
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
IR spectra of cationic 1,5,9-triazacoronene and two of its cationic derivatives†
Jerry
Kamer‡
 a,
Domenik
Schleier‡
ab,
Andy
Jiao
c,
Grégory
Schneider
c,
Jonathan
Martens
d,
Giel
Berden
d,
Jos
Oomens
d and
Jordy
Bouwman
*efg
a,
Domenik
Schleier‡
ab,
Andy
Jiao
c,
Grégory
Schneider
c,
Jonathan
Martens
d,
Giel
Berden
d,
Jos
Oomens
d and
Jordy
Bouwman
*efg
aLaboratory for Astrophysics, Leiden Observatory, Leiden University, 2300 RA Leiden, The Netherlands
bInstitut für Optik und Atomare Physik, Technische Universität Berlin, Hardenbergstrasse 36, Berlin 10623, Germany
cLeiden Institute of Chemistry, Leiden University, 2300 RA Leiden, The Netherlands
dInstitute for Molecules and Materials, FELIX Laboratory, Radboud University, Toernooiveld 7, 6525 ED Nijmegen, The Netherlands
eLaboratory for Atmospheric and Space Physics, University of Colorado, Boulder, CO 80303, USA. E-mail: jordy.bouwman@colorado.edu
fDepartment of Chemistry, University of Colorado, Boulder, CO 80309, USA
gInstitute for Modeling Plasma, Atmospheres and Cosmic Dust (IMPACT), NASA/SSERVI, Boulder, CO 80309, USA
First published on 15th October 2024
Abstract
Infrared emission features are observed towards diverse astronomical objects in the interstellar medium (ISM). Generally, the consensus is that these IR features originate from polycyclic aromatic hydrocarbons (PAHs) and are hence named aromatic infrared bands (AIBs). More recently, it has been suggested that nitrogen substituted PAHs (PANHs) contribute to the AIBs as well and it has even been shown that nitrogen inclusion in PAHs can improve the match with the AIBs, specifically around the 6.2 μm feature. In order to determine which specific molecules or functional groups are at the origin of the AIBs, IR spectra of various PA(N)Hs are experimentally and computationally studied. In this work we expand on the spectroscopic investigation of PANHs by presenting the gas-phase mid-IR spectra of 1,5,9-triazacoronene˙+ (TAC˙+, m/z 303), a threefold nitrogenated congener of coronene˙+, its protonated derivative TACH+ (m/z 304) and the product that forms when water adds to dehydrogenated TAC˙+[TAC–H + H2O]+ (m/z 320). We analyze the mid-IR spectra by comparing them with vibrational modes calculated at the B3LYP/6-311++G(d,p) level a theory and we present a possible geometry for [TAC–H + H2O]+. The TAC˙+ mid-IR spectrum is compared to that of coronene˙+ and is demonstrated to be remarkably similar. We put TAC˙+ and TACH+ into astronomical context by comparing their recorded mid-IR spectra to observed ISM spectra of four spectral classes. From this we conclude that TAC˙+ and TACH+ could both contribute to the AIBs, with TACH+ being a more likely contributor than TAC˙+.
1 Introduction
Prominent infrared (IR) emission features at 3.3, 6.2, 7.7, 8.6, 11.2 and 12.7 μm are observed towards a large variety of astronomical objects in the interstellar medium (ISM), such as protoplanetary disks, HII regions and reflection nebulae.1,2 There is a general consensus that these mid-IR features are caused by polycyclic aromatic hydrocarbon (PAH) molecules that cascade down to the ground state after being excited by ultraviolet (UV) photons from the interstellar radiation field.3–5 Hence, these mid-IR features are commonly referred to as aromatic infrared bands (AIBs).The band positions and shapes of the AIBs are fairly consistent among the various observed astronomical objects, although some variations exist. Based on these variations, the mid-IR spectra are grouped into class A through D, each of which represents an specific spectral type.6–8 Class A, the most commonly observed of the four, is characterized by features which peak at 6.22, 7.6 and 8.6 μm. The 7.6 μm feature also contains a shoulder at 7.8 μm. A shift of these three class A features towards longer wavelengths results in class B, where the peaks are found at 6.24–6.28, 7.8–8.0 and >8.62 μm. Generally, the feature around 6.2 μm is broader for class B, compared to A. Class C contains a peak around 6.3 μm and a broad feature ranging from 7.5 to 9.3 μm with the peak at 8.22 μm. Finally, class D is characterized by a feature at 6.24 μm, and a broad feature ranging from 7.0 to 9.0 μm, peaking around 7.7 μm. Additionally, class D can contain a feature at 6.9 μm. Numerous studies have demonstrated the very strong resemblance between the mid-IR emission of (ionic) homocyclic PAHs and astronomical objects.9–25 However, the experimental and computed IR emission features of cationic homocyclic PAHs around 6.2 μm are consistently redshifted with respect to astronomical observations.6,26
Substitution of a carbon atom in the structure of a PAH cation by a nitrogen, forming a so-called polycyclic aromatic nitrogen heterocycle (PANH) cation, can introduce a spectral shift in the 6.2 μm band.26,27 Based on computational studies, it has been shown that this shift depends strongly on the position of the substituted carbon atom in the PAH structure and size and symmetry of the PAH. Substitution of an endoskeletal carbon atom in the PAH structure (commonly referred to as endo-PANH) results in a drastic change of the mid-IR emission spectrum with respect to its pure PAH congener, with most notably a blue-shift in the 6.2 μm band, resulting in a better agreement with observational data.26–28 Cationic endo-PANHs have, contrary to their cationic PAH congeners, a closed-shell electronic configuration. Recently, Ricca et al.27 estimated an upper limit for the fraction of cationic endo-PANH emission contributing to the astronomically observed 6.2 μm band to be about 12%, by assuming all of the 11.0 μm emission is due to singly ionized endo-PANHs and combining their results with the reported values for NGC 7023.29 Exoskeletal PANHs (exo-PANHs), where a peripheral CH-group has been substituted by a nitrogen, do not show a significant change in the mid-IR range compared to their pure, cationic PAH analogue.26,27,30 This makes exo-PANH cations challenging to distinguish from their PAH counterpart using IR spectra alone. Therefore, exo-PANHs are potential carriers of the observed mid-IR features as well and potentially make up a large fraction of the PAH population.
Although nitrogen-bearing PAHs have been shown to be astronomically relevant,26,27,31 the number of experimental works studying (spectral) characteristics of (ionic) PANHs is limited. Moreover, the works that do report on nitrogen-bearing PAHs are often limited to species containing only a single nitrogen in the aromatic structure. For example, Ricca et al.27 studied PAHs up to 103 carbon atoms while mainly considering species containing a single nitrogen atom. Based on the interstellar abundances of elements, PAHs of astronomically relevant sizes could contain multiple nitrogen atoms as well, and thus spectroscopic studies of such species are needed.
The proton affinity (PA) of especially exo-PANHs is greatly enhanced relative to the analogous homocyclic PAHs. For example, the PA of naphthalene is determined to be 802.9 kJ mol−1,32 while its nitrogenated congeners quinoline and isoquinoline have a PA of 953.2 and 951.7 kJ mol−1,32 respectively. An exposed N-atom is a strong nucleophile that easily binds a proton, which are abundantly present in HII regions, resulting in the formation of very stable, closed-shell, protonated PANH species. Experimental and computational works have shown that protonated PANHs are potential carriers of the 6.2 μm band.27,33 Ricca et al.27 calculated the 6.2 μm band position of protonated PANHs to span the 6.17 to 6.45 μm range, depending on the size and symmetry of the protonated PANH. Singly dehydrogenated exo-PANH cations have been shown to be highly reactive. In a previous study by our group, we showed that after losing a hydrogen atom from the radical cation of 1,5,9-triazacoronene (TAC˙+, m/z 303, see Fig. 1 for the molecular structure), a species is formed that reacts very efficiently with background water vapor in the trap to form a very stable product ion.34 It is important to spectroscopically characterize exo-PANH radical cations, protonated exo-PANHs and even the reaction products formed in the reaction of exo-PANHs and water to determine their potential contribution to the interstellar mid-IR emission features.
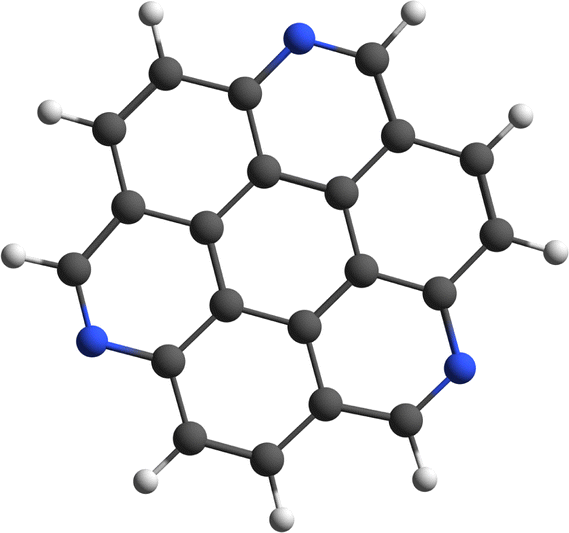 | ||
| Fig. 1 Molecular structure of cationic 1,5,9-triazacoronene (TAC˙+, C21H9N3˙+). | ||
In this work, we study the IR spectra of TAC˙+, protonated TAC (TACH+, m/z 304) and the product that forms when water adds to dehydrogenated TAC˙+[TAC–H + H2O]+ (m/z 320), using a combination of infrared multiple photon dissociation (IRMPD) spectroscopy at the Free Electron Laser for Infrared eXperiments (FELIX) and quantum chemical calculations. TAC is a threefold nitrogenated congener of coronene and its three available nitrogen sites allow for facile TACH+ formation and the formation of [TAC–H + H2O]+ in the presence of H2O. We first present the dissociation mass spectra that arise upon resonantly exciting TAC˙+, TACH+ and [TAC–H + H2O]+ with IR radiation from FELIX. IR spectra are subsequently constructed from the frequency dependent dissociation mass spectra. The experimental spectra are analyzed using density functional theory (DFT) simulated spectra to characterize the vibrational normal modes. Next, we compare the experimental spectrum of TAC˙+ to the coronene cation (Cor˙+) to investigate the effect on the IR characteristics of triple nitrogen substitution in Cor˙+. Lastly, we put the ramification of nitrogen inclusion on the IR spectra in astrophysical context by comparing the TAC˙+ and TACH+ IRMPD spectra to observed astronomical mid-IR spectra of each spectral type class.
2 Methods
2.1 Experimental
All IRMPD spectra were recorded on a commercially available Bruker Amazon Speed ETD 3D quadrupole ion trap mass spectrometer, which is modified to allow the radiation from the free electron laser for infrared experiments, FELIX, to be focused onto the ion cloud inside the trap. A detailed description of the system can be found in previous publications18,35 and a short description of the apparatus and the measurement procedures is provided here.TAC was synthesized following the procedure described by Tan et al.36 and a detailed description is given in a previous publication.34 A glass direct insertion probe (DIP) was coated with the solid sample and subsequently introduced into an atmospheric pressure chemical ionization source (Bruker APCI II) which was connected to the mass spectrometer. The DIP is then gently heated to desorb the sample material. A flow of N2 carries the desorbed molecules into a corona discharge region, where TAC˙+, TACH+ and [TAC–H + H2O]+ are produced. These are subsequently guided through a spray shield into a glass capillary leading to the ion trap. The ions are mass-isolated using the mass spectrometer's tandem mass spectrometry features.
The trapped and mass isolated ions are exposed to five macropulses of mid-infrared (mid-IR) light originating from the free electron laser that operates at a 10 Hz repetition rate. Each macropulse has an average duration of 6 μs with a maximum pulse energy of 75 mJ and a spectral full width at half maximum (FWHM) of about 5–10 cm−1. The IR frequency is tuned from 730 to 1780 cm−1 in steps of 5 cm−1 and at each wavelength five mass spectra are averaged. The precursor ion absorbs multiple IR photons when the laser is tuned to a frequency that is resonant with a vibrational transition, which eventually leads to dissociation of the precursor ion. The resulting fragment ion signals and the remaining precursor ion signal are integrated and an IRMPD dissociation yield is calculated. The wavelength-dependent IRMPD yield is linearly corrected for free electron laser pulse energy, resulting in the IRMPD spectrum of our studied species.
2.2 Computational
Quantum chemical calculations of our species of interest were performed using the Gaussian1637 suite of software. We employed the B3LYP functional38,39 in combination with the 6-311++G(d,p) basis set to calculate harmonic vibrational spectra. All computed normal mode frequencies are uniformly scaled with a factor of 0.9679 to correct for anharmonicity effects.40 The stick spectrum is convolved with a 20 cm−1 FWHM Gaussian function to facilitate comparison with the experimental IRMPD spectra.3 Results
3.1 Mass spectrometry
The mass spectra resulting from irradiating the isolated TAC˙+, TACH+ and [TAC–H + H2O]+ ions are shown in Fig. 2. The top and bottom panel of each sub-figure represent mass spectra resulting from off- and on-resonance irradiation, respectively. From these figures it can be seen that clean isolation of the precursor ion is achieved and significant dissociation is induced when resonantly exciting the ions. Interestingly, the dissociation fragments that arise are significantly different for each of the precursor ions. Surprisingly, both TAC˙+ and TACH+ show m/z 320 as a product after on-resonant irradiation. Likely, this product forms when H2O binds with [TAC–H]+.34 Section 3.4 will describe this in more detail. The mass spectra will not be analyzed further in this work, as it is outside of the scope of this study. The dissociation of TAC˙+ using 630 nm radiation, however, has been described in much detail in a previous work by our group.34 | ||
| Fig. 2 Mass spectra of isolated TAC˙+ (a), TACH+ (b) and [TAC–H + H2O]+ (c) after exposure to off-resonance (top panel) and on-resonance (bottom panel) infrared radiation. The wavenumber of the IR radiation is depicted in each respective panel. The proposed geometry for the [TAC–H + H2O]+ species is discussed in Section 3.4. | ||
3.2 TAC˙+ IRMPD spectroscopy
The IRMPD spectrum that is constructed by plotting the laser power-corrected dissociation yield of TAC˙+ as a function of IR frequency between 750 and 1750 cm−1 is presented as the black trace in Fig. 3a. Shown together with the IRMPD is the scaled B3LYP/6-311++G(d,p) computed vibrational spectrum of TAC˙+, depicted by the blue vertical lines and the convolution in green. Neutral TAC can be optimized at a C3h symmetry and a molecular orbitals (MO) calculation of neutral TAC, reveals that the highest occupied MO (HOMO), with E′′ orbital symmetry, is degenerate. The radical cation is therefore expected to undergo Jahn–Teller distortion. Indeed, optimizing the geometry of TAC˙+ at a C3h symmetry, similar to neutral TAC, resulted in an imaginary frequency. Optimization at the Jahn–Teller distorted Cs symmetry does converge with all real frequencies, and hence the TAC˙+ vibrational modes presented are calculated in this symmetry. | ||
| Fig. 3 Experimental IRMPD (black) and simulated IR (blue vertical lines) spectra of TAC˙+ (a) and TACH+ (b). The B3LYP/6-311++G(d,p) computed spectrum (blue sticks) is uniformly scaled with a factor of 0.9679 and convolved with a 20 cm−1 FWHM Gaussian function (green shaded area). The IRMPD intensity is represented by the left axis and the intensity of the computed spectra by the right axis. All spectra are normalized to their most intense peak. The horizontal grey, dotted line in (b) indicates an apparent threshold of 21 km mol−1. Exciting modes with an intensity below line does not allow the molecule to reach the dissociation threshold. | ||
Six broad features at 803, 932, 1145, 1189, 1352 and 1553 cm−1 can be distinguished from the IRMPD spectrum. The peak at 1352 cm−1 is the broadest and also the most intense feature. According to our calculations (see Table 1 for computed modes), this most intense feature originates mainly from two modes which primarily consist of in-plane CH bending combined with some CC and CN stretching character at 1333 and 1346 cm−1. The computed IR spectrum shows noticeable involvement of at least one of the nitrogen atoms in almost all vibrational modes, of which the most intense are the modes at 781 and 1542 cm−1. This is in contrast with 3-azafluoranthene, where the nitrogen atom was shown to only be involved in a few specific modes.30
| Experimental | Calculatedab | ||
|---|---|---|---|
| Pos. (cm−1) | Pos. (cm−1) | Int. (km mol−1) | Symmetry |
| a The computed band positions are scaled with a factor of 0.9679. b Only computed modes with intensities ≥20 km mol−1 are listed. c A plateau is observed in the experimental data without resolved features. | |||
| 803 | 781 | 224 | A′ |
| 813 | 70 | A′′ | |
| 932 | 931 | 33 | A′′ |
| −c | 983 | 20 | A′ |
| 1003 | 44 | A′ | |
| 1107 | 21 | A′ | |
| 1145 | 1128 | 56 | A′ |
| 1189 | 1178 | 65 | A′ |
| 1191 | 46 | A′ | |
| 1214 | 34 | A′ | |
| 1352 | 1289 | 39 | A′ |
| 1305 | 41 | A′ | |
| 1333 | 147 | A′ | |
| 1346 | 241 | A′ | |
| 1389 | 54 | A′ | |
| 1399 | 39 | A′ | |
| 1429 | 26 | A′ | |
| — | 1467 | 41 | A′ |
| 1492 | 25 | A′ | |
| 1553 | 1512 | 27 | A′ |
| 1528 | 57 | A′ | |
| 1534 | 71 | A′ | |
| 1542 | 127 | A′ | |
The overall shape of the experimental spectrum is quite well replicated by the convolved calculated spectrum, but the relative intensities do not always match. This effect is sometimes observed, especially for systems with high dissociation thresholds, such as PAHs,9,10,22,23,30,33,41,42 and likely is the result of several effects, such as non-linearities in the multiple-photon excitation in combination with the linear power correction applied.22 Furthermore, there may be an apparent intensity cut-off below which the molecules cannot be excited to above the dissociation threshold with the employed laser powers43–45 (see Section 3.3) and moreover, closely spaced bands in the spectrum may give rise to co-operative effects in the excitation that are absent in a (computed) absorption spectrum.23,46 In addition, there may be flaws in the computed spectra, which would require further computational investigation beyond the scope of this study. The scaled harmonic modes in the 1100 to 1400 cm−1 spectral range do match quite well with the experimental spectrum. A plateau can be observed between 970 and 1100 cm−1. Calculations show three harmonic modes at 983, 1003 and 1107 cm−1 that do not correspond to an experimental IRMPD feature. In the observed spectrum, these features are likely blended, thus resulting in a featureless plateau. The IRMPD feature at 803 cm−1 seems to consist mainly of two modes calculated at 781 and 813 cm−1, albeit the feature is slightly blue-shifted with respect to the calculated 781 cm−1 vibration. The first calculated mode originates from overall in-plane CC and CN stretching, and CH bending, while the latter originates mainly from out-of-plane CH bending and minor out-of-plane CC and CN stretching.
3.3 TACH+ IRMPD spectroscopy
The APCI source on the mass spectrometer not only generated TAC˙+, but also protonated TAC, TACH+. The black line in Fig. 3b corresponds to the TACH+ IRMPD spectrum. Due to the high proton affinity of the nitrogen atom in the aromatic system,27,47 it is expected that the proton will bind on one of the three available nitrogen atoms of TAC. We optimized this geometry and calculated the vibrational spectrum and scaled them to account for anharmonicities. The calculated position and intensity of the TACH+ modes are listed in Table 2 and depicted in Fig. 3b by the blue vertical sticks and their convolved spectral profile in green.| Experimental | Calculatedab | ||
|---|---|---|---|
| Pos. (cm−1) | Pos. (cm−1) | Int. (km mol−1) | Symmetry |
| a The computed band positions are scaled with a factor of 0.9679. b Only modes with intensities ≥20 km mol−1 are listed. | |||
| 807 | 801 | 54 | A′′ |
| 814 | 68 | A′′ | |
| 1130 | 1126 | 22 | A′ |
| — | 1155 | 20 | A′ |
| 1296 | 1291 | 54 | A′ |
| 1299 | 43 | A′ | |
| 1335 | 1326 | 27 | A′ |
| 1340 | 109 | A′ | |
| 1357 | 35 | A′ | |
| 1414 | 1421 | 36 | A′ |
| 1432 | 28 | A′ | |
| — | 1461 | 26 | A′ |
| 1546 | 1540 | 36 | A′ |
| 1597 | 1593 | 60 | A′ |
| 1595 | 76 | A′ | |
| 1612 | 133 | A′ | |
Features around 807, 1130, 1296, 1335, 1414, 1546 and 1597 cm−1 can be identified in the IRMPD spectrum of TACH+, the most intense of these being at 1597 cm−1 and 1296 cm−1. According to our calculations the first feature mainly arises from three vibrational modes at 1593, 1595 and 1612 cm−1. The modes at 1593 and 1612 cm−1 originate mostly from in-plane NH and CH bending, with CC stretching and some CN stretching character. Contrarily, the 1595 cm−1 mode involves barely any motion of the NH group and has mostly CC-stretching and in-plane CH bending mode character. The latter feature is calculated to consist of two modes at 1291 and 1299 cm−1. The 1291 cm−1 mode originates from in-plane CC and CN stretching, and CH and NH bending. This is comparable to the 1593 and 1612 cm−1 calculated modes although the CC stretching motion is less pronounced. The 1299 cm−1 mode consists mainly of in-plane CC and CN stretching, and CH bending. Furthermore, this mode involves barely any NH bending.
The TACH+ IRMPD displays no clear features between 820 and 1260 cm−1 (see Fig. S1 in the ESI† for a zoom-in), except for the 1130 cm−1 feature, while modes are predicted to fall in this range. At these frequencies, the calculated intensities of the TACH+ vibrational normal modes are less than 21 km mol−1, which apparently is not enough to drive the molecule to its dissociation threshold using IRMPD. This has been observed in earlier works as well and is not specific to PANHs.43–45TAC˙+ does not seem to have a clear dissociation threshold, likely due to the higher intensity modes compared to TACH+. The intensity reduction in this wavenumber range of the IRMPD spectrum is also observed in an earlier work comparing IRMPD spectra of quinoline˙+ and isoquinoline˙+ to their protonated counterparts,33 with these protonated species showing no distinct bands in the region from ∼820 to ∼1300 cm−1. The calculated modes of TACH+ in the 900 to 980 cm−1 are dominated by out-of-plane CH bending. In-plane CC stretching and, CH and NH bending modes dominate the IR activity between 980 and 1000 cm−1. Within the 1100 to 1260 cm−1 range the calculated modes show mainly in-plane CH and NH bending character with minor CC stretching contribution.
Besides the range spanning 820 to 1260 cm−1, the agreement between the computed (and scaled) normal modes and the IRMPD spectrum is generally good. Especially, the calculated frequencies match the measured IRMPD bands at 807, 1296, 1335, 1546 and 1597 cm−1 well. A discrepancy can be observed for the calculated feature at around 1461 cm−1, which has no distinct experimental counterpart. Also, the calculated spectrum shows a small feature at 762 cm−1, which is not observed in the experimental spectrum, presumably because the dissociation threshold is not reached when exciting the molecule at this frequency.
3.4 [TAC–H + H2O]+ IRMPD spectroscopy
While we attempted to record an IRMPD spectrum of [TAC–H]+ at m/z 302, we found that a peak at m/z 320 dominates the mass spectrum. This indicates that the loss of a hydrogen atom significantly increases the reactivity of the ion, causing it to react with background H2O in the system to form [TAC–H + H2O]+. Removing a hydrogen from the CH group that is located next to the nitrogen atom requires the least amount of energy (see Fig. S2 in the ESI†), and so the H2O will likely bind to that site. Our previous work has shown that the resulting molecule does not dissociate after irradiation with intense light at 630 nm.34 However, the radiation from FELIX did induce dissociation when tuned to a vibrational resonance and, hence, an IRMPD spectrum could be recorded. The resulting spectrum is depicted as black lines in Fig. 4. | ||
| Fig. 4 Experimental IRMPD spectrum (black) and simulated IR spectra of four different isomers of [TAC–H + H2O]+. Also shown are the various structures of [TAC–H + H2O]+ isomers and their relative energies calculated using the B3LYP functional in combination with the 6-311++G(d,p) basis set. The hydrogen atoms that differ between the isomers are highlighted in cyan for clarity. The simulated spectrum in green indicates our tentative assignment that isomer 5 is likely the dominant isomer. The B3LYP/6-311++G(d,p) computed stick spectra are uniformly scaled with a factor of 0.9679. The calculated spectra are also convolved with a 20 cm−1 FWHM Gaussian function. All spectra are normalized with respect to their most intense peak in the wavenumber range 750–1650 cm−1. | ||
Many features can be seen in the IRMPD spectrum of which the more pronounced features are broad bands at 808, 1177, 1319, 1582 and 1718 cm−1. The high wavenumber shoulder of the 1319 cm−1 feature seems to contain subtle features at 1373, 1408, 1452 and 1487 cm−1. Similarly, towards the lower wavenumber side of the 1582 cm−1 feature, a peak can be observed at 1539 cm−1. The broad 1177 cm−1 feature seems to consist of multiple vibrational modes.
A lead towards the most likely geometry of [TAC–H + H2O]+ can be found in the feature at 1718 cm−1, which is indicative of the presence of a carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group.48,49 However, this does not exclude other functional groups, such as a hydroxyl (OH) group, as an IRMPD spectrum can be comprised out of several isomers. A total of 16 plausible isomers were computationally investigated for comparison to the IRMPD spectrum. The geometries of the isomers are optimized and their respective vibrational normal modes are calculated, scaled and convolved with a Gaussian profile. The resulting structures of five plausible isomers, their relative energies and simulated IR spectra calculated using B3LYP/6-311++G(d,p) are shown in Fig. 4. The calculated spectra of the other isomers can be found in Fig. S3 in the ESI.†
O) group.48,49 However, this does not exclude other functional groups, such as a hydroxyl (OH) group, as an IRMPD spectrum can be comprised out of several isomers. A total of 16 plausible isomers were computationally investigated for comparison to the IRMPD spectrum. The geometries of the isomers are optimized and their respective vibrational normal modes are calculated, scaled and convolved with a Gaussian profile. The resulting structures of five plausible isomers, their relative energies and simulated IR spectra calculated using B3LYP/6-311++G(d,p) are shown in Fig. 4. The calculated spectra of the other isomers can be found in Fig. S3 in the ESI.†
Isomer 1, 4 and 5 contain a CO group and in isomers 4 and 5 the amide nitrogen is bound to a hydrogen. The remaining hydrogen is bound to one of the other nitrogen atoms. Isomer 1 has no hydrogen bound to the amide nitrogen, and both hydrogens are bound to the other two nitrogen atoms. Isomer 2 and 3 have an OH group bound to the bare carbon that forms upon hydrogen loss from TAC˙+. Both these isomers have no hydrogen bound to the nitrogen adjacent to the COH moiety and only differ, similar to isomer 4 and 5, by a hydrogen being bound to a different nitrogen atom relative to the OH group. Isomer 5 is found to be the lowest energy isomer and the calculated energies of the other isomers, with respect to this isomer, are 3.1, 18.2, 21.2 and 106.1 kJ mol−1 for isomer 4, 3, 2 and 1, respectively.
Comparing the calculated IR spectra of our isomers to the 750–1650 cm−1 range of the IRMPD spectrum, does not allow for an unambiguous assignment of one specific isomer. Isomers 2, 3 and 5 all match quite well with the experimental spectrum, albeit some intensity mismatches are noted. Only isomers 1, 4 and 5 can account for the CO stretching feature observed at 1718 cm−1. The calculated CO stretching modes for these isomers lay at 1673, 1724 and 1716 cm−1, respectively. Despite having a carbonyl group, isomer 1 does not match the feature at 1718 cm−1, thus indicating that the carbonyl group is likely neighboured by an NH group.
Based on this and the match in the 750–1650 cm−1 range, we tentatively assign isomer 5 to be the most likely candidate for [TAC–H + H2O]+. However, this does not rule out the possibility of several other isomers contributing to the [TAC–H + H2O]+ IRMPD spectrum. Since isomer 4 and 5 are close in energy, we estimated the contribution of both isomers to the IRMPD spectrum at 300 K from the Boltzmann distribution based on their energies obtained from our DFT calculations. This showed a contribution of 22% and 78% isomer 4 and 5, respectively, thus indicating that isomer 4 is a potentially significant component in the IRMPD spectrum. However, this estimation should be treated with care as the energies were calculated using DFT. Perhaps additional measurements could narrow the number of possible contributing isomers. For example, since OH stretching is expected to result in a band around 3600 cm−1,50 an IRMPD measurement in this range might help to confirm or exclude the contribution of the enol isomers.
4 Discussion and astrophysical implication
4.1 Comparison of the TAC˙+ IRMPD to coronene˙+
Comparing the IRMPD spectrum of TAC˙+ to its non-nitrogen bearing analogue coronene˙+ (Cor˙+), measured by Oomens et al.22 in 2001 using the same IRMPD technique on a different ion trap mass spectrometer system gives direct insight into the spectral shift caused by the inclusion of three peripheral nitrogen atoms. Fig. 5 shows the IRMPD spectra of Cor˙+ and TAC˙+.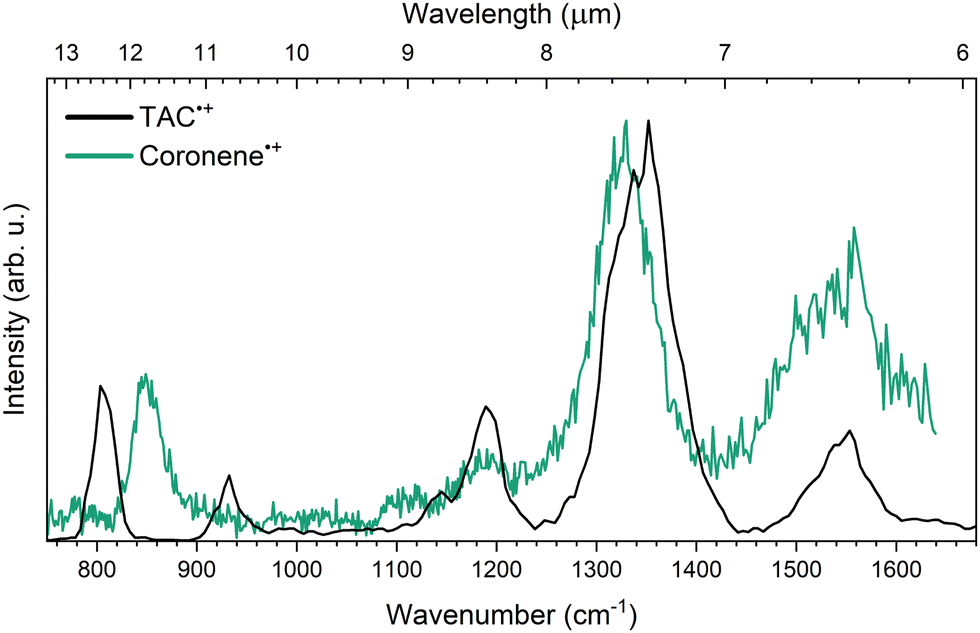 | ||
| Fig. 5 Experimental IRMPD of TAC˙+ (black) and coronene˙+ (green). Both spectra are normalized with respect to their most intense peak. The IRMPD spectrum of coronene˙+ is taken from Oomens et al.22 | ||
From the comparison of the spectra in the spectral region between 1050 and 1700 cm−1 it is clear that there is remarkable similarity in band positions between the experimental TAC˙+ and Cor˙+ spectra. All of the TAC˙+ features in this region fall within about 3% of the position of the Cor˙+ features. Some clear differences between the two spectra are noted in the 750–1050 cm−1 region. The Cor˙+ spectrum only exhibits a single feature at 849 cm−1, which is attributed to the out-of-plane CH bending mode,22,51 while TAC˙+ exhibits two bands at 803 and 932 cm−1. For the 932 cm−1TAC˙+ feature no counterpart can be identified in the Cor˙+ spectrum, as this feature is mainly inherent to a single out-of-plane CH bending mode neighbouring a nitrogen atom. Despite this, the spectra are thus remarkably similar. This is in agreement with earlier works predicting a minor impact on the overall IR spectrum after nitrogen atom incorporating on the periphery of a pure carbon PAH.26,27
4.2 Comparison of the TAC˙+ and TACH+ IRMPD spectra to interstellar emission bands
In order to assess the interstellar relevance of TAC˙+ and TACH+, we compare the IRMPD spectra presented in this work to observed astronomical mid-IR spectra of each of the four spectral classes.6–9 Representative spectra of class A through D in the 6 to 10 μm range are shown together with the experimental IRMPD spectra of TAC˙+ and TACH+ in Fig. 6.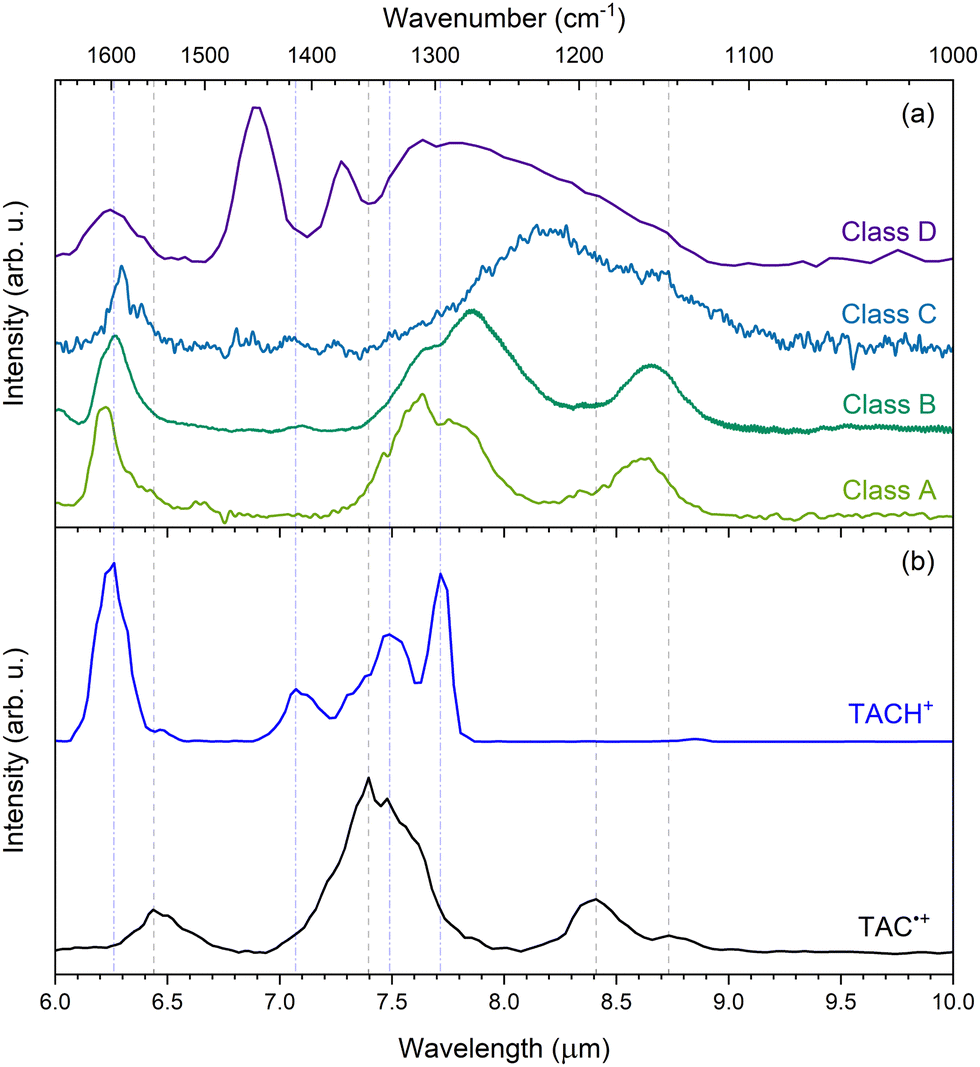 | ||
| Fig. 6 (a): Mid-IR spectra of astronomical representative sources of type A (IRAS 23133 + 6050; olive), type B (HD 44179; green), type C (IRAS 13416-6243; blue), and type D (IRAS 05110-6616; purple). All are normalized to the maximum intensity of the 7–8 μm complex and with continuum subtracted. The type D spectrum was recorded using Spitzer/IRS52,53 and the other classes are obtained from ISO/SWS54,55 observations, as calibrated by Sloan et al.56 (b) Experimental IRMPD of TAC˙+ (black) and TACH+ (blue). Both spectra are normalized with respect to their most intense peak. The vertical grey, dashed and blue, dot-dashed lines represent the peaks of the most clear features in the IRMPD spectra of TAC˙+ and TACH+, respectively. | ||
When comparing the IRMPD of TAC˙+ to the observed astronomical spectra, we find that TAC˙+ does not match well with many of the observed bands in the class A and B spectra, but may still contribute to some extent. Moreover, the modes at 8.41 and 8.73 μm (1189 and 1145 cm−1) can potentially contribute to the shoulders of the broad features in the class C and D spectra. This supports the hypothesis that cationic exo-PANHs can contribute to the AIBs as well. Thus, exo-PANH and PAH cations should both be considered to be contributors to interstellar IR emission bands.
Protonated PANHs have been proposed as possible carriers of the 6.2 μm interstellar emission band.27,33 Ricca et al.27 calculated that these species can give rise to modes in the 6.17 to 6.45 μm range, although, these calculations were performed on molecules containing NC ≥ 51. From our data we find that protonation of TAC results in an IRMPD spectrum with a feature that matches very well with the 6.2 μm interstellar feature in all four classes and suggests medium-sized protonated exo-PANHs may contribute to the 6.2 μm AIB feature. Furthermore, TACH+ has two intense bands at 7.49 and 7.72 μm (1335 and 1296 cm−1), which both match well with the 7.6 μm feature of the class A spectrum. The match with the class A spectrum indicates that TACH+ could be a contributor to the AIBs, therefore suggesting that protonated exo-PANHs are possible carriers of the AIBs.
Based on the match/mismatch with the AIBs it seems that protonated exo-PANHs are more likely contributors than the cationic exo-PANHs. From a physical point of view this is also supported by the fact that exo-PANH radical cations are open-shell species, while protonated exo-PANHs are closed-shell species, making exo-PANH radical cations more reactive with other abundant atoms/molecules such as hydrogen as compared to protonated exo-PANHs. This possibly increases the fraction of interstellar protonated exo-PANH cations at the expense of cationic exo-PANHs.
4.3 Possible astronomical relevance of oxygen-functionalized PA(N)Hs
Weaker emission features between 5.7 and 5.9 μm are observed towards various objects in the ISM.1,2 These features, however, are generally ignored due to their low intensity. The peak in the IRMPD spectrum of [TAC–H + H2O]+ at 5.82 μm (1718 cm−1) shows that the carbonyl stretch of oxygen-functionalized exo-PANH cations is a possible carrier of the weak AIB features between 5.7 and 5.9 μm. Previous studies on oxygen-functionalized PAHs concluded the CO stretch to be at 6.01 μm (1665 cm−1)57 and around 5.88 μm (1700 cm−1).58,59 The precise position of carbonyl stretching is thus influenced by the molecule and its chemical environment. However, it is also argued that these feaures could originate from a blend of CH stretching and in- and out-of-plane CH bending of pure PAHs.60 Possibly, the further (IR) investigation of oxygen-functionalized PA(N)Hs might shed light on a clear origin of the 5.7–5.9 μm AIB features, which could contain valuable information on the atomic and molecular environment of interstellar objects.
5 Conclusions
The gas-phase infrared spectra of 1,5,9-triazacoronene˙+ (TAC˙+), the protonated TAC (TACH+) and the water-addition species [TAC–H + H2O]+, obtained with IRMPD spectroscopy, are presented. TAC˙+ shows six clear features at 803, 932, 1145, 1189, 1352 and 1553 cm−1, with the 1352 cm−1 feature being the broadest and also the most intense. The IRMPD of TACH+ contains seven features at 807, 1130, 1296, 1335, 1414, 1546 and 1597 cm−1. Of which the most intense feature is situated at 1597 cm−1 (6.26 μm), which is akin to the IR spectra of other protonated PANHs.26,27,33 This supports the argument of protonated PANHs being possible carriers of the interstellar 6.2 μm band. [TAC–H + H2O]+ exhibits many features of which the most pronounced are peaks at 808, 1177, 1319, 1582 and 1718 cm−1. From comparing the calculated IR spectra of 16 different isomers to the experimental IRMPD spectrum, the most likely candidate for [TAC–H + H2O]+ is determined to be a ketone.We compared the experimental IRMPD spectrum of TAC˙+ to the IRMPD spectrum of coronene˙+ (Cor˙+) measured by Oomens et al.22 In the 1050 to 1700 cm−1 wavenumber region we find considerable overlap in band positions between the TAC˙+ and Cor˙+. Discrepancies only arise in the 750 to 1050 cm−1 wavenumber region, where TAC˙+ has two features at 803 and 932 cm−1 and Cor˙+ one at 849 cm−1. Despite these features not overlapping, the spectra are remarkably similar. This is in line with the prediction from earlier works that a minor impact on the overall IR spectrum is expected after nitrogen atom incorporation on the periphery of a pure carbon PAH.
To put TAC˙+ and TACH+ into astronomical context, we compared their IRMPD spectra to observed astronomical mid-IR spectra of spectral type class A to D.6–8 The TAC˙+ IRMPD has a few features that have some overlap with the class C and D. This indicates the potential contribution of TAC˙+ to the class C and D spectra. Thus supporting the idea that cationic exo-PANHs can contribute to the AIBs as well. The IR spectra of exo-PANH cations generally do not match better with the AIBs than their unsubstituted PAH congener, due to the similarity of their IR spectra. So, both exo-PANH and PAH cations should be considered to be potential carriers of AIBs. TACH+ produces an IRMPD spectrum that matches well with the 6.2 μm feature in all four classes. Furthermore, the two intense features at 7.49 and 7.72 μm (1335 and 1296 cm−1) have a considerable match with the 7.6 μm feature of the class A spectrum. This match is indicative of TACH+ being a possible contributor to the AIBs, therefore suggesting the possibility of protonated exo-PANHs being carriers of the AIBs.
Author contributions
Jerry Kamer: conceptualization, project administration, resources, investigation, formal analysis, writing – original draft, writing – review and editing. Domenik Schleier: investigation, resources, writing – review and editing, funding acquisition. Andy Jiao: resources, writing – review and editing. Grégory Schneider: resources. Jonathan Martens: investigation. Giel Berden: investigation, writing – review and editing. Jos Oomens: investigation, writing – review and editing. Jordy Bouwman: conceptualization, funding acquisition, project administration, writing – review and editing.Data availability
All available data can be found in this manuscript or has been included in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
JB acknowledges the Netherlands Organisation for Scientific Research (Nederlandse Organisatie voor Wetenschappelijk Onderzoek, NWO) for a Vidi grant (grant number 723.016.006). This work was supported in part by NASAs Solar System Exploration Research Virtual Institute (SSERVI): Institute for Modeling Plasma, Atmosphere, and Cosmic Dust (IMPACT). DS acknowledges support by the PRIME programme of the German Academic Exchange Service (DAAD) with funds from the German Federal Ministry of Education and Research (BMBF). DS ackowledges a grant for computing time at the Paderborn Center for Parallel Computing PC2 under the project PEPICON. DS gratefully acknowledges continued support by Prof. Tina Kasper and funding from DFG under project number KA 3871/3-2 and 270672969. Quantum chemical calculations were carried out on the Dutch national e-infrastructure with the support of SURF Cooperative (EINF-5273).References
- A. G. G. M. Tielens, Annu. Rev. Astron. Astrophys., 2008, 46, 289–337 CrossRef.
- R. Chown, A. Sidhu, E. Peeters, A. G. G. M. Tielens, J. Cami, O. Berné, E. Habart, F. Alarcón, A. Canin, I. Schroetter, B. Trahin, D. Van De Putte, A. Abergel, E. A. Bergin, J. Bernard-Salas, C. Boersma, E. Bron, S. Cuadrado, E. Dartois, D. Dicken, M. El-Yajouri, A. Fuente, J. R. Goicoechea, K. D. Gordon, L. Issa, C. Joblin, O. Kannavou, B. Khan, O. Lacinbala, D. Languignon, R. Le Gal, A. Maragkoudakis, R. Meshaka, Y. Okada, T. Onaka, S. Pasquini, M. W. Pound, M. Robberto, M. Röllig, B. Schefter, T. Schirmer, S. Vicente, M. G. Wolfire, M. Zannese, I. Aleman, L. Allamandola, R. Auchettl, G. A. Baratta, S. Bejaoui, P. P. Bera, J. H. Black, F. Boulanger, J. Bouwman, B. Brandl, P. Brechignac, S. Brünken, M. Buragohain, A. Burkhardt, A. Candian, S. Cazaux, J. Cernicharo, M. Chabot, S. Chakraborty, J. Champion, S. W. J. Colgan, I. R. Cooke, A. Coutens, N. L. J. Cox, K. Demyk, J. D. Meyer, S. Foschino, P. García-Lario, L. Gavilan, M. Gerin, C. A. Gottlieb, P. Guillard, A. Gusdorf, P. Hartigan, J. He, E. Herbst, L. Hornekaer, C. Jäger, E. Janot-Pacheco, M. Kaufman, F. Kemper, S. Kendrew, M. S. Kirsanova, P. Klaassen, S. Kwok, Á. Labiano, T. S.-Y. Lai, T. J. Lee, B. Lefloch, F. Le Petit, A. Li, H. Linz, C. J. Mackie, S. C. Madden, J. Mascetti, B. A. McGuire, P. Merino, E. R. Micelotta, K. Misselt, J. A. Morse, G. Mulas, N. Neelamkodan, R. Ohsawa, A. Omont, R. Paladini, M. E. Palumbo, A. Pathak, Y. J. Pendleton, A. Petrignani, T. Pino, E. Puga, N. Rangwala, M. Rapacioli, A. Ricca, J. Roman-Duval, J. Roser, E. Roueff, G. Rouillé, F. Salama, D. A. Sales, K. Sandstrom, P. Sarre, E. Sciamma-O'Brien, K. Sellgren, S. S. Shenoy, D. Teyssier, R. D. Thomas, A. Togi, L. Verstraete, A. N. Witt, A. Wootten, H. Zettergren, Y. Zhang, Z. E. Zhang and J. Zhen, Astron. Astrophys., 2024, 685, A75 CrossRef.
- A. Leger and J. L. Puget, Astron. Astrophys., 1984, 137, L5–L8 CAS.
- L. J. Allamandola, A. G. G. M. Tielens and J. R. Barker, Astrophys. J., 1985, 290, L25–L28 CrossRef CAS.
- L. J. Allamandola, A. G. G. M. Tielens and J. R. Barker, Astrophys. J., 1989, 71, 733 CAS.
- E. Peeters, S. Hony, C. Van Kerckhoven, A. G. G. M. Tielens, L. J. Allamandola, D. M. Hudgins and C. W. Bauschlicher, Astron. Astrophys., 2002, 390, 1089–1113 CrossRef CAS.
- B. van Diedenhoven, E. Peeters, C. V. Kerckhoven, S. Hony, D. M. Hudgins, L. J. Allamandola and A. G. G. M. Tielens, Astophys. J., 2004, 611, 928–939 CrossRef CAS.
- M. Matsuura, J. Bernard-Salas, T. Lloyd Evans, K. M. Volk, B. J. Hrivnak, G. C. Sloan, Y. Chu, R. Gruendl, K. E. Kraemer, E. Peeters, R. Szczerba, P. R. Wood, A. A. Zijlstra, S. Hony, Y. Ita, D. Kamath, E. Lagadec, Q. A. Parker, W. A. Reid, T. Shimonishi, H. Van Winckel, P. M. Woods, F. Kemper, M. Meixner, M. Otsuka, R. Sahai, B. A. Sargent, J. L. Hora and I. McDonald, Mon. Not. R. Astron. Soc., 2014, 439, 1472–1493 CrossRef CAS.
- J. Bouwman, P. Castellanos, M. Bulak, J. Terwisscha van Scheltinga, J. Cami, H. Linnartz and A. G. G. M. Tielens, Astron. Astrophys., 2019, 621, A80 CrossRef CAS.
- J. Bouwman, C. Boersma, M. Bulak, J. Kamer, P. Castellanos, A. G. G. M. Tielens and H. Linnartz, Astron. Astrophys., 2020, 636, A57 CrossRef CAS.
- J. Bouwman, H. Linnartz and A. G. G. M. Tielens, J. Mol. Spectrosc., 2021, 378, 111458 CrossRef CAS.
- L. J. Allamandola, D. M. Hudgins and S. A. Sandford, Astrophys. J., 1999, 511, L115 CrossRef CAS PubMed.
- D. M. Hudgins and S. A. Sandford, J. Phys. Chem. A, 1998, 102, 329–343 CrossRef CAS.
- D. M. Hudgins and S. A. Sandford, J. Phys. Chem. A, 1998, 102, 344–352 CrossRef CAS.
- J. Zhen, P. Castellanos, J. Bouwman, H. Linnartz and A. G. G. M. Tielens, Astrophys. J., 2017, 836, 28 CrossRef.
- J. Zhen, A. Candian, P. Castellanos, J. Bouwman, H. Linnartz and A. G. G. M. Tielens, Astrophys. J., 2018, 854, 27 CrossRef.
- J. Palotás, J. Martens, G. Berden and J. Oomens, Nat. Astron., 2020, 4, 240–245 CrossRef.
- J. Palotás, J. Martens, G. Berden and J. Oomens, J. Mol. Spectrosc., 2021, 378, 111474 CrossRef.
- A. Ricca, C. W. Bauschlicher, C. Boersma, A. G. G. M. Tielens and L. J. Allamandola, Astrophys. J., 2012, 754, 75 CrossRef.
- A. L. Mattioda, C. W. Bauschlicher, J. D. Bregman, D. M. Hudgins, L. J. Allamandola and A. Ricca, Spectrochim. Acta, Part A, 2014, 130, 639–652 CrossRef CAS PubMed.
- C. J. Mackie, E. Peeters, C. W. Bauschlicher Jr. and J. Cami, Astrophys. J., 2015, 799, 131 CrossRef CAS.
- J. Oomens, B. G. Sartakov, A. G. G. M. Tielens, G. Meijer and G. von Helden, Astrophys. J., 2001, 560, L99 CrossRef CAS.
- J. Oomens, A. G. G. M. Tielens, B. G. Sartakov, G. von Helden and G. Meijer, Astrophys. J., 2003, 591, 968 CrossRef CAS.
- S. Banhatti, J. Palotás, P. Jusko, B. Redlich, J. Oomens, S. Schlemmer and S. Brünken, Astron. Astrophys., 2021, 648, A61 CrossRef CAS.
- J. Gao, G. Berden and J. Oomens, Astrophys. J., 2014, 787, 170 CrossRef.
- D. M. Hudgins, J. Charles, W. Bauschlicher and L. J. Allamandola, Astrophys. J., 2005, 632, 316–332 CrossRef CAS.
- A. Ricca, C. Boersma and E. Peeters, Astophys. J., 2021, 923, 202 CrossRef CAS.
- D. B. Rap, J. G. M. Schrauwen, A. N. Marimuthu, B. Redlich and S. Brünken, Nat. Astron., 2022, 6, 1059–1067 CrossRef.
- C. Boersma, J. D. Bregman and L. J. Allamandola, Astrophys. J., 2013, 769, 117 CrossRef.
- D. Schleier, J. Kamer, J. Martens, G. Berden, J. Oomens and J. Bouwman, ChemPhysChem, 2024, e202300915 CrossRef.
- B. A. McGuire, R. A. Loomis, A. M. Burkhardt, K. L. K. Lee, C. N. Shingledecker, S. B. Charnley, I. R. Cooke, M. A. Cordiner, E. Herbst, S. Kalenskii, M. A. Siebert, E. R. Willis, C. Xue, A. J. Remijan and M. C. McCarthy, Science, 2021, 371, 1265–1269 CrossRef.
- E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef.
- H. Alvaro Galué, O. Pirali and J. Oomens, Astron. Astrophys., 2010, 517, A15 CrossRef.
- D. Schleier, J. Kamer, A. Jiao, G. F. Schneider, H. Linnartz and J. Bouwman, Phys. Chem. Chem. Phys., 2024, 26, 15547–15558 RSC.
- J. Martens, G. Berden, C. R. Gebhardt and J. Oomens, Rev. Sci. Instrum., 2016, 87, 103108 CrossRef PubMed.
- Q. Tan, H. Chen, H. Xia, B. Liu and B. Xu, Chem. Commun., 2016, 52, 537–540 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, 2016, Gaussian Inc., Wallingford CT Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- M. P. Andersson and P. Uvdal, J. Phys. Chem. A, 2005, 109, 2937–2941 CrossRef PubMed.
- J. Bouwman, A. J. de Haas and J. Oomens, Chem. Commun., 2016, 52, 2636–2638 RSC.
- J. Oomens, A. J. A. van Roij, G. Meijer and G. von Helden, Astrophys. J., 2000, 542, 404 CrossRef.
- M. U. Munshi, G. Berden, J. Martens and J. Oomens, Phys. Chem. Chem. Phys., 2017, 19, 19881–19889 RSC.
- M. U. Munshi, J. Martens, G. Berden and J. Oomens, J. Phys. Chem. A, 2020, 124, 2449–2459 CrossRef.
- G. Berden, M. Derksen, K. J. Houthuijs, J. Martens and J. Oomens, Int. J. Mass Spectrom., 2019, 443, 1–8 CrossRef.
- P. Parneix, M. Basire and F. Calvo, J. Phys. Chem. A, 2013, 117, 3954–3959 CrossRef CAS.
- R. G. A. R. Maclagan, S. Gronert and M. Meot-Ner (Mautner), J. Phys. Chem. A, 2015, 119, 127–139 CrossRef CAS.
- S. A. Francis, J. Chem. Phys., 1951, 19, 942–948 CrossRef.
- N. Fuson, M.-L. Josien and E. M. Shelton, J. Am. Chem. Soc., 1954, 76, 2526–2533 CrossRef.
- J. Palotás, J. Martens, G. Berden and J. Oomens, J. Phys. Chem. A, 2022, 126, 2928–2935 CrossRef PubMed.
- D. M. Hudgins and L. J. Allamandola, Astophys. J., 1999, 516, L41 CrossRef PubMed.
- M. W. Werner, T. L. Roellig, F. J. Low, G. H. Rieke, M. Rieke, W. F. Hoffmann, E. Young, J. R. Houck, B. Brandl, G. G. Fazio, J. L. Hora, R. D. Gehrz, G. Helou, B. T. Soifer, J. Stauffer, J. Keene, P. Eisenhardt, D. Gallagher, T. N. Gautier, W. Irace, C. R. Lawrence, L. Simmons, J. E. V. Cleve, M. Jura, E. L. Wright and D. P. Cruikshank, Astrophys. J., Suppl. Ser., 2004, 154, 1 CrossRef.
- J. R. Houck, T. L. Roellig, J. van Cleve, W. J. Forrest, T. Herter, C. R. Lawrence, K. Matthews, H. J. Reitsema, B. T. Soifer, D. M. Watson, D. Weedman, M. Huisjen, J. Troeltzsch, D. J. Barry, J. Bernard-Salas, C. E. Blacken, B. R. Brandl, V. Charmandaris, D. Devost, G. E. Gull, P. Hall, C. P. Henderson, S. J. U. Higdon, B. E. Pirger, J. Schoenwald, G. C. Sloan, K. I. Uchida, P. N. Appleton, L. Armus, M. J. Burgdorf, S. B. Fajardo-Acosta, C. J. Grillmair, J. G. Ingalls, P. W. Morris and H. I. Teplitz, Astrophys. J., Suppl. Ser., 2004, 154, 18 CrossRef.
- M. F. Kessler, J. A. Steinz, M. E. Anderegg, J. Clavel, G. Drechsel, P. Estaria, J. Faelker, J. R. Riedinger, A. Robson, B. G. Taylor and S. Ximénez de Ferrán, Astron. Astrophys., 1996, 315, L27–L31 Search PubMed.
- T. de Graauw, L. N. Haser, D. A. Beintema, P. R. Roelfsema, H. van Agthoven, L. Barl, O. H. Bauer, H. E. G. Bekenkamp, A. J. Boonstra, D. R. Boxhoorn, J. Cote, P. de Groene, C. van Dijkhuizen, S. Drapatz, J. Evers, H. Feuchtgruber, M. Frericks, R. Genzel, G. Haerendel, A. M. Heras, K. A. van der Hucht, T. van der Hulst, R. Huygen, H. Jacobs, G. Jakob, T. Kamperman, R. O. Katterloher, D. J. M. Kester, D. Kunze, D. Kussendrager, F. Lahuis, H. J. G. L. M. Lamers, K. Leech, S. van der Lei, R. van der Linden, W. Luinge, D. Lutz, F. Melzner, P. W. Morris, D. van Nguyen, G. Ploeger, S. Price, A. Salama, S. G. Schaeidt, N. Sijm, C. Smoorenburg, J. Spakman, H. Spoon, M. Steinmayer, J. Stoecker, E. A. Valentijn, B. Vandenbussche, H. Visser, C. Waelkens, L. B. F. M. Waters, J. Wensink, P. R. Wesselius, E. Wiezorrek, E. Wieprecht, J. J. Wijnbergen, K. J. Wildeman and E. Young, Astron. Astrophys., 1996, 315, L49–L54 Search PubMed.
- G. C. Sloan, K. E. Kraemer, S. D. Price and R. F. Shipman, Astrophys. J., Suppl. Ser., 2003, 147, 379 CrossRef.
- M. P. Bernstein, S. A. Sandford, L. J. Allamandola, J. S. Gillette, S. J. Clemett and R. N. Zare, Science, 1999, 283, 1135–1138 CrossRef CAS PubMed.
- Y. J. Pendleton and L. J. Allamandola, Astrophys. J., Suppl. Ser., 2002, 138, 75 CrossRef.
- G. Matrajt, G. M. M. Caro, E. D. L. d'Hendecourt, D. Deboffle and J. Borg, Astron. Astrophys., 2005, 433, 979–995 CrossRef.
- C. Boersma, A. L. Mattioda, C. W. Bauschlicher, E. Peeters, A. G. G. M. Tielens and L. J. Allamandola, Astrophys. J., 2008, 690, 1208 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03004d |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2024 |