
Distinguishing common SARS-CoV2 omicron and recombinant variants with high resolution mass spectrometry†
Henry E.
Lanyon
,
Benjamin P.
Todd
and
Kevin M.
Downard
 *
*
Infectious Disease Responses Laboratory, Prince of Wales Clinical Research Sciences, Sydney, Australia. E-mail: kevin.downard@scientia.org.au
First published on 8th November 2023
Abstract
A selected ion monitoring (SIM) approach combined with high resolution mass spectrometry is employed to identify and distinguish common SARS-CoV2 omicron and recombinant variants in clinical specimens. Mutations within the receptor binding domain (RBD) within the surface spike protein of the virus result in a combination of peptide segments of unique sequence and mass that were monitored to detect BA.2.75 (including CH.1.1) and XBB (including 1.5) variants prevalent in the state's population in early 2023. SIM detection of pairs of peptides unique to each variant were confidently detected and differentiated in 57.3% of the specimens, with a further 10 or 17.5% (for a total of 74.8%) detected based on a single peptide biomarker. The BA.2.75 sub-variant was detected in 18.7%, while recombinant variants XBB and XBB.1.5 were detected in 13.3% and 25.3% of the specimens respectively, consistent with circulating levels in the population characterised by RT-PCR. Virus was detected in 75 SARS-CoV2 positive specimens by mass spectrometry down to the low or mid 104 copy level, with a single false positive and no false negative identified. This article is the first paper to characterise recombinant strains of the SARS-CoV2 virus by this, or any other, MS method.
Introduction
Recombination is found to occur frequently in viruses and has a major impact on their evolution.1 It is common in RNA viruses due to the physical proximity of the genomes during the replication cycle, when host cells are infected with two or more strains. Recombination is facilitated in respiratory viruses due to the rapid rates of infection.Viral recombination plays a major role in increasing the variability of RNA viruses, enhancing their viral fitness, and accelerating their adaptation within new hosts. It can occur in both segmented and non-segmented RNA viruses such as SARS-CoV22 and type A influenza respectively. Both in silico3 and in vivo4 studies have presented evidence for recombination within SARS-CoV-2 strains.
Recombinant strains generally emerge when a major strain, associated with a wave of infections, begins to decline in the population at the same time that a new variant emerges.5 Strains associated with the recombination of delta and omicron variants (so-called deltacron) were of particular concern given their widespread circulation in the world's population and that these variants represented deadliest and most mutated forms respectively.6 An analysis of SARS-CoV2 genome sequences collected from late 2019 to mid 2022 showed a substantial increase in the emergence of such SARS-CoV-2 recombinant lineages during the omicron wave.7
In early 2022, the World Health Organisation (WHO) reported8 three recombinant variants of SARS-CoV-2 that posed the greatest threat to public health. These were denoted XD, derived from both delta (AY.4) and omicron BA.1 variants, XE from two omicron variants (BA.1 and BA.2), and XF from a delta and omicron BA.1 variant. As of March 2023, the WHO declared the need to monitor additional common XBB and XBF recombinant variants. The XBB variants are a recombinant of BA.2.10.1 and BA.2.75 sub-lineages, while the XBF variant combines BA.5.2.3 and BA.2.75.3 sub-lineages (Fig. 1 and Table 1).
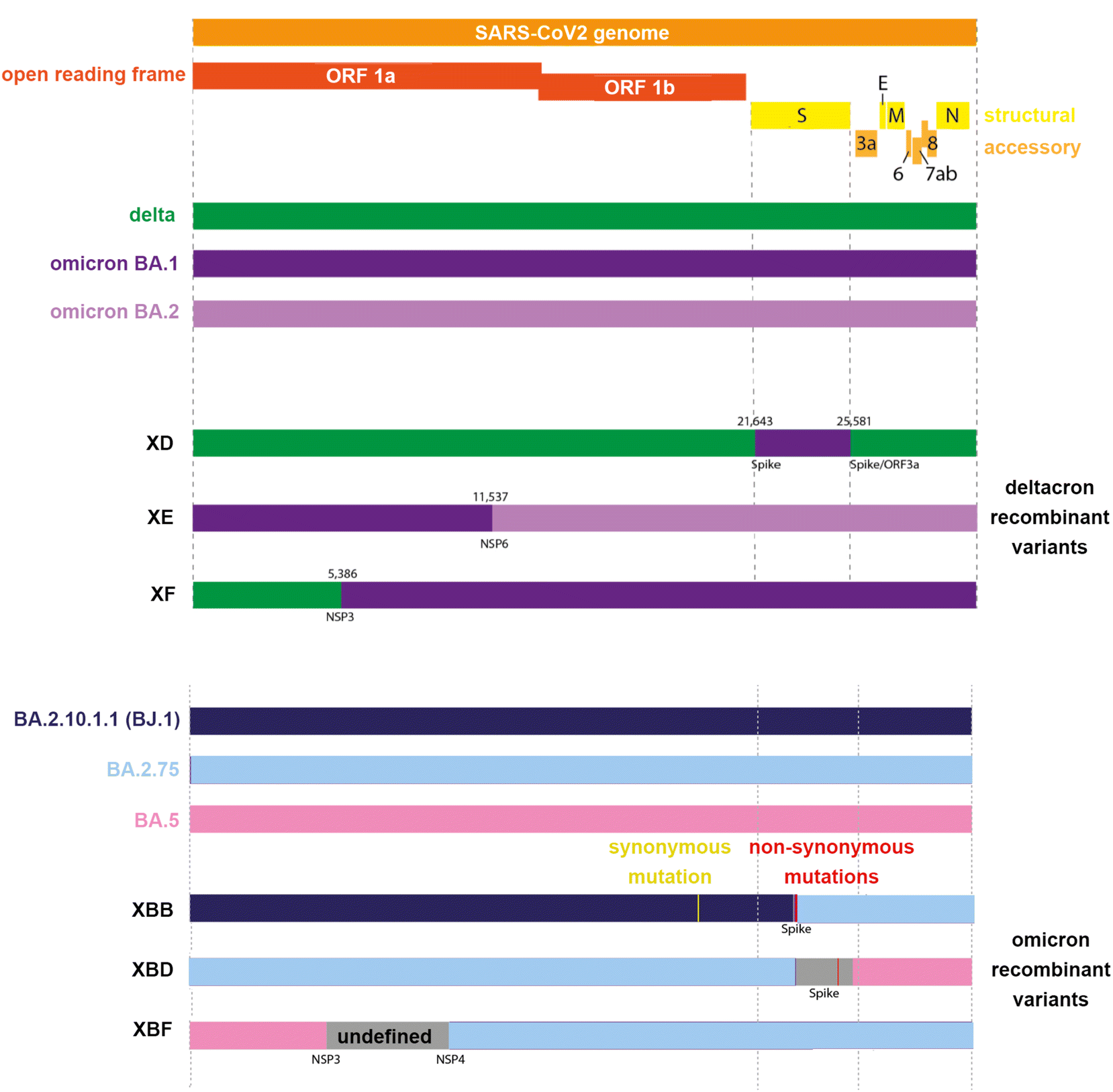 | ||
| Fig. 1 Representation of the SARS-CoV2 genome of common delta-omicron (“deltacron”) and omicron recombinant variants. | ||
| Recombinant designation | Originating strains | Spike protein mutations | Date first identified | |
|---|---|---|---|---|
| Sources: Pavan et al. 2022;28 World Health Organisation (https://www.who.int/activities/tracking-SARS-CoV-2-variants), and NSW Government Agency for Clinical Innovation (https://aci.health.nsw.gov.au/covid-19/critical-intelligence-unit/sars-cov-2-variants). | ||||
| Delta-omicron recombinants | XD | Delta AY.4 and BA.1; omicron S gene incorporated into a Delta genome | T19R; A27S; T95I; G142D; R158G; L212I; G339D; S371L; S373P; S375F; K417N; N440K; G446S; S477N; T478K; E484A | 01/2022 |
| XF | Delta AY.4 and BA.1 | A67 V; T95I; Y145D; L212I; G339D; S371L; S373P; S375F; K417N; N440K; G446S; S477N; T478K; E484A | 02/2022 | |
| Omicron recombinants | XE | BA.1 and BA.2; majority of S gene from BA.2 | T19R; A27S; G142D; V213G; G339D; S371L; S373P; S375F; T376A; D405N; R408S; K417N; N440K; S477N; T478K; E484A | 03/2022 |
| XBB | BA.2.10.1 and BA.2.75 sublineages | G339H, R346T, L368I, V445P, G446S, N460K, F486S, F490S | 08/2022 | |
| XBB1.5 | BA.2.10.1 and BA.2.75 sublineages | XBB + F486P | 01/2022 | |
| XBD | BA.5 and BA.2.75 sublineages | R346T; G446S: N450L; N460K; F486S; R493Q | 08/2022 | |
| XBF | BA.5.2.3 and CJ.1 (a distant descendant of BA.2.75) | BA.5 + K147E, W152R, F157L, I210 V, G257S, G339H, R346T, G446S, N460K, F486P, F490S | Late 2022 | |
Viral recombination can be difficult to detect whenever sub-lineages have minimal mutational differences.9 A typical strategy combines whole-genome sequencing with phylogenetics to detect unusual combinations of single nucleotide polymorphisms (SNPs) which are phylogenetically diverged.10 However, the limited numbers of SNPs that distinguish certain clades are also often clustered within short regions of the genome that, together with reversion events, restrict the ability to identify potential recombinant forms. Algorithms designed to detect recombination from such genetic data assess all SNPs equally, regardless of how phylogenetically significant they are, and thus are prone to misassignments.11
Other strategies to identify recombination events focus on the identification of mutations in the receptor binding domain (RBD) within the surface spike protein.12,13 Recombination events have been inferred to occur disproportionately in the 3′ portion of the genome,14 which contains the spike protein gene. The accrual of variant-defining spike protein mutations has been attributed to recombination events.9 Multiple amino acid mutations in the spike protein have been used to identify recombination events15
In this laboratory, recombinant influenza viruses have been detected at the protein level with high resolution mass spectrometry using specifically-designed, in-house software. Two algorithms, known as FluShuffle and FluResort, were shown to be able to identify reassorted influenza viruses from protein mass map data generated from whole virus digests.16 FluShuffle considers different combinations of viral protein identities that match the mass map data using a Gibbs sampling algorithm. FluResort maps those identities onto phylogenetic trees, constructed from viral protein sequence alignments, to calculate the weighted distance of each across two or more different trees. Each weighted mean distance value is normalized by conversion to a Z-score that is used to establish the probability of a reassorted strain. A combination of two strains represents a reassorted virus from one reassortment event, a combination of three strains represents a reassorted virus from two reassortment events and so on. Minimum composite Z-scores were compared across differing numbers of reassortment events to determine whether or not the virus was reasserted.16
In parallel, we advanced a proteotyping strategy for typing and subtyping viruses17,18 to identify reassortment in 2009 pandemic strains of influenza19 through the detection of unique human and swine host-specific signatures co-circulating in viruses during the same period.20 Given the SARS-CoV2 virus contains a single genome segment, rather than a segmented one in the case of influenza, evidence for reassortment can be uncovered using this simpler viral protein strategy that identifies unique peptide biomarkers. These derive from the accumulation of multiple spike protein mutations that originate from different variants21,22 and combine into a single recombinant form. Those that are ionised and detected in expressed spike protein forms can be used to detect and distinguish recombinant viruses in proteolytically digested whole virus clinical specimens. This article represents the first paper to characterise recombinant SARS-CoV2 virus strains by this, or any other, MS method.
Experimental section
Recombinant protein digestion
Expressed recombinant forms of the SARS-CoV2 spike protein comprising residues 16–185 (of 77 kDa.) for omicron variants BA.2.75 and XBB.1.5 were purchased (Acro Biosystems, Newark, DE, USA) and used without further purification. Each recombinant protein (100 μg) was dissolved in digestion buffer (100 μL of 50 mM ammonium bicarbonate, containing 10% acetonitrile, 2 mM dithiothreitol), incubated for 2.5 hours at 37 °C and digested overnight (∼15 hours) through the addition of 1 μl of proteomics-grade trypsin (Merck, Sydney, NSW, Australia).Clinical specimen processing and whole virus digestion
The collection and preparation of human clinical specimens was carried out in accordance with the Communicable Diseases Network Australia (CDNA) national guidelines for Coronavirus Disease 2019 and State of NSW Health restrictions and protocols. The specimens were sourced from the Prince of Wales or Westmead Hospitals, Sydney NSW, as nasopharyngeal swabs in the first 3 months of 2023. They were obtained with consent from SARS-CoV-2 infected patients (75 specimens), anonymised, and together with separate negative controls (15 specimens), were individually diluted in a saline solution and stored at −70 °C. To minimise errors and biases, reproducible pre-analytical sample handling and pre-purification steps were employed for all specimens. RNA was extracted from one half of the solution and quantified by real-time PCR according to a reported procedure.23 Virus titres were determined to be within 104–106 copies per mL in the positive specimens, though without identification a specific variant.Virus was precipitated from the remainder of the solution through the addition of 95% ethanol solution cooled to −20 °C, collected on a 300 K molecular weight cut-off (MWCO) filter (Pall Corporation, Cheltenham, Victoria, Australia), washed with purified water, to recover the retentate. The filter recovered virus was resuspended in a digestion buffer solution (100 μL of 50 mM ammonium bicarbonate, 10% acetonitrile (99.9% purity), 2 mM dithiothreitol, and 5 mM octyl β-D-glucopyranoside) at pH 7.5, the solution sonicated for 15 min, incubated for 2 h at 37 °C, and digested overnight following the addition of 15 ng μL−1 sequencing-grade modified trypsin (Promega Corporation, Sydney, Australia).
High-resolution MALDI-FT-ICR mass spectrometry
Viral peptide solutions (1 μl) were diluted with a matrix solution (10 μl) of containing 5 mg ml−1 α-cyano-4-hydroxycinnaminic acid in 50% acetonitrile (99.9% purity) with 0.1% trifluoroacetic acid. A portion of the mixed solutions (1 μl) were spotted onto a Matrix-Assisted Laser Desorption Ionization (MALDI) sample plate and spectra acquired on a Bruker SolariX 7T XR Fourier-Transform Ion Cyclotron Resonance (FT-ICR) mass spectrometer (Bruker Daltonics, Preston Victoria, Australia). Spectra were acquired over a mass-to-charge ratio range of m/z 800–5500 using a broadband excitation or narrow band range of 20 mDa. for selected ion monitoring (SIM). Samples were ablated from the MALDI plate held at 450 V, following 200 laser shots, an ion accumulation time of 0.1 s, a time of flight of 1.500 ms, and spectra acquired and summed across 512 K data points. A maximum ion accumulation time of 250 ms was used in the SIM mode.BLAST (protein) search of the human proteome with variant-specific peptides
Sequences for all six omicron variant-specific peptides associated with the spike S-protein, that were used to identify recombinant variants, were accessed as unique (>95–100%) to the SARS-CoV2 virus among all known proteins, across all organisms, employing a BLASTp (Basic Local Alignment Search Tool Protein) search (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins; conducted on 18/07/23) of the non-redundant (nr) protein sequence database.Results and discussion
Prevalence of omicron subvariant and inter-lineage recombinant forms
Omicron subvariant and inter-lineage recombinant forms still cause the vast majority of SARS-CoV2 infections worldwide, including in Australia. As of late 2022 in Australia, the major omicron subvariants in circulation were the BA.2.75 subvariant, and various forms of BA.5. By mid-March 2023, the variants of concern (VOCs) identified by whole genome sequencing of virus recovered from patients in the state of New South Wales (NSW) who tested positive for SARS CoV-2 by PCR, were 38.3% XBB.1.5, 21% XBB, 10.7% XBF, and a combined 19.6% being omicron BA.2.75 lineages (BA.2.75 and CH.1.1; the latter containing two additional spike mutations L452R, and F486S).24 Thus some 79% of, or 4 out of 5, sequenced strains correspond to strains of the either BA.2.75 or XBB lineage (ESI Fig. S1†).XBB is the most widespread inter-lineage omicron recombinant variant. It represents a recombination of two BA.2 lineages; BJ.1 (or BA.2.10.1.1) and a BA.2.75 variant (namely BA.2.75.3.1.1.1). XBB inherited the 5′ part of its genome from BJ.1 and the 3′ end of its genome from BA.2.75, with a single breakpoint within the receptor binding domain (RBD) of the surface spike protein. This breakpoint provides potent antigenic RBD mutations from both BJ.1 and BA.2.75.
Sequences comprising the RBD of the spike protein (residues 320–541, numbered according to the NCBI reference sequence, accession QHD43416.1) for the BA.2.75 subvariants and recombinant forms XBB.1 and XBB.1.5 are shown in Fig. 2. Because of the associated different mutations, and the propensity of proline to prevent tryptic cleavage where it resides C-terminal to lysine or arginine residues, a number of tryptic peptides of unique mass are produced. These are shown both boxed and shaded (each with a unique colour) together with their protonated monoisotopic masses located above or below their sequence.
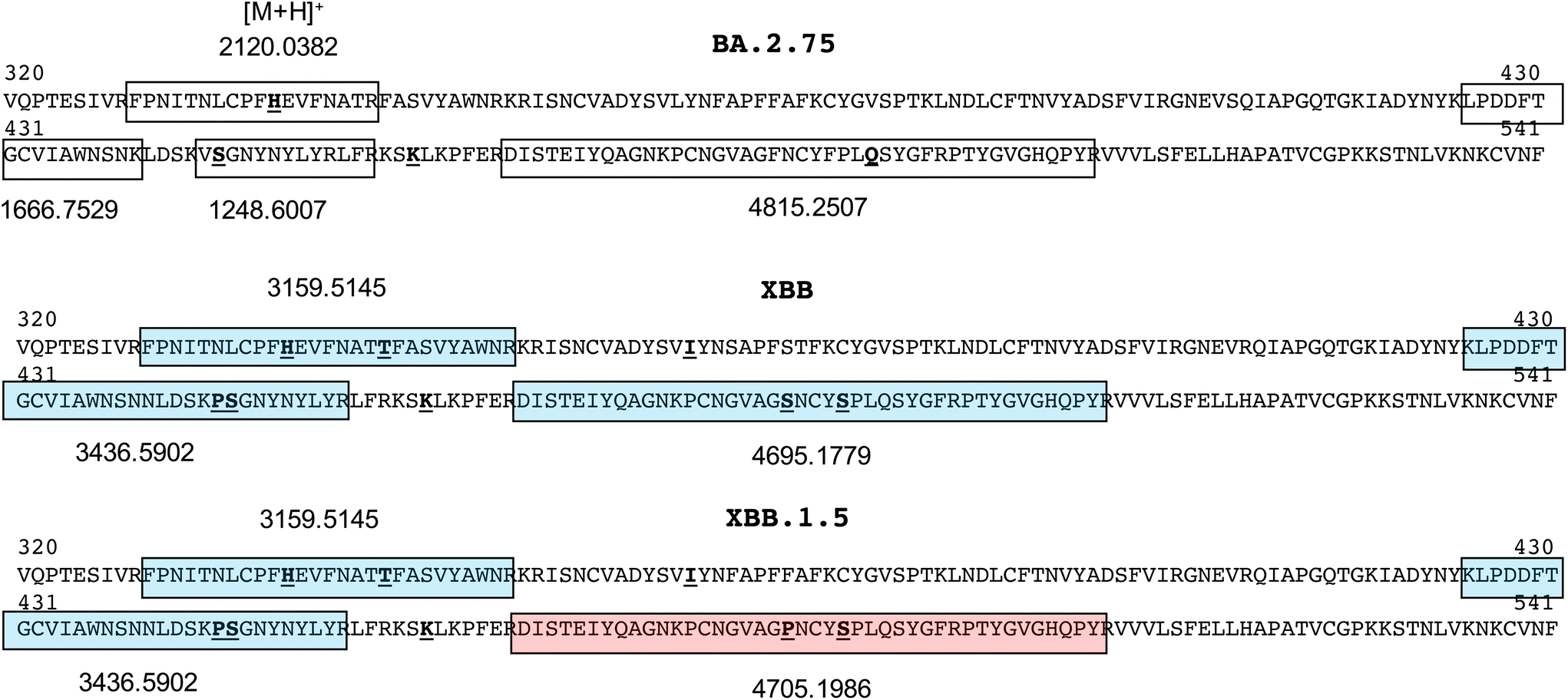 | ||
| Fig. 2 Receptor binding domain sequences (residues 320–541) of spike protein of common omicron and recombinant SARS-CoV2 variants showing peptide segments of unique sequence and mass. | ||
Those that exclusively distinguish the BA.2.75 subvariant and recombinant XBB forms span residues 329–346 in the BA.2.75 subvariant at m/z 2120.0382 and residues 329–355 in the XBB recombinant forms at m/z 3159.5145, and residues 467–509 at m/z 4815.2507, 4695.1779 and 4705.1986 in the BA.2.75, XBB.1 and XBB.1.5 forms respectively. Subsequent MALDI-MS analyses on the trypsin-digested, recombinant spike protein for each variant were used to establish the ionisability and detectability of these ions.
Analysis of recombinant spike protein of BA.2.75 and XBB 1.5 variants
Recombinant protein forms of the S1 subunit of spike protein of the BA.2.75 and XBB 1.5 variants were digested with trypsin and their high resolution MALDI mass spectrum recorded (Fig. 3 and 4). The former of the BA.2.75 variant contains protonated ions of tryptic peptides comprising residues 329–346 and 467–509 at m/z 2120.0367 and 4815.2498 respectively are labelled in bold and their isotopic clusters expanded in the lower boxed sections of the figure.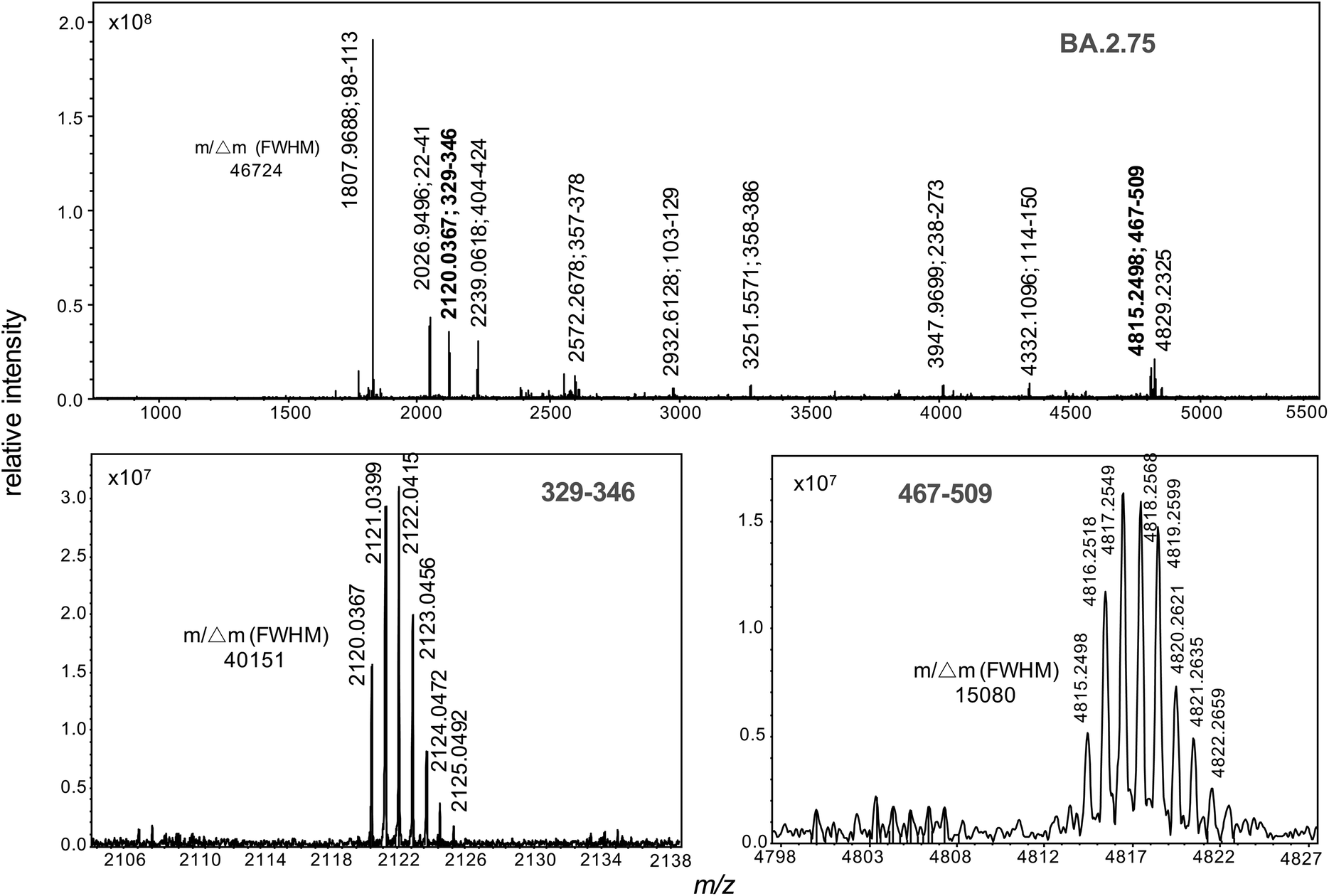 | ||
| Fig. 3 High resolution MALDI mass spectrum of trypsin-digested spike S1 of the omicron BA.2.75 sub-variant. Residue numbering is in accordance with the NCBI reference sequence, accession QHD43416.1. Mass resolutions (m/Δm) at full-width half maximum for select monoisotopic ions are shown. | ||
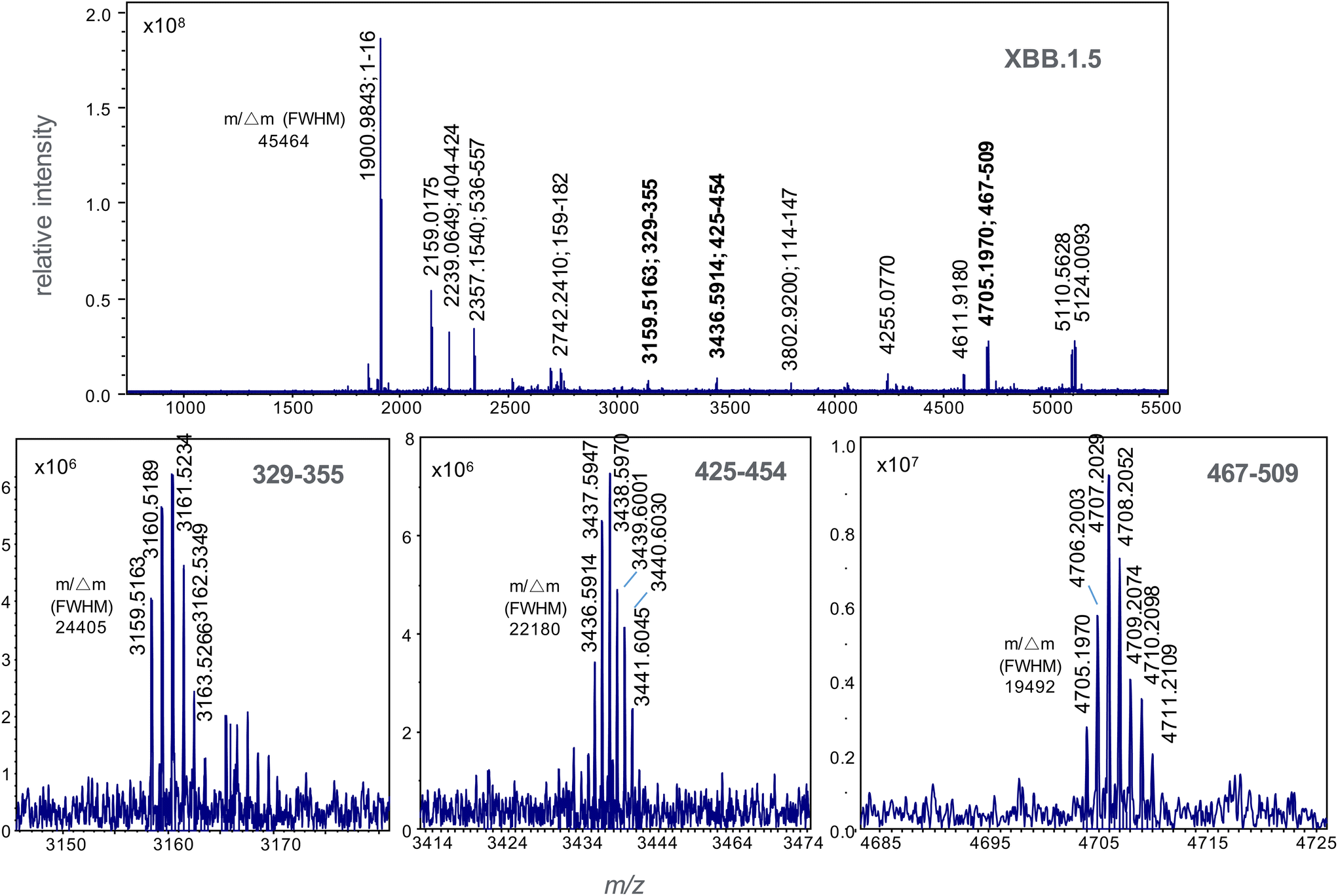 | ||
| Fig. 4 High resolution MALDI mass spectrum of trypsin-digested spike S1 protein of the omicron recombinant variant XBB.1.5. Residue numbering is in accordance with the NCBI reference sequence, accession QHD43416.1. Mass resolutions (m/Δm) at full-width half maximum for select monoisotopic ions are shown. | ||
These ions not appear in the spectrum for XBB 1.5 (Fig. 4). This spectrum displays variant-specific peptide ions at m/z 3159.5163, 3436.5914 and 4705.0770 (shown in bold); the latter unique to the XBB.1.5 form. These ions corresponding to tryptic peptide residues 329–355, 425–454 and 467–509.
Subsequent narrow-band excitations, in selected ion monitoring (SIM) experiments, were performed to detect these ions in clinical specimens for diagnostic purposes. SIM analysis reduces the total spectral acquisition time, and improves sensitivity, by focussing only on the detection of the variant-specific peptide markers of interest. This improves the sensitivity of analysis, to aid peptide detection at low virus titre levels, compared with full-scan MS measurements.25
A narrow mass range of 20 mDa. (±0.01 m/z) centred on the theoretical monoisotopic values (Fig. 2) for each of the respective protonated peptide ions. The detection of at least two variant-specific ions, and not only a single peptide marker, was adopted for diagnostic purposes. This helps reduce, if not completely eliminate, the chance that another single viral peptide, unique to a particular strain, or specimen contaminant, is mistaken for one of the variant-specific mass markers. The high mass accuracy, employed in these high mass resolution experiments, also significantly reduces this possibility.
SIM analysis of BA.2.75, XBB and BA.5 variants in clinical specimens
The presence of both ions at m/z 2120.0367 and 4815.2498 were used as biomarkers for BA.2.75 variants, while the presence of both ions at m/z 3159.5163 and 3436.5914 were used to identify XBB variants. The detection of ions at m/z 4695.1779 and 4705.0770 were then used to distinguish XBB from XBB.1.5. Where only one single marker is detected, the strain under analysis is declared negative to any of the variants (BA.2.75, XBB or XBB.1.5) under analysis. In all cases, the peptide biomarkers must be detected at a signal-to-noise ratio that exceeds the highest value in the SARS-CoV2 negative control specimens.The data is represented in box-plot format (Fig. 5), as employed in a recent study of SARS-CoV2 sub-variants.22 The box edges indicate the first and third quartile of intensities, and the central line indicates the median value. Of the 75 SARS-CoV2 RT-PCR positive specimens, ions at both m/z 2120 and 4815 (nominal mass for BA.2.75) were detected within 2 ppm of the theoretical values, with intensities above the 15 negative control specimens, in 14 (or 18.7%) of the samples. Ions with intensities above the controls at both m/z 3159 and 3436 (for XBB) were detected in 10 (or 13.3%) of the samples, while those at m/z 4695 and 4705 (for XBB.1.5) in 19 (or 25.3%) of the samples. Virus titres across all samples were quantified at between 104 to 106 copies per mL by RT-PCR and this is reflected in the significant deviation in the ion intensities to the box edges in Fig. 5.
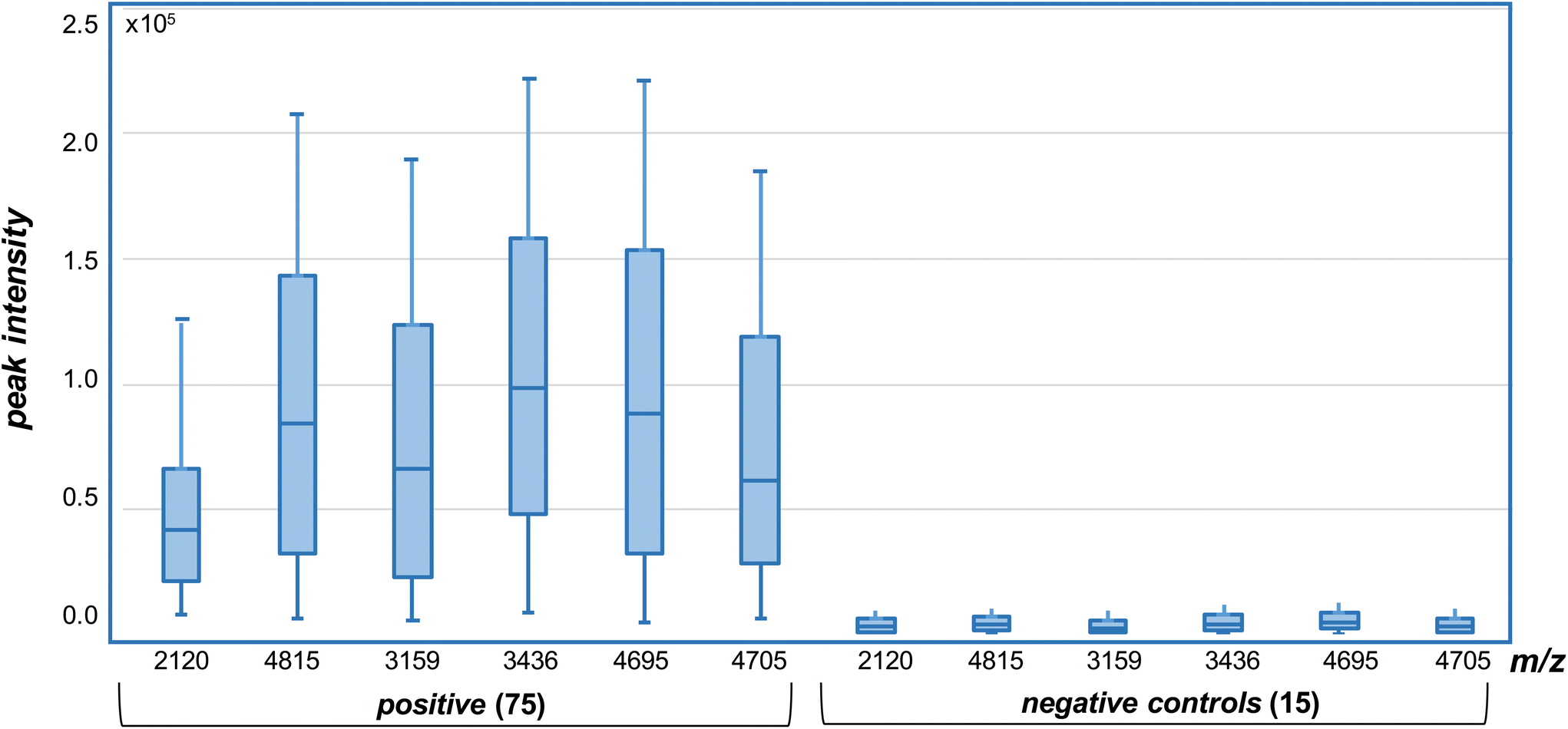 | ||
| Fig. 5 Selected ion monitoring (SIM) MALDI-MS analysis of variant specific peptide ion biomarkers in trypsin digested, whole virus SARS-CoV2 positive and negative (control) samples. Intensities measured above those in the negative (controls) for each of the ions, detected within 2 ppm of the theoretical values (of Fig. 2), are deemed positive. | ||
The results indicate that the BA.2.75, XBB and XBB.1.5 variants could be confidently detected and differentiated in 57.3% of the clinical specimens, while a further 10 or 17.5% (for a total of 74.8%) were identified based on the detection of one (and not two) of the peptide biomarkers. The remaining 25.2% contain either other variants, or were positive for BA.2.75, XBB and XBB.1.5 variants but at levels lower than could be detected by mass spectrometry. A single false positive result (1.3%) was obtained for one of the control samples, based on RT-PCR analysis, with a single m/z 4695 ion associated with the XBB.1.5 variant detected above background control levels. No false negative results were found.
Contaminants unique to the unassigned specimens, or at higher concentration levels in these specimens, may also mask the detection of the variants under study by suppressing the ionization and detection of the peptide biomarkers of interest. Further, some additional strain-specific mutations within the segments studied may alter the masses of peptides, just as mutations at the nucleotide level can confound PCR based hybridization assays.
These, and separate studies, have revealed that mass spectrometry approaches can detect virus components to the low or mid 104 copies per mL level, but are challenged below this level.18,25,26 A SARS-CoV-2 RNA quantitation PCR assay, in contrast, has a limit of detection (LoD) of some 25 copies per mL These limits remain a challenge for mass spectrometry based studies which directly detect virus present in clinical specimens without the benefit of amplification afforded by PCR analytical approaches. However, given that a viral load greater than approximately 103 copies per mL is typically required for a patient to be infectious,27 mass spectrometry has a role to play as a diagnostic tool as infection takes hold. Indeed, the ability to reliably detect and distinguish the most common BA.2.75, XBB and XBB.1.5 SARS-CoV2 variants in up to 75% of positive specimens analysed in this study demonstrate the very real and powerful applicability of high resolution mass spectrometry in the analysis of SARS-CoV2 viruses within clinical specimens and settings.
Conclusions
Mass spectrometry represents a useful and complementary approach for the molecular detection of infectious disease causing viruses. In particular, the role of high resolution mass spectrometry in the differentiation of SARS-CoV2 variants and sub-variants with high confidence21,22 provide a broad framework for the implementation of a clinical analytic approach that offers benefits over lower resolution, low mass accuracy conventional MALDI-TOF or QTOF, and time and higher sample consuming LC-ESI-MS strategies26 where liquid chromatographic separation and tandem MS/MS sequencing steps are necessary.29 Even when so-called selective reaction monitoring (SRM) or multiple reaction monitoring (MRM) is employed, establishing precursor and fragment ion pairs that are efficiently produced and detected must be determined a priori, and the fragmentation (reaction) time and efficiency of peptide fragmentation remains a limitation.30,31 The latter reduces the sensitivity of analysis over MS-only analyses, irrespective of the MS instrumentation deployed.Here a mass-only detection approach, with high mass accuracy and assignment confidence, are employed. Any high resolution MALDI based mass spectrometer, such as extended-path TOF32,33 or Orbitrap (Kingdon trap)34 instrument, could be used over an ICR for this purpose. This is coupled to SIM analysis versus full-scan experiments to improve sensitivity. The SIM of multiple peptides associated with specific variants21 and sub-variants22 can also be conducted to differentiate these from recombinant forms. The pre-MS analysis steps are minimal, with the virus specimen filtered, to remove extraneous contaminants, then precipitated and proteolytically digested. The digestion step could be accelerated using immobilized enzymes35 or other reported approaches.36
The confident detection of BA.2.75, XBB and XBB.1.5 variants in the majority of SARS-CoV2 positive clinical specimens analysed in this study, at percentages consistent with those characterised by RT-PCR sequencing that are in circulation in the local state (NSW) population, demonstrate the real power and benefits of a mass-based detection approach to follow variant-specific mutations in the surface spike protein. The ability to confidently detect and differentiate omicron sub-variants and emerging recombinant forms, without the need and considerable time (days) required for gene or peptide and protein sequencing, is of vital importance to manage outbreaks of the virus and develop effective clinical responses. The approach overcomes limitations of RT-PCR approaches where limited numbers of SNPs and reversion events, restrict the ability to identify potential recombinant forms.
Ethical statement
All procedures for collection, preparation, and transport of the clinical specimens were carried out in accordance with the Communicable Diseases Network Australia (CDNA) national guidelines for Coronavirus Disease 2019 and NSW Health restrictions and protocols. Clinical specimens were obtained from the Prince of Wales or Westmead Hospitals from SARS-CoV2 infected patients with consent with specimens anonymised to remove patient data.Conflicts of interest
The authors report no conflicts or competing interests.Acknowledgements
Author Downard received support from Clinical Research Fund (CRF21) and donours to this study.References
- M. Pérez-Losada, M. Arenas, J. C. Galán, F. Palero and F. González-Candelas, Recombination in viruses: mechanisms, methods of study, and evolutionary consequences, Infect., Genet. Evol., 2015, 30, 296–307, DOI:10.1016/j.meegid.2014.12.022.
- D. Focosi and F. Maggi, Recombination in Coronaviruses, with a Focus on SARS-CoV-2, Viruses, 2022, 14, 1239, DOI:10.3390/v14061239.
- N. Najeeb, A. B. Murukan, A. Renjitha, M. Jayaram, A. A. Jabbar, H. Haridasan, A. Prijikumar, S. Baiju, A. A. Nixon, P. A. Krishnan, S. Rodriguez, S. Kumar, S. K. Polipalli, K. K. Singh, B. G. Nair, S. D. Ghate, R. S. P. Rao, P. B. K. Kishor, A. Aloor, R. Suravajhala, G. Chaubey and P. Suravajhala, Inferring Recombination Events in SARS-CoV-2 Variants In Silico, Adv. Exp. Med. Biol., 2023, 1412, 253–270, DOI:10.1007/978-3-031-28012-2_14.
- H. L. Wells, C. M. Bonavita, I. Navarrete-Macias, B. Vilchez, A. L. Rasmussen and S. J. Anthony, The coronavirus recombination pathway, Cell Host Microbe, 2023, 31, 874–889, DOI:10.1016/j.chom.2023.05.003.
- H. Yi, 2019 Novel Coronavirus Is Undergoing Active Recombination, Clin. Infect. Dis., 2002, 71, 884–887, DOI:10.1093/cid/ciaa219.
- Y. Wang, Y. Long, F. Wang, C. Li and W. Liu, Characterization of SARS-CoV-2 recombinants and emerging Omicron sublineages, Int. J. Med. Sci., 2023, 20, 151–162 CrossRef CAS PubMed.
- R. Shiraz and S. Tripathi, Enhanced recombination among Omicron subvariants of SARS-CoV-2 contributes to viral immune escape, J. Med. Virol., 2023, 95, e28519, DOI:10.1002/jmv.28519.
- WHO Tracking SARS-CoV-2 variants. 2022. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (Accessed: April, 5 2022).
- B. Jackson, M. F. Boni, M. J. Bull, A. Colleran, R. M. Colquhoun, A. C. Darby, S. Haldenby, V. Hill, A. Lucaci, J. T. McCrone, S. M. Nicholls, A. O'Toole, N. Pacchiarini, R. Poplawski, E. Scher, F. Todd, H. J. Webster, M. Whitehead and C. Wierzbicki, COVID-19 Genomics UK (COG-UK) Consortium, N.J. Loman, T.R. Connor, D.L. Robertson, O.G. Pybus, A. Rambaut, Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic, Cell, 2021, 184, 5179–5188, DOI:10.1016/j.cell.2021.08.014.
- A. Lohrasbi-Nejad, Detection of homologous recombination events in SARS-CoV-2, Biotechnol. Lett., 2022, 44, 399–414, DOI:10.1007/s10529-021-03218-7.
- D. P. Martin, B. Murrell, M. Golden, A. Khoosal and B. Muhire, RDP4: Detection and analysis of recombination patterns in virus genomes, Virus Evol., 2015, 1, vev003, DOI:10.1093/ve/vev003.
- J. A. Patiño-Galindo, I. Filip, R. Chowdhury, C. D. Maranas, P. K. Sorger, M. AlQuraishi and R. Rabadan, Recombination and lineage-specific mutations linked to the emergence of SARS-CoV-2, Genome Med., 2021, 13, 124, DOI:10.1186/s13073-021-00943-6.
- F. Scarpa, D. Sanna, I. Azzena, M. Casu, P. Cossu, P. L. Fiori, D. Benvenuto, E. Imperia, M. Giovanetti, G. Ceccarelli, R. Cauda, A. Cassone, S. Pascarella and M. Ciccozzi, Genome-based comparison between the recombinant SARS-CoV-2 XBB and its parental lineages, J. Med. Virol., 2023, 95, e28625, DOI:10.1002/jmv.28625.
- Y. Turakhia, B. Thornlow, A. Hinrichs, J. McBroome, N. Ayala, C. Ye, K. Smith, N. De Maio, D. Haussler, R. Lanfear and R. Corbett-Detig, Pandemic-scale phylogenomics reveals the SARS-CoV-2 recombination landscape, Nature, 2022, 609, 994–997, DOI:10.1038/s41586-022-05189-9.
- J. Ou, W. Lan, X. Wu, T. Zhao, B. Duan, P. Yang, Y. Ren, L. Quan, W. Zhao, D. Seto, J. Chodosh, Z. Luo, J. Wu and Q. Zhang, Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events, Signal Transduction Targeted Ther., 2022, 7, 138, DOI:10.1038/s41392-022-00992-2.
- A. T. Lun, J. W. Wong and K. M. Downard, FluShuffle and FluResort: new algorithms to identify reassorted strains of the influenza virus by mass spectrometry, BMC Bioinf., 2012, 13, 208, DOI:10.1186/1471-2105-13-208.
- A. B. Schwahn, J. W. Wong and K. M. Downard, Subtyping of the influenza virus by high resolution mass spectrometry, Anal. Chem., 2009, 81, 3500–3506, DOI:10.1021/ac900026f.
- K. M. Downard, Proteotyping for the rapid identification of influenza virus and other biopathogens, Chem. Soc. Rev., 2013, 42, 8584–8595, 10.1039/c3cs60081e.
- N. D. Fernandes and K. M. Downard, Origins of the reassortant 2009 pandemic influenza virus through proteotyping with mass spectrometry, J. Mass Spectrom., 2014, 49, 93–102, DOI:10.1002/jms.3310.
- J. W. Ha, A. B. Schwahn and K. M. Downard, Proteotyping to establish gene origin within reassortant influenza viruses, PLoS One, 2011, 6, e15771, DOI:10.1371/journal.pone.0015771.
- C. Mann, J. H. Griffin and K. M. Downard, Detection and Evolution of SARS-CoV2 Coronavirus Variants of Concern with Mass Spectrometry, Anal. Bioanal. Chem., 2021, 413, 7241–7249, 10.1039/d2an00028h.
- H. E. Lanyon, J. S. Hoyle and K. M. Downard, Resolving Omicron Sub-Variants of SARS CoV-2 Coronavirus with MALDI Mass Spectrometry, Analyst, 2023, 148, 966–972, 10.1039/d2an01843h.
- E. Keyaerts, L. Vijgen, P. Maes, G. Duson, J. Neyts and M. V. Ransta, Viral load quantitation of SARS-coronavirus RNA using a one-step real-time RT-PCR, Int. J. Infect. Dis., 2006, 10, 32–37, DOI:10.1016/j.ijid.2005.02.003.
- NSW COVID-19 WEEKLY DATA OVERVIEW Epidemiological week 11, ending 18 March 2023. Source: https://www.health.nsw.gov.au/coronavirus.
- N. L. Dollman, J. H. Griffin and K. M. Downard, Detection, mapping, and proteotyping of SARS-CoV-2 coronavirus with high resolution mass spectrometry, ACS Infect. Dis., 2020, 6, 3269–3276, DOI:10.1021/acsinfecdis.0c00664.
- J. H. Griffin and K. M. Downard, Mass spectrometry analytical responses to the SARS-CoV2 coronavirus in review, Trends Anal. Chem., 2021, 142, 116328, DOI:10.1016/j.trac.2021.116328.
- A. Viloria Winnett, R. Akana, N. Shelby, H. Davich, S. Caldera, T. Yamada, J. R. B. Reyna, A. E. Romano, A. M. Carter, M. K. Kim, M. Thomson, C. Tognazzini, M. Feaster, Y. Y. Goh, Y. C. Chew and R. F. Ismagilov, Extreme differences in SARS-CoV-2 viral loads among respiratory specimen types during presumed pre-infectious and infectious periods, PNAS Nexus, 2023, 2, pgad033, DOI:10.1093/pnasnexus/pgad033.
- M. Pavan, D. Bassani, M. Sturlese and S. Moro, From the Wuhan-Hu-1 strain to the XD and XE variants: is targeting the SARS-CoV-2 spike protein still a pharmaceutically relevant option against COVID-19?, J. Enzyme Inhib. Med. Chem., 2022, 37, 1704–1714, DOI:10.1080/14756366.2022.2081847.
- S. Rajoria, A. Halde, I. Tarnekar, P. Pal, P. Bansal and S. Srivastava, Detection of Mutant Peptides of SARS-CoV-2 Variants by LC/MS in the DDA Approach Using an In-House Database, J. Proteome Res., 2023, 22, 1816–1827, DOI:10.1021/acs.jproteome.2c00819.
- Y. Shen, N. Tolić, F. Xie, R. Zhao, S. O. Purvine, A. A. Schepmoes, R. J. Moore, G. A. Anderson and R. D. Smith, Effectiveness of CID, HCD, and ETD with FT MS/MS for degradomic-peptidomic analysis: comparison of peptide identification methods, J. Proteome Res., 2011, 10, 3929–3943 CrossRef CAS PubMed.
- A. F. Jarnuczak, D. C. H. Lee, C. Lawless, S. W. Holman, C. E. Eyers and S. J. Hubbard, Analysis of Intrinsic Peptide Detectability via Integrated Label-Free and SRM-Based Absolute Quantitative Proteomics, J. Proteome Res., 2016, 15, 2945–2959 CrossRef CAS PubMed.
- T. Satoh, T. Sato and J. Tamura, Development of a high-performance MALDI-TOF mass spectrometer utilizing a spiral ion trajectory, J. Am. Soc. Mass Spectrom., 2007, 18, 1318–1323 CrossRef CAS PubMed.
- T. Yoshinari, K. Hayashi, S. Hirose, K. Ohya, T. Ohnishi, M. Watanabe, S. Taharaguchi, H. Mekata, T. Taniguchi, T. Maeda, Y. Orihara, R. Kawamura, S. Arai, Y. Saito, Y. Goda and Y. Hara-Kudo, Matrix-Assisted Laser Desorption and Ionization Time-of-Flight Mass Spectrometry Analysis for the Direct Detection of SARS-CoV-2 in Nasopharyngeal Swabs, Anal. Chem., 2022, 94, 4218–4226 CrossRef CAS PubMed.
- K. Strupat, V. Kovtoun, H. Bui, R. Viner, G. Stafford and S. Horning, MALDI Produced Ions Inspected with a Linear Ion Trap-Orbitrap Hybrid Mass Analyzer, J. Am. Soc. Mass Spectrom., 2009, 8, 1451–1463 CrossRef.
- K. Duong, S. D. Maleknia, D. Clases, A. Minett, M. P. Padula, P. A. Doble and R. Gonzalez de Vega, Immunoaffinity extraction followed by enzymatic digestion for the isolation and identification of proteins employing automated μSPE reactors and mass spectrometry, Anal. Bioanal. Chem., 2023, 415, 4173–4184 CrossRef CAS PubMed.
- L. Switzar, M. Giera and W. M. A. Niessen, Protein Digestion: An Overview of the Available Techniques and Recent Developments, J. Proteome Res., 2013, 12, 1067–1077 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an01376f |
| This journal is © The Royal Society of Chemistry 2023 |