
A rotational spectroscopy study of microsolvation effects on intramolecular proton transfer in trifluoroacetylacetone–(H2O)1–3†
Dingding
Lv
 ,
Xinlei
Chen
,
Ningjing
Jiang
,
Guanjun
Wang
,
Xiaoqing
Zeng
,
Wei
Fang
*,
Weixing
Li
* and
Mingfei
Zhou
*
,
Xinlei
Chen
,
Ningjing
Jiang
,
Guanjun
Wang
,
Xiaoqing
Zeng
,
Wei
Fang
*,
Weixing
Li
* and
Mingfei
Zhou
*
Department of Chemistry, Collaborative Innovation Center of Chemistry for Energy Materials, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Fudan University, Songhu Rd. 2005, 200438 Shanghai, China. E-mail: wei_fang@fudan.edu.cn; weixingli@fudan.edu.cn; mfzhou@fudan.edu.cn
First published on 2nd April 2024
Abstract
Trifluoroacetylacetone (TFAA) has two enol forms, which can switch to each other via proton transfer. While much attention has been paid to their conformational preferences, the influence of microsolvation on regulating the proton position remains unexplored. Herein, we report the rotational spectra of trifluoroacetylacetone–(water)n (n = 1–3) investigated by chirped pulse Fourier transform microwave spectroscopy in the 2–8 GHz frequency range. Two conformers were identified for both TFAA–H2O and TFAA–(H2O)2, while only one conformer was characterized for TFAA–(H2O)3. The results indicate that water binding on the CH3 side stabilizes the enolF form, whereas water binding on the CF3 side stabilizes the enolH form. The enolF form predominates over the enolH form in these hydrated complexes, which contrasts with the fact that only enolH exists in isolated TFAA. EnolH becomes preferred only when water inserts itself into the intramolecular hydrogen bond. Instanton theory calculations reveal that the proton transfer reaction is dominated by quantum tunneling at low temperatures, leading to the stable existence of only one enol form in each configuration of the hydrated clusters.
Introduction
Proton transfer (PT) is an ubiquitous process in biological and chemical reactions,1,2 commonly occurring in solution and markedly influenced by environmental molecules, particularly water.3–6 The influence of water on the PT process is well established in various biological contexts, such as DNA mutations7,8 and enzymatic reactions in proteins.9 In these cases, water facilitates PT by forming a water bridge, known as Grotthuss mechanism.10 This process reduces PT barriers,11–14 representing a primary means by which water engages in and facilitates PT. When water serves as an environmental molecule without direct participation in the water bridge, its impact is generally weak and insufficient to enable PT due to high PT barriers.15 Herein, we report a spectroscopic investigation of the effect of water as an environmental molecule on the PT process of the intramolecular hydrogen bond (HB) in trifluoroacetylacetone.Trifluoroacetylacetone exists in two tautomeric forms, namely 1,1,1-trifluoro-4-hydroxy-3-penten-2-one (enolH) and 5,5,5-trifluoro-4-hydroxy-3-penten-2-one (enolF), as illustrated in Scheme 1. These isomers undergo interconversion that involves PT processes with a low barrier height,16 rendering TFAA a prototypical model system for the study of PT. The enolH form was reported to be more stable than the enolF form. Only the enolH form was observed in the gas phase by rotational spectroscopy and in a low temperature solid argon matrix by infrared absorption spectroscopy.17,18 In contrast, both forms were confirmed to coexist in a low temperature solid nitrogen matrix and in aqueous solution by Fourier transform infrared absorption spectroscopy.18 The influence of water on the structure of trifluoroacetylacetone has also been studied by matrix isolation infrared spectroscopy. The most abundant isomer among trifluoroacetylacetone–water complexes was found to be the enolF–water form with water attached to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of TFAA in both the argon and nitrogen matrices. The other isomer of the enolH–water complex was also observed in smaller amounts.18
O group of TFAA in both the argon and nitrogen matrices. The other isomer of the enolH–water complex was also observed in smaller amounts.18
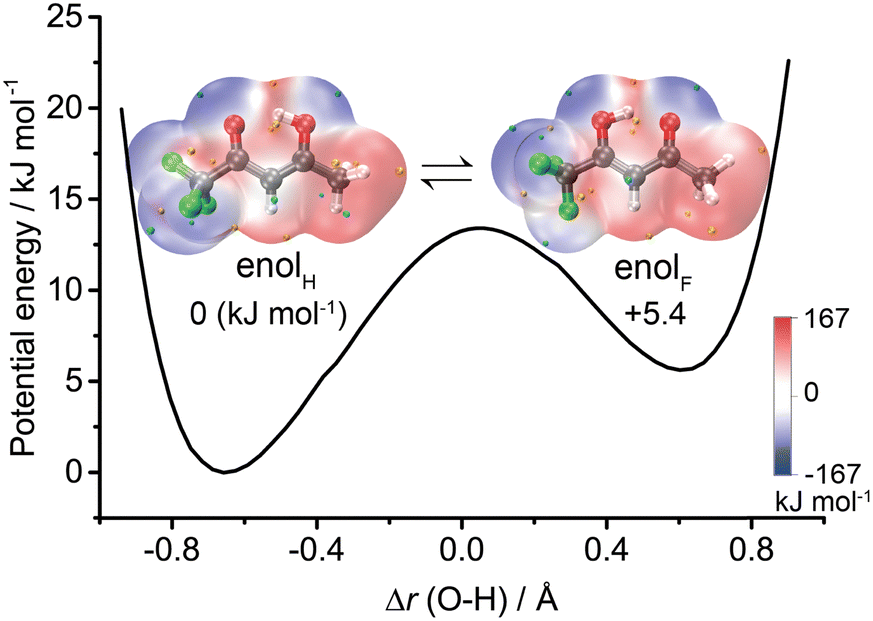 | ||
| Scheme 1 Schematic illustration of the double minimum potential governing proton transfer along the intramolecular HB between the enolH and enolF forms of trifluoroacetylacetone and the molecular electrostatic potential surfaces. | ||
Rotational spectroscopy has inherently high resolution and is exceptionally sensitive to molecular mass distribution and geometry, making it capable of providing precise experimental evidence for tautomerism equilibrium.19–21 This technique has been widely used to characterize the structural and/or dynamic properties of microsolvated clusters.15,22–24 In this work, the structures and dynamic properties of PT reactions in the TFAA–(H2O)1–3 clusters were investigated using chirped-pulse Fourier transform microwave (CP-FTMW) spectroscopy24–27 complemented with theoretical calculations. The results demonstrate that the direction of PT is contingent upon the quantity and specific docking sites of water molecules, elucidating an indirect influence of water on the PT process.
Experimental and theoretical methods
A chirped pulse Fourier transform microwave (CP-FTMW) spectrometer operating in the 2–8 GHz range was used to investigate the TFAA–(water)n complexes. Commercial samples of TFAA (Aladdin, anhydrous 99.9%), H218O and D2O (Sigma-Aldrich LLC., 97%) were used without further purification. TFAA and water were mixed in a gas tank with helium. Natural water and isotopically enriched water samples containing 33% H218O or D2O were used in the measurements. The TFAA–water complexes were produced by supersonic expansion of helium seeded with about 0.1% TFAA and 0.1% water, respectively, at a backing pressure of about 0.3 MPa using a pulsed solenoid valve, which was oriented perpendicular to the axis of microwave propagation. After a delay of 860 μs, a chirped pulse of 4 μs was broadcasted into the vacuum chamber through a horn antenna. The free induction decay signal (FID) due to molecular relaxation was collected using another horn antenna, amplified, and recorded in a digital oscilloscope in the time domain. The signal was then transformed into the frequency domain by the application of Fourier transform. A fast frame setup of six excitation–emission cycles per supersonic expansion was used to reduce the measurement time and sample consumption. The spectra have an accuracy in the frequency measurement better than 15 kHz and a resolution power better than 25 kHz. The FID data were collected 1.2 million times. The rotational spectrum was firstly assigned to a semi-rigid (DJ corrected) asymmetric rotor Hamiltonian using our homemade automatic fitting program, then the fitted parameters were refined using the assignment and analysis of broadband spectra (AABS) package28 and pickett's SPFIT.29The search for stable isomers was performed with the CREST (conformer-rotamer ensemble sampling tool) software,30,31 an efficient scheme employing the meta-dynamics algorithm combined with semiempirical tight-binding methods. The resulting structures were further optimized at the B3LYP-D4/def2-TZVP level of theory, and their single-point energies were evaluated at the DLPNO-CCSD(T)/def2-TZVP level using the Orca 4 program package.32 The zero-point energy (ZPE) and basis set superposition error (BSSE) of the stable conformers were calculated at the B3LYP-D4/def2-TZVP level of theory. Additionally, the relaxed potential energy surfaces of proton transfer along the intramolecular HB in the TFAA monomer and observed TFAA–(H2O)1–3 complexes were calculated at the B3LYP-D4/def2-TZVP level as well. The plots were generated by Chimera software.33
Results and discussion
The conformers predicted to lie within 15 kJ mol−1 for each cluster are listed in Fig. S1 (ESI†) (the conformations are abbreviated by the form of TFAA (enolH or enolF), the number of waters (1W, 2W, 3W), and the water(s) docking site at CH3 (WCH), between HB OH⋯O (WOH), or at CF3 (WCF), respectively. Their calculated rotational constants are listed in Table S1 (ESI†). Fig. 1 shows the experimental spectrum after removing the spectral lines arising from the TFAA monomer17 and pure water clusters.34–36 Guided by the spectral parameters predicted by theoretical calculations, five sets of rotational transitions are assigned to five isomers of TFAA–(H2O)1–3. The representative transitions for each isomer are shown in the bottom panel of Fig. 1. All observed transitions are listed in the ESI.† Their experimental rotational constants are in good agreement with the calculated values (Table 1). The relative intensities of the a-, b-, and c-type transitions are consistent with the theoretical dipole moment components.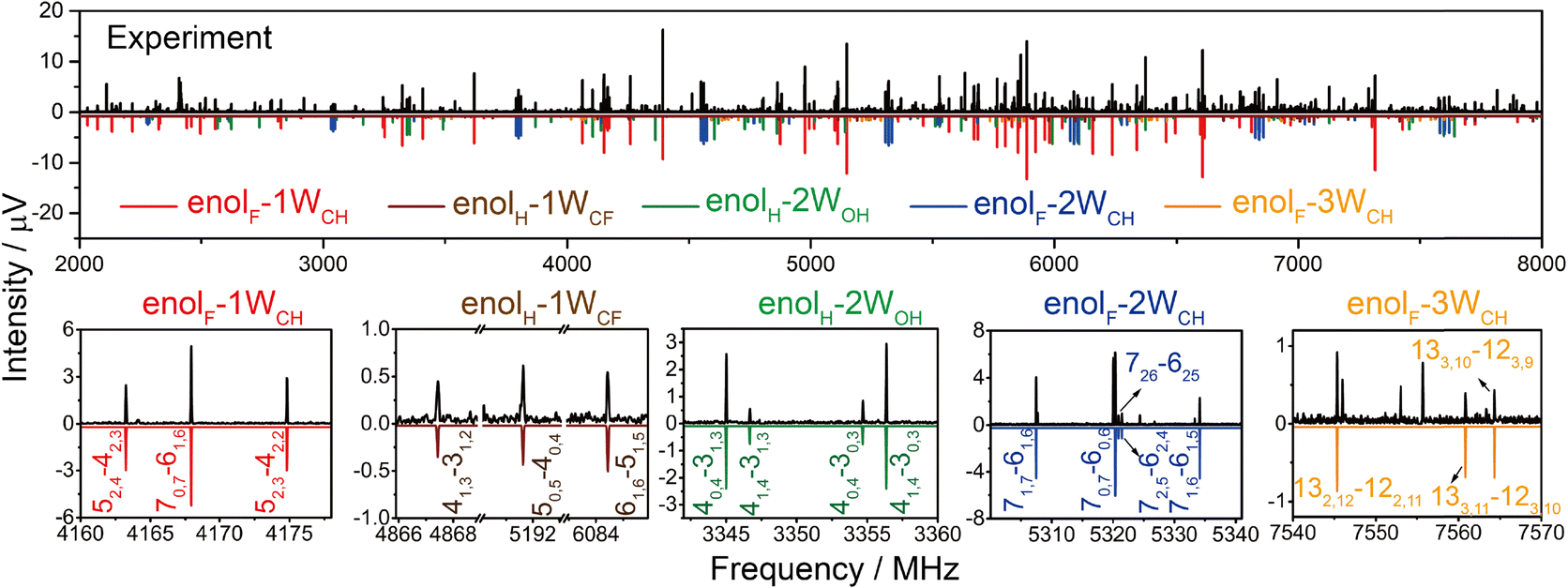 | ||
| Fig. 1 The rotational spectra of the TFAA–(H2O)1–3 complexes. The black upper trace corresponds to the experimental spectrum after removing the spectral lines arising from the TFAA monomer and pure water clusters. The lower traces in different colors represent the simulated spectra of TFAA–(H2O)1–3 complexes from the experimental spectroscopic parameters, at a rotational temperature of 1 K, and the theoretical dipole moment components for the respective complexes. The bottom panel shows some representative transitions for the TFAA–(H2O)1–3 complexes. | ||
| enolF–1WCH | enolH–1WCF | enolH–2WOH | enolF–2WCH | enolF–3WCH | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Exp.a | Cal. | Exp. | Cal. | Exp. | Cal. | Exp. | Cal. | Exp. | Cal. | |
a Error in parentheses in units of the last digit.
b Planar moment of inertia 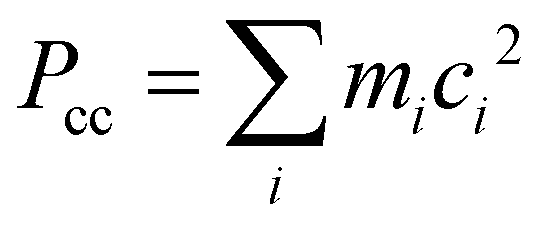 .
c Number of lines in the fit.
d Standard deviation of the fit. .
c Number of lines in the fit.
d Standard deviation of the fit.
|
||||||||||
| A/MHz | 2429.2774(7) | 2446.2 | 1266.877(2) | 1278.3 | 786.5539(4) | 809.1 | 1461.932(6) | 1443.6 | 1188.53(8) | 1171.1 |
| B/MHz | 436.1908(3) | 439.7 | 668.600(1) | 664.7 | 640.3761(3) | 630.1 | 381.9927(4) | 373.5 | 297.5584(2) | 304.0 |
| C/MHz | 396.7171(2) | 400.0 | 475.9664(8) | 475.6 | 377.7953(3) | 380.9 | 378.1846(4) | 365.4 | 283.6020(2) | 293.3 |
| D J/kHz | 0.011(2) | 0.039(7) | 0.12(1) | 0.200(2) | 0.0425(6) | |||||
P
cc/uÅ2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
46.3764(8) | 46.3 | 46.499(2) | 46.5 | 47.0040(9) | 49.9 | 166.185(2) | 160.0 | 170.82(2) | 185.4 |
| μ a/μb/μc/D | yes/yes/no | 0.4/1.2/0.0 | yes/yes/no | 4.6/3.8/0.2 | yes/yes/no | 1.5/1.6/1.0 | yes/no/yes | 2.0/1.3/1.6 | yes/no/no | 2.3/1.0/1.2 |
| N | 97 | 27 | 57 | 36 | 51 | |||||
| σ /kHz | 8.6 | 7.3 | 8.2 | 9.3 | 7.3 | |||||
To achieve straightforward and unequivocal identification, we performed two experiments using isotopically enriched water samples containing 33% H218O (2.1 million FID acquisitions) and 33% D2O (1.7 million FID acquisitions), respectively. The spectra of H218O mono-substituted species for enolF–1WCH, enolH–1WCF, and enolH–2WOH, and the spectra of DOH substituted species for enolF–1WCH and enolH–2WOH have been obtained. Their fitted rotational constants are listed in Tables S2–S4 (ESI†), respectively. Based on these rotational constants of isotopologues, the experimental rs coordinates of these substituted atoms are determined using Kraitchman equations and are listed in Tables S5–S10 (ESI†).37 The superimposition between the rs and the calculated coordinates confirms the isomer assignments.
For the TFAA–H2O complexes, theoretical calculations predict six conformers within a relative energy of 10 kJ mol−1 (Fig. S1, ESI†). In four of them, the water molecule is anchored to the oxygen atom of TFAA through HB OH···O as a HB donor (Fig. 2) and interacts with CH3 or CF3via a secondary weak HB (wHB). Two of the four isomers (enolF–1WCH and enolH–1WCF) have been characterized in our spectra. One is the global minimum (enolF–1WCH) in which water interacts with the oxygen atom of the CO group as the HB donor and with the CH3 group as the wHB acceptor, respectively. The other one (enolH–1WCF) is the third energetic minimum with 5.8 kJ mol−1 higher in energy than the global minimum, which is in agreement with the observation that the spectral intensity of enolF–1WCH is stronger than that of enolH–1WCF. In isomer enolH–1WCF, water donates both of its hydrogen atoms to form two HBs as the donor. One HB is with the oxygen atom of the CO group and the other is with the fluorine atom of the CF3 group. The planar moment of inertia 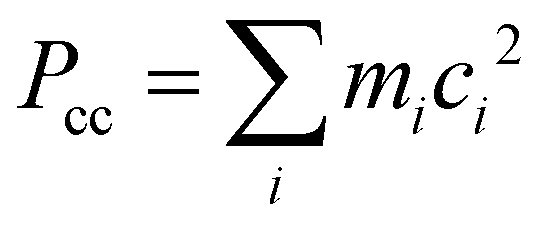 , which is deduced from the rotational constants, indicates that the mass distribution lies out of the ab inertial plane of a molecule. The experimental value of Pcc of the TFAA monomer was determined to be 46.72275(6) uÅ2.18 For the TFAA–H2O complexes, the experimental value is 46.3764(8) uÅ2 for enolF–1WCH and 46.499(2) uÅ2 for enolH–1WCF, which are close to the value of the monomer. This result indicates that the entire water molecule lies in the ab plane of TFAA.
, which is deduced from the rotational constants, indicates that the mass distribution lies out of the ab inertial plane of a molecule. The experimental value of Pcc of the TFAA monomer was determined to be 46.72275(6) uÅ2.18 For the TFAA–H2O complexes, the experimental value is 46.3764(8) uÅ2 for enolF–1WCH and 46.499(2) uÅ2 for enolH–1WCF, which are close to the value of the monomer. This result indicates that the entire water molecule lies in the ab plane of TFAA.
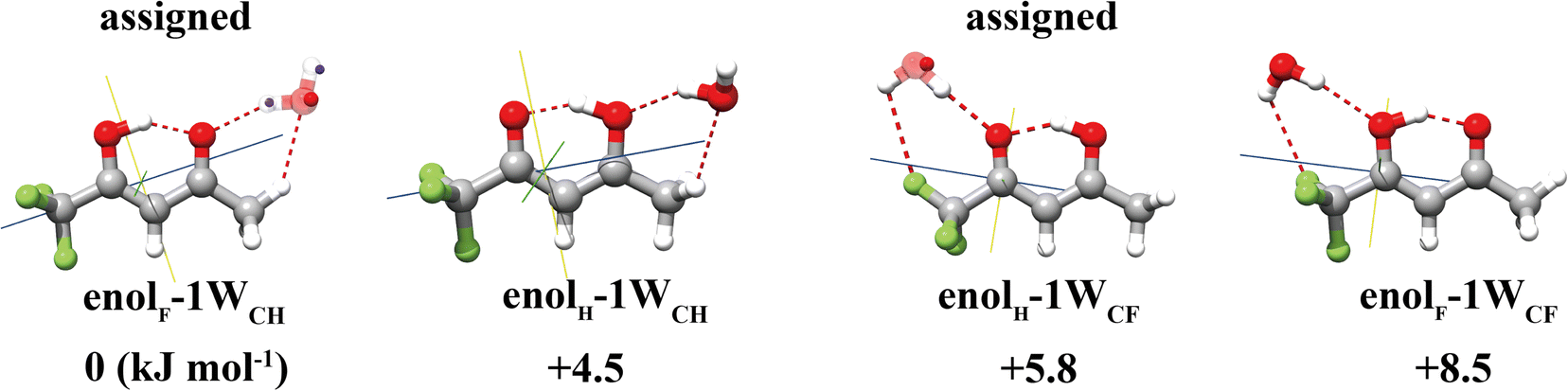 | ||
| Fig. 2 Four energetically low-lying isomers of TFAA–H2O with two of them being assigned experimentally. The blue and red spheres superimposed in the calculated structures are the rs coordinates of the respective atoms. | ||
For the TFAA–(H2O)2 clusters, the four most stable isomers theoretically predicted are shown in Fig. 3 and Fig. S1 (ESI†). Once again, the global and the third energy minima are experimentally assigned. The global minimum adopts an arrangement where the water dimer inserts itself into the intramolecular HB of TFAA. All the atoms of the water dimer lie nearly in the ab plane of TFAA, as evidenced by its Pcc value of 47.0040(9) uÅ2, which is close to that of the TFAA monomer. This water dimer arrangement is structurally different from the pure water dimer arrangement, where the planes of the two water molecules are perpendicular to each other.38 With the insertion of water into its intramolecular HB, the O⋯O distance of TFAA dramatically increases to 2.85 Å from a distance of 2.57 Å in the TFAA monomer. The O⋯O distance of the water dimer in this isomer is experimentally determined to be 2.79(4) (rs), which is much shorter than the O⋯O distance (2.98(4) Å) in the pure water dimer.38 For the other assigned isomer, enolF–2WCH, the HB acceptor of the water dimer forms a HB with the oxygen atom of the CO group in TFAA as the HB donor, and the HB donor of the water dimer forms a secondary wHB with the CH3 group as the HB acceptor. The second water molecule of the water dimer sits above the ab plane of TFAA rather than in the ab plane. As a result, it can retain more room to accommodate additional water molecules and grow into larger microsolvated clusters.
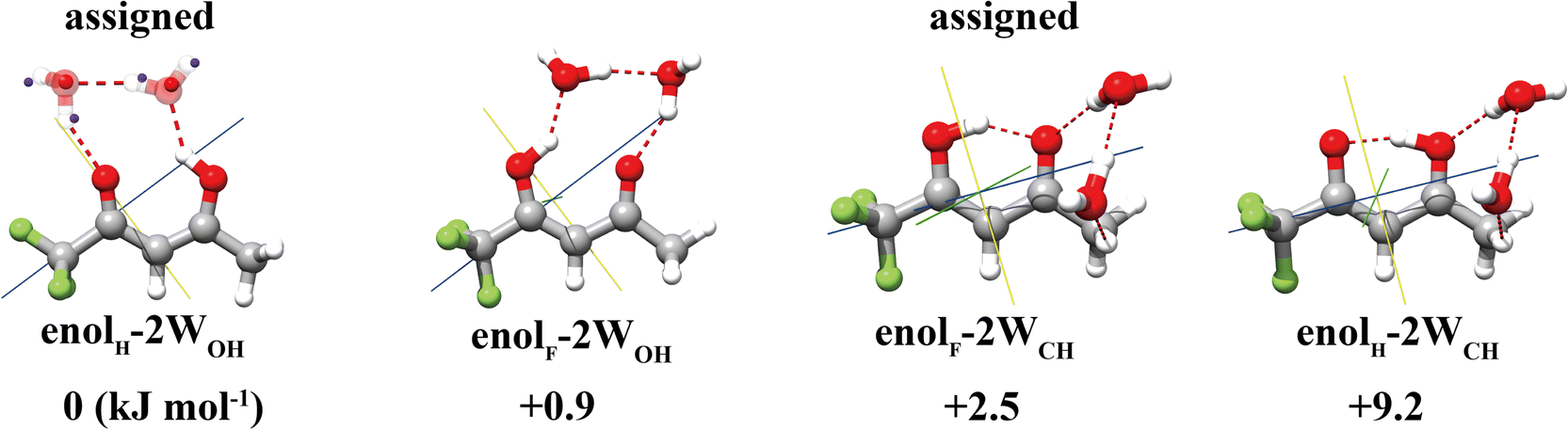 | ||
| Fig. 3 Four energetically low-lying isomers of TFAA–(H2O)2 with two of them being assigned experimentally. The blue and red spheres superimposed in the calculated structures are the rs coordinates of the respective atoms. | ||
For the TFAA–(H2O)3 clusters, only one isomer, enolF–3WCH, was spectroscopically identified. The structure appears to have grown stepwise from the smaller cluster enolF–2WCH, with the third water molecule anchored at one end of the water chain of enolF–2WCHvia OH⋯O as a HB donor (Fig. 4). The orientations of the free hydrogens are flipped by the added water. Its oxygen atom forms bifurcated interactions with the adjacent CH bond and the carbon atom of the CO group. The interaction between the oxygen of water and the carbonyl carbon is well known as the Bürgi–Dunitz interaction and plays an important role in the stabilization of the protein structure.22,39,40 The Bürgi–Dunitz (αBD) angle and the Flippin–Lodge (αFL) angle are calculated to be 97.2° and 3.9°, respectively. Natural bond orbital (NBO) analysis can provide insight into this interaction which is derived from the lp(O) → π*(CO) electron delocalization, as shown in Fig. 5.
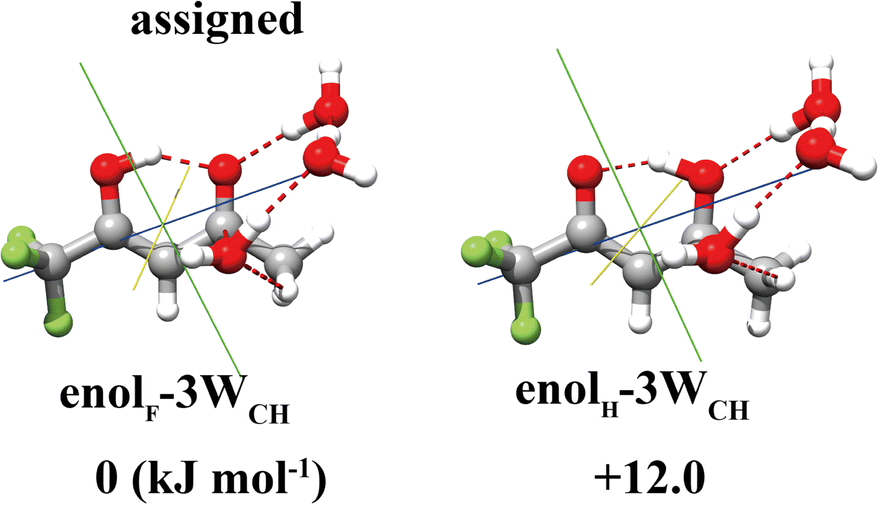 | ||
| Fig. 4 Two calculated stable isomers of TFAA–(H2O)3 with one of them being assigned experimentally. | ||
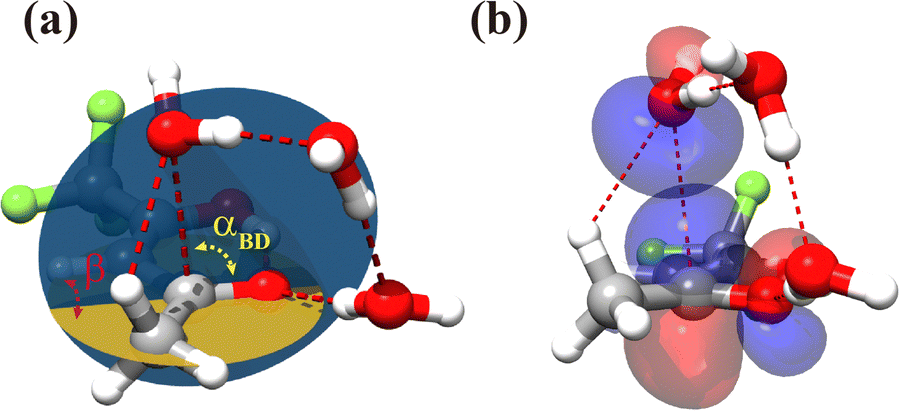 | ||
Fig. 5 (a) Bürgi–Dunitz angle αBD = 97.2°, Flippin–Lodge angle αFL = 3.9° (αFL = a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) sin(−sin(αBD)·cos(β))), where β is the dihedral angle defined by the yellow and blue planes, and a distance of nucleophilic 2.99 Å calculated at the B3LYP-D4/def2-TZVP level of theory; (b) NBO representation of the lone pair (lp) orbital of H2O and the antibonding (π*) orbital of TFAA (isosurface = 0.03 a.u.). sin(−sin(αBD)·cos(β))), where β is the dihedral angle defined by the yellow and blue planes, and a distance of nucleophilic 2.99 Å calculated at the B3LYP-D4/def2-TZVP level of theory; (b) NBO representation of the lone pair (lp) orbital of H2O and the antibonding (π*) orbital of TFAA (isosurface = 0.03 a.u.). | ||
The results indicate that solvation significantly affects the relative stability of the two enol conformers of TFAA. The relaxed potential energy surfaces (PESs) of PT between enolH and enolF show that in the observed clusters where water acts as an environmental molecule, water binding on the CH3 side stabilizes the enolF form, whereas binding on the CF3 side stabilizes the enolH form, as shown in Fig. S2 (ESI†). The enolF form dominates over the enolH form in these hydrated complexes, which differs from the fact that only enolH exists in isolated TFAA.
The theoretical results agree well with the experimental results, showing that only one enol form is experimentally characterized in each configuration of the hydrated clusters. As shown in Fig. 2, in the TFAA–H2O system, the enolH–1WCH and enolF–1WCF isomers were predicted to be 4.5 and 2.7 kJ mol−1 higher in energy than the corresponding enolF–1WCH and enolH–1WCF isomers, respectively. The enolH–1WCH and enolF–1WCF conformers can isomerize to the more stable enolF–1WCH and enolH–1WCF conformers via proton transfer reactions through a transition state, as shown in Fig. S2 (ESI†). Despite the barrier being relatively small, the reaction rate based on the transition state theory (TST)41 decreases to less than 8 × 10−42 s-1 at 5 K. This suggests that the barriers would be sufficient to trap the enolH–1WCH and enolF–1WCF isomers, allowing them to be observed experimentally. However, neither enolH–1WCH nor enolF–1WCF is observed, despite that the enolH–1WCH conformer is even more stable than the experimentally observed enolH–1WCF conformer. This suggests that there is a mechanism that allows fast conversion of the metastable enolH–1WCH and enolF–1WCF conformers to the more stable enolF–1WCH and enolH–1WCF conformers. Due to the light mass of hydrogen, quantum tunneling could play an important role in these PT processes. We therefore used ring-polymer instanton theory42–46 to understand the proton transfer process from enolH–1WCH to enolF–1WCH (computational details are given in the ESI†). As shown in Fig. 6, the instanton theory calculation results show that the rate constant becomes almost temperature independent below 120 K, indicating that the reaction is dominated by quantum tunneling at low temperatures. The tunneling rate is very fast (9 × 1010 s−1) even at low temperatures, suggesting that the less stable enolH–1WCH conformer can rapidly be converted to the more stable enolF–1WCH conformer even at low temperatures.
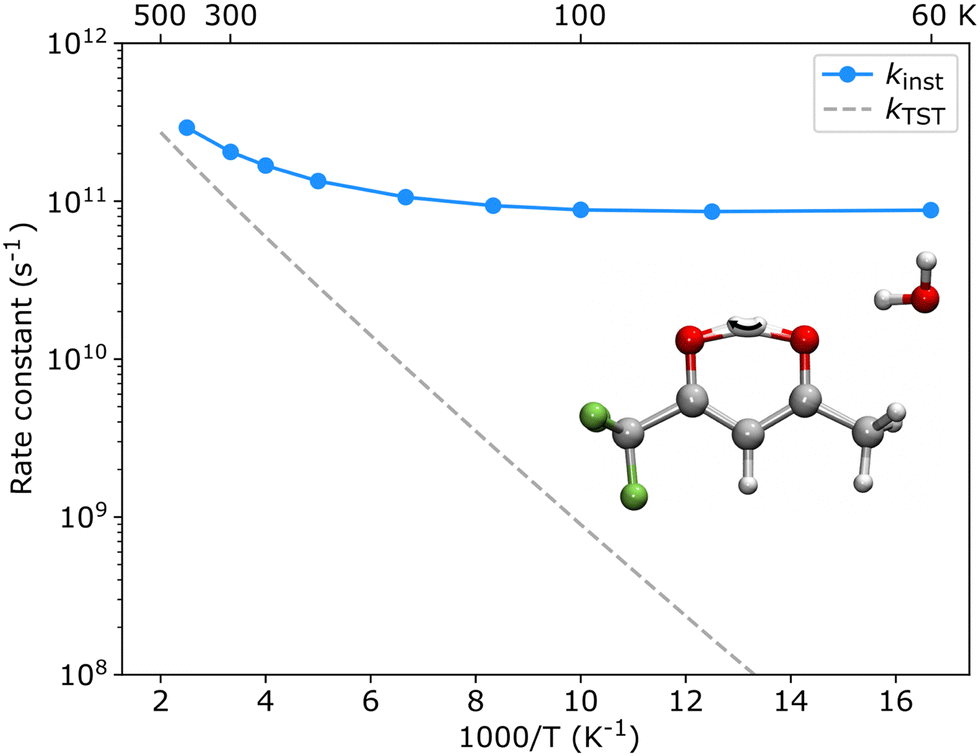 | ||
| Fig. 6 Instanton rates for the enolH–1WCH to enolF–1WCH isomerization reaction. The inset shows the instanton optimized at 80 K. The Eyring transition state theory rate (kTST) is shown for comparison. | ||
Conclusions
In summary, the rotational spectroscopy study of TFAA–(H2O)1–3 clusters provides experimental evidence that water, as an environmental molecule, facilitates the proton transfer reaction without direct participation in the process. Two conformers for both TFAA–H2O and TFAA–(H2O)2, and one conformer for TFAA–(H2O)3 were identified in the rotational spectra guided by theorical calculations. The results demonstrate that the number and binding sites of water molecules influence the prototropic tautomerism. Water binding on the CH3 side stabilizes the enolF form, whereas water binding on the CF3 side stabilizes the enolH form. The enolF form emerges in TFAA–(H2O)1–3 and dominates over the enolH form in TFAA–H2O based on the relative intensity of their spectra, which differs from the fact that only the enolH form exists in the TFAA monomer. Instanton theory calculations show that the proton transfer reaction is dominated by quantum tunneling at low temperatures, so that only one enol form can stably exist in each configuration of the hydrated clusters.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant number 22321003).References
- A. Mueller, E. Diemann, H. Ratajczak and W. Junge, Electron and Proton Transfer in Chemistry and Biology, 1992, vol. 78 Search PubMed.
- P. Gilli, V. Bertolasi, L. Pretto, A. Lyčka and G. Gilli, The Nature of Solid-State N–H⋯O/O–H⋯N Tautomeric Competition in Resonant Systems. Intramolecular Proton Transfer in Low-Barrier Hydrogen Bonds Formed by the ⋯OC–CN–NH⋯ ⇄ ⋯HO–CC–NN⋯Ketohydrazone–Azoenol System. A Variable-Temperature X-Ray Crystallographic and Dft Computational Study, J. Am. Chem. Soc., 2002, 124, 13554–13567 CrossRef CAS PubMed.
- A. Douhal, F. Lahmani and A. H. Zewail, Proton-Transfer Reaction Dynamics, Chem. Phys., 1996, 207, 477–498 CrossRef CAS.
- P. C. Gómez and L. F. Pacios, Environmental Effects on Proton Transfer in a Strong Hydrogen Bond Dimer: The 4-Methyl-Imidazole-Aspartate Case, Phys. Chem. Chem. Phys., 2005, 7, 1374–1381 RSC.
- B. Das, A. Chakraborty and S. Chakraborty, Experimental and Theoretical Investigation of Ground State Intramolecular Proton Transfer (Gsipt) in Salicylideneaniline Schiff Base Derivatives in Polar Protic Medium, Spectrochim. Acta, Part A, 2020, 225, 117443 CrossRef CAS PubMed.
- G. J. Kearley, F. Fillaux, M. H. Baron, S. Bennington and J. Tomkinson, A New Look at Proton Transfer Dynamics Along the Hydrogen Bonds in Amides and Peptides, Science, 1994, 264, 1285–1289 CrossRef CAS PubMed.
- L. Gorb and J. Leszczynski, Intramolecular Proton Transfer in Mono- and Dihydrated Tautomers of Guanine: An Ab Initio Post Hartree−Fock Study, J. Am. Chem. Soc., 1998, 120, 5024–5032 CrossRef CAS.
- M. Hanus, F. Ryjáček, M. Kabeláč, T. Kubař, T. V. Bogdan, S. A. Trygubenko and P. Hobza, Correlated Ab Initio Study of Nucleic Acid Bases and Their Tautomers in the Gas Phase, in a Microhydrated Environment and in Aqueous Solution. Guanine: Surprising Stabilization of Rare Tautomers in Aqueous Solution, J. Am. Chem. Soc., 2003, 125, 7678–7688 CrossRef CAS PubMed.
- F. Garczarek and K. Gerwert, Functional Waters in Intraprotein Proton Transfer Monitored by Ftir Difference Spectroscopy, Nature, 2006, 439, 109–112 CrossRef CAS PubMed.
- N. Agmon, The Grotthuss Mechanism, Chem. Phys. Lett., 1995, 244, 456–462 CrossRef CAS.
- D. Li and H. Ai, Catalysis Effects of Water Molecules and of Charge on Intramolecular Proton Transfer of Uracil, J. Phys. Chem. B, 2009, 113, 11732–11742 CrossRef CAS PubMed.
- C. Kinz-Thompson and E. Conwell, Proton Transfer in Adenine−Thymine Radical Cation Embedded in B-Form DNA. J. Phys, Chem. Lett., 2010, 1, 1403–1407 CAS.
- A. Michalkova, D. Kosenkov, L. Gorb and J. Leszczynski, Thermodynamics and Kinetics of Intramolecular Water Assisted Proton Transfer in Na+-1-Methylcytosine Water Complexes, J. Phys. Chem. B, 2008, 112, 8624–8633 CrossRef CAS PubMed.
- J. P. Cerón-Carrasco, A. Requena, C. Michaux, E. A. Perpète and D. Jacquemin, Effects of Hydration on the Proton Transfer Mechanism in the Adenine−Thymine Base Pair, J. Phys. Chem. A, 2009, 113, 7892–7898 CrossRef PubMed.
- S. Sérgio, R. Domingos and M. Schnell, Wet Sunscreens in the Gas Phase: Structures of Isolated and Microsolvated Oxybenzone. J. Phys, Chem. Lett., 2018, 9, 46 Search PubMed.
- A. Nowroozi, H. Roohi, M. S. Sadeghi Ghoogheri and M. Sheibaninia, The Competition between the Intramolecular Hydrogen Bond and Π-Electron Delocalization in Trifluoroacetylacetone—a Theoretical Study, Int. J. Quantum Chem., 2011, 111, 578–585 CrossRef CAS.
- L. B. Favero, L. Evangelisti, B. Velino and W. Caminati, Morphing the Internal Dynamics of Acetylacetone by CH3 → CF3 Substitutions. The Rotational Spectrum of Trifluoroacetylacetone, J. Phys. Chem. A, 2014, 118, 4243–4248 CrossRef CAS PubMed.
- R. Platakyte, A. Gutiérrez-Quintanilla, V. Sablinskas and J. Ceponkus, Influence of Environment and Association with Water, to Internal Structure of Trifluoroacetylacetone. Matrix Isolation FTIR Study, Low Temp. Phys., 2019, 45, 615–626 CrossRef CAS.
- L. D. Hatherley, R. D. Brown, P. D. Godfrey, A. P. Pierlot, W. Caminati, D. Damiani, S. Melandri and L. B. Favero, Gas-Phase Tautomeric Equilibrium of 2-Pyridinone and 2-Hydroxypyridine by Microwave Spectroscopy, J. Phys. Chem., 1993, 97, 46–51 CrossRef CAS.
- J. L. Alonso, I. Peña, J. C. López and V. Vaquero, Rotational Spectral Signatures of Four Tautomers of Guanine, Angew. Chem., Int. Ed., 2009, 48, 6141–6143 CrossRef CAS PubMed.
- D. Lv, W. Li, L. Evangelisti, I. Usabiaga, C. Calabrese, A. Maris, S. Melandri, G. Wang and M. Zhou, Rotational Spectroscopy Probes Lone Pair···Π-Hole Interactions in Hexafluorobenzene-Tertiary Alkylamines Complexes, J. Phys. Chem. Lett., 2023, 14, 5335–5342 CrossRef CAS PubMed.
- W. Li, M. M. Quesada-Moreno, P. Pinacho and M. Schnell, Unlocking the Water Trimer Loop: Isotopic Study of Benzophenone-(H2O)1–3 Clusters with Rotational Spectroscopy, Angew. Chem., Int. Ed., 2021, 60, 5323–5330 CrossRef CAS PubMed.
- S. Blanco, J. C. López, A. Lesarri and J. L. Alonso, Microsolvation of Formamide: A Rotational Study, J. Am. Chem. Soc., 2006, 128, 12111–12121 CrossRef CAS PubMed.
- W. Li, C. Pérez, A. L. Steber, M. Schnell, D. Lv, G. Wang, X. Zeng and M. Zhou, Evolution of Solute–Water Interactions in the Benzaldehyde-(H2O)1–6 Clusters by Rotational Spectroscopy, J. Am. Chem. Soc., 2023, 145, 4119–4128 CrossRef CAS PubMed.
- G. G. Brown, B. C. Dian, K. O. Douglass, S. M. Geyer, S. T. Shipman and B. H. Pate, A Broadband Fourier Transform Microwave Spectrometer Based on Chirped Pulse Excitation, Rev. Sci. Instrum., 2008, 79, 53103 CrossRef PubMed.
- J. L. Neill, S. T. Shipman, L. Alvarez-Valtierra, A. Lesarri, Z. Kisiel and B. H. Pate, Rotational Spectroscopy of Iodobenzene and Iodobenzene–Neon with a Direct Digital 2–8 GHz Chirped-Pulse Fourier Transform Microwave Spectrometer, J. Mol. Spectrosc., 2011, 269, 21–29 CrossRef CAS.
- D. Schmitz, V. A. Shubert, T. Betz and M. Schnell, Multi-Resonance Effects within a Single Chirp in Broadband Rotational Spectroscopy: The Rapid Adiabatic Passage Regime for Benzonitrile, J. Mol. Spectrosc., 2012, 280, 77–84 CrossRef CAS.
- Z. Kisiel, L. Pszczółkowski, I. R. Medvedev, M. Winnewisser, F. C. De Lucia and E. Herbst, Rotational Spectrum of Trans–Trans Diethyl Ether in the Ground and Three Excited Vibrational States, J. Mol. Spectrosc., 2005, 233, 231–243 CrossRef CAS.
- H. M. Pickett, The Fitting and Prediction of Vibration-Rotation Spectra with Spin Interactions, J. Mol. Spectrosc., 1991, 148, 371–377 CrossRef CAS.
- S. Grimme, Exploration of Chemical Compound, Conformer, and Reaction Space with Meta-Dynamics Simulations Based on Tight-Binding Quantum Chemical Calculations, J. Chem. Theory Comput., 2019, 15, 2847–2862 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Automated Exploration of the Low-Energy Chemical Space with Fast Quantum Chemical Methods, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, The Orca Quantum Chemistry Program Package, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, Ucsf Chimera - a Visualization System for Exploratory Research and Analysis, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- J. A. Odutola and T. R. Dyke, Partially Deuterated Water Dimers: Microwave Spectra and Structure, J. Chem. Phys., 1980, 72, 5062–5070 CrossRef CAS.
- C. Pérez, M. T. Muckle, D. P. Zaleski, N. A. Seifert, B. Temelso, G. C. Shields, Z. Kisiel and B. H. Pate, Structures of Cage, Prism, and Book Isomers of Water Hexamer from Broadband Rotational Spectroscopy, Science, 2012, 336, 897–901 CrossRef PubMed.
- C. Pérez, S. Lobsiger, N. A. Seifert, D. P. Zaleski, B. Temelso, G. C. Shields, Z. Kisiel and B. H. Pate, Broadband Fourier Transform Rotational Spectroscopy for Structure Determination: The Water Heptamer, Chem. Phys. Lett., 2013, 571, 1–15 CrossRef.
- J. Kraitchman, Determination of Molecular Structure from Microwave Spectroscopic Data, Am. J. Phys., 1953, 21, 17–24 CrossRef CAS.
- T. R. Dyke and J. S. Muenter, Microwave Spectrum and Structure of Hydrogen Bonded Water Dimer, J. Chem. Phys., 1974, 60, 2929–2930 CrossRef CAS.
- H. B. Burgi, J. D. Dunitz and E. Shefter, Geometrical Reaction Coordinates. Ii. Nucleophilic Addition to a Carbonyl Group, J. Am. Chem. Soc., 1973, 95, 5065–5067 CrossRef CAS.
- R. W. Newberry and R. T. Raines, The N → π* Interaction, Acc. Chem. Res., 2017, 50, 1838–1846 CrossRef CAS PubMed.
- H. Eyring, The Theory of Absolute Reaction Rates, Trans. Faraday Soc., 1938, 34, 41–48 RSC.
- J. O. Richardson and S. C. Althorpe, Ring-Polymer Molecular Dynamics Rate-Theory in The Deep-Tunneling Regime: Connection with Semiclassical Instanton Theory, J. Chem. Phys., 2009, 131, 214106 CrossRef PubMed.
- J. O. Richardson, Ring-Polymer Instanton Theory, Int. Rev. Phys. Chem., 2018, 37, 171–216 Search PubMed.
- W. Fang, P. Winter and J. O. Richardson, Microcanonical Tunneling Rates from Density-of-States Instanton Theory, J. Chem. Theory Comput., 2021, 17, 40–55 CrossRef CAS PubMed.
- J. Kästner, Theory and Simulation of Atom Tunneling in Chemical Reactions, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 158–168 Search PubMed.
- S. Andersson, G. Nyman, A. Arnaldsson, U. Manthe and H. Jónsson, Comparison of Quantum Dynamics and Quantum Transition State Theory Estimates of the H + CH4 Reaction Rate, J. Phys. Chem. A, 2009, 113, 4468–4478 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp01061b |
| This journal is © the Owner Societies 2024 |