
Pb6Ba3Si2S8I10: a new thiohalide with a quasi-two-dimensional structure and wide band gap†
Wang
Zhao
ab,
Jiazheng
Zhou
a,
Linan
Wang
ab,
Wenqi
Jin
ab,
Yingying
Kong
a,
Yu
Chu
 *ab and
Junjie
Li
*ab
*ab and
Junjie
Li
*ab
aResearch Center for Crystal Materials; CAS Key Laboratory of Functional Materials and Devices for Special Environments, Xinjiang Technical Institute of Physics & Chemistry, CAS, Urumqi 830011, China. E-mail: lijunjie@ms.xjb.ac.cn; chuy@ms.xjb.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 27th September 2024
Abstract
Pb-based chalcogenides display abundant structural diversity and distinguished properties. Based on a mixed anion and dimensional reduction combined strategy, a wide band gap Pb-based thiohalide, Pb6Ba3Si2S8I10, has been rationally designed and synthesized experimentally by the flux method. The compound crystallizes in the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c space group with cell parameters a = 9.7925(2) Å, b = 9.7925(2) Å, and c = 70.628(3) Å and is composed of [SiS4] tetrahedra and unprecedented [PbI5S2] polyhedral units, resulting in a unique quasi-two-dimensional structure, which enriches the chemical and structural diversity of Pb-based thiohalides. The experimental band gap of Pb6Ba3Si2S8I10 was determined to be 2.80 eV. Based on statistical analyses and to the best of our knowledge, it is the largest experimental optical band gap among the known Pb-based thiohalides. The results demonstrate the feasibility of using highly electropositive Ba atoms to regulate the dimensions of the structural framework of thiohalides and give new insights into the structure and property modifications of thiohalides by the mixed anion and dimensional reduction combined strategy.
c space group with cell parameters a = 9.7925(2) Å, b = 9.7925(2) Å, and c = 70.628(3) Å and is composed of [SiS4] tetrahedra and unprecedented [PbI5S2] polyhedral units, resulting in a unique quasi-two-dimensional structure, which enriches the chemical and structural diversity of Pb-based thiohalides. The experimental band gap of Pb6Ba3Si2S8I10 was determined to be 2.80 eV. Based on statistical analyses and to the best of our knowledge, it is the largest experimental optical band gap among the known Pb-based thiohalides. The results demonstrate the feasibility of using highly electropositive Ba atoms to regulate the dimensions of the structural framework of thiohalides and give new insights into the structure and property modifications of thiohalides by the mixed anion and dimensional reduction combined strategy.
Introduction
The development of functional materials and structural chemistry is highly dependent on the discovery of new compounds with distinctive crystal structures and physicochemical properties.1–8 Introducing cations with stereochemically active lone-pair electrons, such as Pb2+, Sn2+, Bi3+, Sb3+, and As3+, has been demonstrated as an effective strategy to enhance the optical anisotropy of optoelectronic functional materials.9–13 Among them, Pb2+-containing compounds with stereochemically active lone-pair electrons are conducive to inducing a significant second-harmonic generation (SHG) response, but they usually exhibit an adverse effect on the band gap, such as PbGa2GeSe6 (1.96 eV, 5 × AgGaS2 (AGS)), Pb0.65Mn2.85Ga3S8 (1.68 eV, 1.5 × AGS), Pb0.72Mn2.84Ga2.95Se8 (1.65 eV, 4.4 × AGS), Pb4Ga4GeS12 (2.18 eV, 2 × AGS), and PbGa4Se7 (2.1 eV, 3.3 × AGS).14–20 As a critical parameter of optoelectronic functional materials, a wide band gap can effectively suppress two-photon/multi-photon absorption in IR nonlinear optical materials. For instance, a material with a band gap wider than 2.33 eV can only be effectively pumped by mature 1064 nm laser sources in the absence of two-photon absorption and free carrier absorption. Moreover, a larger band gap is advantageous for the compound to achieve a higher laser-induced damage threshold.21–27 Thus, it is important to overcome the drawback of the narrow band gap in Pb-based chalcogenides.Recently, introducing highly electronegative halogens has been proved to be a feasible strategy to increase the band gap of Pb-based chalcogenides,28–37 and a series of Pb-based salt-inclusion chalcogenides have been developed, such as [K2PbI][Ga7S12] (2.41 eV), [K2PbBr][Ga7S12] (2.49 eV), [K2PbCl][Ga7S12] (2.54 eV), [Na2PbI][Ga7S12] (2.53 eV), Pb3S3Cl2 (2.02 eV), Pb3SBrI3 (2.16 eV), Pb2SbS2I3 (2.19 eV), Pb4SeBr6 (2.62 eV), Pb3.5GeS4Br3 (2.6 eV), etc.38–50 In addition, reducing the dimensions of the crystal structure using highly electro-positive/negative elements is also helpful in increasing the optical band gap of inorganic compounds, and this has been demonstrated in many systems, such as sulfides,51,52 selenides,53 tellurides and carbonates.54
In this work, based on a mixed anion and dimensional reduction (introducing highly electropositive Ba atoms) combined strategy, a new Pb-based thiohalide Pb6Ba3Si2S8I10 with unprecedented [PbI5S2] octahedra has been rationally designed and synthesized by the flux method. The compound crystallizes in the centrosymmetric Rc space group and shows a quite large cell parameter c = 70.628(3) Å (due to the presence of six distinct filling modes of Ba and I atoms in the layers), resulting in a quasi-two-dimensional (quasi-2D) crystal structure composed of tetrahedral [SiS4] and polyhedral [PbI5S2] units. The formed quasi-2D structure is different from the three-dimensional (3D) frameworks in most of the Pb-based thiohalides. Moreover, due to the introduction of alkaline-earth metal (AEM) Ba and I atoms into the compound simultaneously, a relatively wide band gap (among the thiohalides) of 2.80 eV in the title compound is achieved.
Experimental section
Reagents
BaS (Aladdin, 99.7%), Pb (Aladdin, 99.9%), PbI2 (Aladdin, 99.99%), Si (Aladdin, 99.99%), and S (Sinopharm, 99.9%) were purchased commercially and used as the starting materials without further purification.Synthesis
The single crystal of the title compound for structural determination (Table 1) was grown by the flux method55,56 with PbCl2 as the flux in a sealed quartz tube at ∼950 °C. The details for the crystal growth are shown as follows: (1) the mixtures of BaS (0.094 g), Pb (0.038 g), Pbl2 (0.427 g), Si (0.030 g) and S (0.010 g) were ground and loaded into a graphite crucible with an inner diameter of 7 mm; (2) the crucible was transferred into a 10 mm (inner diameter) quartz tube, and it was further sealed with a methane-oxygen flame under a high vacuum of 10−3 Pa; and (3) the sealed tube was placed into a computer-controlled furnace and then heated to 950 °C for 24 h and kept at this temperature for 48 h, cooled to 650 K at a rate of 1 °C h−1, and then cooled to room temperature naturally. The pure phase powder samples were prepared in sealed quartz tubes with the starting materials of BaS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pb:PbI2:Si:S = 3:1:5:2:5 (molar ratio). The temperature program for the chemical synthesis involved quickly heating up to 950 °C and maintaining that temperature for 48 h, and then cooling to room temperature rapidly. Finally, the polycrystalline powder samples of Pb6Ba3Si2S8I10 were harvested at the bottom with some impurities on the top of the tubes.
:Pb:PbI2:Si:S = 3:1:5:2:5 (molar ratio). The temperature program for the chemical synthesis involved quickly heating up to 950 °C and maintaining that temperature for 48 h, and then cooling to room temperature rapidly. Finally, the polycrystalline powder samples of Pb6Ba3Si2S8I10 were harvested at the bottom with some impurities on the top of the tubes.
| a R 1 = ∑||Fo| − |Fc||/∑|Fo| and wR2 = [∑w(Fo2 − Fc2)2/∑wFo4]1/2 for Fo2 > 2σ(Fo2). | |
|---|---|
| Empirical formula | Pb6Ba3Si2S8I10 |
| Formula weight | 3236.76 |
| Temperature | 298.15 K |
| Crystal system, space group | Trigonal, Rc |
| Unit cell dimensions | a = 9.7925(2) Å |
| b = 9.7925(2) Å | |
| c = 70.628(3) Å | |
| Volume | 5865.4(3) Å3 |
| Z, calculated density | 6, 5.498 g cm−3 |
| Absorption coefficient | 37.085 mm−1 |
| F(000) | 8076 |
| Data/restraints/parameters | 1870/0/47 |
| Index ranges | −13 ≤ h ≤ 12, −8 ≤ k ≤ 9, −89 ≤ l ≤ 98 |
| Completeness | 99.0% |
| Independent reflections | 1870 [Rint = 0.0465, Rsigma = 0.0403] |
| GOF on F2 | 1.074 |
| 2θ range for data collection | 4.94° to 60.068° |
| Reflections collected | 8103 |
| Final R indices [Fo2 > 2σ(Fo2)]a | R 1 = 0.0434, wR2 = 0.0869 |
| R indices (all data)a | R 1 = 0.0540, wR2 = 0.0911 |
| Largest diff. peaks and holes | 2.11/−2.19 e Å−3 |
Structural determination
A yellow and transparent Pb6Ba3Si2S8I10 single crystal with a size of 0.15 × 0.05 × 0.04 mm3 was selected for the structural characterization. The data were collected on a Bruker Smart APEX III. The crystal structure was solved by direct methods and then refined by full-matrix least squares on F2 with the SHELXTL-14 program.57,58 The PLATON program was used to check the possible missing symmetry elements and no higher symmetry was suggested in the structure.59 The crystallographic data are available at CCDC 2369466† in CIF format.Powder X-ray diffraction (PXRD)
PXRD data of Pb6Ba3Si2S8I10 were collected on a Bruker D2 PHASER X-ray diffractometer equipped with Cu Kα radiation (λ = 1.54056 Å) at room temperature and the diffraction patterns were taken from 10° to 70° (2θ).60,61 Then, powder Rietveld refinement was performed using GSAS software.Energy dispersive X-ray spectroscopy (EDS)
The EDS spectrum of the title compound was recorded on a field emission scanning electron microscope (FE-SEM, JEOL JSM-7610F Plus, Japan) with an energy dispersive X-ray spectrometer (Oxford, X-Max 50) at room temperature. It was operated at 5 kV.UV-vis-NIR diffuse reflectance and infrared (IR) spectra
The UV-vis-NIR diffuse reflectance spectrum of Pb6Ba3Si2S8I10 was characterized on a Shimadzu SolidSpec-3700DUV spectrophotometer at room temperature within a wavelength range from 180 to 2600 nm. The reflectance spectrum was converted to pseudo-absorption data based on the Kubelka–Munk function: F(R) = (1 − R)2/2R = K/S (R = reflectance; K = absorption; and S = scattering).62 Before recording the IR spectrum, the polycrystalline Pb6Ba3Si2S8I10 powder sample was made into a pellet with KBr powder. The IR spectrum of the compound was recorded on a Shimadzu IR Affinity-1 Fourier transform infrared spectrometer within a range of 4000–400 cm−1.Refractive index difference (RID)
The RID of the Pb6Ba3Si2S8I10 single crystal was tested on a polarizing microscope equipped (ZEISS Axio Scope. 5 pol) with a Berek compensator. The wavelength of the used light source was 546 nm. The difference in the optical path (D) for one direction was determined according to the interference color with the maximum value of the crystal under polarized light.63 The RID can be calculated from the following equation: D = |N2 − N1| × T = Δn × T, where Δn represents the difference in the refractive index and T is the thickness of the crystal.64Computational methods
The first-principles calculations were performed by the plane wave pseudopotential method implemented in the CASTEP package.65,66 The generalized gradient approximation (GGA) and Perdew–Burke–Ernzerhof (PBE) functional were adopted for the calculations, and the norm-conserving pseudopotentials (NCPs) were used to calculate the electronic structure and optical properties.67 The cutoff energy was set to 820 eV. In order to model the effective interactions between valence electrons and the atom cores in Pb6Ba3Si2S8I10, the valence electrons were set as Ba-5s25p66s2, Pb-5s25p65d106s26p2, Si-3s23p2, S-3s23p4, and I-5s25p5. The Monkhorst–Pack k-point grid was set to 3 × 3 × 3 in the Brillouin zone (BZ). The residual calculation parameters and convergent criteria were set to the default values in the CASTEP package.Results and discussion
The results of single crystal XRD show that Pb6Ba3Si2S8I10 crystallizes in the trigonal Rc space group with cell parameters a = 9.7925(2) Å, b = 9.7925(2) Å, c = 70.628(3) Å, V = 5865.36(35) Å3 and Z = 6. The crystallographic data and structure refinement information for Pb6Ba3Si2S8I10 are provided in Table 1, and the atomic coordinates, bond distances, angles, and isotropic displacement parameters are given in Tables S1 and S2.† The calculated bond valence sums (Pb2+: 1.83, Ba2+: 2.31, Si4+: 3.94, S2−: 2.12–2.15, and I−: 0.79–1.55 for Pb6Ba3Si2S8I10) and small global instability indices GII = 0.178 (Table S1†) verify the reasonability of the crystal structure. The results of EDS spectroscopy and mapping (Fig. S1†) confirm the presence of Pb, Ba, Si, S, and I in the structure.
In the asymmetric unit of Pb6Ba3Si2S8I10, there is one crystallographically independent Pb atom, one Ba atom, one Si atom, two S atoms and four I atoms. The Si atoms are coordinated with four S atoms to form [SiS4] tetrahedral units with the bond lengths of d(Si–S) = 2.112–2.152 Å (Fig. 1a). The Pb atoms are coordinated with two S and six I atoms to build [PbI5S2] polyhedral units (Fig. 1b) with d(Pb–I) = 3.344–3.548 Å and d(Pb–S) = 2.821–2.883 Å. The formed [PbI5S2] mixed anionic unit is totally different from the formed [PbS5Cl2] in Pb5Sn3S10Cl2,47 [PbS4Br4], [PbS5Br3], [PbS6Br2] in Pb4S3Br2,41 [PbSI4] in Pb3SBrI3,46 and [PbS3I5] and [PbSBr2I5] in Pb3SBrI3.45 Based on statistical investigations using the Inorganic Crystal Structure Database (ICSD-5.2.0, the latest release of ICSD-2024/04/10) and to the best of our knowledge, [PbI5S2] is for the first time observed in Pb-based thiohalide compounds. Furthermore, the six [PbI5S2] octahedra are further interconnected with each other by edge-sharing to construct a [Pb6I13S12] polymer (Fig. 1c). The formed [Pb6I13S12] polymers are linked by tetrahedral [SiS4] units in a vertex-sharing way to produce 2D Pb–Si–S–I layers (Fig. 1d). The Ba and partial I atoms are located in the gaps of 2D Pb–Si–S–I layers to balance the valence states, resulting in the final quasi-2D structure of the title compound.
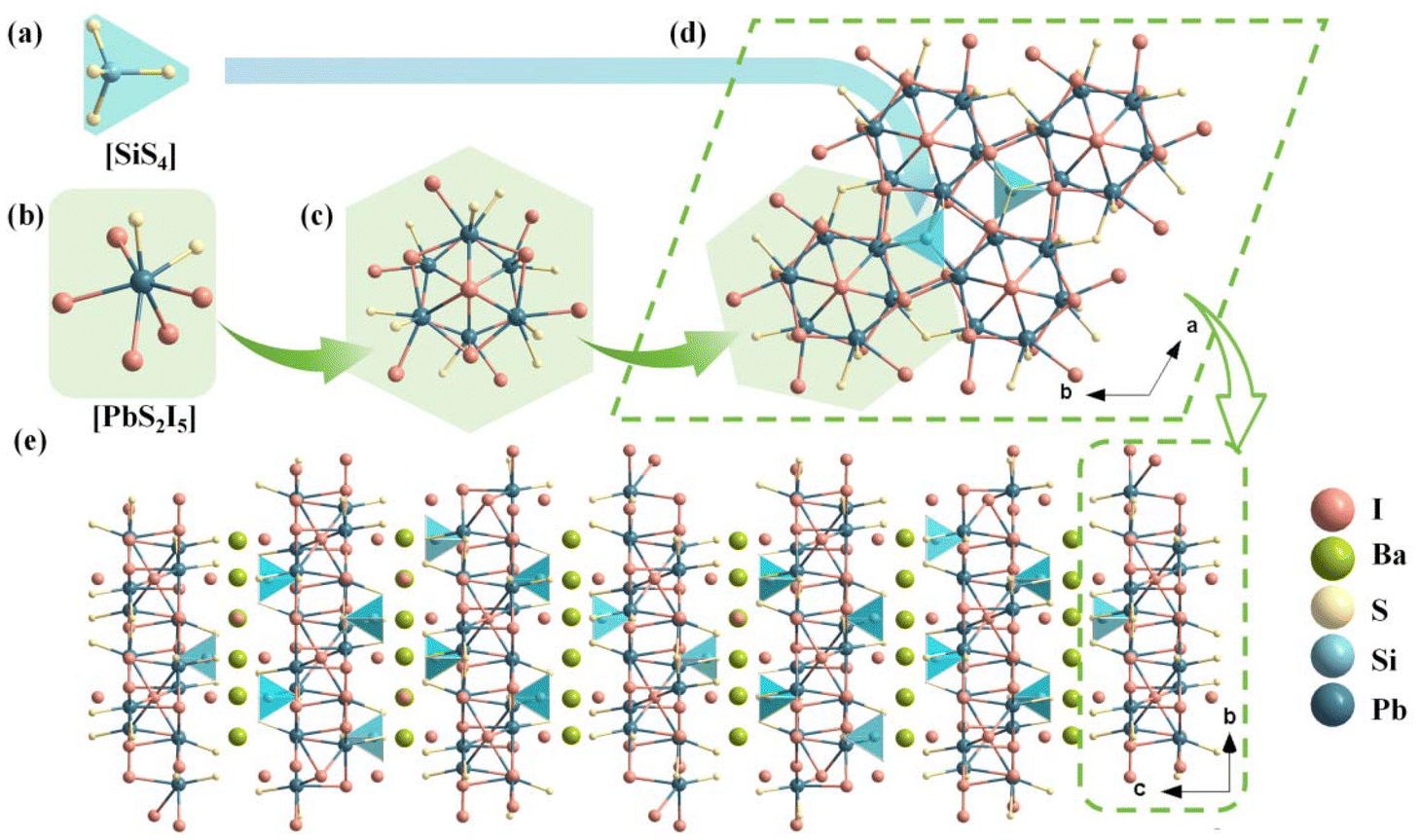 | ||
| Fig. 1 Crystal structure of Pb6Ba3Si2S8I10. (a and b) The coordination mode of Si (a) and Pb atoms (b); (c) the formed [Pb6I13S12] group; (d) the resultant 2D Pb–Si–S–I layer; and (e) the quasi-2D structure of Pb6Ba3Si2S8I10. | ||
Compared to the 3D Pb–S–Cl and Pb–S–Br frameworks in Pb5Sn3S10Cl247 and Pb4S3Br2,48 respectively, the Pb–S–I framework is reduced to a 2D structure in Pb6Ba3Si2S8I10, due to the introduction of highly electropositive Ba atoms with a high atomic proportion. In addition, from the perspective of Ba–I layers, Ba atoms exhibit three distinct arrangements, referred to as A, B, and C in Fig. S2.† The arrangements give rise to A′, B′, and C′ through a combination of secondary screw axes and glide symmetry operations along the c-direction, preventing the Ba atom layer from repeating within the unit cell via translational operations. The interleaved arrangements of Ba and I atoms ultimately result in six unique Ba–I configurations along the c-axis. Hence, due to the presence of six distinct filling modes of Ba and I atoms between the inter-layers of 2D Pb–Si–S–I, a large cell parameter c = 70.628(3) Å is observed in Pb6Ba3Si2S8I10. To confirm the chemical bonding and optical absorption regions, the IR spectrum of Pb6Ba3Si2S8I10 was recorded on a Shimadzu IR Affinity-1 Fourier transform infrared spectrometer, and it shows three strong absorption peaks at 501 cm−1, 534 cm−1 and 1084 cm−1, which can be attributed to the characteristic vibrations of Si–S bonding.68 Besides, the absorption peak at 785 cm−1 originates from the in-plane bending vibration of carbon dioxide (Fig. S3†).69
To elucidate the influence of highly electropositive cations on the structural dimension in Pb-based thiohalides, we extended the investigation to the all-known alkali metal (AM)- and/or AEM-containing Pb-based thiohalides in the ICSD, and 16 Pb-based thiohalides without C, H, O, and N elements are summarized in Table S3.† It can be seen that, except Pb6Ba3Si2S8I10 with a quasi-2D structure, all the known compounds including [Na2PbI][Ga7S12], [K2PbI][Ga7S12], [K2PbBr][Ga7S12], and [K2PbCl][Ga7S12] show 3D frameworks. Previous investigations indicate that the atomic ratio between electropositive AM/AEM cations and other central atoms capable of forming polyhedral units plays an important role in determining the dimensions of polyhedron-based frameworks.70 Based on statistical investigations, when the A/(Pb + M) ratio (where A represents highly electropositive AM or AEM atoms, while M denotes other central atoms capable of forming polyhedral units) is smaller than about 0.3, the compounds show 3D frameworks (Fig. 2a). Taking [Na2PbI][Ga7S12] as an example (Fig. 2c, where Pb6Ba3Si2S8I10 is used as a reference in Fig. 2b), the Na/(Pb + Ga) atomic ratio is 0.25, smaller than the Ba/(Pb + Si) ratio of 0.375 in Pb6Ba3Si2S8I10, resulting in a 3D pore-like structure in [Na2PbI][Ga7S12], and the Na atoms are placed in the pores, different from the interlayered Ba atoms in Pb6Ba3Si2S8I10 (Fig. 2b). The results highlight that the proportion of highly electropositive cations affects the crystal structures in thiohalides.
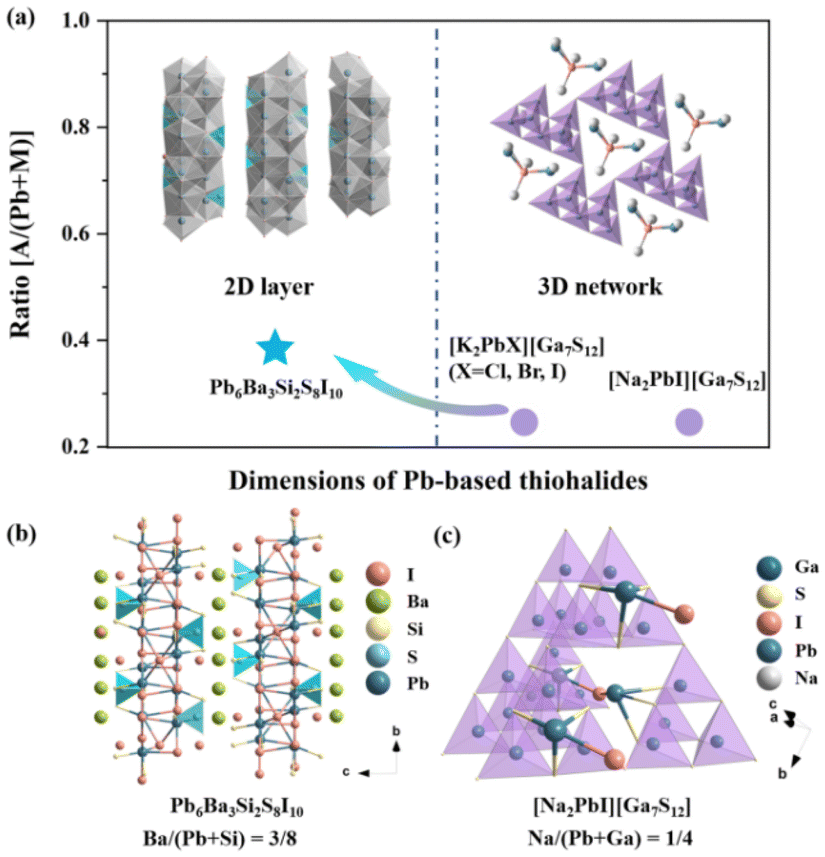 | ||
| Fig. 2 (a) Relationship between the A/(Pb + M) ratio (A = Na, K, and Ba; M = Ga and Si) and the dimensions of Pb-based thiohalides; structural comparison between quasi-2D Pb6Ba3Si2S8I10 (b) and 3D [Na2PbI][Ga7S12] (c). | ||
To detect the optical properties, the pure phase polycrystalline powder samples of the title compound were synthesized and characterized. As shown in Fig. 3a, the PXRD pattern of Pb6Ba3Si2S8I10 matches well the theoretical result derived from the CIF file using Mercury. Moreover, to assess the purity of the powder samples, the results of powder Rietveld refinement (RWP = 4.00% and RP = 2.98%) have been analysed, which indicate high purity for the obtained polycrystalline samples and confirm the results of single crystal XRD. In addition, to check the thermal stability, XRD characterization before and after melting has been carried out, as shown in Fig. S5.† The results imply that Pb6Ba3Si2S8I10 could be a congruently melting compound, which is favorable for the growth of large-sized single crystals. To detect the experimental optical band gap, the UV-vis-NIR diffuse reflectance spectrum of the title compound was recorded by using the pure phase polycrystalline powder samples. Moreover, based on the [(kνF(R))]1/2 spectrum (Fig. S6†), the band gap of Pb6Ba3Si2S8I10 was determined to be 2.13 eV, while it was 2.80 eV based on the [(kνF(R))]2 spectrum (Fig. 3b). The latter shows good agreement with the color of Pb6Ba3Si2S8I10 (yellow). It is worth noting that the value is larger than those of most of the Pb-based thiohalides, such as Pb5Sn3Se10Cl2 (1.44 eV), Pb5Sn3S10Cl2 (1.72 eV), Pb5S2I6 (1.73 eV), Pb4S3I2 (1.76 eV), Pb4S3Br2 (1.91 eV), [K2PbI][Ga7S12] (2.41 eV), [K2PbBr][Ga7S12] (2.49 eV), [K2PbCl][Ga7S12] (2.54 eV), [Na2PbI][Ga7S12] (2.53 eV), Pb3S3Cl2 (2.02 eV), Pb3SBrI3 (2.16 eV), Pb2SbS2I3 (2.19 eV), Pb4SeBr6 (2.62 eV), and Pb3.5GeS4Br3 (2.6 eV) (Fig. 3c).38–50 The reason for the relatively wide band gap in Pb6Ba3Si2S8I10 could be related to the low-dimensional quasi-2D structure, similar to the cases of AIIHgMIVS4 (AII = Sr and Ba; MIV = Si and Ge) (2.94–3.06 eV),71 PbFIO3 (3.87 eV),72 and AEGe2O4Se (AE = Sr and Ba) (3.57 and 3.81 eV).73 Moreover, the RID of Pb6Ba3Si2S8I10 was determined to be 0.006 at 546 nm (Fig. S7†).
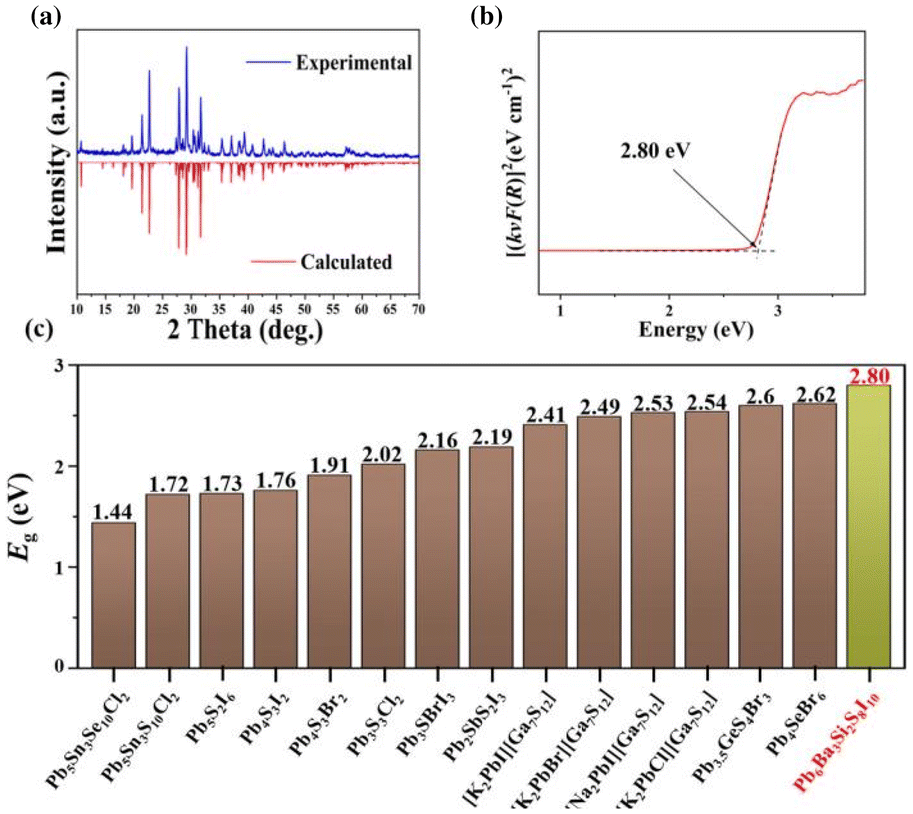 | ||
| Fig. 3 (a) Experimental and theoretical XRD patterns; (b) diffuse reflectance spectrum of Pb6Ba3Si2S8I10 plotted as [(kνF(R))]2 (direct band gap), where F(R) is the Kubelka–Munk function. Dotted lines show the fit used to extract the band gap; (c) the statistical investigations on the band gaps of typical Pb-based thiohalides (brown: 3D structured compounds and green: quasi-2D structured compounds). | ||
To detect the origin of optical properties in Pb6Ba3Si2S8I10, density functional theory (DFT) calculations were performed. The band structure indicates that Pb6Ba3Si2S8I10 is an indirect band gap semiconductor with a calculated GGA band gap of 2.341 eV (Fig. 4a), smaller than the experimental value of 2.80 eV, because of the discontinuity of the exchange–correlation energy functional.74 From the density of states (DOS) and partial DOS (PDOS) diagram in Fig. 4b, it can be seen that the top of the valence band (VB) is mainly derived from the S-3p and I-5p orbitals, while the bottom of the conduction band (CB) is occupied by the Pb-6p orbital (Fig. 4b). It indicates that the optical band gap in the title compound is mainly determined by the rare [PbI5S2] units, indicating the positive contribution of the mixed anions.30
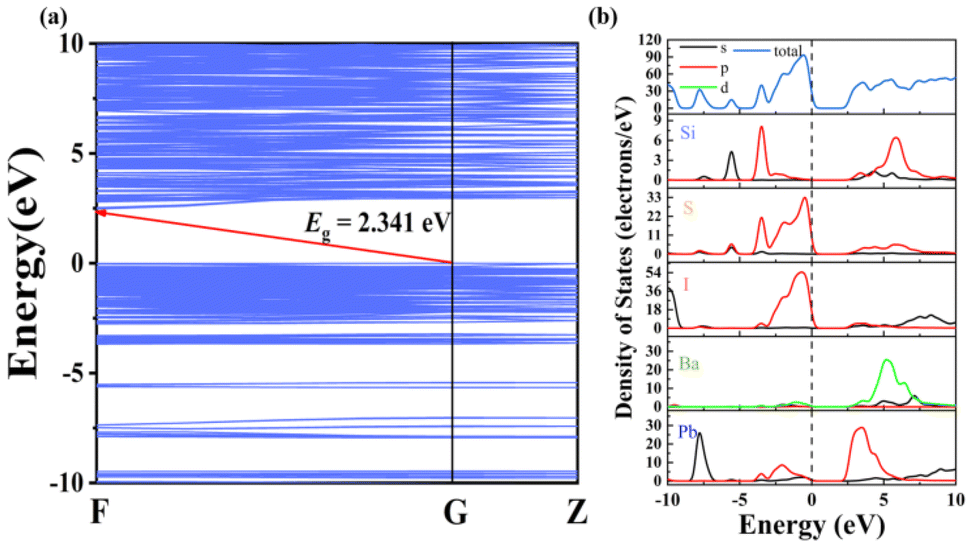 | ||
| Fig. 4 (a) The band structure of Pb6Ba3Si2S8I10 and (b) the total and partial densities of states of Pb6Ba3Si2S8I10. | ||
Conclusions
In summary, a new Pb-based thiohalide Pb6Ba3Si2S8I10 with unprecedented [PbI5S2] mixed anionic units has been designed by a mixed anion and dimensional reduction combined strategy and synthesized experimentally. The crystal structure of the compound was determined by single crystal XRD, and it shows a quasi-2D structure that is composed of polyhedral [PbI5S2] and tetrahedral [SiS4] units. More importantly, the introduction of highly electropositive Ba atoms with a high atomic proportion reduces the structural dimension, resulting in a relatively wide band gap of 2.80 eV in Pb6Ba3Si2S8I10, which is higher than those of known Pb-based thiohalides. DFT calculations indicate that Pb6Ba3Si2S8I10 is an indirect band gap semiconductor and that the band gap mainly originates from the interaction of the S-3p and I-5p orbitals in the unique [PbI5S2] mixed-anionic units. Experimental and theoretical results imply that introducing mixed anions and highly electropositive cations simultaneously is a feasible strategy to develop wide band gap Pb-based thiohalides.Data availability
Supporting data for this article are presented in the ESI.† The raw data of this article can be obtained from the corresponding author upon reasonable request.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the Natural Science Foundation of the Xinjiang Uygur Autonomous Region (2024D01E30), the Open Fund of the Anhui Key Laboratory of Photonic Materials and Devices (AHKL2024KF02), and the National Natural Science Foundation of China (22475234, 52402017, 22335007, and 52002398).References
- M. Mutailipu, K. R. Poeppelmeier and S. Pan, Chem. Rev., 2021, 121, 1130–1202 Search PubMed.
- J. Li and F. L. Deepak, Chem. Rev., 2022, 122, 16911–16982 Search PubMed.
- M. Mutailipu, J. Han, Z. Li, F. Li, J. Li, F. Zhang, X. Long, Z. Yang and S. Pan, Nat. Photonics, 2023, 17, 694–701 CrossRef.
- Z. Gong, X. Wang, W. Pi, N. Yao, Z. Fang, H. Bao and Q. Wu, Mater. Today Phys., 2024, 43, 101399 Search PubMed.
- Y.-G. Chen, X. Hu, Y. Guo, S. Zhao, B. Zhang, X. Zhang and X.-M. Zhang, Chem. Mater., 2024, 36, 4775–4781 CrossRef.
- X. Fan, Crit. Rev. Environ. Sci. Technol., 2022, 52, 2227–2269 CrossRef.
- G. Shi, Y. Wang, F. Zhang, B. Zhang, Z. Yang, X. Hou, S. Pan and K. R. Poeppelmeier, J. Am. Chem. Soc., 2017, 139, 10645–10648 CrossRef.
- G. Deokar, N. S. Rajput, J. Li, F. L. Deepak, W. Ou-Yang, N. Reckinger, C. Bittencourt, J.-F. Colomer and M. Jouiad, Beilstein J. Nanotechnol., 2018, 9, 1686–1694 CrossRef PubMed.
- Z.-X. Zheng, Z.-X. Qiu, C.-H. Xie, Y.-P. Zhang, X.-M. Jiang, B.-W. Liu and G.-C. Guo, Sci. China Mater., 2023, 66, 2795–2802 CrossRef.
- L. Wang, Q. Sun and J. Li, Chin. J. Struct., 2023, 42, 100013 CrossRef CAS.
- T. K. Bera, J.-H. Song, A. J. Freeman, J. I. Jang, J. B. Ketterson and M. G. Kanatzidis, Angew. Chem., Int. Ed., 2008, 47, 7828–7832 CrossRef CAS.
- J. Wang, B. Xiong, H. Wu, H. Yu, Z. Hu, J. Wang and Y. Wu, Inorg. Chem. Front., 2021, 8, 344–351 RSC.
- M. Yan, H.-G. Xue and S.-P. Guo, Cryst. Growth Des., 2021, 21, 698–720 CrossRef CAS.
- Z.-Z. Luo, C.-S. Lin, H.-H. Cui, W.-L. Zhang, H. Zhang, H. Chen, Z.-Z. He and W.-D. Cheng, Chem. Mater., 2015, 27, 914–922 CrossRef CAS.
- M. Zhou, X. Jiang, Y. Guo, Z. Lin, J. Yao and Y. Wu, Inorg. Chem., 2017, 56, 8454–8461 CrossRef CAS.
- W. Yin, A. K. Iyer, C. Li, X. Lin, J. Yao and A. Mar, J. Solid State Chem., 2016, 241, 131–136 CrossRef CAS.
- X. Chen, H. Jo and K. M. Ok, Angew. Chem., Int. Ed., 2020, 59, 7514–7520 CrossRef CAS.
- A.-Y. Wang, S.-H. Zhou, M.-Y. Ran, B. Li, X.-T. Wu, H. Lin and Q.-L. Zhu, Inorg. Chem. Front., 2024, 11, 3744–3754 RSC.
- J.-H. Zhang, S. S. Stoyko, A. J. Craig, P. Grima, J. W. Kotchey, J. I. Jang and J. A. Aitken, Chem. Mater., 2020, 32, 10045–10054 CrossRef CAS.
- G. Peng, C. Lin, H. Fan, K. Chen, B. Li, G. Zhang and N. Ye, Angew. Chem., 2021, 133, 17555–17558 CrossRef.
- L. Wang, D. Chu, Z. Yang, J. Li and S. Pan, Chem. Sci., 2024, 15, 6577–6582 RSC.
- H. Qiu, F. Li, C. Jin, Z. Yang, J. Li, S. Pan and M. Mutailipu, Angew. Chem., Int. Ed., 2024, 63, e202316194 CrossRef CAS PubMed.
- H. Wang, X. Pan, W. Zhao, Y. Chu and J. Li, Inorg. Chem. Front., 2023, 10, 6253–6261 RSC.
- H. Wang, Y. Chu, X. Pan, Z. Yang, S. Pan and J. Li, Mater. Today Phys., 2023, 38, 101243 CrossRef CAS.
- P. Wang, Y. Chu, A. Tudi, C. Xie, Z. Yang, S. Pan and J. Li, Adv. Sci., 2022, 9, 2106120 CrossRef CAS.
- X. Zhang, H. Wu, Z. Hu, J. Wang, Y. Wu and H. Yu, Adv. Opt. Mater., 2024, 12, 2301735 CrossRef CAS.
- J. Zhou, K. Hou, Y. Chu, Z. Yang, J. Li and S. Pan, Small, 2024, 20, 2308806 CrossRef CAS.
- B.-W. Liu, X.-M. Jiang, H.-Y. Zeng and G.-C. Guo, J. Am. Chem. Soc., 2020, 142, 10641–10645 CrossRef CAS PubMed.
- B.-W. Liu, X.-M. Jiang, B.-X. Li, H.-Y. Zeng and G.-C. Guo, Angew. Chem., Int. Ed., 2020, 59, 4856–4859 CrossRef CAS.
- L. Wang, C. Tu, J. Zhou, Y. Chu, Z. Yang, S. Pan and J. Li, Adv. Opt. Mater., 2023, 12, 2301634 CrossRef.
- J. Zhou, L. Wang, Y. Chu, H. Wang, S. Pan and J. Li, Adv. Opt. Mater., 2023, 11, 2300736 CrossRef.
- B.-W. Liu, H.-Y. Zeng, X.-M. Jiang, G.-E. Wang, S.-F. Li, L. Xu and G.-C. Guo, Chem. Sci., 2016, 7, 6273–6277 RSC.
- H. Chen, Y.-Y. Li, B. Li, P.-F. Liu, H. Lin, Q.-L. Zhu and X.-T. Wu, Chem. Mater., 2020, 32, 8012–8019 CrossRef.
- B.-W. Liu, S.-M. Pei, X.-M. Jiang and G.-C. Guo, Mater. Horiz., 2022, 9, 1513–1517 RSC.
- M. Yan, C.-L. Hu, R.-L. Tang, W.-D. Yao, W. Liu and S.-P. Guo, Chem. Sci., 2024, 15, 8500–8505 RSC.
- J. Xu and K. Wu, Coord. Chem. Rev., 2023, 486, 215139 CrossRef.
- X. Liu, Y.-C. Yang, M.-Y. Li, L. Chen and L.-M. Wu, Chem. Soc. Rev., 2023, 52, 8699–8720 RSC.
- Z.-X. Wu, W.-F. Chen, X.-M. Jiang, B.-W. Liu and G.-C. Guo, Chem. Mater., 2024, 36, 3444–3451 CrossRef CAS.
- W.-F. Chen, B.-W. Liu, S.-M. Pei, X.-M. Jiang and G.-C. Guo, Adv. Sci., 2023, 10, 2207630 CrossRef CAS.
- J. Zhou, H. Wang, J. Liu, X. Su, Y. Chu, J. Qu and X. Jiang, Inorg. Chem. Front., 2024, 11, 2681–2689 RSC.
- S. Toso, Q. A. Akkerman, B. Martín-García, M. Prato, J. Zito, I. Infante, Z. Dang, A. Moliterni, C. Giannini, E. Bladt, I. Lobato, J. Ramade, S. Bals, J. Buha, D. Spirito, E. Mugnaioli, M. Gemmi and L. Manna, J. Am. Chem. Soc., 2020, 142, 10198–10211 CrossRef CAS.
- J. Wang, H. Wu, H. Yu, Z. Hu, J. Wang and Y. Wu, Adv. Opt. Mater., 2022, 10, 2102673 CrossRef CAS.
- D. Ni, S. Guo, K. M. Powderly, R. Zhong and R. J. Cava, J. Solid State Chem., 2019, 280, 120982 CrossRef CAS.
- L. Bindi, A. Garavelli, D. Pinto, G. Pratesi and F. Vurro, J. Solid State Chem., 2008, 181, 306–312 CrossRef CAS.
- A. N. Roth, Y. Chen, A. Santhiran, J. Opare-Addo, E. Gi, E. A. Smith, A. J. Rossini and J. Vela, Chem. Sci., 2023, 14, 12331–12338 RSC.
- M. Yan, R.-L. Tang, W. Zhou, W. Liu and S.-P. Guo, Dalton Trans., 2022, 51, 12921–12927 RSC.
- L.-T. Jiang, M.-Z. Li, X.-M. Jiang, B.-W. Liu and G.-C. Guo, Dalton Trans., 2022, 51, 6638–6645 RSC.
- R. Nie, B. Kim, S.-T. Hong and S. I. Seok, ACS Energy Lett., 2018, 3, 2376–2382 CrossRef CAS.
- H. Wang, G. Chen, J. Xu, Y. Xu and Q. Yang, Cryst. Growth Des., 2018, 18, 1987–1994 CrossRef CAS.
- Z. Lin, K. Feng, H. Tu, L. Kang, Z. Lin, J. Yao and Y. Wu, J. Alloys Compd., 2014, 611, 422–426 CrossRef CAS.
- J. Androulakis, S. C. Peter, H. Li, C. D. Malliakas, J. A. Peters, Z. Liu, B. W. Wessels, J.-H. Song, H. Jin, A. J. Freeman and M. G. Kanatzidis, Adv. Mater., 2011, 23, 4163–4167 CrossRef PubMed.
- J.-X. Zhang, P. Feng, M.-Y. Ran, X.-T. Wu, H. Lin and Q.-L. Zhu, Coord. Chem. Rev., 2024, 502, 215617 CrossRef.
- J. Chen, C. Lin, X. Jiang, G. Yang, M. Luo, X. Zhao, B. Li, G. Peng, N. Ye, Z. Hu, J. Wang and Y. Wu, Mater. Horiz., 2023, 10, 2876–2882 RSC.
- Y. Guo, Y. Deng, T. Zheng, L. Huang, D. Gao, J. Bi and G. Zou, Inorg. Chem. Front., 2022, 9, 440–447 RSC.
- J. Li, J. Shen, Z. Li, X. Li, Z. Sun, Z. Hu and S. Huang, Mater. Lett., 2013, 92, 330–333 CrossRef.
- X. Wang, J. Li, Z. Zhao, S. Huang and W. Xie, J. Appl. Phys., 2012, 112, 023701 CrossRef.
- F. van den Akker and W. G. J. Hol, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 206–218 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- S. K. Kurtz and T. T. Perry, J. Appl. Phys., 1968, 39, 3798–3813 CrossRef CAS.
- H. Qiu, F. Li, Z. Li, Z. Yang, S. Pan and M. Mutailipu, J. Am. Chem. Soc., 2023, 145, 24401–24407 CrossRef CAS PubMed.
- Y. Chu, H. Wang, Q. Chen, X. Su, Z. Chen, Z. Yang, J. Li and S. Pan, Adv. Funct. Mater., 2024, 34, 2314933 CrossRef CAS.
- Y. Chu, H. Wang, T. Abutukadi, Z. Li, M. Mutailipu, X. Su, Z. Yang, J. Li and S. Pan, Small, 2023, 19, 2305074 CrossRef CAS.
- J. Zhou, Z. Fan, K. Zhang, Z. Yang, S. Pan and J. Li, Mater. Horiz., 2023, 10, 619–624 RSC.
- L. Wang, D. Chu, D. Yin, C. Xie, Z. Yang, J. Li and S. Pan, Mater. Today Phys., 2023, 38, 101245 CrossRef CAS.
- C. Shen, F. Zhang, T. Sasaki, C. Eerdun, M. Hayashi, H.-W. Wang, K. Tominaga, M. Mutailipu and S. Pan, Angew. Chem., Int. Ed., 2024, 63, e202319121 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- M. Gerwig, M. Schwarz, E. Brendler and E. Kroke, Eur. J. Inorg. Chem., 2016, 2016, 4557–4560 CrossRef.
- A. B. Delpeuch, F. Maillard, M. Chatenet, P. Soudant and C. Cremers, Appl. Catal., B, 2016, 181, 672–680 CrossRef.
- L. Luo, L. Wang, J. Chen, J. Zhou, Z. Yang, S. Pan and J. Li, J. Am. Chem. Soc., 2022, 144, 21916–21925 CrossRef CAS.
- X. Zhang, H. Wu, Z. Hu, J. Wang, Y. Wu and H. Yu, Adv. Opt. Mater., 2024, 12, 2301735 CrossRef CAS.
- Y. Xu, C. Lin, D. Zhao, B. Li, L. Cao, N. Ye and M. Luo, Scr. Mater., 2022, 208, 114347 CrossRef CAS.
- M.-Y. Ran, S.-H. Zhou, W.-B. Wei, A.-Y. Wang, X.-T. Wu, H. Lin and Q.-L. Zhu, Inorg. Chem. Front., 2024, 11, 1890–1898 RSC.
- A. Abudurusuli, J. Li and S. Pan, Dalton Trans., 2021, 50, 3155–3160 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Related figures and tables. CCDC 2369466. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02315c |
| This journal is © The Royal Society of Chemistry 2024 |