
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in catalytic enantioselective carbometallation of cyclopropenes and cyclobutenes
Chuiyi
Lin
a,
Qianghui
Wu
a,
Yu
Wang
a,
Qinglei
Chong
*a and
Fanke
Meng
 *abc
*abc
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, 345 Lingling Road, Shanghai, 200032, China. E-mail: chongql@sioc.ac.cn; mengf@sioc.ac.cn
bSchool of Chemistry and Material Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, 1 Sub-lane Xiangshan, Hangzhou, 310024, China
cBeijing National Laboratory for Molecular Sciences, Beijing, 100871, China
First published on 1st October 2024
Abstract
Enantioenriched small carbocycles are key structures in numerous natural products and pharmaceutically important molecules as well as vital intermediates in organic synthesis. Although various catalytic approaches for the construction of such molecules from acyclic precursors have been developed, direct enantioselective functionalization of preformed three-membered and four-membered rings represents the most straightforward and modular strategy, enabling rapid and diversified synthesis of enantioenriched cyclopropanes and cyclobutanes from a single set of starting materials without the need for the incorporation of specific functional groups. In this Feature Article, we have summarized the recent advances in catalytic enantioselective functionalization of cyclopropenes and cyclobutenes through carbometallation. The plausible mechanisms of such reactions and future of this field are also discussed.
1. Introduction
Enantioenriched small carbocycles, namely cyclopropanes and cyclobutanes, are crucial structures in natural products and pharmaceutically important molecules (Scheme 1).1 Moreover, such moieties represent useful and versatile building blocks and intermediates in organic synthesis.2 However, it is challenging to construct enantioenriched cyclopropanes and cyclobutanes due to their inherent high energy arising from the ring strain. It is also nontrivial for the chiral catalysts to accurately control stereoselectivity. Although catalytic approaches for the synthesis of enantioenriched small carbocycles from acyclic starting materials through cyclization have been developed, such methods suffer from the requirement of specific functional groups and significant limitations on substrate scope.3 An alternative strategy is catalytic enantioselective functionalization of preformed three- and four-membered rings. The advantage is that a high diversity of functional groups can be directly introduced from a single set of starting materials without the requisite of pre-installation of a specific functional group.4 In this context, C–H functionalization of cyclopropanes5 and cyclobutanes6 constitutes a straightforward approach in the presence of a directing group. However, the substituents that can be incorporated have remained limited. A more versatile strategy is catalytic enantioselective functionalization of small cyclic alkenes that enable the introduction of a broader scope of functional groups. A higher diversity of reaction types can be achieved as well.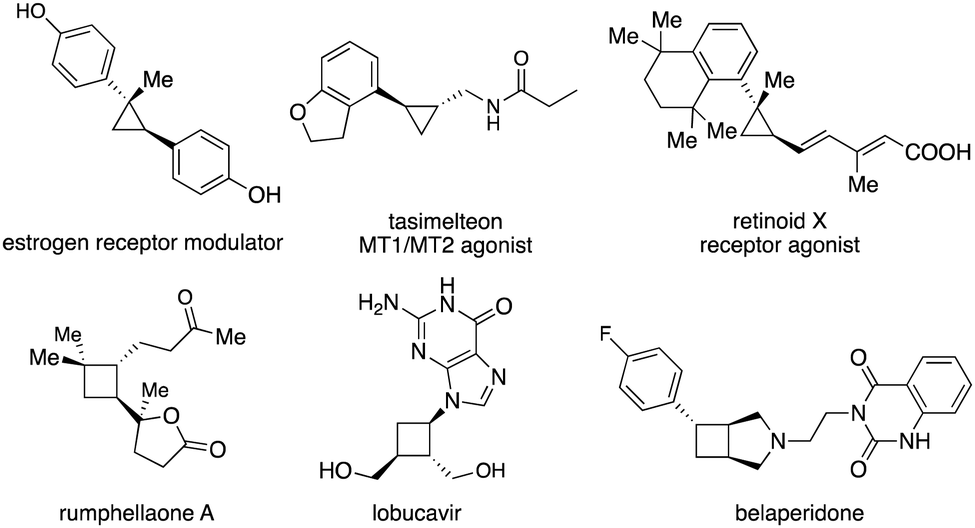 | ||
| Scheme 1 Selected biologically active chiral cyclopropanes and cyclobutanes. | ||
Carbometallation of alkenes represents one of the most important elementary steps for carbon–carbon bond formation. Taking advantage of the higher reactivity activated by the ring strain, it is an attractive strategy for incorporating carbon-based substituents onto the small cyclic scaffolds through catalytic enantioselective carbometallation (Scheme 2). However, there are huge challenges to achieve such processes, as the possible oxidative cleavage of the strained small carbocycles induced by the metal-based catalysts has to be suppressed. A suitable choice of chiral ligand is crucial for the efficiency of carbometallation, particularly for nonpolar alkenes, as well as accurate control of stereoselectivity. It should be noted that an elegant review on regio- and diastereoselective carbometallation of cyclopropenes has been disclosed by Marek and co-workers.7 In this Feature Article, we have summarized recent advances in catalytic enantioselective carbometallation of cyclopropenes and cyclobutenes, particularly focusing on enantioselective transformations and incorporating progress on enantioselective reactions of cyclobutenes. The plausible mechanisms and future of this field are discussed.
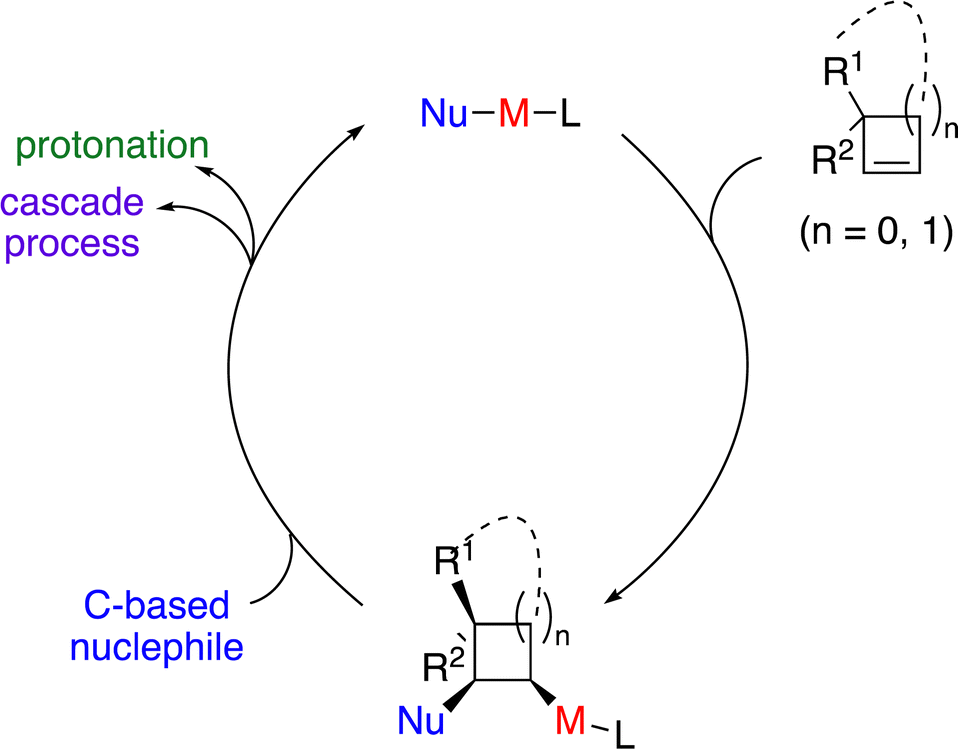 | ||
| Scheme 2 Strategy for catalytic enantioselective carbometallation of small cyclic alkenes. | ||
2. Catalytic enantioselective carbometallation of cyclopropenes
Enantioenriched cyclopropanes are common motifs in biologically active molecules and natural products, as well as important intermediates in organic synthesis (Scheme 1). Therefore, approaches for enantioselective incorporation of carbon-based substituents onto the three-membered rings to access enantioenriched cyclopropanes are in high demand. Although pioneering works on catalytic enantioselective carbometallation of cyclopropenes with highly reactive organometallic reagents have been reported, only simple alkyl and phenyl groups can be introduced due to the requirement of stoichiometric sensitive organometallic reagents that require multistep processes and are difficult to handle.8 Such limitation prompted research on enantioselective carbometallation with more robust carbon-based precursors and catalytically in situ generated organometallic species.Organoboron compounds represent a class of more robust reagents with high diversity and functional group tolerance albeit with lower reactivity. Marek and co-workers reported the first example of Rh-catalyzed enantioselective carbometallation of cyclopropenes with aryl boronic acids (Scheme 3).9 A wide range of aryl groups can be introduced, albeit with the incompatibility of heteroaryls. Mechanistic studies revealed that the reaction proceeded through carbometallation of cyclopropenes followed by protonation with H2O as a co-solvent (Scheme 4a). The proposed catalytic cycle is shown in Scheme 4b. Transmetallation with the aryl boronic acid generated Rh–Ar species, which underwent enantioselective carbometallation of cyclopropenes to afford a cyclopropyl–Rh intermediate and subsequent protonation to release the product and regenerate the catalyst.
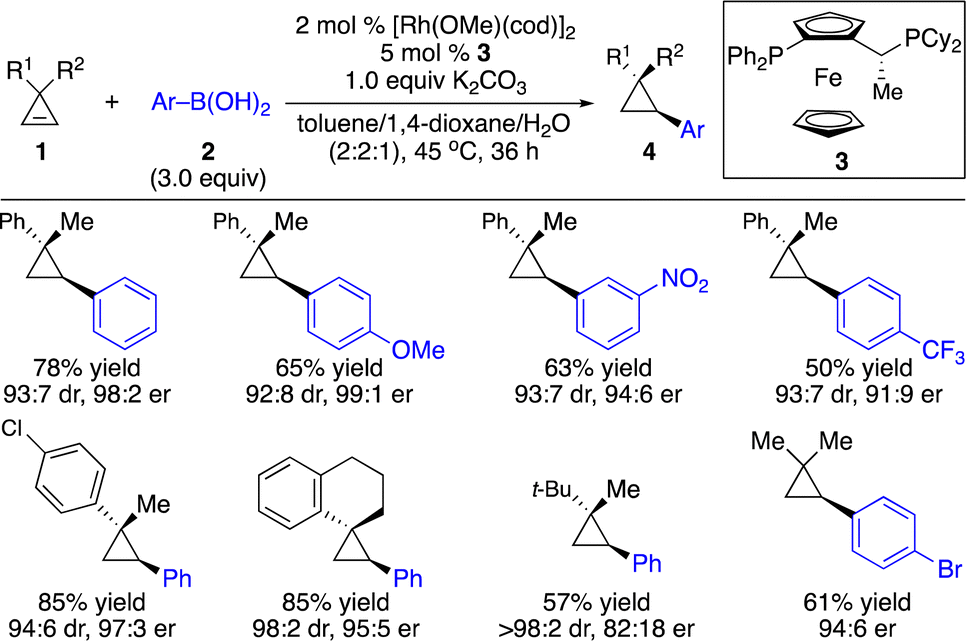 | ||
| Scheme 3 Rh-catalyzed enantioselective hydroarylation of cyclopropenes. | ||
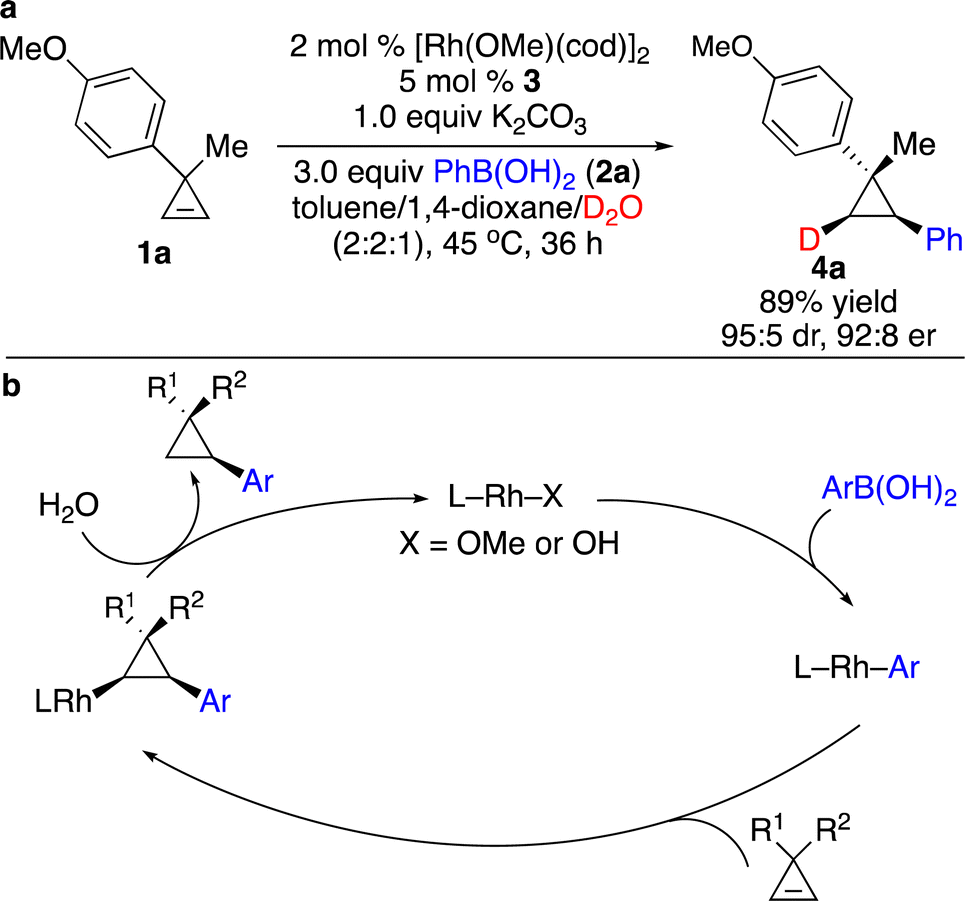 | ||
| Scheme 4 Mechanistic studies and proposed catalytic cycle. (a) Reaction with D2O. (b) The proposed catalytic cycle. | ||
Subsequently, we developed a complementary Co-catalyzed protocol for enantioselective hydroarylation of cyclopropenes (Scheme 5).10 2-Furyl and 2-benzofuryl boronic acids were well tolerated. Unlike the Rh-catalyzed process, performing the reaction in the presence of CD3OD resulted in no deuterium incorporation, indicating that the proton source for the protonation of the C–Co bond came from the boronic acid.
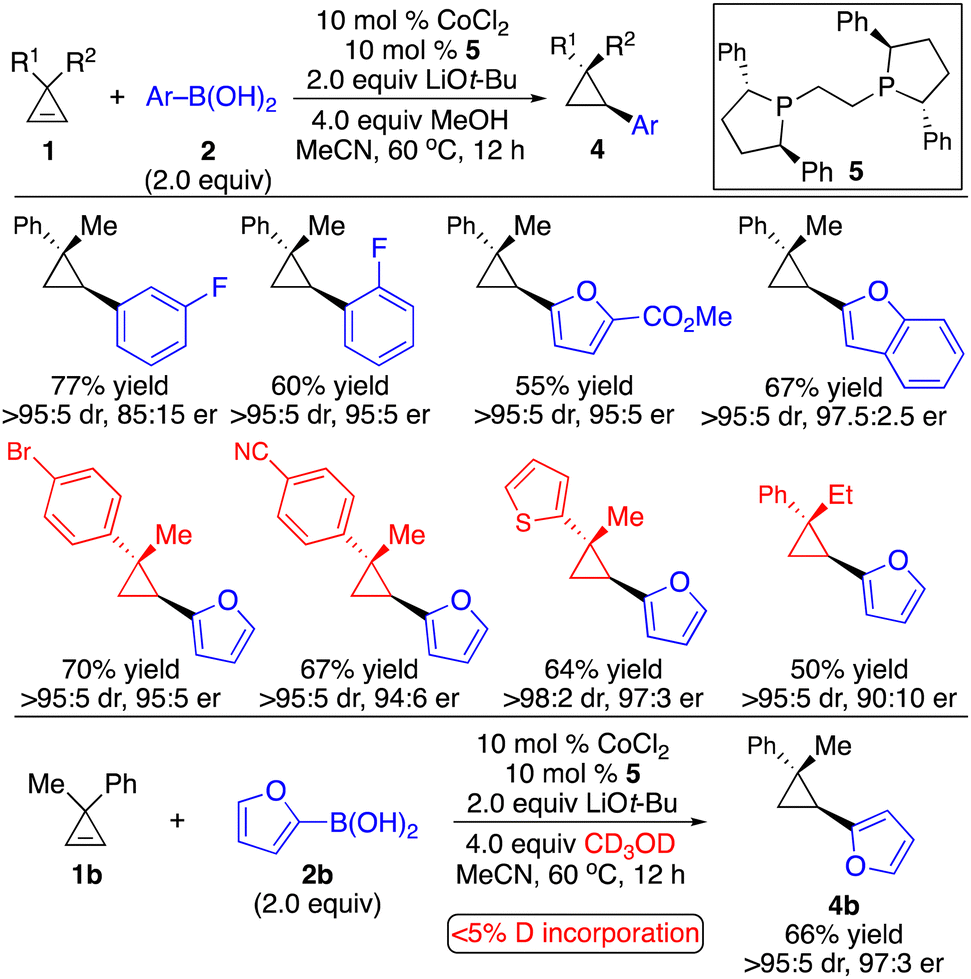 | ||
| Scheme 5 Co-catalyzed enantioselective hydroarylation of cyclopropenes. | ||
In contrast to hydroarylation of cyclopropenes, enantioselective hydroalkenylation of cyclopropenes is more challenging due to the difficulty to generate alkenyl–metal reagents and instability of such species. A pioneering Cu-catalyzed enantioselective hydroalkenylation of cyclopropenes with alkenyl–Al reagents was disclosed, albeit with significant limitations for the substrate scope and inefficient control of stereoselectivity (Scheme 6).11 As the alkenyl–Al reagents were prepared through hydroalumination of alkynes, the substitution pattern was limited to (E)-1,2-disubstituted alkenyls. Inseparable side products from direct alkyl addition were observed in most cases. Therefore, enantioselective hydroalkenylation of cyclopropenes with alkenylborons is in high demand.
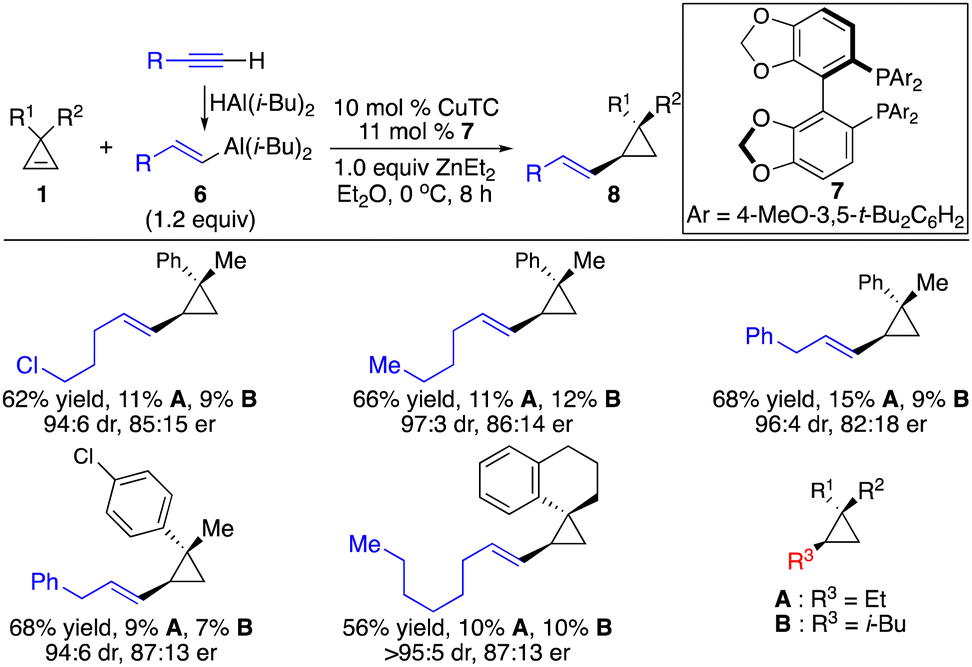 | ||
| Scheme 6 Cu-catalyzed enantioselective hydroalkenylation of cyclopropenes with alkenyl–Al reagents. | ||
We applied our Co-catalyzed system to transformations with alkenylboron reagents 9, and found that the Co complex derived from bisphosphine 10 promoted the reaction, enabling the incorporation of (E)-, (Z)-disubstituted and trisubstituted alkenyl groups that are otherwise difficult to access with other organometallic reagents in high efficiency and stereoselectivity (Scheme 7).12 Highly functionalized cyclopropane 1c and dienylboron 12 can be fused followed by hydrolysis of the ester to afford an analogue of retinoid X receptor agonist 13 with 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 er as a single diastereomer (Scheme 7).
:3 er as a single diastereomer (Scheme 7).
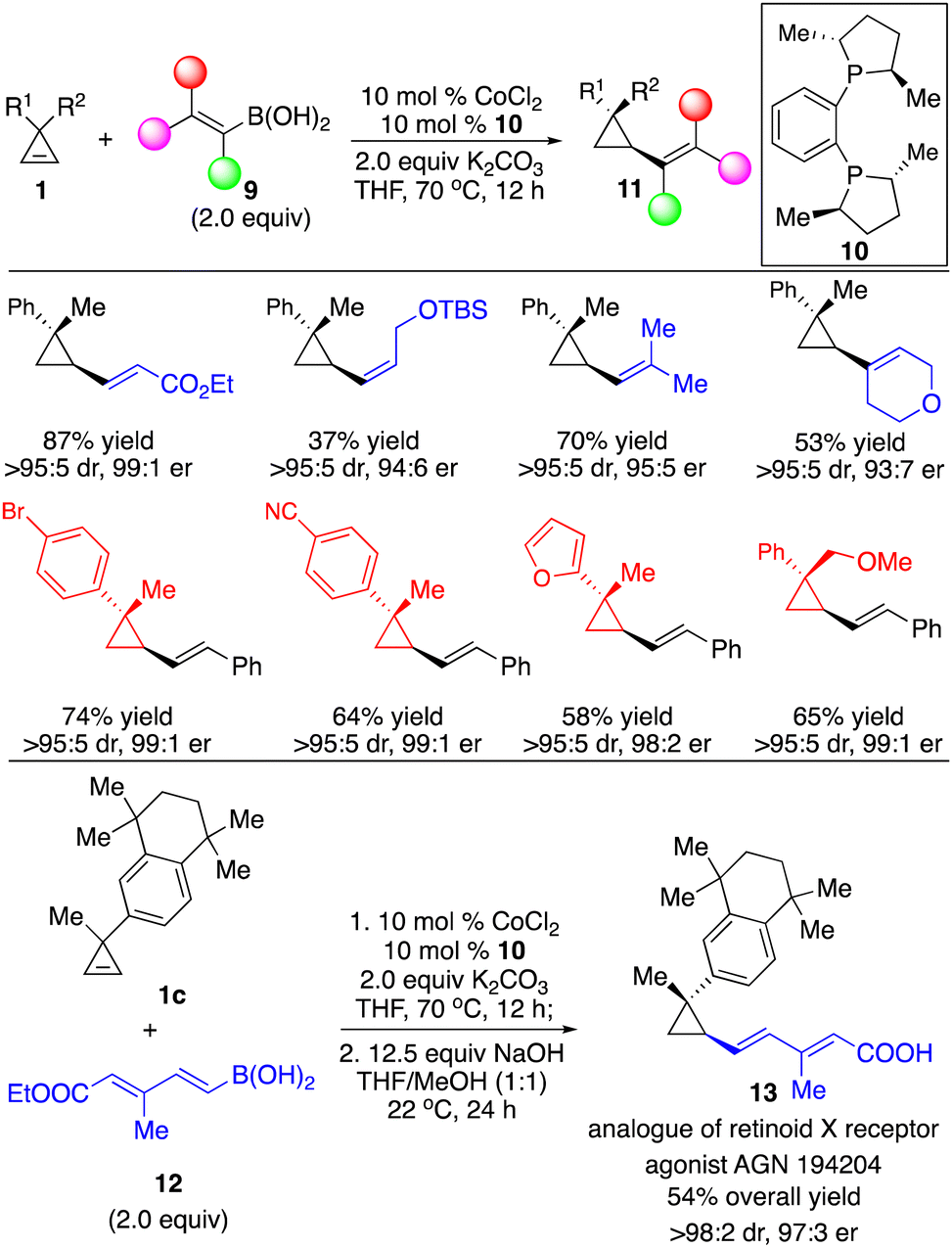 | ||
| Scheme 7 Co-catalyzed enantioselective hydroalkenylation of cyclopropenes with alkenylboronic acids. | ||
A series of preliminary mechanistic studies were conducted. Reaction of deuterated cyclopropene 1d delivered a single diastereomer of the product 14, suggesting that syn-addition of alkenyl–Co species to the cyclopropene occurred and protonation of the stereogenic C–Co bond was stereoretentive (Scheme 8a). Competitive kinetic isotope effect experiments revealed that cleavage of the O–H bond might be the rate-determining step (Scheme 8b). Based on all the observations, we proposed the possible catalytic cycle (Scheme 8c). Transmetallation of the alkenylboronic acid 9a with the phosphine–Co(II) complex assisted by K2CO3 generated alkenyl–Co(II) species II, which underwent enantioselective carbometallation with cyclopropene 1b to afford cyclopropyl–Co intermediate III. Protonation of the C–Co bond with the boronic acid provided the hydroalkenylation product 17 and regenerated the catalyst.
 | ||
| Scheme 8 Mechanistic studies and proposed catalytic cycle for Co-catalyzed enantioselective hydroalkenylation of cyclopropenes. (a) Reaction with deuterated cyclopropene. (b) Competitive kinetic isotope effect experiments. (c) The proposed catalytic cycle. | ||
Besides protonation of the cyclopropyl–metal formed through transmetallation with organoboron reagents, transformations with another electrophile that enabled catalytic enantioselective difunctionalization of cyclopropenes are more attractive and challenging. Although processes involving highly sensitive organometallic reagents, such as Grignard, organozinc, or organoaluminum reagents, to generate cyclopropyl–metal species followed by trapping with an extra operation or step for addition of electrophiles have been revealed, direct three-component reactions of more robust organoboron reagents with the electrophiles in the same vessel provided better functional group tolerance and incorporated higher diversity of substituents. Zhang and co-workers reported a Cu-catalyzed three-component protocol for diastereo- and enantioselective difunctionalization of cyclopropenes with arylboron reagents and electrophilic O-benzoyl hydroxylamines (Scheme 9a).13 The reactions proceeded through transmetallation with arylboron reagents to form aryl–Cu intermediates followed by carbocupration of the cyclopropenes, delivering cyclopropyl–Cu species that reacted with O-benzoyl hydroxylamines. An aryl and an amino group can be installed simultaneously in a single transformation with high diastereo- and enantioselectivity. Extension of this system to alkenylboron reagents resulted in a Cu-catalyzed three-component diastereo- and enantioselective alkenylamination of cyclopropenes (Scheme 9b).14 A variety of (E)-1,2-disubstituted alkenyl and aminyl groups can be introduced onto the cyclopropane scaffold in high diastereo- and enantioselectivity.
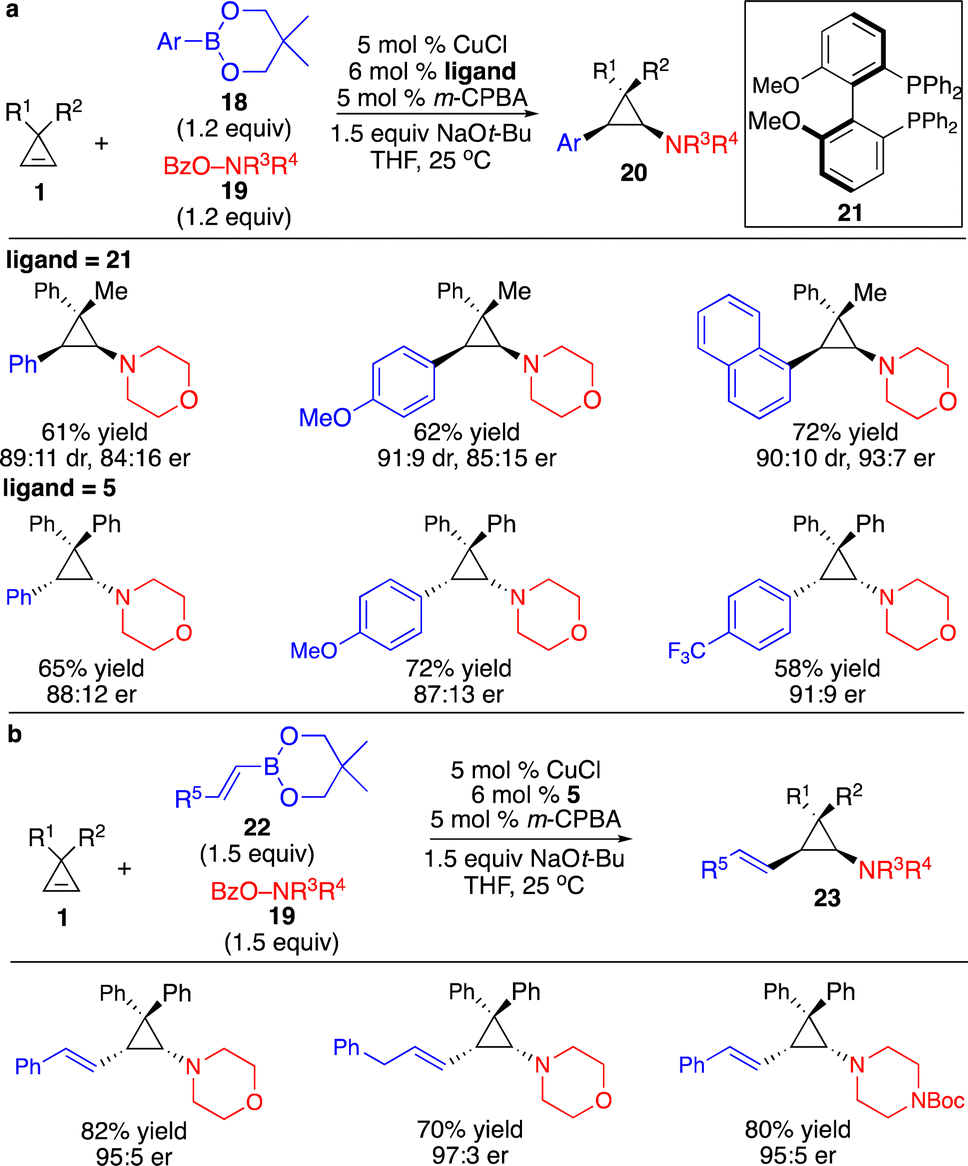 | ||
| Scheme 9 Cu-catalyzed three-component diastereo- and enantioselective difunctionalization of cyclopropenes. (a) Cu-catalyzed diastereo- and enantioselective arylamination of cyclopropenes. (b) Cu-catalyzed diastereo- and enantioselective alkenylamination of cyclopropenes. | ||
Catalytic in situ generation of organometallic species provided another attractive strategy for carbometallation of cyclopropenes in mild conditions with broad substrate scope and high functional group tolerance. Inspired by the precedence on Co-induced ring-opening of cyclopropanols to form Co-homoenolates in situ,15 we developed a Co-catalyzed protocol for enantioselective hydroalkylation of cyclopropenes with in situ generated Co-homoenolates from cyclopropanols, furnishing a wide range of enantioenriched cyclopropanes and enabling the introduction of functionalized alkyl groups in high efficiency, diastereo- and enantioselectivity (Scheme 10).16 The transformations proceeded without the need for any additive with 100% atom economy. Basic anion OAc− was crucial for the reactivity, working as a proton shuttle to facilitate coordination of the cyclopropanols to the Co center.
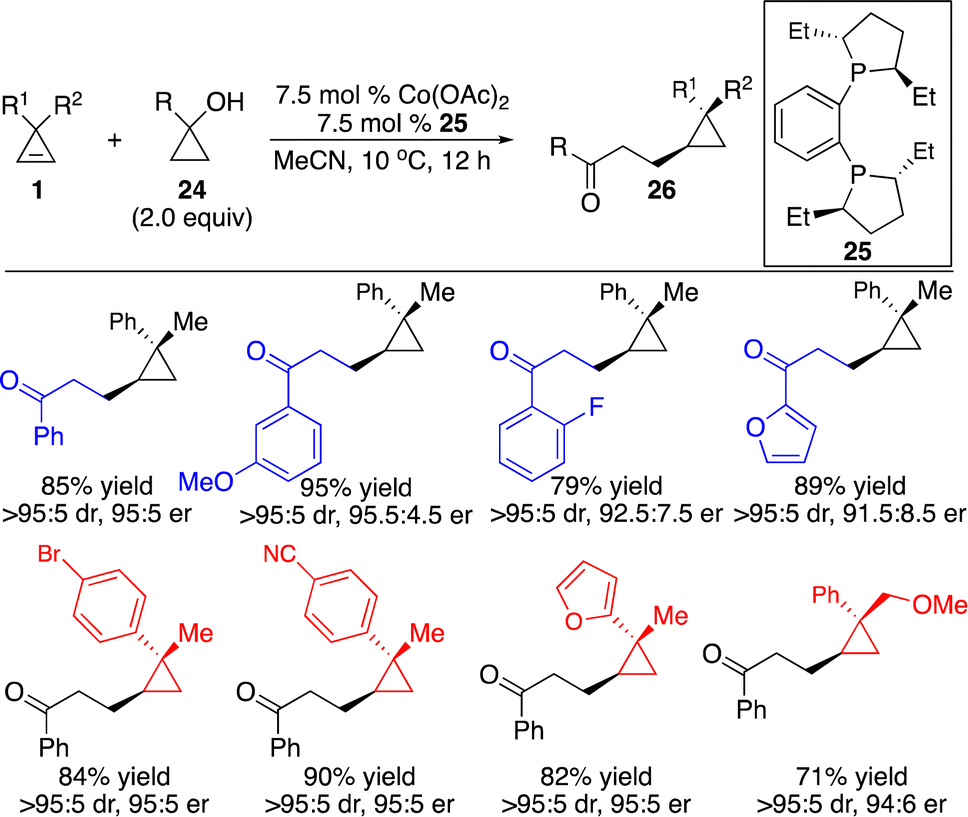 | ||
| Scheme 10 Co-catalyzed enantioselective hydroalkylation of cyclopropenes with Co homoenolates. | ||
To gain some insight into the mechanism, a series of experiments were performed. Treatment of deuterated cyclopropane 1d with cyclopropanol 24a in the presence of Co complex derived from phosphine 25 afforded 27 as a single diastereomer, suggesting that similar to our Co-catalyzed enantioselective hydroarylation and hydroalkenylation processes,10,12syn addition of the cyclopropene occurred and protonation of the C–Co bond was stereoselective (Scheme 11a). Competitive kinetic isotope experiments revealed that cleavage of the O–H bond might be the rate-determining step (Scheme 11b). Reaction of deuterated cyclopropanol 24b-D delivered ketone 29 with the incorporation of deuterium at the α position of the ketone, implying that enolization of the Co homoenolate intermediate occurred (Scheme 11c). The possible catalytic cycle was proposed, as shown in Scheme 11d. The basic OAc− facilitated deprotonation of the cyclopropanol and ring-opening to form Co homoenolate VII, which underwent carbometallation of cyclopropene to furnish cyclopropyl–Co species X. Protonation of the C–Co bond provided the product and regenerated the catalyst.
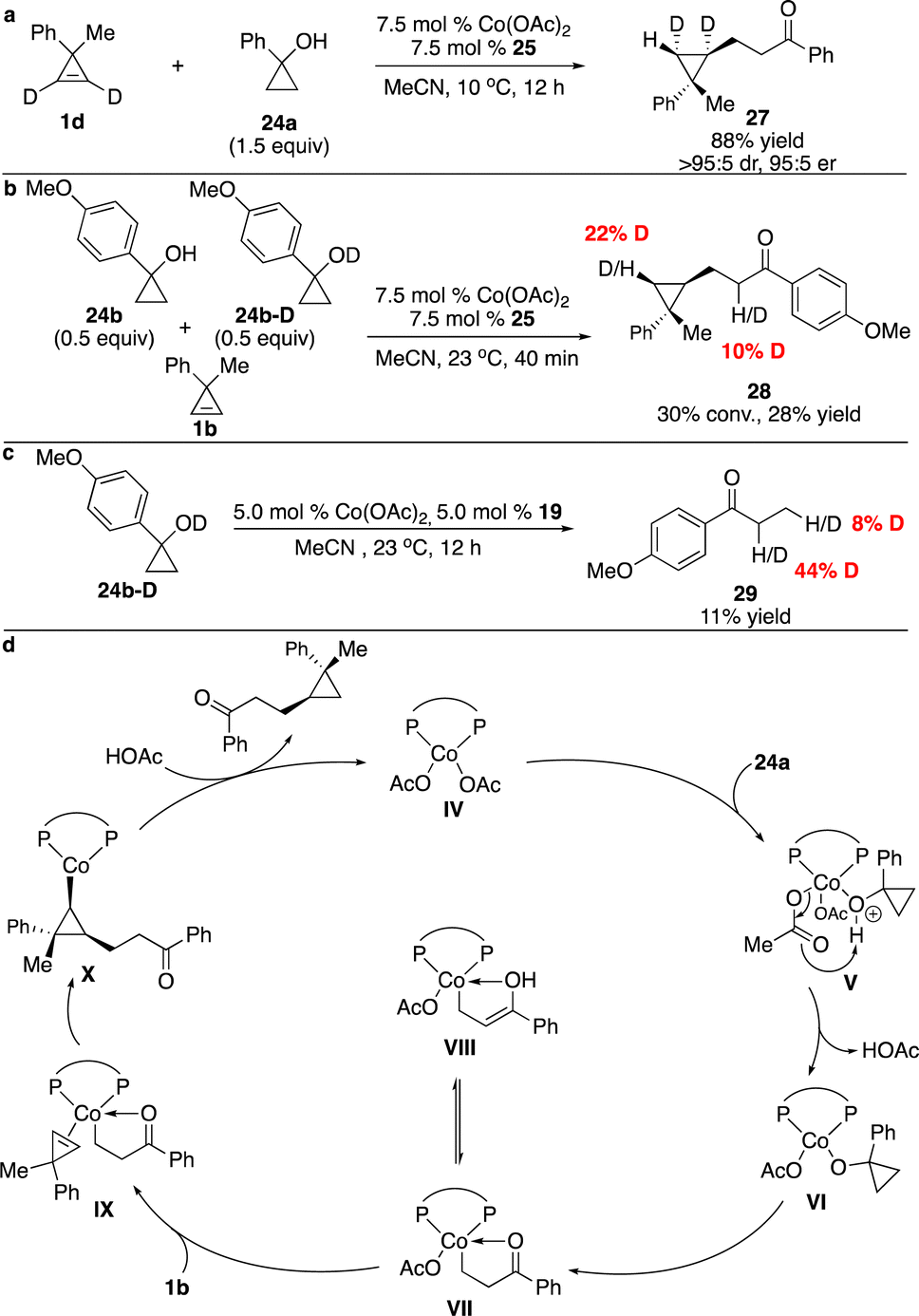 | ||
| Scheme 11 Mechanistic studies and proposed catalytic cycle for Co-catalyzed enantioselective hydroalkylation of cyclopropenes with Co homoenolates. (a) Reaction with deuterated cyclopropene. (b) Competitive kinetic isotope effect experiments. (c) Control experiments. (d) The proposed catalytic cycle. | ||
Based on the same concept, in situ deprotonation of terminal alkynes to generate alkynyl–metal species followed by enantioselective carbometallation of cyclopropenes provided an atom-economical strategy for the enantioselective introduction of an alkynyl group onto three-membered rings. Hou and co-workers described a chiral half-sandwich Gd-based catalyst 31 that promoted enantioselective hydroalkynylation of cyclopropenes through alkynyl–Gd addition to cyclopropenes followed by protonation of the C–Gd bond (Scheme 12).17 A broad scope of aryl-, alkyl-and silyl-substituted terminal alkynes can be incorporated to furnish the enantioenriched alkynyl cyclopropanes.
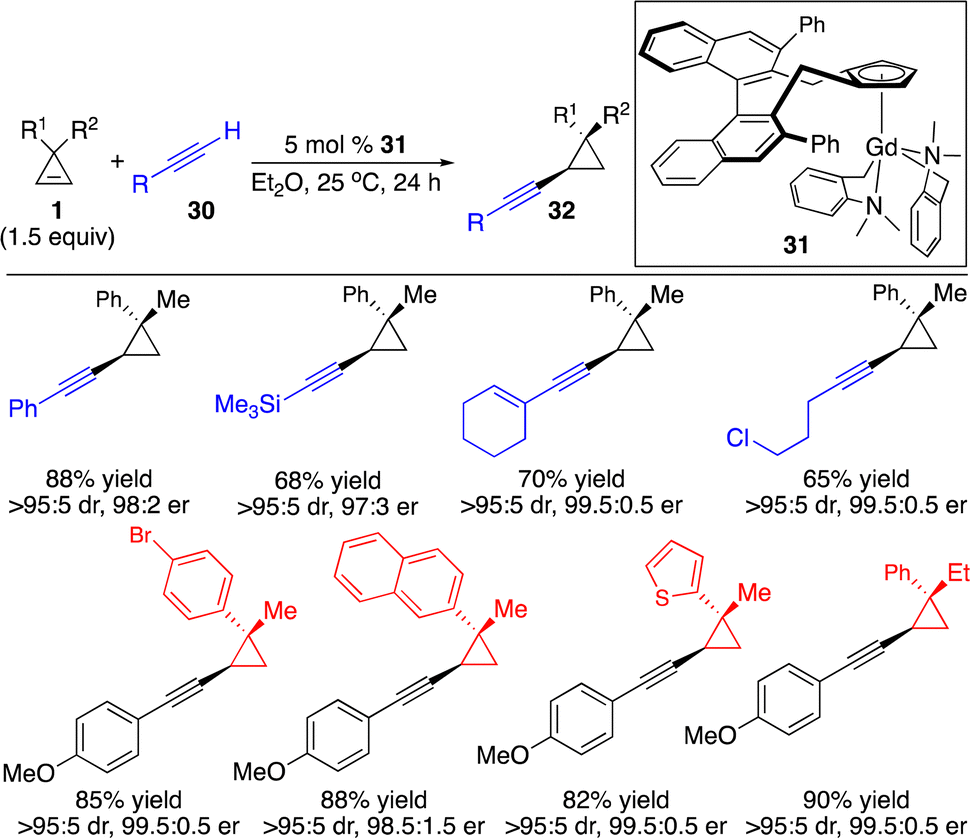 | ||
| Scheme 12 Gd-catalyzed enantioselective hydroalkynylation of cyclopropenes. | ||
Subsequently, Marek and co-workers disclosed a Pd-catalyzed protocol for enantioselective alkynylation of cyclopropenes with terminal alkynes (Scheme 13).18 A wide range of aryl-substituted alkynes, 1,3-diynes and enynes can be converted in high efficiency, diastereo- and enantioselectivity.
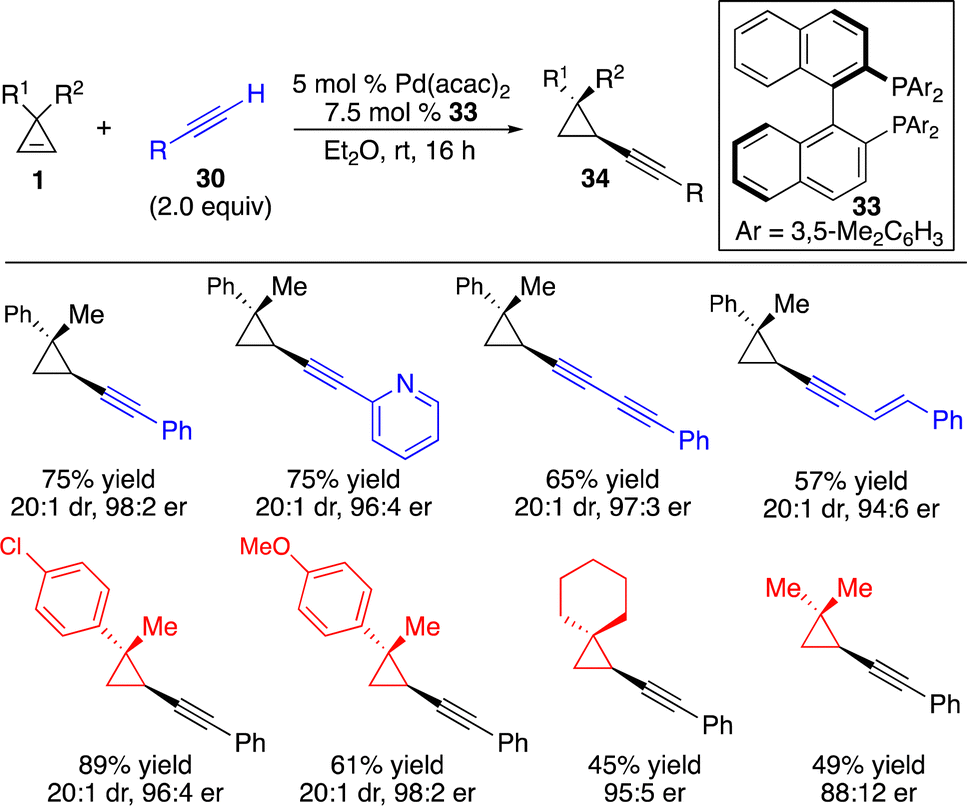 | ||
| Scheme 13 Pd-catalyzed enantioselective hydroalkynylation of cyclopropenes. | ||
3. Catalytic enantioselective carbometallation of cyclobutenes
Enantioenriched cyclobutanes widely exist in natural products and pharmaceutically important molecules, and are useful building blocks in organic synthesis (Scheme 1). Therefore, the development of a general and modular strategy for the enantioselective introduction of highly diversified and functionalized units onto the four-membered scaffolds is sought after. Enantioselective carbometallation of cyclobutenes represents a straightforward approach for direct incorporation of a carbon-based substituent onto the cyclobutanes. However, in contrast to cyclopropenes, cyclobutenes are much less reactive. The sluggish reactivity arose from relatively lower olefinic strain (1.9 kcal mol−1) compared with that in cyclopropenes (27.7 kcal mol−1) and bicyclic olefins (norbornene: 4.8 kcal mol−1).9,19 Although few protocols for Cu-catalyzed enantioselective activated cyclobutenes with dialkylzinc reagents have been disclosed,20 approaches for catalytic enantioselective introduction of other carbon-based substituents with high diversity and functional group tolerance and transformations of unactivated cyclobutenes have remained far less developed.Enantioselective carbometallation of cyclobutenes with arylboron reagents was challenging due to the relatively low reactivity of arylborons and cyclobutenes. Pioneering work on Rh-catalyzed enantioselective arylation of ester-activated cyclobutenes with arylboronic acids to afford a broad scope of enantioenriched disubstituted cyclobutanes was reported by Lin and co-workers (Scheme 14).21
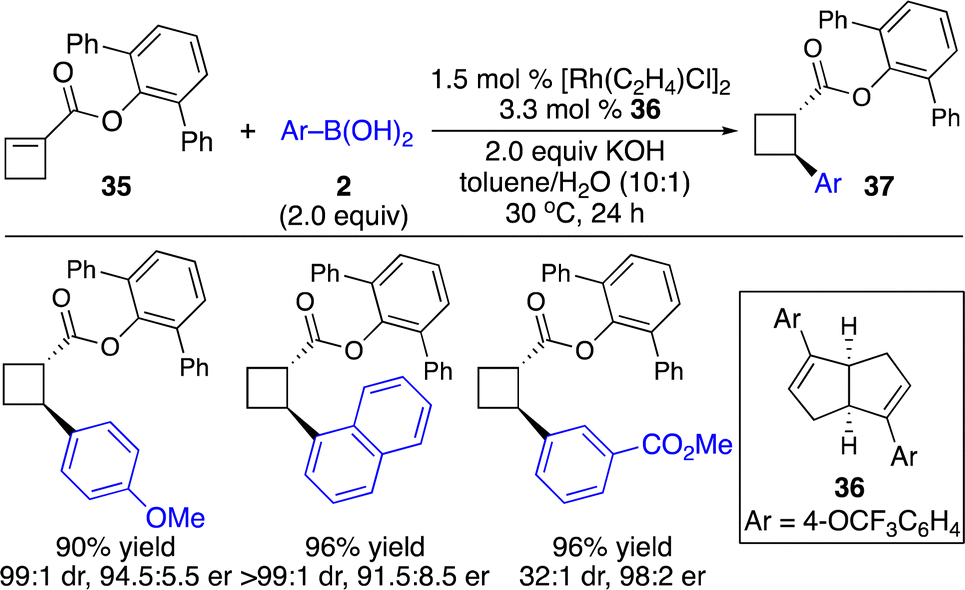 | ||
| Scheme 14 Rh-catalyzed enantioselective arylation of ester-activated cyclobutenes. | ||
More recently, Fletcher and co-workers disclosed a series of Rh-catalyzed enantioselective transformations of unactivated cyclobutenes initiated by carbometallation with arylboronic acids followed by several different pathways (Scheme 15).22 Reactions of cyclobutenes fused by a five-membered ring with a wide range of arylboronic acids afforded arylation products through aryl–Rh addition followed by cascade β-H elimination and reinsertion with subsequent protonation (Scheme 15a and b). Transformations of cyclobutenes with arylboronic acids proceeded through enantioselective carbometallation followed by a chain-walking process terminated by β-O elimination (pathway a, Scheme 15c). When electron-deficient boronic acids were used, an alternative pathway for carbometallation followed by 1,4-Rh migration and protonation was observed (pathway b, Scheme 15c).
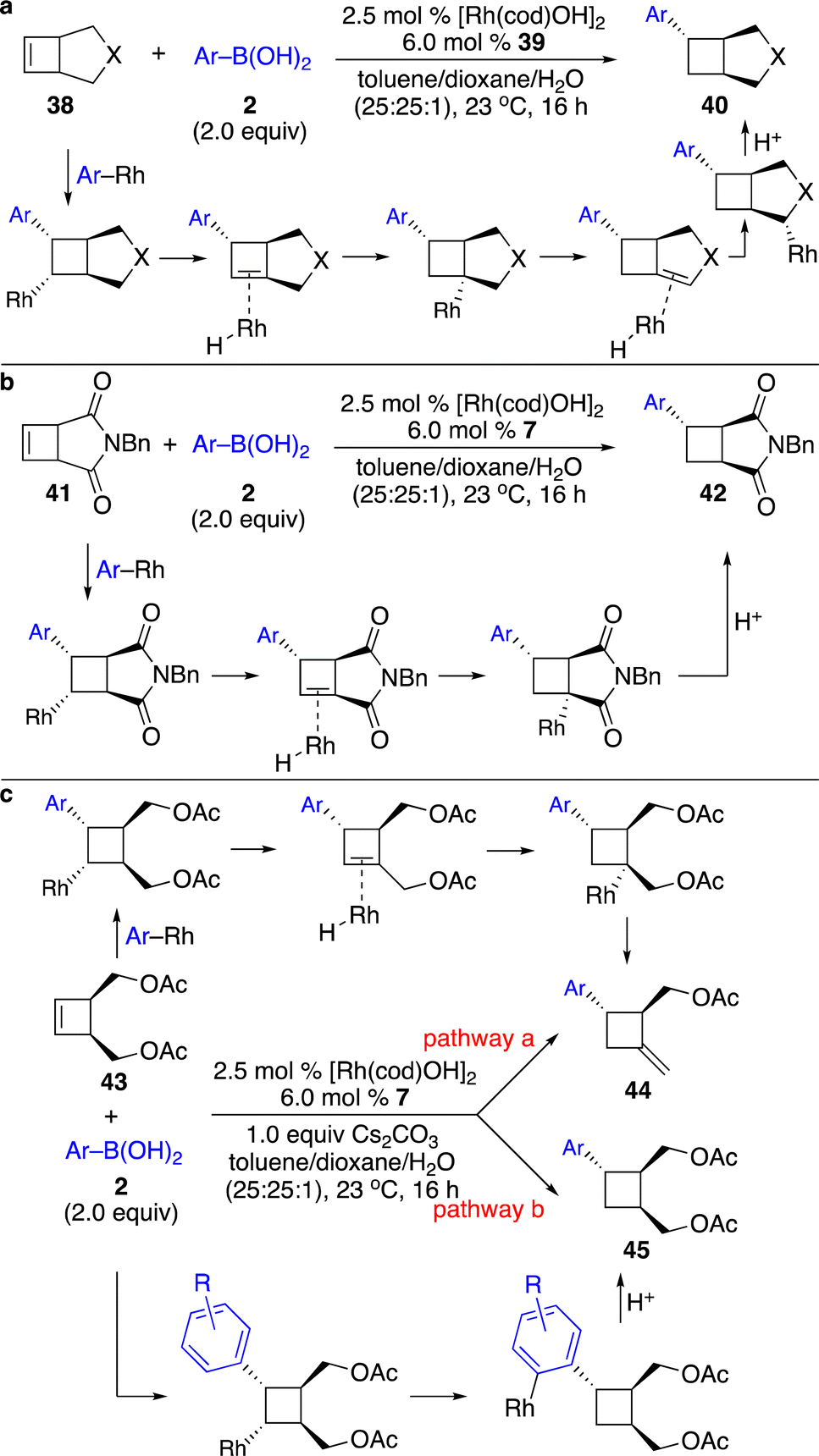 | ||
| Scheme 15 Rh-catalyzed enantioselective cascade arylation of cyclobutenes. (a) Rh-catalyzed enantioselective cascade arylation of bicyclic cyclobutenes. (b) Rh-catalyzed enantioselective 1,5-addition to a non-conjugated β,γ-unsaturated carbonyl compound. (c) Remote substitution. | ||
The same group also extended the concept to enantioselective arylation of cyclobutanone ketals (Scheme 16).23 Ring-opening products were generated through regio- and enantioselective carbometallation followed by β-O elimination.
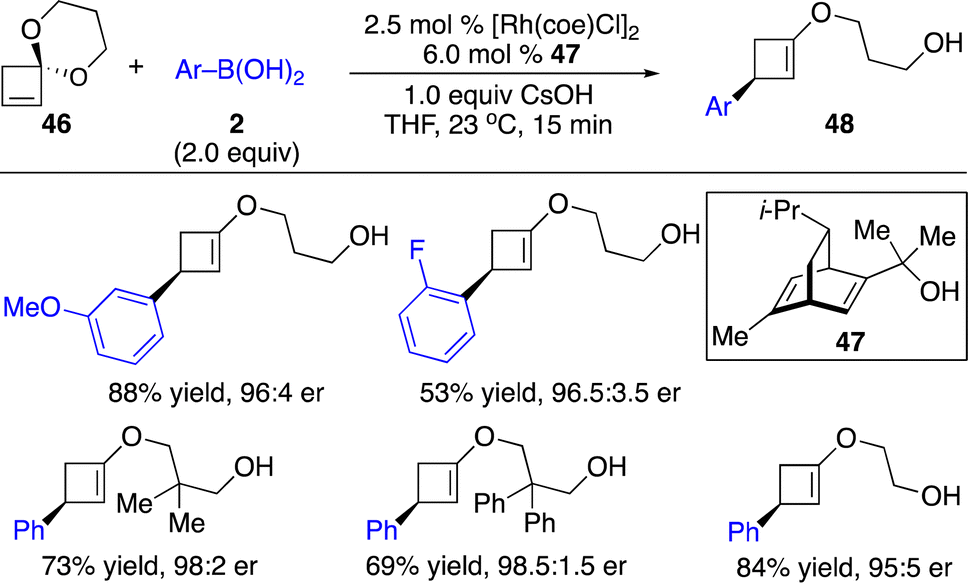 | ||
| Scheme 16 Rh-catalyzed enantioselective arylation of cyclobutanone ketals. | ||
Lu and co-workers reported a Pd-catalyzed protocol for divergent enantioselective functionalization of cyclobutenes triggered by aryl–Pd addition with aryl iodides (Scheme 17).24 A new monodentate P-stereogenic phosphine ligand 50 was developed. In the presence of HCOONa, β-H elimination followed by re-insertion and protonation delivered formal hydroarylation products 51, whereas 1,3-diarylation occurred with arylboronic acids through chain-walking followed by transmetallation and transformations with tetraarylborates provided 1,2-diarylation products 53 without chain-walking. The authors also demonstrated that deuterated cyclobutanes with deuterium at different sites could be generated by proper combination of deuterated reagents.
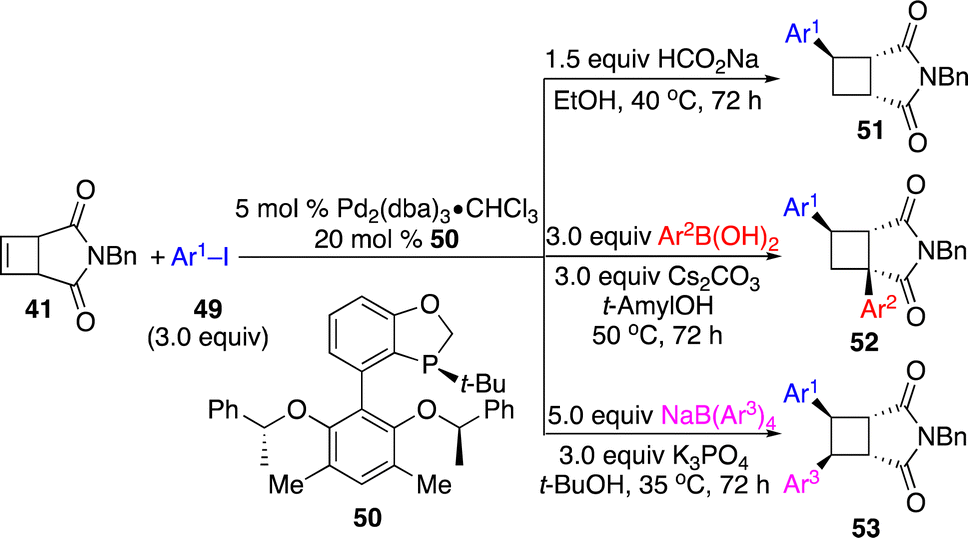 | ||
| Scheme 17 Pd-catalyzed divergent enantioselective arylation of cyclobutenes. | ||
Besides all the precedents on enantioselective carbometallation of cyclobutenes with aryl nucleophiles, we developed a series of cobalt-catalyzed protocols for enantioselective carbometallation of cyclobutenes with cobalt homoenolates, alkynyl and allyl nucleophiles.25 Unlike reactions of cobalt homoenolates with cyclopropenes that provided high efficiency even at cryogenic conditions,16 elevated temperature was required for transformations of ester-activated cyclobutenes (Scheme 18a). Addition of Zn to reduce the Co(II) complex to Co(I) species was crucial for the reactivity, as the Co(I) complex had higher affinity to the alkenes, assisting coordination of the substrate to the Co center. Phosphinooxazoline ligand 55 was found to give high enantioselectivity. A broad scope of cyclopropanols bearing aryl, alkenyl and alkyl groups were able to undergo the reactions with high diastereo- and enantioselectivity, enabling the incorporation of a high diversity of densely functionalized alkyl groups onto the cyclobutane scaffold. Ligands with stronger σ-donation were required for transformations with unactivated cyclobutenes. Only the Co complex derived from bisphosphine 57a was able to promote the reaction of cyclobutene fused with succinimide 41 (Scheme 18b). We modified the ligand as we previously reported, and revealed that phosphine 57b that contains electron-deficient aryls retarded the reaction, whereas transformation in the presence of bisphosphine 57c bearing phenyls substituted with a strong electron-donating group slightly improved the enantioselectivity. We further found that more sterically congested unactivated cyclobutenes containing a spirocycle could be converted regioselectively in the presence of the Co complex formed in situ from imidazolinium salt 60 (Scheme 18c). As expected, only the strong σ-donor N-heterocyclic carbene ligand provided the reactivity. The regioselectivity might result from the longer distance between the carbon and cobalt than that between carbon and the homoenolate chain in the transition state of carbometallation with cobalt homoenolate, as the C–Co bond is longer than the C–C bond. The Co terminus is less sensitive to steric hindrance, and locating the Co center adjacent to the quaternary carbon is favored.
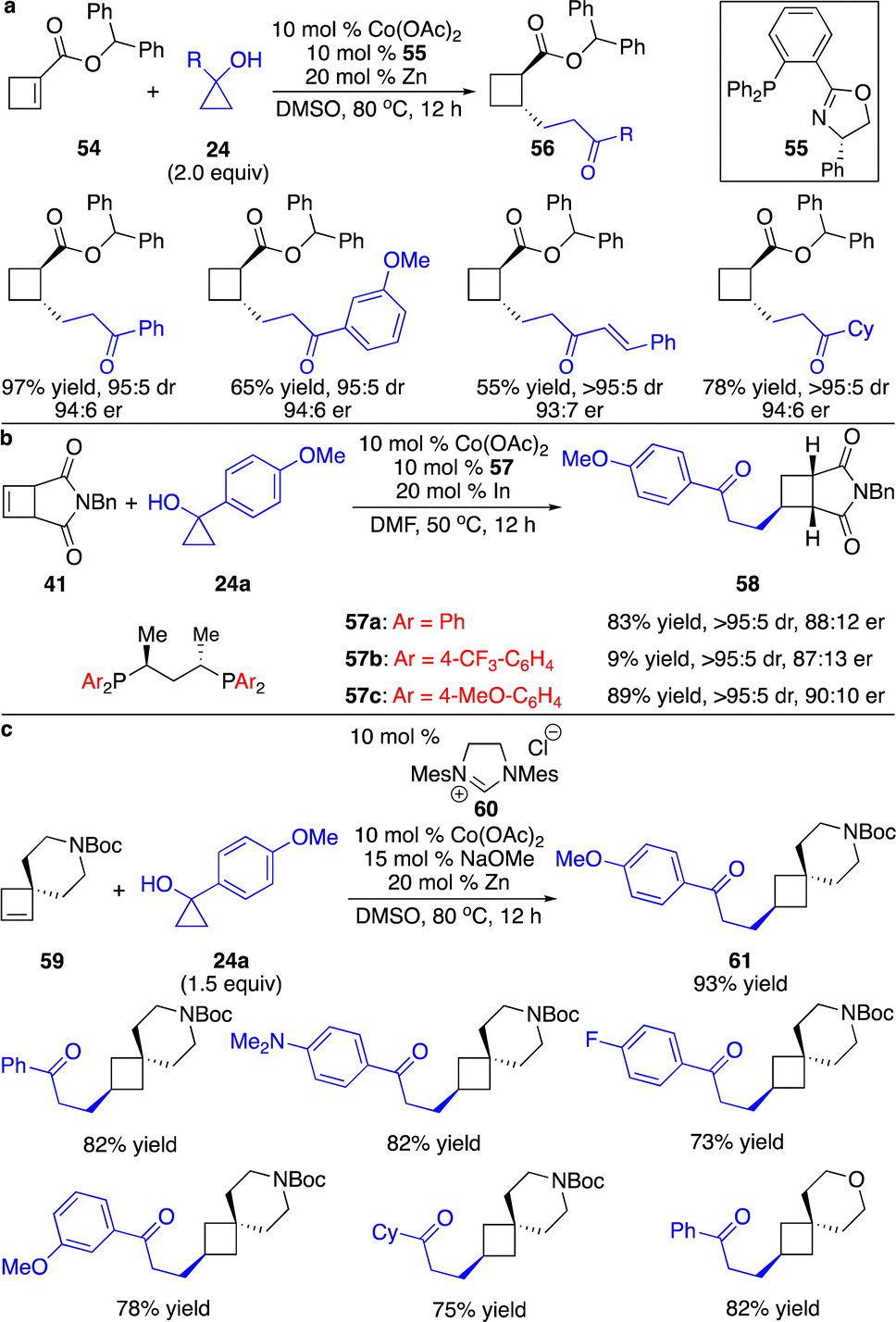 | ||
| Scheme 18 Co-catalyzed hydroalkylation of cyclobutenes with Co homoenolates. (a) Enantioselective 1,4-addition to ester-activated cyclobutenes. (b) Reaction with the nonpolar cyclobutene. (c) Reactions with cyclobutenes bearing a quaternary center. | ||
Parallel kinetic isotope effect experiments implied that proton transfer might not be the rate-determining step (Scheme 19a). Similar to reactions with cyclopropenes,16 the proposed catalytic cycle is shown in Scheme 19b. Coordination of cyclopropanol 24b to the Co center and deprotonation assisted by basic counterion (OAc−) generated the Co homoenolate XIV, which underwent enantioselective carbometallation followed by protonation to afford the product and regenerate the catalyst.
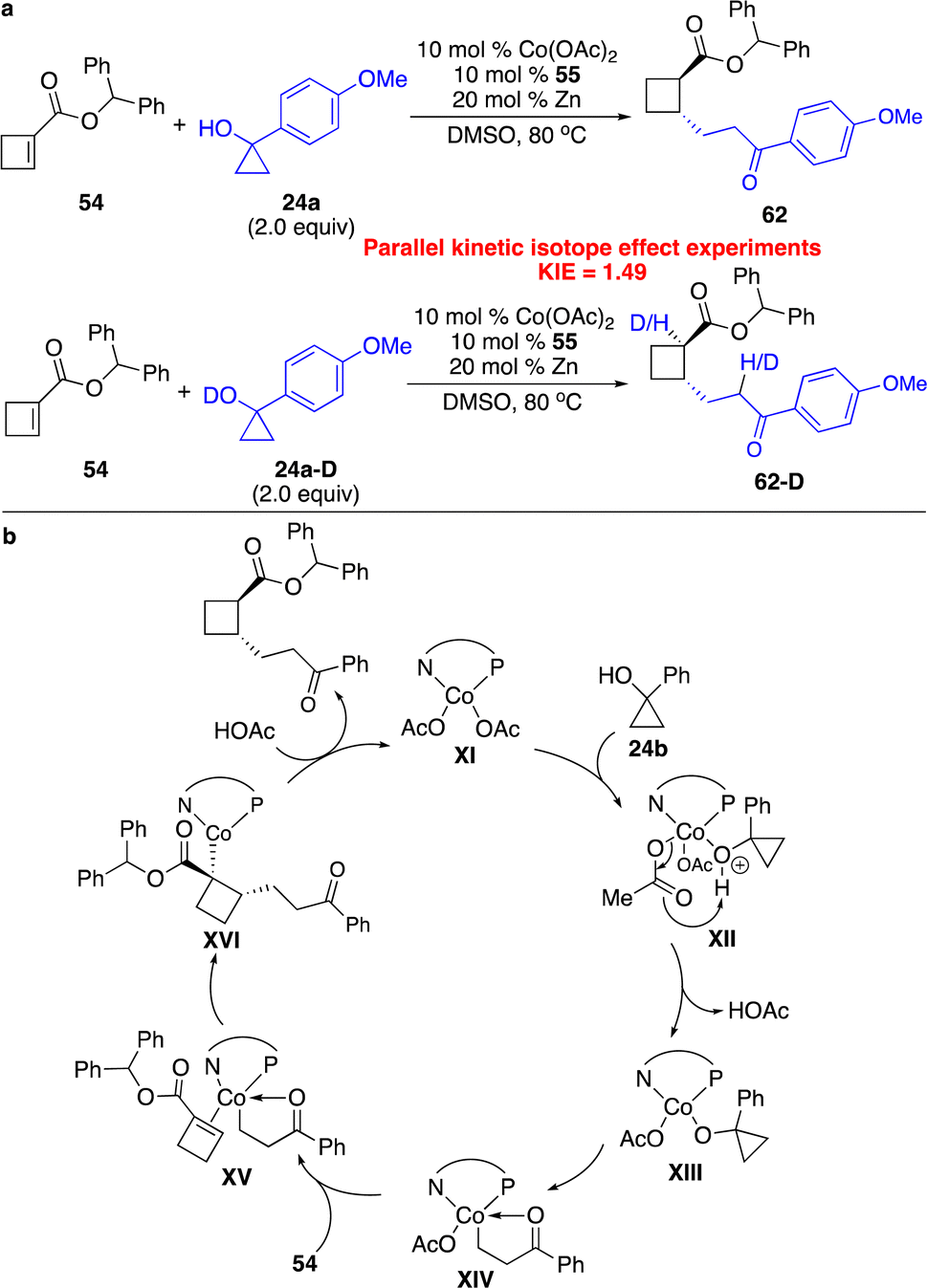 | ||
| Scheme 19 Mechanistic studies and proposed catalytic cycle for Co-catalyzed enantioselective hydroalkylation of cyclobutenes with Co homoenolates. (a) Parallel kinetic isotope effect experiments. (b) The proposed catalytic cycle. | ||
We next attempted to incorporate an alkynyl group onto the four-membered rings. The facile coordination of the Co(I) center to the alkyne enabled deprotonation of the alkyne by a basic counterion (OAc−) to generate an alkynyl–Co intermediate. Although enantioselective cobalt-catalyzed alkynyl addition to conjugated dienes,26 allenes,27 bridged bicyclic alkenes,28 and enones29 has been developed, metal-catalyzed hydroalkynylation of less strained cyclobutenes in the absence of an activated group remained unknown. By taking advantage of this nature of the Co(I) complex, we have disclosed the first example of enantioselective alkynylation of unactivated cyclobutenes promoted by a Co-based catalyst (Scheme 20).25 It was found that only electron-rich bisphosphine 63 bearing P-stereogenic centers induced high enantioselectivity for reactions of cyclobutenes fused with succinimide. Cyclobutenes containing a wide range of aryls and alkyls can be transformed with silyl-substituted terminal alkynes with high efficiency and stereoselectivity.
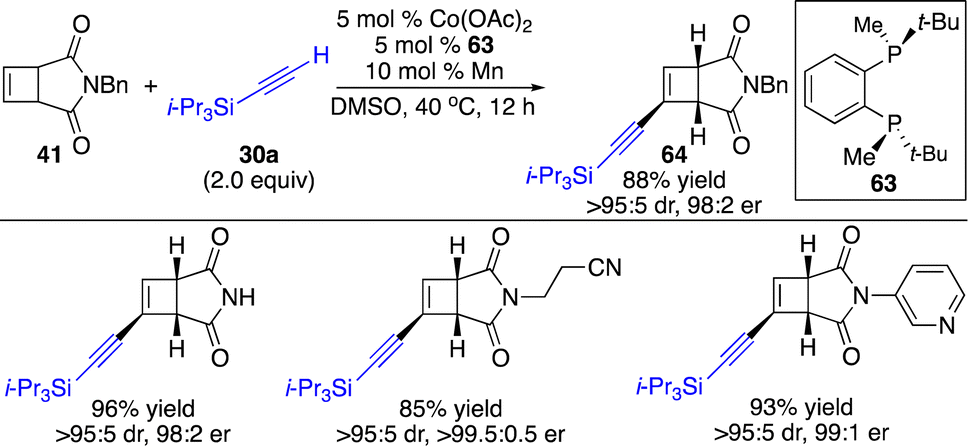 | ||
| Scheme 20 Co-catalyzed enantioselective hydroalkynylation of cyclobutenes with terminal alkynes. | ||
We also conducted a series of preliminary mechanistic studies. Reaction with deuterated alkyne 30a-D afforded enantioenriched cyclobutene 64-D with a deuterated stereogenic center, indicating that syn alkynyl–Co addition occurred and protonation of the resulting C–Co bond was stereoretentive (Scheme 21a). Parallel competitive isotope effect experiments suggested that the proton transfer process might not be the rate-determining step (Scheme 21b). The proposed catalytic cycle is shown in Scheme 21c. Activation of the C–H bond of the terminal alkyne by the Co(I) complex to form the alkynyl–Co species XIX followed by addition to cyclobutenes furnished cyclobutyl–Co intermediate XXI, which underwent stereoselective protonation to release the product 64 and regenerate the catalyst.
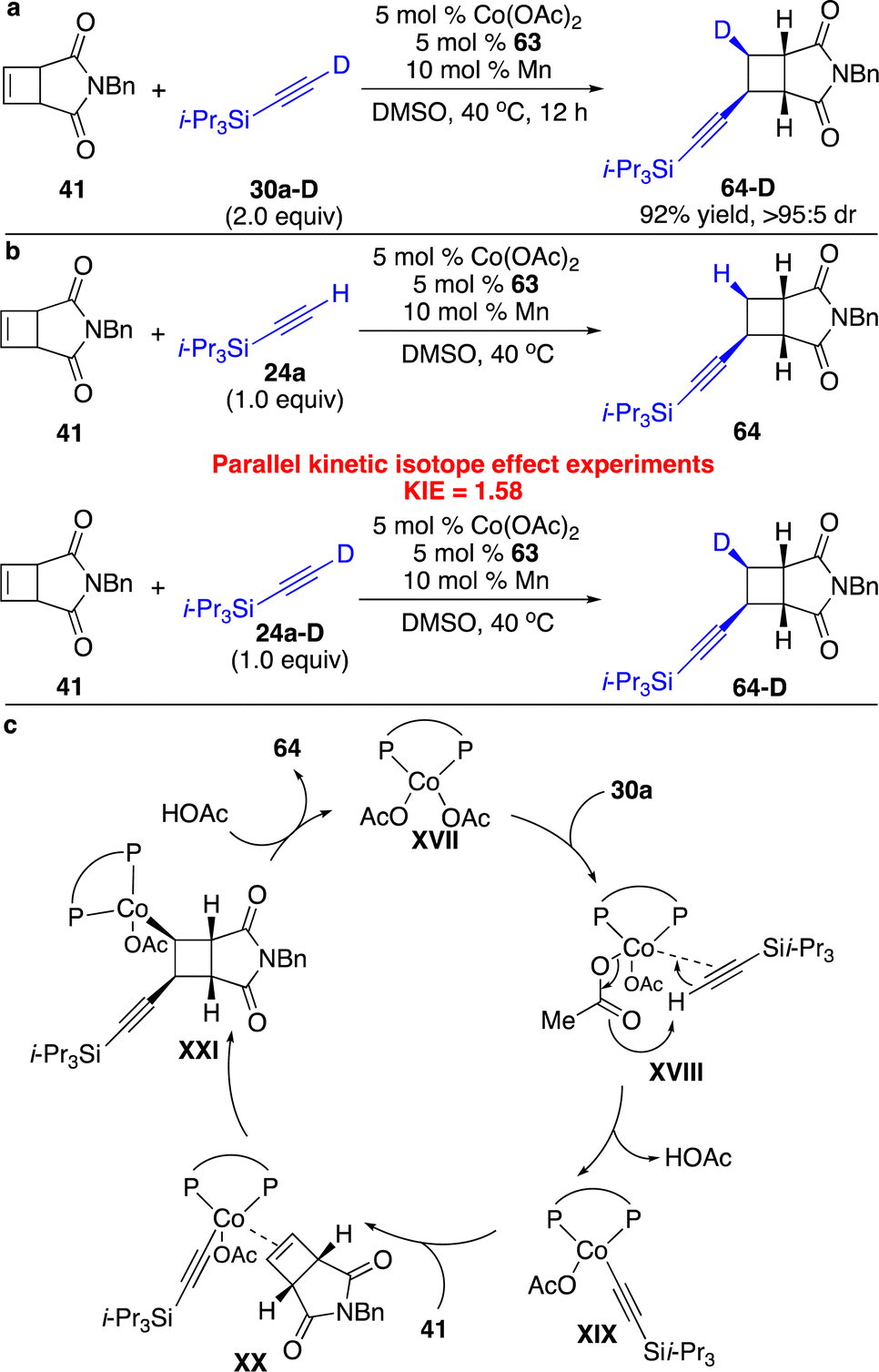 | ||
| Scheme 21 Mechanistic studies and proposed catalytic cycle for Co-catalyzed enantioselective hydroalkynylation of cyclobutenes. (a) Reaction with deuterated alkyne. (b) Parallel kinetic isotope effect experiments. (c) The proposed catalytic cycle. | ||
We also investigated the enantioselective functionalization of cyclobutenes initiated by allyl–Co addition. Only one pioneering instance of Co-catalyzed enantioselective allyl–Co addition to the more strained bridged bicyclic alkenes has been reported by Zhao and co-workers.30 For the less reactive cyclobutenes, this process remained challenging and elusive. Unexpectedly, application of the phosphine–Co complex derived from 63 to the transformation of cyclobutene with potassium trifluoroborate 65 furnished the ring-opening product 66 in 79% yield with 98.5:1.5 er (Scheme 22). The cyclobutane generated from hydroallylation was not observed. A wide range of cyclobutenes containing alkyls, and aryls can be converted to the ring-opening products in high efficiency and stereoselectivity. Functional groups, such as cyano, ketone, and heteroaryls, are well tolerated. Similar to the alkynylation, the succinimide moiety was necessary for the reactivity.
 | ||
| Scheme 22 Co-catalyzed enantioselective cascade allyl addition to cyclobutenes. | ||
To investigate the mechanism for the ring-opening process, we have conducted a series of experiments. Reaction of 41 with 65 in the presence of CD3OD enabled D incorporation at the α-position of the amide, suggesting that protonation of the C–Co bond occurred at this position (Scheme 23a). Treatment of 41 with deuterated allylboron 65-D furnished 68, implying that the allylic C–H bond was cleaved followed by the formation of a C–Co bond at the allylic site and simultaneously the cleaved C–H bond migrated to the four-membered ring skeleton (Scheme 23b). The initial allyl–Co addition to the cyclobutene 41 with subsequent 1,3-Co shift on the sp3-hybridized carbons might account for the observations. The addition of BHT didn’t retard the reaction, indicating that a radical process might not be involved in the 1,3-Co migration process (Scheme 23c). As shown in the proposed catalytic cycle (Scheme 23d), allyl–Co species XXIII generated from transmetallation with allylboron 65 underwent carbometallation of cyclobutene 41 to produce intermediate XXV. 1,3-Co migration followed by β-carbon elimination enabled a ring-opening process and delivered intermediate XXVII, which was protonated with MeOH to release the product 66 and regenerate the catalyst.
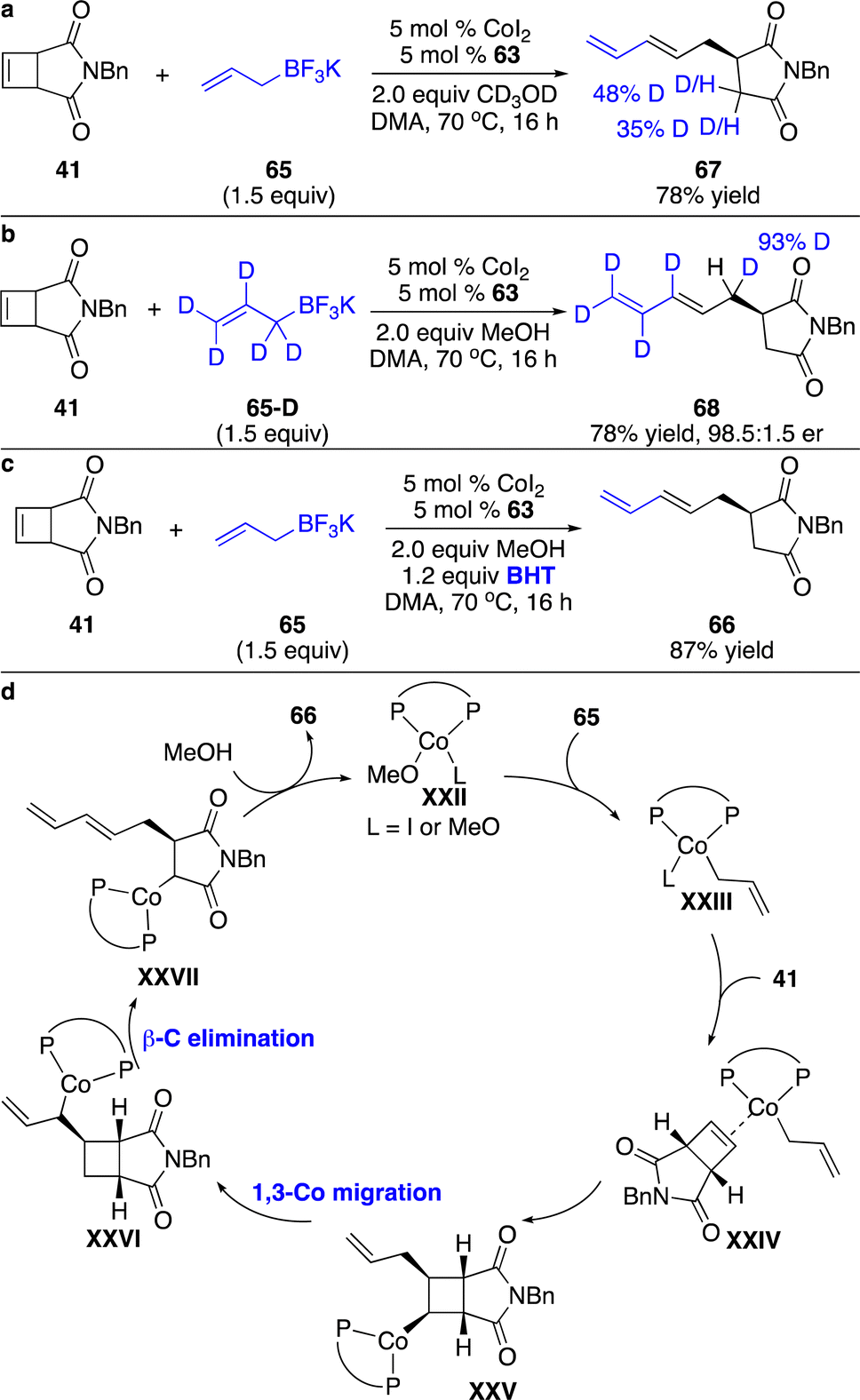 | ||
| Scheme 23 Mechanistic studies and proposed catalytic cycle for Co-catalyzed enantioselective cascade allyl addition to cyclobutenes. (a) Reaction in the presence of CD3OD. (b) Reaction with deuterated allylboron reagent. (c) Radical trapping experiment. (d) The proposed catalytic cycle. | ||
4. Conclusions
Catalytic enantioselective functionalization of small cyclic alkenes has emerged as an important strategy for the construction of enantioenriched cyclopropanes and cyclobutanes. Carbometallation of cyclopropenes and cyclobutenes provided a straightforward and versatile approach to incorporate a high diversity of carbon-based nucleophiles into the small ring scaffolds. Different transition metal-based catalysts have been developed, enabling highly efficient and stereoselective synthesis of a broad scope of small cyclic building blocks. Although promising, there are still many crucial challenges unaddressed. For example, high catalyst loadings are always required in these processes. It would be desirable to develop a more efficient catalytic system with higher turnover number. Moreover, transformation of the C–metal bond generated from carbometallation of small cyclic alkenes is limited to protonation. Difunctionalization of cyclopropenes and cyclobutenes through direct conversion of the C–metal bond remained far less developed. In addition, there is a lack of general strategy for enantioselective establishment of a quaternary stereogenic center. We believe that the concept of enantioselective desymmetrization of cyclopropenes and cyclobutenes will continue to show its unique advantages and attract more and more interest from both academia and industry.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2022YFA1506100), the National Natural Science Foundation of China (Grant No. 22371294, and 21821002), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB0610000), the Beijing National Laboratory for Molecular Sciences (BNLMS202304), and the Shanghai Science and Technology Committee (Grant No. 20XD1425000).Notes and references
- (a) V. Vuligonda, S. M. Thacher and R. A. S. Chandraratna, J. Med. Chem., 2001, 44, 2298 CrossRef PubMed; (b) R. Faust, Angew. Chem., Int. Ed., 2001, 40, 2251 CrossRef; (c) H. R. Sonawane, N. S. Bellur, D. G. Kulkarni and J. R. Ahuja, Synlett, 1993, 875 CrossRef; (d) M. Yoshida, M. Ezaki, M. Hashimoto, M. Yamashita, N. Shigematsu, M. Okuhara, M. Kohsaka and K. Horikoshi, J. Antibiot., 1990, 43, 748 CrossRef PubMed; (e) E. Lee-Ruff and G. Mladenova, Chem. Rev., 2003, 103, 1449 CrossRef PubMed; (f) M. Wang and P. Lu, Org. Chem. Front., 2018, 5, 254 RSC; (g) V. M. Dembitsky, Phytomedicine, 2014, 21, 1559 CrossRef CAS PubMed.
- (a) V. Ganesh and S. Chandrasekaran, Synthesis, 2016, 4347 CAS; (b) S. Krüger and T. Gaich, Beilstein J. Org. Chem., 2014, 10, 163 CrossRef PubMed; (c) T. Hudlicky and J. Reed, Angew. Chem., Int. Ed., 2010, 49, 4864 CrossRef CAS PubMed; (d) L. Jiao and Z.-X. Yu, J. Org. Chem., 2013, 78, 6842 CrossRef CAS PubMed; (e) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (f) S. C. Wang and D. J. Tantillo, J. Organomet. Chem., 2006, 691, 4386 CrossRef CAS; (g) J. C. Namyslo and D. E. Kaufmann, Chem. Rev., 2003, 103, 1485 CrossRef CAS PubMed; (h) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef CAS PubMed.
- (a) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed; (b) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS PubMed; (c) H. Shitama and T. Katsuki, Angew. Chem., Int. Ed., 2008, 47, 2450 CrossRef CAS PubMed; (d) A. B. Charette, C. Molinaro and C. Brochu, J. Am. Chem. Soc., 2001, 123, 12168 CrossRef CAS PubMed; (e) A. B. Charette, H. Juteau, H. Lebel and C. Molinaro, J. Am. Chem. Soc., 1998, 120, 11943 CrossRef CAS; (f) S. E. Denmark and S. P. O’Connor, J. Org. Chem., 1997, 62, 584 CrossRef CAS PubMed; (g) M. Montesinos-Magraner, M. Costantini, R. Ramírez-Contreras, M. E. Muratore, M. J. Johansson and A. Mendoza, Angew. Chem., Int. Ed., 2019, 58, 5930 CrossRef CAS PubMed; (h) M.-C. Lacasse, C. Poulard and A. B. Charette, J. Am. Chem. Soc., 2005, 127, 12440 CrossRef CAS PubMed; (i) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS PubMed.
- L. Dian and I. Marek, Chem. Rev., 2018, 118, 8415 CrossRef CAS PubMed.
- (a) R. Giri, X. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112 CrossRef PubMed; (b) M. Wasa, K. M. Engle, D. W. Lin, E. J. Yoo and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 19598 CrossRef PubMed; (c) K. S. L. Chan, H.-Y. Fu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 2042 CrossRef PubMed; (d) J. Pedroni, T. Saget, P. A. Donets and N. Cramer, Chem. Sci., 2015, 6, 5164 RSC; (e) J. Pedroni and N. Cramer, Angew. Chem., Int. Ed., 2015, 54, 11826 CrossRef PubMed; (f) T. Lee and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 8723 CrossRef PubMed; (g) T. Saget and N. Cramer, Angew. Chem., Int. Ed., 2012, 51, 12842 CrossRef CAS PubMed; (h) J. Kim, M. Sim, N. Kim and S. Hong, Chem. Sci., 2015, 6, 3611 RSC; (i) Y. Shi, Q. Gao and S. Xu, J. Am. Chem. Soc., 2019, 141, 10599 CrossRef CAS PubMed; (j) Y. Shi, Y. Yang and S. Xu, Angew. Chem., Int. Ed., 2022, 61, e202201463 CrossRef CAS PubMed.
- (a) K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 8138 CrossRef PubMed; (b) J. He, Q. Shao, Q. Wu and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 3344 CrossRef PubMed; (c) Q.-F. Wu, X.-B. Wang, P.-X. Shen and J.-Q. Yu, ACS Catal., 2018, 8, 2577 CrossRef PubMed; (d) L.-J. Xiao, K. Hong, F. Luo, L. Hu, W. R. Ewing, K.-S. Yeung and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 9594 CrossRef PubMed; (e) J. Rodrgalvarez, L. A. Reeve, J. Miró and M. J. Gaunt, J. Am. Chem. Soc., 2022, 144, 3939 CrossRef PubMed; (f) L. Hu, P.-X. Shen, Q. Shao, K. Hong, J. X. Qiao and J.-Q. Yu, Angew. Chem., Int. Ed., 2019, 58, 2134 CrossRef PubMed; (g) X. Chen, L. Chen, H. Zhao, Q. Gao, Z. Shen and S. Xu, Chin. J. Chem., 2020, 38, 1533 CrossRef CAS.
- Y. Cohen and I. Marek, Acc. Chem. Res., 2022, 55, 2848 CrossRef CAS PubMed.
- (a) D. S. Müller and I. Marek, J. Am. Chem. Soc., 2015, 137, 15414 CrossRef PubMed; (b) L. Dian, D. S. Müller and I. Marek, Angew. Chem., Int. Ed., 2017, 56, 6783 CrossRef CAS PubMed; (c) M. Simaan and I. Marek, Angew. Chem., Int. Ed., 2018, 57, 1543 CrossRef CAS PubMed; (d) K. Krämer, P. Leong and M. Lautens, Org. Lett., 2011, 13, 819 CrossRef PubMed; (e) M. Nakamura, A. Hirai and E. Nakamura, J. Am. Chem. Soc., 2000, 122, 978 CrossRef CAS.
- L. Dian and I. Marek, Angew. Chem., Int. Ed., 2018, 57, 3682 CrossRef CAS PubMed.
- J. Zhang, L. Wang, Q. Chong and F. Meng, Asian J. Org. Chem., 2023, 12, e202200720 CrossRef CAS.
- D. S. Müller, V. Werner, S. Akyol, H.-G. Schmalz and I. Marek, Org. Lett., 2017, 19, 3970 CrossRef PubMed.
- H. Zhang, W. Huang, T. Wang and F. Meng, Angew. Chem., Int. Ed., 2019, 58, 11049 CrossRef CAS PubMed.
- Z. Li, M. Zhang, Y. Zhang, S. Liu, J. Zhao and Q. Zhang, Org. Lett., 2019, 21, 5432 CrossRef CAS PubMed.
- Y. Zhang, Y. Li, W. Zhou, M. Zhang, Q. Zhang, R. Jia and J. Zhao, Chem. Commun., 2020, 56, 12250 RSC.
- (a) J. Yang, Y. Shen, Y. J. Lim and N. Yoshikai, Chem. Sci., 2018, 9, 6928 RSC; (b) J. Yang, Q. Sun and N. Yoshikai, ACS Catal., 2019, 9, 1973 CrossRef CAS; (c) J. Yang, Y. Sekiguchi and N. Yoshikai, ACS Catal., 2019, 9, 5638 CrossRef CAS.
- W. Huang and F. Meng, Angew. Chem., Int. Ed., 2021, 60, 2694 CrossRef CAS PubMed.
- H.-L. Teng, Y. Ma, G. Zhan, M. Nishiura and Z. Hou, ACS Catal., 2018, 8, 4705 CrossRef CAS.
- L. Dian and I. Marek, ACS Catal., 2020, 10, 1289 CrossRef CAS PubMed.
- (a) K. B. Wiberg, Angew. Chem., Int. Ed. Engl., 1986, 25, 312 CrossRef; (b) F. Menard and M. Lautens, Angew. Chem., Int. Ed., 2008, 47, 2085 CrossRef CAS PubMed; (c) J. Bexrud and M. Lautens, Org. Lett., 2010, 12, 3160 CrossRef CAS PubMed.
- C. Zhong, Y. Huang, H. Zhang, Q. Zhou, Y. Liu and P. Lu, Angew. Chem., Int. Ed., 2020, 59, 2750 CrossRef CAS PubMed.
- Y.-J. Chen, T.-J. Hu, C.-G. Feng and G.-Q. Lin, Chem. Commun., 2015, 51, 8773 RSC.
- F. W. Goetzke, A. M. L. Hell, L. van Dijk and S. P. Fletcher, Nat. Chem., 2021, 13, 880 CrossRef CAS PubMed.
- D. Egea-Arrebola, F. W. Goetzke and S. P. Fletcher, Angew. Chem., Int. Ed., 2023, 62, e202217381 CrossRef CAS PubMed.
- Z. Wang, J. Zhu, M. Wang and P. Lu, J. Am. Chem. Soc., 2024, 146, 12691 CrossRef CAS PubMed.
- Z. Liang, L. Wang, Y. Wang, L. Wang, Q. Chong and F. Meng, J. Am. Chem. Soc., 2023, 145, 3588 CrossRef CAS PubMed.
- (a) T. Sawano, A. Ashouri, T. Nishimura and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 18936 CrossRef CAS PubMed; (b) M. Shirakura and M. Suginome, Angew. Chem., Int. Ed., 2010, 49, 3827 CrossRef CAS PubMed.
- (a) T. Sawano, K. Ou, T. Nishimura and T. Hayashi, J. Org. Chem., 2013, 78, 8986 CrossRef CAS PubMed; (b) T. Nishimura, X.-X. Guo and T. Hayashi, Chem. – Asian J., 2008, 3, 1505 CrossRef CAS PubMed.
- (a) T. Sawano, K. Ou, T. Nishimura and T. Hayashi, Chem. Commun., 2012, 48, 6106 RSC; (b) Q. Yang, P. Y. Choy, B. Fan and F. Y. Kwong, Adv. Synth. Catal., 2015, 357, 2345 CrossRef.
- (a) T. Nishimura, T. Sawano, K. Ou and T. Hayashi, Chem. Commun., 2011, 47, 10142 RSC; (b) Y. Zhi, J. Huang, N. Liu, T. Lu and X. Dou, Org. Lett., 2017, 19, 2378 CrossRef PubMed.
- Y. Huang, C. Ma, Y. X. Lee, R.-Z. Huang and Y. Zhao, Angew. Chem., Int. Ed., 2015, 54, 13696 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |