
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyrene™ as a green alternative to N,N′-dimethylformamide (DMF) in the synthesis of MLCT-emissive ruthenium(II) polypyridyl complexes for biological applications†
Steffan D.
James‡
a,
Christopher E.
Elgar‡
 a,
Dandan
Chen
b,
Matthew I.
Lewis
a,
Elias T. L.
Ash
a,
Dominic S.
Conway
a,
Benjamin J.
Tuckley
a,
Leigh E.
Phillips
a,
Natália
Kolozsvári
a,
Xiaohe
Tian
*b and
Martin R.
Gill
*a
a,
Dandan
Chen
b,
Matthew I.
Lewis
a,
Elias T. L.
Ash
a,
Dominic S.
Conway
a,
Benjamin J.
Tuckley
a,
Leigh E.
Phillips
a,
Natália
Kolozsvári
a,
Xiaohe
Tian
*b and
Martin R.
Gill
*a
aDepartment of Chemistry, Faculty of Science and Engineering, Swansea University, Swansea, UK. E-mail: m.r.gill@swansea.ac.uk
bState Key Laboratory of Biotherapy, Department of Radiology and National Clinical Research Center for Geriatrics, Huaxi MR Research Center (HMRRC), Frontiers Science Center for Disease-Related Molecular Network, West China Hospital of Sichuan University, Sichuan University, Chengdu 610000, Sichuan Province, China. E-mail: xiaohe.t@wchscu.cn
First published on 31st October 2024
Abstract
Ruthenium(II) polypyridyl complexes (RPCs) that emit from triplet metal-to-ligand charge transfer (MLCT) states find a wide variety of uses ranging from luminophores to potential anti-cancer or anti-bacterial therapeutics. Herein we describe a greener, microwave-assisted synthetic pathway for the preparation of homoleptic [Ru(N^N)3]2+ and bis-heteroleptic [Ru(N^N)2(N'^N')]2+ type complexes. This employs the bio-renewable solvent Cyrene™, dihydrolevoglucosenone, as a green alternative to N,N′-dimethylformamide (DMF) in the synthesis of Ru(N^N)2Cl2 intermediate complexes, obtaining comparable yields for N^N = 2,2′-bipyridine, 1,10-phenanthroline and methylated derivatives. Employing these intermediates, a range of RPCs were prepared and we verify that the ubiquitous luminophore [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) can be prepared by this two-step green pathway where it is virtually indistinguishable from a commercial reference. Furthermore, the novel complexes [Ru(bpy)2(10,11-dmdppz)]2+ (10,11-dmdppz = 10,11-dimethyl-dipyridophenazine) and [Ru(5,5′-dmbpy)2(10,11-dmdppz)]2+ (5,5′-dmbpy = 5,5′-dimethyl-bpy) intercalate duplex DNA with high affinity (DNA binding constants, Kb = 5.7 × 107 and 1.0 × 107 M−1, respectively) and function as plasma membrane and nuclear DNA dyes for confocal and STED microscopies courtesy of their long-lived MLCT luminescence.
Introduction
Due to their attractive photophysical, electrochemical and redox properties, combined with relative ease of synthesis, octahedral ruthenium(II) polypyridyl complexes (RPCs) find a wide variety of uses in modern inorganic chemistry.1–3 As a result, RPCs have been utilised in a plethora of applications, ranging from sensitizers for dye-sensitized solar cells,4 water oxidation catalysts,5 biomolecular probes6 and cellular imaging agents7 to chemotherapeutics,8 anti-microbials,9 and even radiopharmaceuticals.10 Common classes of RPCs encountered are homoleptic [Ru(N^N)3]2+ type complexes and bis-heteroleptic [Ru(N^N)2(N′^N′)]2+ type complexes, where N^N and N′^N′ are bidentate chelating ligands. Perhaps the most famous example is [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine), a common luminophore that emits from a long-lived metal-to-ligand charge transfer (MLCT) state.11 Based on [Ru(bpy)3]2+, researchers have developed derivatives for specific applications. Noteworthy examples include [Ru(4-carboxy-4′-carboxylate-2,2′-bipyridine)2(NCS)2] (N719), a benchmark sensitizer for dye-sensitized solar cells,12 the luminescent DNA probe [Ru(bpy)2(dppz)]2+ (dppz = dipyridophenazine),13 the photodynamic therapy photosensitizer [Ru(4,4′-dmbpy)2(2-(2′,2′′:5′′,2′′′-terthiophene)-imidazo[4,5-f][1,10]phenanthroline)]2+ (4,4′-dmbpy = 4,4′-dimethyl-bpy), TLD1433,14 and the antibiotic KLS-219, [Ru(tmpphen)2(tpphz)]2+ (tmphen = 3,4,7,8-tetramethyl-1,10-phenanthroline, tpphz = tetrapyrido[3,2-a:2′,3′-c:3′′,2′′-h:2′′′,3′′′-j]phenazine)15 (Fig. S1 in the ESI†).The starting point for RPC synthesis is most commonly the preparation of the cis-Ru(N^N)2Cl2 (hereafter described as Ru(N^N)2Cl2) intermediate complex from RuCl3·3H2O (Scheme 1). The synthesis of Ru(N^N)2Cl2 complexes was developed by the groups of Dwyer and Meyer in the 60s and 70s:16–19 efforts that culminated in the seminal paper by Sullivan et al. describing the use of the polar aprotic solvent N,N′-dimethylformamide (DMF) in this reaction.20 The precursor Ru(N^N)2Cl2 complex can then be reacted with the final bidentate ligand (or 2 equivalents of a monodentate ligand) to generate the required homo- or heteroleptic complex of interest. This offers great opportunity for functionalisation, where the second N′^N′ may be a functional ligand, e.g. designed for DNA or other biomolecule binding. In these cases, the N^N ligands are often viewed as ancillary and employed to fine-tune molecular properties such as emission intensity, emission maxima, cellular uptake, intracellular localisation etc.21
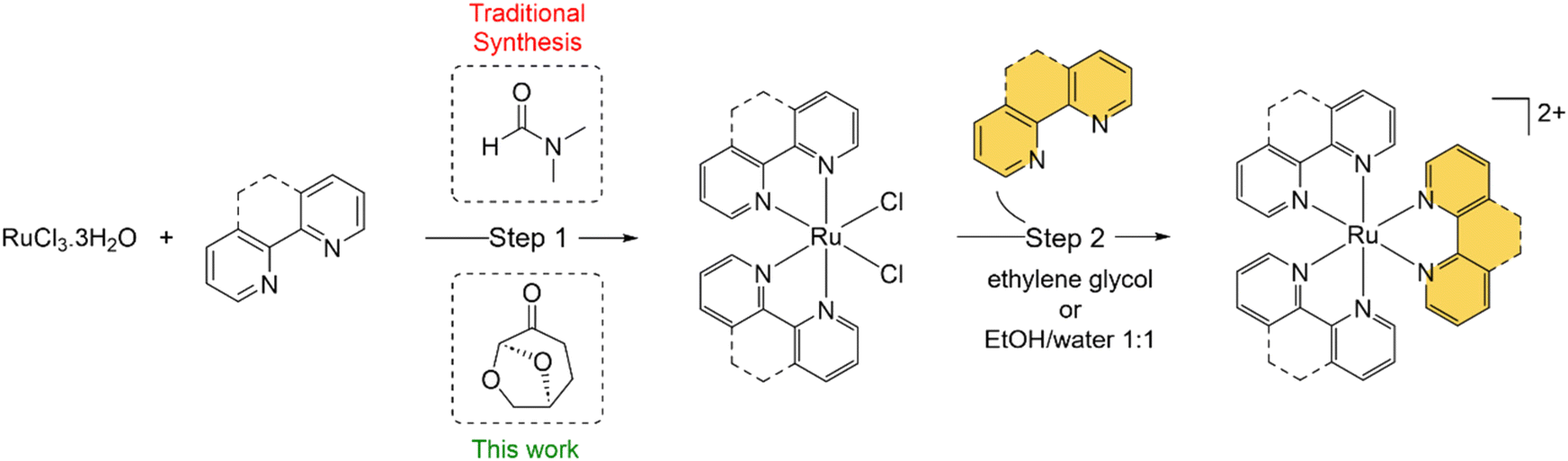 | ||
| Scheme 1 General synthesis of homoleptic [Ru(N^N)3]2+ or heteroleptic [Ru(N^N)2(N^N)]2+ RPCs. | ||
This reaction has proven suitable for a wide range of N^N groups of varying hydrophobicity, size and reactivity and the precursors generated are used for the synthesis of a wide range of mono-, di- and poly-nuclear RPCs.22,23 As such, RPC chemistry still relies heavily on this reaction and, as a result, DMF. However, DMF is highly toxic solvent, classified as a substance of very high concern (SVHC) by European Union Registration, Evaluation, Authorization, and Restriction of Chemicals (REACH) legislation,24 and has significant thermal decomposition risks.25 As of December 2023, the EU implemented stricter regulations on the purchasing of DMF,24 resulting in greater levels of safety considerations to justify its selection within reaction schemes. Thus, replacing DMF with a safer, biocompatible solvent would improve the green chemistry credentials of this reaction to align it with the “12 principles of green chemistry”.26
It is now accepted need that pursuing greener or more sustainable solvents is an essential requirement within modern synthetic chemistry.27 Cyrene™, dihydrolevoglucosenone, is a bio-available and biodegradable compound derived from waste cellulose and has been proposed as a green alternative for dipolar aprotic solvents.28 Cyrene™ may be used in place of DMF for numerous organic reactions (reviewed in ref. 29 and 30), including the synthesis of ureas31 or amides,32,33 and palladium-catalysed cross coupling reactions.34 Other work has explored Cyrene™ within lignin fractionation35 and MOF preparation.36 However, we are not aware that Cyrene™ has been tested within small molecule inorganic synthesis to date. There is also a general paucity of work that has examined greener pathways to prepare RPCs, although Vierucci et al. reported a green synthesis of N719.37 Herein, we report the substitution of DMF for Cyrene™ in the synthesis of Ru(N^N)2Cl2 precursor complexes and utilised these intermediates to prepare MLCT-emissive [Ru(N^N)3]2+ and [Ru(N^N)2(N′^N′)]2+ final complexes (Fig. 1), showing two of these to function as novel DNA dyes for light microscopy and a third to exhibit potent cytotoxicity towards human cancer cells.
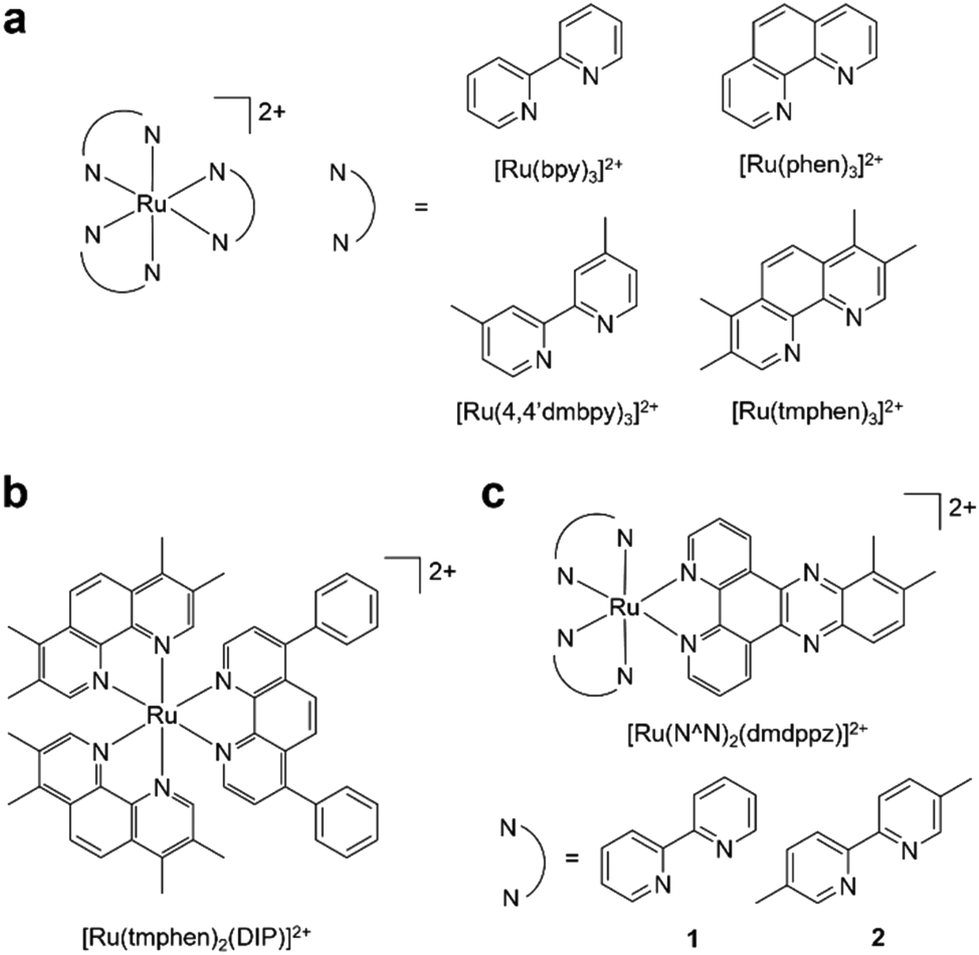 | ||
| Fig. 1 (a) Homoleptic and (b and c) heteroleptic RPCs synthesised in this study. Complexes were synthesised as a mixture of enantiomers. | ||
Results and discussion
Optimisation of the reaction conditions
The initial investigation focused on the reaction to synthesise the archetypal compound, cis-bis(2,2′-bipyridine)bischlororuthenium(II) employing Cyrene™ as the solvent in place of DMF (Table 1). This reaction involves the addition of RuCl3·3H2O to 2,2′-bipyridine (2 molar equivalence) in the presence of excess chloride anions. In addition to being the solvent, DMF also acts as a mild reducing agent to facilitate the reduction of Ru(III) to Ru(II). DMF conditions were employed to serve as a direct comparison, where a yield of 68% Ru(bpy)2Cl2 was obtained after the standard reaction time of 8 h, consistent with literature data (Table 1).20 Initial reactions utilising Cyrene™ first trialled reaction conditions similar to the DMF synthesis, with temperatures tested from 140 °C to reflux. However, this resulted in solvent degradation and the formation of a black tar-like substance. This was a surprising outcome, although a recent technical support comment states Cyrene™ to have a safe operating temperature of below 140 °C,38 and the instability of Cyrene™ in basic conditions39 and in 2 M HCl40 has been noted.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temperature (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: RuCl3·3H2O (0.38 mmol), 2,2-bipyridine (0.76 mmol), LiCl (2.67 mmol) and solvent (1 mL). b Isolated yield. c Solvent degradation was observed in these conditions. MW = 150 W microwave irradiation. | ||||
| 1 | DMF | 150/Reflux | 8 | 68 |
| 2 | DMF | 150/Reflux | 5 | 31 |
| 3 | DMF | 150/Reflux | 3 | 39 |
| 4 | Cyrene | 60 | 8 | 33 |
| 5 | Cyrene | 60 | 5 | 38 |
| 6 | Cyrene | 60 | 3 | 18 |
| 7 | Cyrene | 80 | 8 | 45 |
| 8 | Cyrene | 80 | 5 | 33 |
| 9c | Cyrene | 110 | 8 | 89 |
| 10 | Cyrene | 110 | 5 | 79 |
| 11 | Cyrene | 110 | 3 | 68 |
| 12 | Cyrene | 110 | 1.5 | 61 |
| 13 | Cyrene | 100/MW | 0.08 | 61 |
Employing reduced temperatures to avoid solvent degradation, performing the reaction at 110 °C yielded the desired black/dark purple solid in good yields with a reaction time of 8 hours achieving an 89% yield (Table 1). However, one drawback was at this length of reaction some solvent degradation during repeats was again seen. Reduced reaction times of 3 or 5 h generated yields of 68 and 79% for 3 and 5 h in 110 °C Cyrene™ respectively and, importantly, no solvent degradation was observed in this timeframe. In addition to conventional heating, this reaction was also optimised for a microwave reactor, where 100 °C, 5 min under 150 W temperature-controlled microwave irradiation achieved comparable yields to conventional heating. This is one of the few examples employing Cyrene™ in microwave chemistry41 and demonstrates the compatibility of the reaction with this technique.
The product generated in Cyrene™ was characterised by elemental analysis, where values within ±0.4% of the calculated values indicated a purity approaching 95%. High-resolution mass spectrometry (HRMS) indicated a major species at 449 m/z, corresponding to [M − Cl]+ with minor peaks at 477 [M − Cl + Na]+ and 507 [M + Na]+ also observed (Fig. S2†). Both elemental analysis and HRMS results were directly comparable to results achieved employing DMF. No evidence of Cyrene™ coordination to the Ru centre or other side-products were detected and FT-IR analysis showed successful removal of Cyrene™ from the products in the wash steps (Fig. S3†). The successful synthesis of Ru(bpy)2Cl2 from RuCl3·3H2O also suggests that Cyrene™ acts as a reducing agent in a similar manner to DMF; a finding supported by recent work describing Cyrene™ in the synthesis of metal nanoparticles.42
Scope of reaction
We next assessed whether the reaction was compatible with other bidentate polypyridyl ligands with increasing hydrophobicity. For this purpose, we selected phen (1,10-phenanthroline), 4,4′-dmbpy, 5,5′-dmbpy (5,5′-dimethyl-2,2′-bipyridine) and tmphen, thereby generating a polypyridyl ligand series with calculated octanol/water partition coefficients, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, ranging from 1.44 (bpy) to 4.12 (tmphen). Employing optimised conditions that included the green reducing agent ascorbic acid, comparable yields in Cyrene™ to preparation via the DMF pathway were achieved for all ligands tested (Table 2). Elemental analysis indicated all complexes were of good purity, with values approaching 0.4% of the predicted content, and HRMS confirmed the formation of each Ru(N^N)2Cl2 species as the major peak (Table 2, Fig. S4–S7†). All results were comparable to complexes prepared via the DMF pathway. The poor solubility of the complexes was a barrier to 1H and 13C NMR analysis, however, HRMS and elemental analysis characterisation of these intermediate complexes is standard within RPC synthetic workflows and they are used in subsequent reactions without further purification.20,22,43,44
P, ranging from 1.44 (bpy) to 4.12 (tmphen). Employing optimised conditions that included the green reducing agent ascorbic acid, comparable yields in Cyrene™ to preparation via the DMF pathway were achieved for all ligands tested (Table 2). Elemental analysis indicated all complexes were of good purity, with values approaching 0.4% of the predicted content, and HRMS confirmed the formation of each Ru(N^N)2Cl2 species as the major peak (Table 2, Fig. S4–S7†). All results were comparable to complexes prepared via the DMF pathway. The poor solubility of the complexes was a barrier to 1H and 13C NMR analysis, however, HRMS and elemental analysis characterisation of these intermediate complexes is standard within RPC synthetic workflows and they are used in subsequent reactions without further purification.20,22,43,44
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Ligand | logPb |
Conditions | Yieldc (%) | HRMS major peak | Assignment | AEc (%) | AEf (%) |
|
a Reaction conditions: RuCl3·3H2O (0.38 mmol), N^N (2 eq.), LiCl (2.67 mmol) and solvent (5 mL).
b LogP values calculated using Molinspiration Cheminformatics Software.
c Isolated yield.
d Bpy data duplicated from Table 1 for completeness and reaction was performed without ascorbic acid. MW = 150 W microwave irradiation. AEc = atom economy. AEf = atom efficiency.
|
|||||||
| bpy | 1.44 | Cyrened | 79 | 449 | [M − Cl]+ | 55.8 | 44.1 |
| DMFd | 68 | 449 | [M − Cl]+ | 55.8 | 38.0 | ||
| phen | 1.90 | Cyrene | 67 | 497 | [M − Cl]+ | 53.0 | 35.5 |
| DMF | 80 | 554 | [M + Na]+ | 58.1 | 46.5 | ||
| 4,4′-dmbpy | 2.24 | Cyrene | 49 | 505 | [M − Cl]+ | 53.4 | 26.2 |
| DMF | 54 | 505 | [M − Cl]+ | 58.5 | 31.6 | ||
| 5,5′-dmbpy | 2.35 | Cyrene | 67 | 505 | [M − Cl]+ | 53.4 | 35.8 |
| DMF | 40 | 505 | [M − Cl]+ | 58.5 | 23.4 | ||
| tmphen | 4.22 | Cyrene | 55 | 644 | [M]+ | 57.8 | 31.8 |
| DMF | 81 | 644 | [M]+ | 62.7 | 50.8 | ||
In terms of green chemistry, atom economies ranged from 53.0–57.8% with atom efficiencies 26.2–44.1% (Table 2). In two cases, N^N = bpy or 5,5′-dmbpy, the reaction in Cyrene™ out-performed the reaction in DMF. However, despite the general success of this reaction, a decrease in yield compared to DMF conditions was evident for the most hydrophobic ligand, tmphen. We can also report that attempts employing the hydrophobic ligand DIP (4,7-diphenyl-1,10-phenanthroline, logP = 5.95) generated low yields (<20%) of product with unreacted RuCl3·3H2O as an impurity. To combat the decreased reactivity of larger, more hydrophobic polypyridyl ligands such as DIP, extended reaction times in DMF are typically employed; for example, 24 h reflux in DMF is standard in the synthesis of Ru(DIP)2Cl2.45 Unfortunately, the degradation of Cyrene™ at extended reaction times prevented us similarly testing whether an increased reaction time would improve reaction yields.
Preparation of homo- and heteroleptic RPCs
To demonstrate the functionality of the Ru(N^N)2Cl2 intermediate complexes synthesised, four commercially available homoleptic [Ru(N^N)3]2+ (N^N = bpy, phen, 4,4′-dmbpy or tmphen) complexes were prepared (Fig. 1a). To show that these conditions are compatible with the preparation of bis-heteroleptic [Ru(N^N)2(N′^N′)]2+ type complexes, we also synthesised the novel complex [Ru(tmphen)2(DIP)]2+ (Fig. 1b). To maintain a green credential, ethylene glycol, which can be produced from biomass,46 was used as the solvent for the addition of the final bipyridine ligand. MW conditions were adapted from Luis et al.47 All complexes were characterised by 1H, 13C NMR and HRMS and purity determined by HPLC (Experimental section and Fig. S8–S24 in the ESI†). Of particular note is [Ru(bpy)3](PF6)2 prepared by this two-step green pathway achieved >99% purity and near-identical HPLC and 1H peaks compared to a commercial reference (Fig. 2 and 3).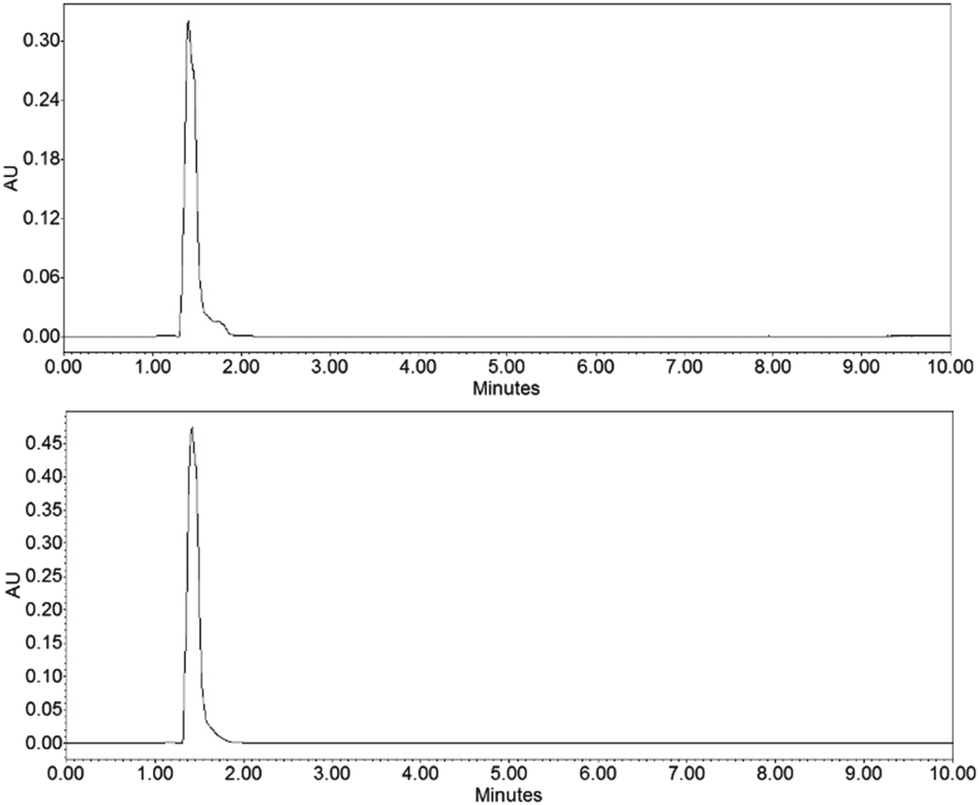 | ||
| Fig. 2 HPLC chromatograms of [Ru(bpy)3]2+ (top) compared to a commercial reference (bottom). | ||
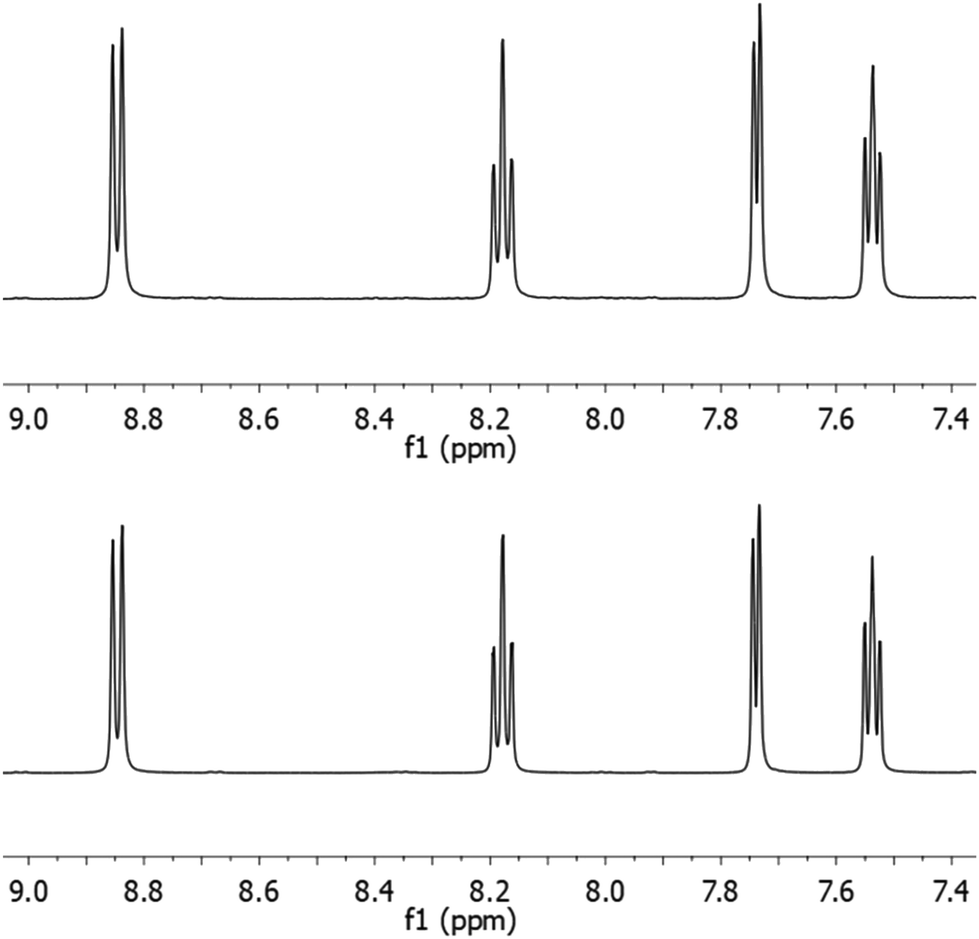 | ||
| Fig. 3 1H NMR spectra of [Ru(bpy)3]2+ (top) compared to a commercial reference (bottom). Aromatic region shown. | ||
All complexes were emissive in acetonitrile from MLCT states with 450 nm excitation resulting in characteristic orange/red emission with ns lifetimes (Table 3, and Fig. S25†). The excitation, absorption and emission spectra of [Ru(bpy)3]2+ displayed the same peaks as a commercial reference and molar extinction coefficients and relative quantum yield (Φrel) were within 5% of published values (Table 3, ref. 48 and 49). Inspection of the results show that the relative quantum yield for [Ru(phen)3]2+ was 59% of [Ru(bpy)3]2+, in agreement with published data,50 and the reduced quantum yields of [Ru(4,4′-dmbpy)3]2+ and [Ru(tmphen)3]2+ show that methylation of the bpy or phen ligand substantially decrease emission intensity. Compared to [Ru(tmphen)3]2+, [Ru(tmphen)2(DIP)]2+ demonstrates a slight red-shift in emission along with increased lifetimes due to the extended aromatic system of DIP (Table 3, and Fig. S25†).
| λ abs (nm) ( [M−1 cm−1]) | λ em (max)a (nm) |
Φ
relb |
τ 1 (ns) (%) | τ 2 (ns) (%) | |
|---|---|---|---|---|---|
| a λ ex = 450 nm. b Relative quantum yield in CH3CN compared to commercial [Ru(bpy)3]2+ (Φ = 0.018, ref. 49). | |||||
| [Ru(bpy)3]2+ | 268 (106000), 453 (15275) |
615 | 0.017 | 149.6 (90.9) | 757 (9.1) |
| [Ru(phen)3]2+ | 261 (96533), 447 (16101) |
600 | 0.010 | 96.5 (90.8) | 828.4 (9.2) |
| [Ru(4,4′-dmbpy)3]2+ | 287 (104413), 458 (16398) |
608 | 0.007 | 101 (89.5) | 816 (10.5) |
| [Ru(tmphen)3]2+ | 268 (142133), 437 (21400) |
596 | 0.003 | 53 (86.8) | 606 (13.2) |
| [Ru(tmphen)2(DIP)]2+ | 268 (128200), 431 (23267) |
602 | 0.003 | 123 (88.1) | 751 (11.9) |
| [Ru(bpy)2(dppz)]2+ | 281 (99195), 360 (21045), 448 (16629) |
622 | 0.008 | 145 (89.8) | 715 (10.2) |
| 1 | 288 (130790), 368 (20480), 450 (21120) |
600 | 0.012 | 173 (85.6) | 524 (14.4) |
| 2 | 292 (136510), 369 (22230), 435 (19660) |
615 | 0.012 | 141 (88.5) | 634 (11.5) |
Application of RPCs: DNA binding and cellular imaging
[Ru(bpy)2(dppz)]2+ and related molecules have been the subject of much attention for their DNA binding properties, where such molecules often show quenched emission in aqueous media accompanied by a dramatic increase in MLCT emission when bound to DNA.13 The basis for this effect is that DNA binding (commonly via intercalation) shields the dppz ligand from water, preventing molecular quenching by water molecules and activating luminescence as a result.51 This has inspired the development of numerous [Ru(bpy)2(dppz)]2+ derivatives52 where, for example, methylation of the dppz ligand increases MLCT intensity and lifetimes.44,53–56 We therefore prepared [Ru(bpy)2(dppz)]2+ and two novel methylated derivatives, [Ru(bpy)2(10,11-dmdppz)]2+ (1) and [Ru(5,5′-dmbpy)2(10,11-dmdppz)]2+ (2), where the 10,11-dmdppz ligand is dppz dimethylated at the 10- and 11-positions (Fig. 1c). Unfortunately, employing ethylene glycol in Step 2 of Scheme 1 was unsuccessful for preparation of dppz-based complexes and so conventional reflux in EtOH:H2O was employed instead. As EtOH:H2O is also a green solvent system,57 this adjustment did not affect the green credentials of this step. Complexes were characterised by 1H, 13C NMR and HRMS and >95% purity was confirmed by HPLC (Fig. S26–42†).
1 and 2 are emissive in acetonitrile, where they display a blue-shifted emission compared to the parent complex [Ru(bpy)2(dppz)]2+ due to methylation of the dppz ligand (Table 3, and Fig. S43†). As for [Ru(bpy)2(dppz)]2+, both 1 and 2 are non-emissive in aqueous environments but MLCT emission is activated by the addition of calf-thymus DNA (Fig. S44†). Strikingly, 1 showed a much greater (>10 fold) maximum MLCT intensity than [Ru(bpy)2(dppz)]2+ (Fig. 4a, and Table 4). Examining the lifetimes of the emissions, 1 showed two very long-lived components of 2.6 and 6.9 μs, substantially greater than 110 ns and 598 ns observed for [Ru(bpy)2(dppz)]2+ (Fig. 4b, and Table 4). A similar enhancement in MCLT intensity and lifetimes was observed for 2, although at reduced intensity and lifetimes compared to 1 due to methylation of the bpy ligand (Fig. 4a, b, and Table 4). Quantifying binding affinity by emission titration and the McGhee von Hippel binding model,58 binding affinities of 5.67 × 107 M−1 and 1.04 × 107 M−1 were obtained for 1 and 2, respectively, a notable increase compared to the non-methylated dppz analogues (Kb = 2.5 × 106 M−1 for [Ru(bpy)2(dppz)]2+, Table 4, and Kb = 2.63 × 106 M−1 for [Ru(5,5′dmbpy)2(dppz)]2+, ref. 59). Viscosity experiments confirmed that each complex acts to increase the viscosity of DNA, thereby indicating that each molecule binds DNA by intercalation (Fig. S45†). As such, we conclude that dimethylation of dppz at the 10- and 11-positions has acted to substantially increase duplex DNA binding affinity. In addition to duplex DNA, we also examined the ability of 1 and 2 to interact with G-quadruplex DNA (G4) using a recently developed MLCT-Cy5.5 FRET binding assay.59 Based on MLCT and Cyanine 5.5 spectral overlap (Fig. 4c, and Table 4), the resultant FRET with a Cy5.5-labelled G4 structure (Fig. 4d) generated binding curves and dissociation constants, Kd, to show a binding order of [Ru(bpy)2(dppz)]2+ > 1 > 2 for G4 DNA (Fig. 4e).
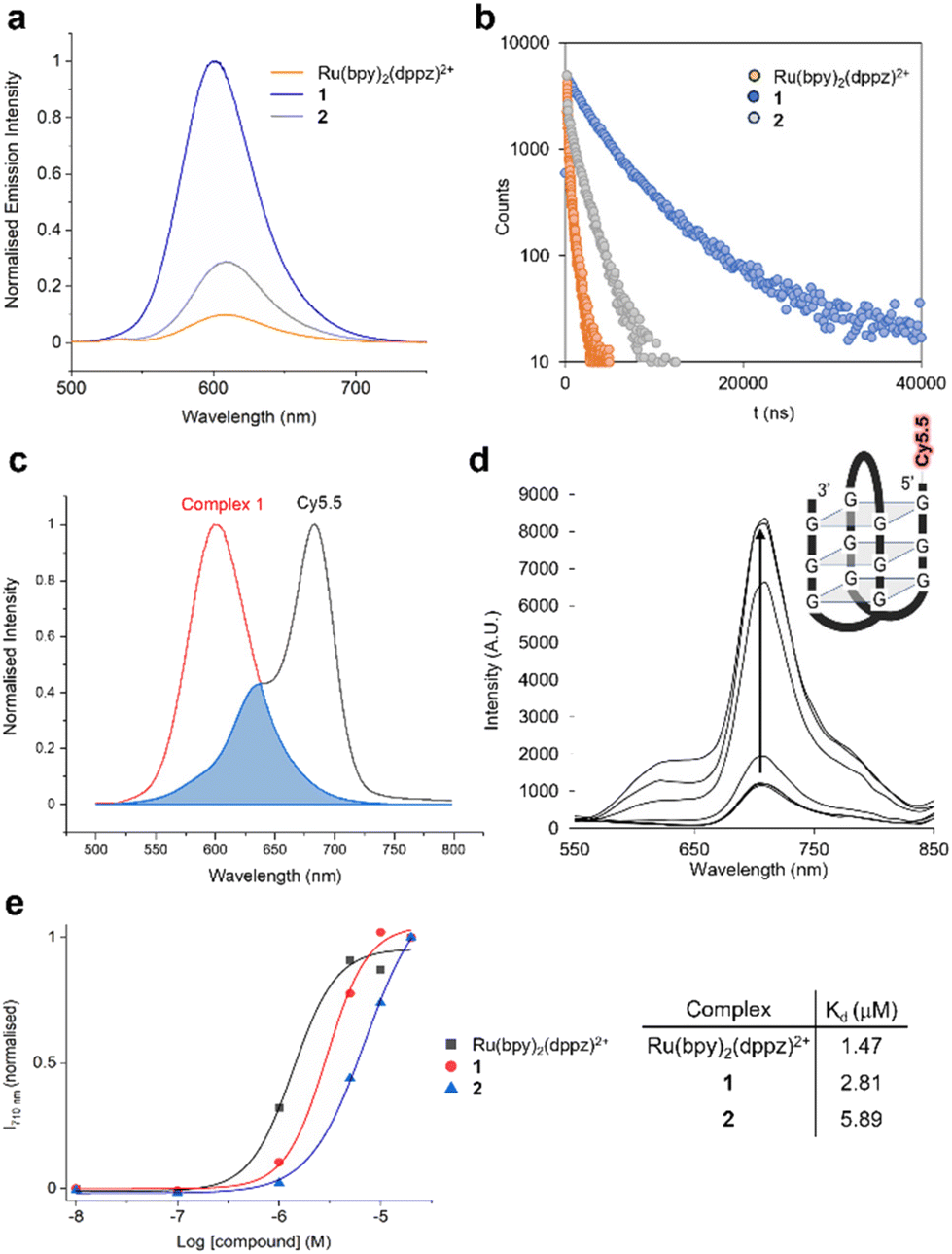 | ||
| Fig. 4 (a) Relative MLCT emission intensities of 1, 2 and [Ru(bpy)2(dppz)]2+ when bound to DNA (λex = 450 nm). Results normalised to 1. (b) Lifetime of DNA-bound MLCT emission of 1, 2 and [Ru(bpy)2(dppz)]2+. (c) Spectral overlap of MLCT emission of DNA-bound 1 and Cy5.5 absorption. (d) Emission spectra of Cy5.5-labelled G4-quadruplex DNA (1 μM, inset) with increasing concentrations of 1 (0.1–20 μM, λex = 450 nm). (e) FRET binding curves and Kd values derived from addition of 1, 2 or [Ru(bpy)2(dppz)]2+ (0.1–20 μM) to Cy5.5-labelled G4 DNA (1 μM). λex = 450 nm, λem = 710 nm. FRET intensity was background corrected and normalised. Duplex DNA buffer: 5 mM Tris, 200 mM NaCl, pH 7.5. G4 buffer: 100 mM KCl, 10 mM Tris·HCl, pH 7.4. | ||
| K b | n | λ em (max) | Φ DNA | τ 1 (ns) (%) | τ 2 (ns) (%) | J (λ) (M−1 cm−1 nm4) | R 0 (Å) | |
|---|---|---|---|---|---|---|---|---|
| K b = binding constant n = site size in base pairs, derived from luminescence titration data. J (λ) = spectral overlap integral, R0 = Förster radius as FRET pair with Cy5.5. Buffer: 5 mM Tris, 200 mM NaCl, pH 7.5.a Data from ref. 59. | ||||||||
| [Ru(bpy)2(dppz)]2+ | 2.5 × 106 | 1.26 | 620 | 0.008 | 110 (48.7) | 597.7 (51.3) | 1.50 ± 0.23 × 1016a | 38.9 ± 1.02a |
| 1 | 5.7 × 107 | 3.1 | 610 | 0.085 | 2600 (57.8) | 6900 (42.2) | 1.02 × 1016 | 54.07 |
| 2 | 1.0 × 107 | 2.07 | 620 | 0.033 | 607 (29.8) | 1762 (70.2) | 1.27 × 1016 | 47.85 |
Based on the bright MLCT emission of 1 and 2 when bound to DNA, their cellular uptake and localization properties were examined by light microscopy. Low cytotoxicity of 1 and 2 was observed towards a panel of human cancer cell lines (half inhibitory concentrations, IC50s, >100 μM, Fig. S46†). Examining live cells treated with 1 or 2 showed MLCT emission from the plasma membrane of cells, as indicated by co-localisation with the plasma membrane stain CellMask Deep Red (Fig. 5a). Clear evidence of nuclear staining was provided in fixed cells, where both 1 and 2 functioned as excellent dyes for cell nuclei compatible with both confocal microscopy and STED (stimulated emission depletion microscopy) (Fig. 5b). Co-staining with the DNA dye DAPI confirms nuclear DNA is being targeted (Fig. S47†). This behaviour contrasts to [Ru(tmphen)2(DIP)]2+, where instead cytosolic cellular distribution is observed (Fig. S48†). Thus, 1 and 2 join the emerging collection of RPCs suitable for super-resolution microscopy techniques,60,61 where their relatively high quantum yield when bound to DNA, large Stokes shifts, aqueous solubility and low cytotoxicity are advantageous properties as luminescent dyes. One obvious disadvantage of 1 and 2 is their poor uptake in live cells. Although at first glance this finding is similar to that reported for [Ru(bpy)2(dppz)]2+,62 a clear difference between the complexes is that 1 and 2 generate plasma membrane foci, indicating that each molecule targets membrane structures in addition to DNA. Whilst the precise nature of the foci visualised by 1 and 2 is unknown, it is likely that the addition of multiple methyl groups to the [Ru(bpy)2(dppz)]2+ scaffold has achieved this dual-localisation effect, an intriguing outcome considering that neither 1 nor 2 are strongly hydrophobic (calculated logP values of −1.7 and 0.12 for 1 and 2, respectively). This contrasts to other membrane-targeting RPCs, which typically employ highly hydrophobic or lipophilic ligands to achieve this outcome.63,64 It is also significant that [Ru(tmphen)2(DIP)]2+ demonstrates high cytotoxicity towards HeLa cells (IC50 = 8.4 μM, Fig. S48†). Numerous cytotoxic RPCs have been explored as anti-cancer agents8 and considering that the closely related molecule [Ru(bpy)2(DIP)]2+ shows promising activity towards pancreatic cancer cells,65 this shows that our synthetic pathway can prepare molecules of therapeutic interest in addition to cell imaging agents.
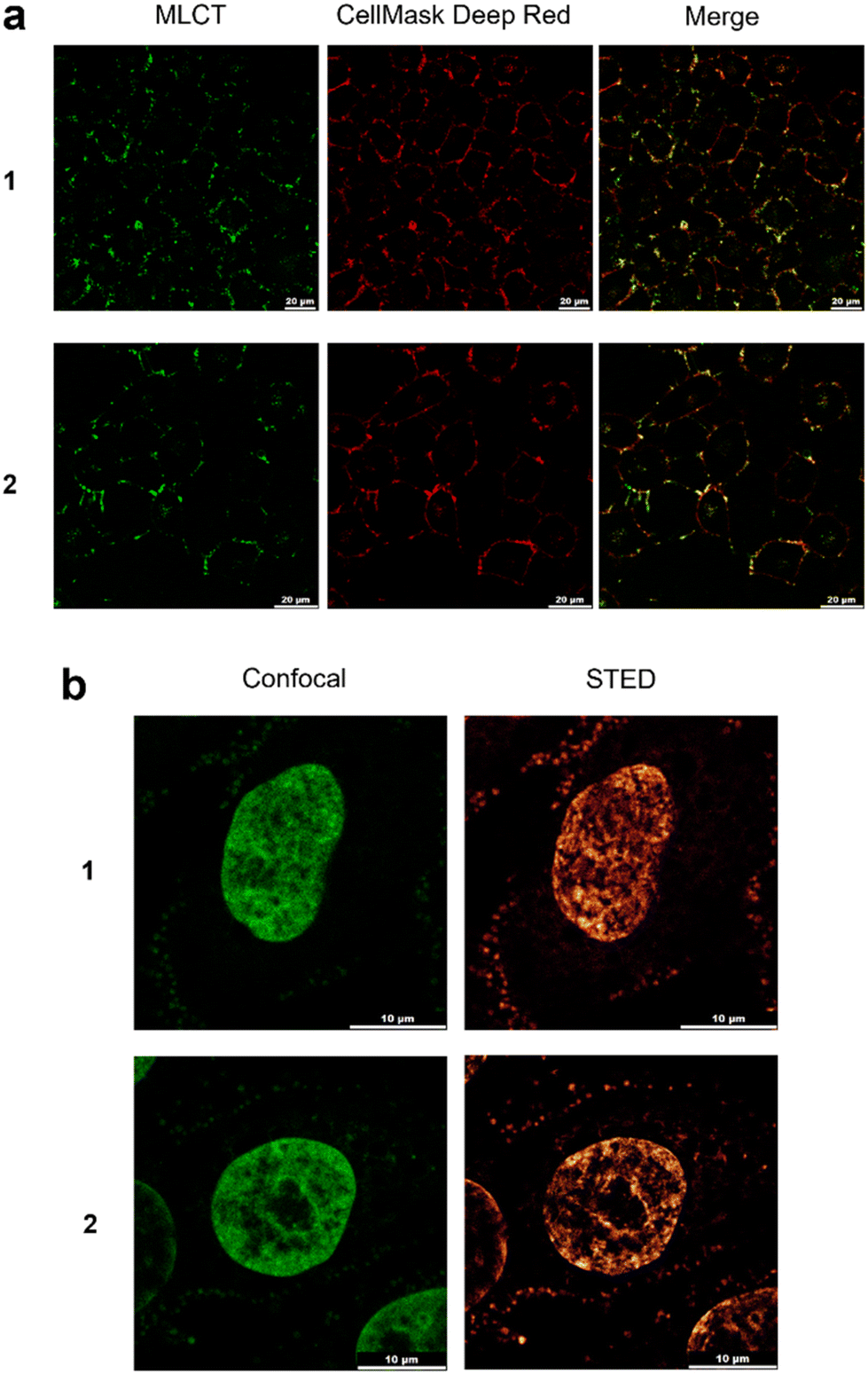 | ||
| Fig. 5 (a) Co-localisation of MLCT emission of HeLa cells treated with 1 or 2 (50 μM, 30 min) and the plasma membrane dye CellMask Deep Red. (b) Fixed Hep G2 cells stained with 1 or 2 (5 μM) and visualised by confocal (left) or STED (right) microscopy showing nuclear staining in addition to plasma membrane foci. | ||
In terms of the wider scope for the use of Cyrene™ in coordination and organometallic chemistry, other aprotic, polar solvents that it could be a candidate to replace include N-methyl-2-pyrrolidone, dioxane and tetrahydrofuran. This would therefore make Cyrene™ of interest for Pd coupled cross coupling reactions, as may be demonstrated by a recent study showing Cyrene™ to be a viable option for Heck-coupling reactions.66 However, the significant miscibility of Cyrene™ with water and that Cyrene™ cannot easily be distilled would largely exclude it from moisture- and air-sensitive reactions, which are particularly commonplace in coordination and organometallic chemistry. A final consideration is hydrophobic complexes with low aqueous solubility are often dissolved in DMSO before dilution in aqueous buffer or media for biological application. Thus, Cyrene™ may find an additional application in medicinal inorganic chemistry, where Cyrene™ would offer reduced cytotoxicity and environmental impact compared to DMSO.67 Moreover, as DMSO can act as a ligand for a variety of metal centres, and considering that we have not seen any evidence of Cyrene™ -Ru(II) coordination in our own work, the use of Cyrene™ in this capacity would also have the advantage of reducing the risk of solvent interference in bio-assays.
Conclusions
In summary, we have shown that Cyrene™, dihydrolevoglucosenone, may replace DMF in the synthesis of Ru(N^N)2Cl2 complexes from RuCl3·3H2O, improving the green credentials of this classic coordination chemistry reaction used heavily in the synthesis of ruthenium polypyridyl complexes. We successfully demonstrated that the precursor Ru(N^N)2Cl2 complexes can be used to prepare homoleptic and heteroleptic MLCT-emissive complexes, including two novel methylated [Ru(bpy)2(dppz)]2+ derivatives that are suitable for cellular imaging in confocal and STED microscopies. Whilst this work shows that replacing DMF with Cyrene™ is well-suited to N^N = bpy, phen and methylated derivatives, it may not be viable for hydrophobic polypyridyl ligands with extended aromatic systems. This, in part, is due to the poor compatibility of Cyrene with longer reaction times at elevated temperatures and constitutes its main disadvantage along with cost.Author contributions
MG: conceptualization, investigation, data curation, writing – review & editing, writing – original draft, supervision, funding acquisition. XT: supervision, funding acquisition. CE: methodology, investigation, data curation, writing – original draft. CE, SJ, DC, ML, EA, DS, BT, LP and NK: investigation, data curation.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Royal Society of Chemistry (RSC) Research Fund and Research Enablement grants (R20-8717 and E21-9540096197) and the National Natural Science Foundation of China (32171361). We thank the James Pantyfedwen Foundation for a scholarship to SJ.References
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef.
- J. G. Vos and J. M. Kelly, Dalton Trans., 2006, 4869–4883 RSC.
- J. Shum, P. K.-K. Leung and K. K.-W. Lo, Inorg. Chem., 2019, 58, 2231–2247 CrossRef PubMed.
- M. K. Nazeeruddin, C. Klein, P. Liska and M. Grätzel, Coord. Chem. Rev., 2005, 249, 1460–1467 CrossRef.
- L. Tong and R. P. Thummel, Chem. Sci., 2016, 7, 6591–6603 RSC.
- B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 4565–4579 RSC.
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662–667 CrossRef CAS PubMed.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- K. L. Smitten, H. M. Southam, J. B. de la Serna, M. R. Gill, P. J. Jarman, C. G. W. Smythe, R. K. Poole and J. A. Thomas, ACS Nano, 2019, 13, 5133–5146 CrossRef CAS PubMed.
- M. R. Gill, M. G. Walker, S. Able, O. Tietz, A. Lakshminarayanan, R. Anderson, R. Chalk, A. H. El-Sagheer, T. Brown, J. A. Thomas and K. A. Vallis, Chem. Sci., 2020, 11, 8936–8944 RSC.
- E. M. Kober and T. J. Meyer, Inorg. Chem., 1984, 23, 3877–3886 CrossRef CAS.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Graetzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960–4962 CrossRef CAS.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- K. L. Smitten, E. J. Thick, H. M. Southam, J. Bernardino de la Serna, S. J. Foster and J. A. Thomas, Chem. Sci., 2020, 11, 8828–8838 RSC.
- B. Bosnich and F. P. Dwyer, Aust. J. Chem., 1966, 19, 2229–2233 CrossRef CAS.
- F. P. Dwyer, H. A. Goodwin and E. C. Gyarfas, Aust. J. Chem., 1963, 16, 42–50 CrossRef CAS.
- T. J. Meyer and J. B. Godwin, Inorg. Chem., 1971, 10, 471–474 CrossRef CAS.
- G. M. Brown, T. R. Weaver, F. R. Keene, T. J. Meyer and W. R. Kenan, Inorg. Chem., 1976, 15, 190–196 CrossRef CAS.
- B. P. Sullivan, D. J. Salmon and T. J. Meyer, Inorg. Chem., 1978, 17, 3334–3341 CrossRef.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC.
- J. Bolger, A. Gourdon, E. Ishow and J.-P. Launay, Inorg. Chem., 1996, 35, 2937–2944 CrossRef.
- A. Börje, O. Köthe and A. Juris, J. Chem. Soc., Dalton Trans., 2002, 843–848 RSC.
- A. Jordan, C. G. J. Hall, L. R. Thorp and H. F. Sneddon, Chem. Rev., 2022, 122, 6749–6794 CrossRef PubMed.
- Q. Yang, M. Sheng and Y. Huang, Org. Process Res. Dev., 2020, 24, 1586–1601 CrossRef.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef PubMed.
- J. Sherwood, M. De bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650–9652 RSC.
- J. E. Camp, ChemSusChem, 2018, 11, 3048–3055 CrossRef.
- N. A. Stini, P. L. Gkizis and C. G. Kokotos, Green Chem., 2022, 24, 6435–6449 RSC.
- L. Mistry, K. Mapesa, T. W. Bousfield and J. E. Camp, Green Chem., 2017, 19, 2123–2128 RSC.
- T. W. Bousfield, K. P. R. Pearce, S. B. Nyamini, A. Angelis-Dimakis and J. E. Camp, Green Chem., 2019, 21, 3675–3681 RSC.
- K. L. Wilson, J. Murray, C. Jamieson and A. J. B. Watson, Org. Biomol. Chem., 2018, 16, 2851–2854 RSC.
- K. L. Wilson, J. Murray, C. Jamieson and A. J. B. Watson, Synlett, 2018, 650–654 Search PubMed.
- A. Duval and L. Avérous, Green Chem., 2022, 24, 338–349 RSC.
- J. Zhang, G. B. White, M. D. Ryan, A. J. Hunt and M. J. Katz, ACS Sustainable Chem. Eng., 2016, 4, 7186–7192 CrossRef.
- S. Vierucci, S. Muzzioli, P. Righi, V. Borzatta, G. Gorni and I. Zama, RSC Adv., 2016, 6, 55768–55777 RSC.
- https://www.sigmaaldrich.com/GB/en/product/sial/807796 . Accessed 27/07/2024.
- H. A. Le Phuong, L. Cseri, G. F. S. Whitehead, A. Garforth, P. Budd and G. Szekely, RSC Adv., 2017, 7, 53278–53289 RSC.
- https://circa-group.com/cyrene (accessed 22 November 2021).
- R. J. I. Tamargo, P. Y. M. Rubio, S. Mohandoss, J.-J. Shim and Y. R. Lee, ChemSusChem, 2021, 14, 2133–2140 CrossRef PubMed.
- E. Hernández-Pagán, A. Yazdanshenas, D. J. Boski, J. Bi, H. R. Lacey, O. J. Moreno Piza and C. C. Sanchez Sierra, J. Nanopart. Res., 2024, 26, 200 CrossRef.
- J. Cao, Q. Wu, W. Zheng, L. Li and W. Mei, RSC Adv., 2017, 7, 26625–26632 RSC.
- N. A. Yusoh, P. R. Tiley, S. D. James, S. N. Harun, J. A. Thomas, N. Saad, L.-W. Hii, S. L. Chia, M. R. Gill and H. Ahmad, J. Med. Chem., 2023, 66, 6922–6937 CrossRef.
- R. Caspar, C. Cordier, J. B. Waern, C. Guyard-Duhayon, M. Gruselle, P. Le Floch and H. Amouri, Inorg. Chem., 2006, 45, 4071–4078 CrossRef.
- Y.-Q. Jiang, J. Li, Z.-W. Feng, G.-Q. Xu, X. Shi, Q.-J. Ding, W. Li, C.-H. Ma and B. Yu, Adv. Synth. Catal., 2020, 362, 2609–2614 CrossRef.
- E. T. Luis, G. E. Ball, A. Gilbert, H. Iranmanesh, C. W. Newdick and J. E. Beves, J. Coord. Chem., 2016, 69, 1686–1694 CrossRef.
- S. P. Pitre, C. D. McTiernan, W. Vine, R. DiPucchio, M. Grenier and J. C. Scaiano, Sci. Rep., 2015, 5, 16397 CrossRef PubMed.
- H. Ishida, S. Tobita, Y. Hasegawa, R. Katoh and K. Nozaki, Coord. Chem. Rev., 2010, 254, 2449–2458 CrossRef.
- K. Shinozaki and T. Shinoyama, Chem. Phys. Lett., 2006, 417, 111–115 CrossRef.
- C. Turro, S. H. Bossmann, Y. Jenkins, J. K. Barton and N. J. Turro, J. Am. Chem. Soc., 1995, 117, 9026–9032 CrossRef.
- G. Li, L. Sun, L. Ji and H. Chao, Dalton Trans., 2016, 45, 13261–13276 RSC.
- R. M. Hartshorn and J. K. Barton, J. Am. Chem. Soc., 1992, 114, 5919–5925 CrossRef.
- J. Olofsson, L. M. Wilhelmsson and P. Lincoln, J. Am. Chem. Soc., 2004, 126, 15458–15465 CrossRef PubMed.
- J. P. Hall, H. Beer, K. Buchner, D. J. Cardin and C. J. Cardin, Organometallics, 2015, 34, 2481–2486 CrossRef.
- A. K. F. Mårtensson and P. Lincoln, Phys. Chem. Chem. Phys., 2018, 20, 11336–11341 RSC.
- C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC.
- J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 469–489 CrossRef PubMed.
- C. E. Elgar, N. A. Yusoh, P. R. Tiley, N. Kolozsvári, L. G. Bennett, A. Gamble, E. V. Péan, M. L. Davies, C. J. Staples, H. Ahmad and M. R. Gill, J. Am. Chem. Soc., 2023, 145, 1236–1246 CrossRef PubMed.
- A. Byrne, C. S. Burke and T. E. Keyes, Chem. Sci., 2016, 7, 6551–6562 RSC.
- S. Sreedharan, M. R. Gill, E. Garcia, H. K. Saeed, D. Robinson, A. Byrne, A. Cadby, T. E. Keyes, C. Smythe, P. Pellett, J. Bernardino de la Serna and J. A. Thomas, J. Am. Chem. Soc., 2017, 139, 15907–15913 CrossRef PubMed.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2007, 129, 46–47 CrossRef PubMed.
- M. R. Gill, D. Cecchin, M. G. Walker, R. S. Mulla, G. Battaglia, C. Smythe and J. A. Thomas, Chem. Sci., 2013, 4, 4512–4519 RSC.
- Y. Wang, Y.-T. Hu, H.-L. Zhang, Y.-Y. Chen, H.-D. Shi, J.-G. Liu and Q.-L. Zhang, Dalton Trans., 2023, 52, 8051–8057 RSC.
- Y. Zhou, Y. Xu, L. Lu, J. Ni, J. Nie, J. Cao, Y. Jiao and Q. Zhang, Theranostics, 2019, 9, 6665–6675 CrossRef.
- Z. Medgyesi and L. T. Mika, ChemPlusChem, 2024, e202400379 CrossRef PubMed.
- A. Citarella, A. Amenta, D. Passarella and N. Micale, Int. J. Mol. Sci., 2022, 23, 15960 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and Supplementary Fig. S1–S47. See DOI: https://doi.org/10.1039/d4dt02676d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |