz and N. Konforti, J. Hosp. Infect., 1994, 28, 49–56 CrossRef CAS PubMed.
- R. O. Darouiche, M. Priebe and J. E. Clarridge, Spinal Cord, 1997, 35, 534–539 CrossRef CAS PubMed.
- G. Croxall, V. Weston, S. Joseph, G. Manning, P. Cheetham and A. McNally, J. Med. Microbiol., 2011, 60, 102–109 CrossRef PubMed.
- C. E. Armbruster, A. L. Brauer, M. S. Humby, J. Shao and S. Chakraborty, JCI Insight, 2021, 6, e144775 CrossRef PubMed.
- M. Melzer and C. Welch, Postgrad. Med. J., 2013, 89, 329–334 CrossRef PubMed.
- M. Yogesha, K. Chawla, A. Bankapur, M. Acharya, J. S. D'Souza and S. Chidangil, Anal. Bioanal. Chem., 2019, 411, 3165–3177 CrossRef PubMed.
- K. Kline and A. Lewis, Microbiol. Spectr., 2016, 4, UTI-0012-2012 Search PubMed.
- S. Kloss, B. Kampe, S. Sachse, P. Rosch, E. Straube, W. Pfister, M. Kiehntopf and J. Popp, Anal. Chem., 2013, 85, 9610–9616 CrossRef CAS PubMed.
- W. Zhang, H. Sun, S. He, X. Chen, L. Yao, L. Zhou, Y. Wang, P. Wang and W. Hong, Front. Microbiol., 2022, 13, 874966 CrossRef PubMed.
- D. Galvan and Q. Yu, Adv. Healthcare Mater., 2018, 7, 1701335 CrossRef PubMed.
- S. Stöckel, J. Kirchhoff, U. Neugebauer, P. Rösch and J. Popp, J. Raman Spectrosc., 2016, 47, 89–109 CrossRef.
- A. Tannert, R. Grohs, J. Popp and U. Neugebauer, Appl. Microbiol. Biotechnol., 2019, 103, 549–566 CrossRef CAS PubMed.
- K. Mlynarikova, O. Samek, S. Bernatova, F. Ruzicka, J. Jezek, A. Haronikova, M. Siler, P. Zemanek and V. Hola, Sensors, 2015, 15, 29635–29647 CrossRef PubMed.
- H. Hu, J. Wang, X. Yi, K. Lin, S. Meng, X. Zhang, C. Jiang, Y. Tang, M. Wang, J. He, X. Xu and Y. Song, Anal. Methods, 2022, 14, 4014–4020 RSC.
- U.-C. Schröder, A. Ramoji, U. Glaser, S. Sachse, C. Leiterer, A. Csaki, U. Hübner, W. Fritzsche, W. Pfister, M. Bauer, J. Popp and U. Neugebauer, Anal. Chem., 2013, 85, 10717–10724 CrossRef PubMed.
- Z. Pilat, S. Bernatova, J. Jezek, J. Kirchhoff, A. Tannert, U. Neugebauer, O. Samek and P. Zemanek, Sensors, 2018, 18, 1623 CrossRef PubMed.
- R Core Team, R Studio (version 4.0.2) R Foundation for Statistical Computing, 2020 Search PubMed.
- T. W. Bocklitz, T. Dörfer, R. Heinke, M. Schmitt and J. Popp, Spectrochim. Acta, Part A, 2015, 149, 544–549 CrossRef CAS PubMed.
- M. Morhac, Peaks. Peaks https://CRAN.R-project.org/package=Peaks2012.
- C. Ryan, E. Clayton, W. Griffin, S. Sie and D. Cousens, Nucl. Instrum. Methods Phys. Res., Sect. B, 1988, 34, 396–402 CrossRef.
- S. Guo, T. Bocklitz, U. Neugebauer and J. Popp, Anal. Methods, 2017, 9, 4410–4417 RSC.
- K. Rebrošová, S. Bernatová, M. Šiler, M. Uhlirova, O. Samek, J. Ježek, V. Holá, F. Růžička and P. Zemanek, Anal. Chim. Acta, 2022, 1191, 339292 CrossRef PubMed.
- U.-C. Schröder, J. Kirchhoff, U. Hübner, G. Mayer, U. Glaser, T. Henkel, W. Pfister, W. Fritzsche, J. Popp and U. Neugebauer, J. Biophotonics, 2017, 10, 1547–1557 CrossRef PubMed.
- A. Nakar, A. Wagenhaus, P. Rösch and J. Popp, Analyst, 2022, 147, 3938–3946 RSC.
- J. Kirchhoff, U. Glaser, J. A. Bohnert, M. W. Pletz, J. Popp and U. Neugebauer, Anal. Chem., 2018, 90, 1811–1818 CrossRef CAS PubMed.
- M. Paret, S. Sharma, L. Green and A. Alvarez, Appl. Spectrosc., 2010, 64, 433–441 CrossRef CAS PubMed.
- K. Rebrosova, M. Siler, O. Samek, F. Ruzicka, S. Bernatova, V. Hola, J. Jezek, P. Zemanek, J. Sokolova and P. Petras, Sci. Rep., 2017, 7, 14846 CrossRef PubMed.
- C. Wichmann, M. Chhallani, T. Bocklitz, P. Rosch and J. Popp, Anal. Chem., 2019, 91, 13688–13694 CrossRef CAS PubMed.
- F. Oliveira, A. da Silva, M. Pacheco, H. Giana and L. Silveira, Lasers Med. Sci., 2021, 36, 289–302 CrossRef PubMed.
- C.-S. Ho, N. Jean, C. A. Hogan, L. Blackmon, S. S. Jeffrey, M. Holodniy, N. Banaei, A. A. E. Saleh, S. Ermon and J. Dionne, Nat. Commun., 2019, 10, 4927 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an00679d |
| ‡ Present address of KAD: Faculty of Nuclear Sciences and Physical Engineering, Czech Technical University in Prague, Brehova 7, 11519 Prague, Czech Republic. |
| This journal is © The Royal Society of Chemistry 2023 |