
Ruthenium, copper and ruthenium–copper complexes of an unsymmetrical phosphino pyridyl 1,8-naphthyridine PNNN ligand†
Jingyun
Wu
 ,
Michael A.
Stevens
,
Michael G.
Gardiner
and
Annie L.
Colebatch
*
,
Michael A.
Stevens
,
Michael G.
Gardiner
and
Annie L.
Colebatch
*
Research School of Chemistry, Australian National University, Canberra, ACT 2601, Australia. E-mail: annie.colebatch@anu.edu.au
First published on 15th October 2024
Abstract
A new unsymmetrical dinucleating phosphino pyridyl 1,8-naphthyridine ligand PNNN is reported. Reaction with CuCl gave the dicopper complex [Cu2(μ-Cl)2(PNNN)] (1). In contrast, complexation of [RuCl2(cymene)]2 yielded a monometallic species [RuCl(cymene)(PNNN)]Cl ([2]Cl) in which the Ru is bound to the κ2-N,N, rather than κ2-P,N, binding pocket. The selective formation of the monoruthenium complex [2]Cl enabled synthesis of heterobimetallic complexes [RuCuCl3(cymene)(PNNN)] (3) and [RuCuCl2(cymene)(PNNN)]2[PF6]2 ([4]2[PF6]2), which both exhibit κ1-P coordination of Cu. Complexes 1 and [4]2[PF6]2 exhibit reversible dearomatisation–aromatisation behaviour at the metal–ligand cooperative methylene site upon sequential treatment with base (KOtBu) and acid (HCl). Notably, deprotonation of [4]2[PF6]2 induces a shift in the coordination mode of Cu to κ2-P,N.
Introduction
Interest in controlled bimetallic architectures has grown significantly in recent years, inspired in part by the prevalence of multinuclear active sites in enzymes and heterogeneous catalytic materials. This has led to significant innovation in ligand design to develop synthetic bimetallic systems with control over the metal pairings, coordination environments and metal–metal distances.1–4 While a main focus remains the impact of potential metal–metal cooperative behaviour, interest extends to combined aspects of metal–metal, metal–ligand, and metal–metal–ligand cooperativity.5–8Within the ligand designs that are encountered in the literature, 2,7-disubstituted-1,8,-naphthyridine ligands have become very popular for supporting bimetallic complexes with close metal–metal distances. One of the main categories is the tetradentate expanded pincer family of ENNE type ligands, such as dipyridyl (NNNN),9,10 diimine (NNNN)11,12 and diphosphine (PNNP)13–15 examples. Despite the widespread occurrence of unsymmetrical mononucleating pincer ligands, examples of unsymmetrical ligands of the expanded pincer ENNE′ format are very rare.16,17
We have been exploring naphthyridine architectures incorporating pyridyl10,18 and phosphorus donors.19–21 Herein, we present a new unsymmetrical phosphino pyridyl expanded pincer ligand PNNN and examine its coordination chemistry towards copper and ruthenium. The PNNN ligand design is inspired by Milstein's archetypal PNN pincer design,22,23 which contains a non-innocent methylene linker that facilitates dearomatisation–aromatisation metal–ligand cooperativity.
Results and discussion
Ligand synthesis
The preparation of the PNNN ligand was achieved in two steps from 2-chloro-7-methyl-1,8-naphthyridine (Scheme 1).24,25 Adaptation of the procedure for the synthesis of 2,7-dipyridyl-1,8-naphthyridines10,26 enabled installation of the 2-pyridyl substituent via Stille cross-coupling. Subsequent deprotonation with two equivalents LiNiPr2 followed by addition of PCltBu2 yielded the desired product PNNN in 50% isolated yield.27 It is notable that we found quenching the reaction with degassed H2O led to purification problems that could be obviated by quenching with EtOH instead.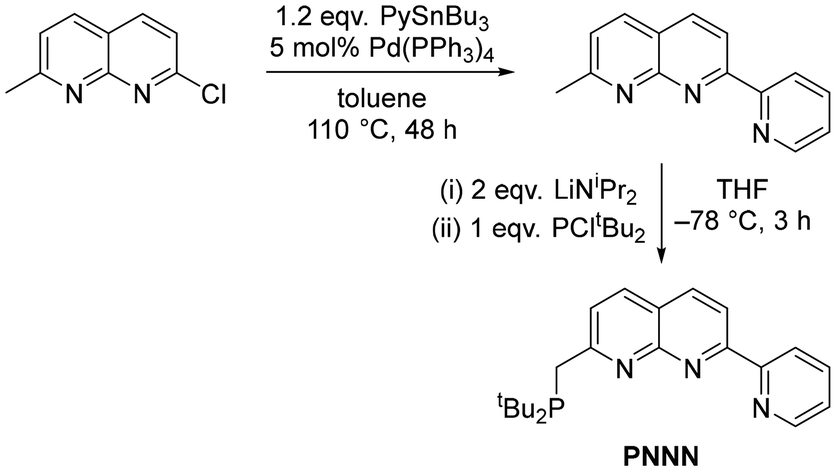 | ||
| Scheme 1 Synthesis of PNNN. | ||
The targeted ligand PNNN was isolated as green powder in 50% yield and characterised by multinuclear NMR spectroscopy, high-resolution mass spectrometry and X-ray crystallography (ESI). A resonance is observed in the 31P{1H} NMR spectrum at 35.2 ppm (CDCl3), very similar to that observed in the symmetric PNNP ligand (35.8 ppm, CD2Cl2)13 and the pentadentate PNNNN ligand dpepn (33.9 ppm, C6D6).27 In the 1H NMR spectrum, the CH2 moiety appears as a doublet at 3.37 ppm (2JPH = 3.9 Hz), which collapses to a singlet in the 1H{31P} NMR spectrum.
Copper complexation
The complexation of copper(I) halide precursors is well-documented among 2,7-disubstituted-1,8-naphthyridine ligands to produce stable Cu2(μ-X)2 binding.13,15,28,29 Reaction of two equivalents of CuCl with PNNN in THF led to precipitation of [Cu2(μ-Cl)2(PNNN)] (1) as an air-stable red solid in 80% yield (Scheme 2). NMR analysis revealed a new broad 31P{1H} NMR resonance at 21.9 ppm due to the quadrupolar nature of Cu (I(63Cu) = I(65Cu) = 3/2). The CH2–P 2JPH coupling constant increases from 3.9 Hz in PNNN to 7.3 Hz upon complexation.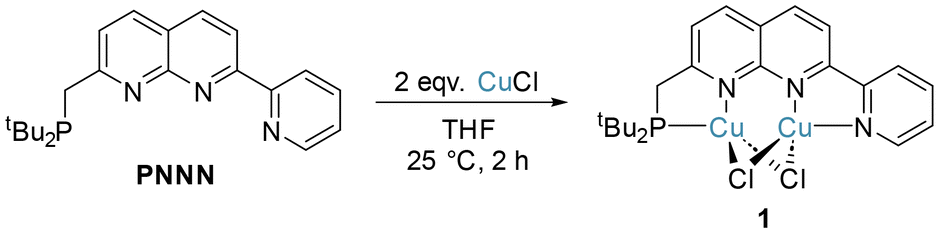 | ||
| Scheme 2 Synthesis of [Cu2Cl2(PNNN)] (1). | ||
The solid-state structure of 1 (Fig. 1) shows the expected Cu2 coordination within the P,N and N,N bidentate binding sites. The distance between the copper centres (2.6202(7) Å) is within the range of other related Cu2(μ-Cl)2 naphthyridine complexes.13–15,28,29 To accommodate the size discrepancy between the P,N and N,N binding pockets, displacement of the P1 and Cu1 atoms from the N2C8 naphthyridine plane of 0.912 and 0.724 Å, respectively, is accompanied by a larger P1–Cu1–N1 bite angle 85.46(7)° in comparison to the N2–Cu2–N3 bite angle of 80.51(11)°.
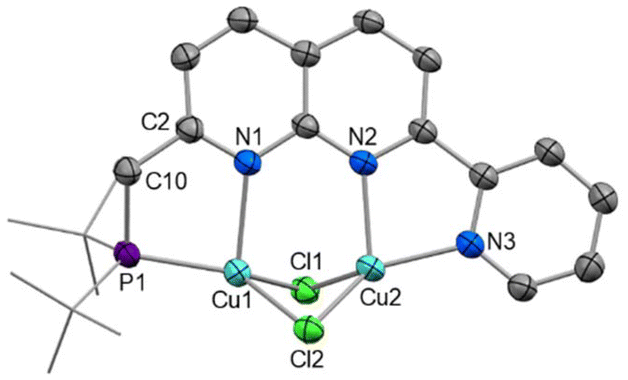 | ||
| Fig. 1 X-ray crystal structure of 1 (50% displacement ellipsoids, H atoms and DCM solvent omitted). Selected bond lengths (Å) and angles (°): Cu1⋯Cu2 2.6202(7), Cu1–P1 2.1994(9), Cu1–N1 2.221(3), Cu2–N2 2.077(2), Cu2–N3 2.011(3), C2–C10 1.509(4), P1–Cu1–N1 85.46(7), N2–Cu2–N3 80.51(11), P1–C10–C2 112.5(2). | ||
Ruthenium complexation
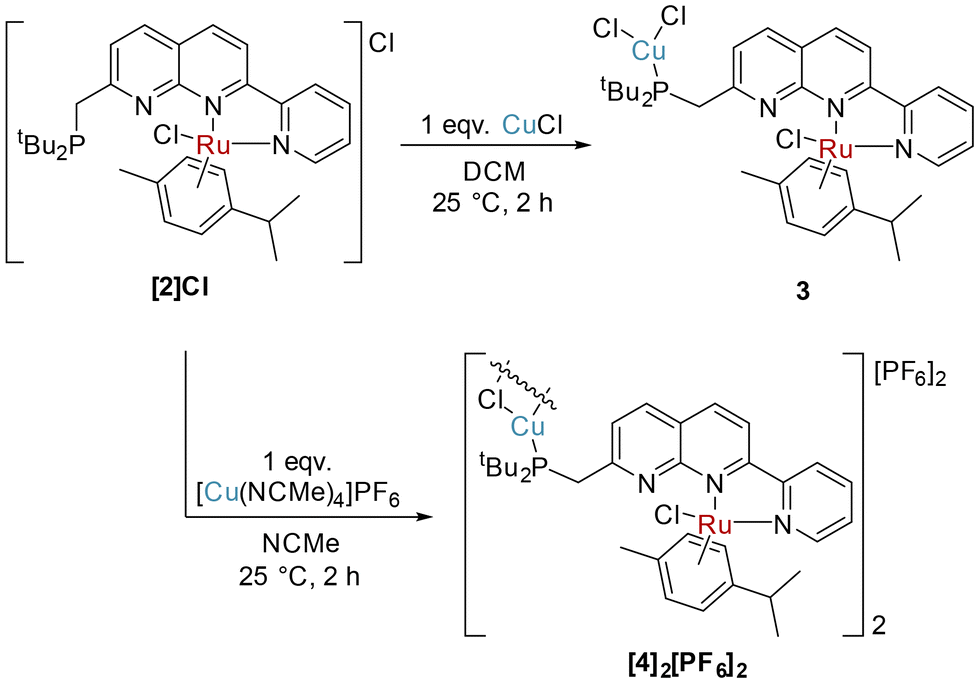
We were in pursuit of Ru complexes inspired by their widespread utility in monometallic metal–ligand cooperative complexes. Reaction of PNNN with one equivalent of [RuCl2(cymene)]2 at 85 °C did not yield a dinuclear complex, but rather the salt [RuCl(cymene)(PNNN)][RuCl3(cymene)] ([2][RuCl3(cymene)]). By instead using 0.45 equivalents of [RuCl2(cymene)]2 as the limiting reagent, the chloride salt [2]Cl could be isolated in 74% yield (Scheme 3). Alternatively, [2][PF6] could be obtained by conducting the reaction in the presence of NaPF6.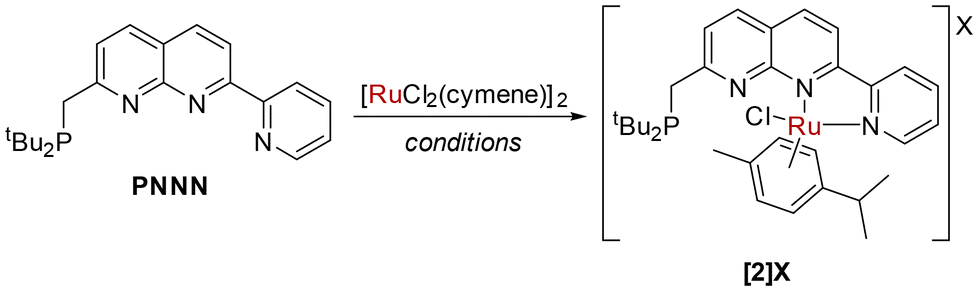 | ||
| Scheme 3 Synthesis of [2]X. Conditions: 1 eq. [RuCl2(cymene)]2, DCE, 85 °C, 18 h, X = RuCl3(cymene); 0.45 eq. [RuCl2(cymene)]2, DCM, 25 °C, 2 h, X = Cl; 0.5 eq. [RuCl2(cymene)]2, 1 eq. NaPF6, DCM, 25 °C, 2 h, X = PF6. | ||
Only a minor shift is observed in the 31P NMR spectrum upon complexation from 35.2 ppm in PNNN to 39.1 ppm in [2]+, whereas a noticeable downfield shift of 1.22 ppm is observed for the pyridyl H6 hydrogen in the 1H NMR spectrum. This is in line with the solid state structure of [2]Cl (Fig. 2), which shows N,N coordination of the RuCl(cymene) fragment, likely due to steric requirements. The structural parameters closely resemble those of the related complexes [RuCl(cymene)(κ2-N,N′-L)]Cl (L = 7-pyrazolyl-1,8-naphthyridine-2-carboxylic acid24 or 2-pyridyl-1,8-naphthyridine).30 The loss of the mirror plane is evident from the appearance of two distinct tBu environments in the 1H NMR spectrum at 1.27 and 1.16 ppm.
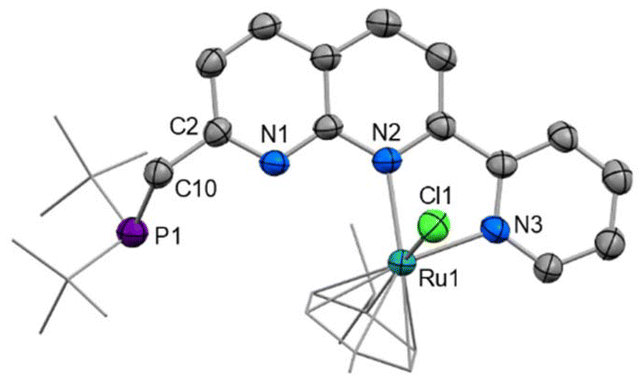 | ||
| Fig. 2 X-ray crystal structure of [2]Cl (50% displacement ellipsoids, H atoms and non-coordinating Cl− anion omitted). Selected bond lengths (Å) and angles (°): Ru1–N2 2.128(3), Ru1–N3 2.084(3), N2–Ru1–N3 76.72(12). | ||
Diruthenium and monoruthenium complexes of 1,8-naphthyridine-based ligands are known.11,16,17,31–35 However, instances of expanded pincer type 1,8-naphthyridine ligands in which selective monometallic complexation of ruthenium occurs are, to the best of our knowledge, restricted to Liu's 7-pyrazolyl-1,8-naphthyridine-2-carboxylic acid system, which similarly forms monometallic complexes [RuCl(arene)(κ2-N,N′-L)]Cl (although in this case only a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 L:Ru reaction stoichiometry was explored).24 Monometallic complexation in the related 2,7-dipyridyl-1,8-naphthyridine expanded pincer ligands has been reported with Mn, Re and Zn.10,18,36–38
:1 L:Ru reaction stoichiometry was explored).24 Monometallic complexation in the related 2,7-dipyridyl-1,8-naphthyridine expanded pincer ligands has been reported with Mn, Re and Zn.10,18,36–38
Heterobimetallic complexes
Since selective monometallic complexation was observed in [2]Cl, this complex was selected to investigate the synthesis of heterobimetallic complexes. Copper precursors were chosen as the second metal based on their demonstrated suitability for both hetero- and homobimetallic naphthyridine bimetallic complexes.13,15,27–29,39–41Addition of one equivalent of CuCl to [2]Cl in DCM resulted in precipitation of the heterobimetallic complex [RuCuCl3(cymene)(PNNN)] (3) as an orange powder. A shift of only 0.5 ppm is observed in the 31P NMR spectrum of 3, however, the diagnostic broadening of the 31P NMR signal is indicative of P-coordination by Cu. The solid state structure (Fig. 3) demonstrates monodentate coordination of a CuCl2 fragment to the phosphine moiety, rather than the targeted P,N bidentate coordination, while the RuCl(cymene)(NN) fragment remains unperturbed.
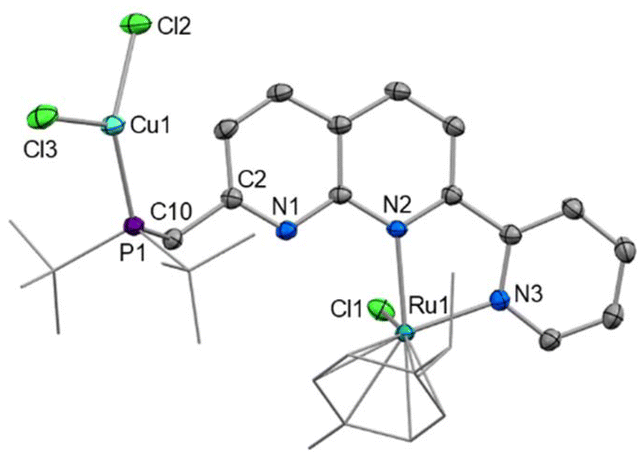 | ||
| Fig. 3 X-ray crystal structure of 3 (50% displacement ellipsoids, H atoms and MeCN solvent omitted). Selected bond lengths (Å) and angles (°): Cu1–P1 2.2091(6), Cu1–Cl2 2.2426(6), Cu1–Cl3 2.3110(6), Ru1–N2 2. 1376(17), Ru1–N3 2.0828(18), N2–Ru1–N3 76.95(7). | ||
When the reaction of [2]Cl with one equivalent of [Cu(NCMe)4]PF6 was conducted, monodentate P-complexation of Cu was observed again in the product [RuCuCl2(cymene)(PNNN)]2[PF6]2 ([4]2[PF6]2, Scheme 4). In this case, the replacement of a coordinating Cl− anion with a weakly-coordinating PF6− anion leads to a chloride-bridged dimer linked by a planar Cu2(μ-Cl)2 fragment. Single crystals demonstrating the cation structure were obtained from a test scale reaction (Fig. 4), in which anion disorder modelled as [4]2[PF6]1.655[CuCl2]0.345 was observed in the lattice. The structural metrics for [42]2+ closely resemble those of 2, with Cu–Cl distances of 2.2917(19) and 2.303(2) Å. Because of the anion co-crystallisation observed in the single crystal structure, elemental analysis was conducted which confirmed the formulation of the bulk sample as [4]2[PF6]2.
 | ||
| Scheme 4 Synthesis of 3 and [4]2[PF6]2. | ||
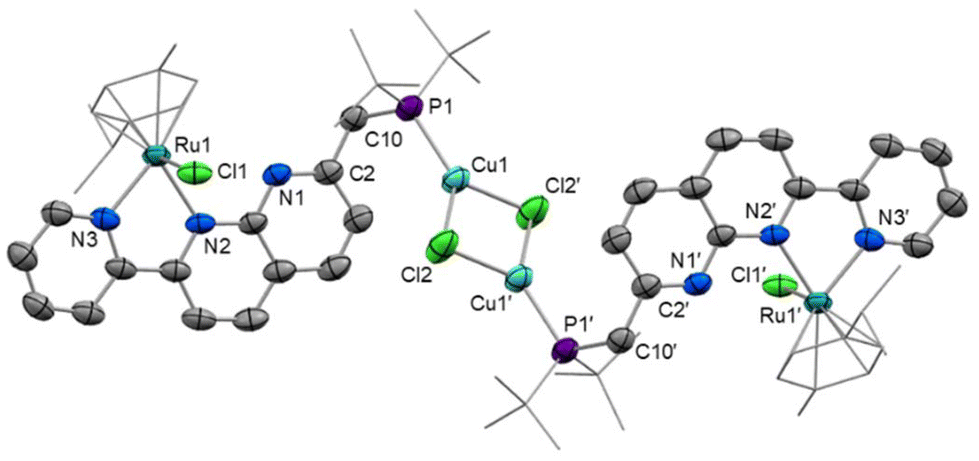 | ||
| Fig. 4 X-ray crystal structure of [4]2[PF6]1.655[CuCl2]0.345 (50% displacement ellipsoids, H atoms, PF6− anions, CuCl2− anion and MeCN solvent omitted). Selected bond lengths (Å) and angles (°): Cu1–P1 2.1788(12), Cu1–Cl2 2.2909(13), Cu1–Cl2′ 2.3006(14), Cu1⋯Cu1′ 2.9775(12), Ru1–N2 2.111(3), Ru1–N3 2.085(3), Ru1–Cl1 2.3882(11), P1–C10 1.852(4), C2–C10 1.497(6), Cu1–Cl2–Cu1 80.85(4), Cl2–Cu1–Cl2′ 99.15(5), N2–Ru1–N3 76.58(14), P1–C10–C2 115.5(3). | ||
Dearomatisation of 1 and [4]2[PF6]2
The PNNN ligand possesses a potential proton-responsive CH2 site, rendering it capable of metal–ligand cooperativity via dearomatisation. Reversible aromatisation–dearomatisation processes have been widely utilised in monometallic catalysis,42 particularly in hydrogenative and dehydrogenative processes.6 Broere and Khusnutdinova have shown that 1,8-naphthyridine complexes are similarly capable of dearomatisation and rearomatisation processes, albeit typically with less reversibility than has been reported in monometallic systems.13,34,39 For example, the symmetric diphosphine complex [Cu2(μ-Cl)2(PNNP)] could be reversibly dearomatised with KOtBu (yielding [Cu2(μ-OtBu)(PNNP*)]) and rearomatised using HNEt3Cl.13Akin to this, treatment of complex 1 with two equivalents of KOtBu in THF gave the dearomatised complex [Cu2(μ-OtBu)(PNNN*)] (5, PNNN* denotes the deprotonated, dearomatised form of PNNN) as a deep purple solid (Scheme 5). Dearomatisation is evident from the 13C{1H} NMR data for the CH linker. A 50 ppm upfield shift and a dramatic increase in the 1JPC value is observed, from 33.3 ppm (CH2–P, 1JPC 11.6 Hz) for the CH2 linker in 1 to 82.9 ppm (CH–P, 1JPC 41.8 Hz) for the CH linker in 5, reflecting the change in hybridisation at C upon deprotonation. These changes closely resemble those observed in Broere's diphosphine system [Cu2(μ-OtBu)(PNNP*)].13 The reversibility of the aromatisation–dearomatisation process was demonstrated by treatment of 5 with two equivalents of HCl (2 M in Et2O). This resulted in an immediate colour change from purple to red-brown, and precipitation of the rearomatised complex 1 (Scheme 5).
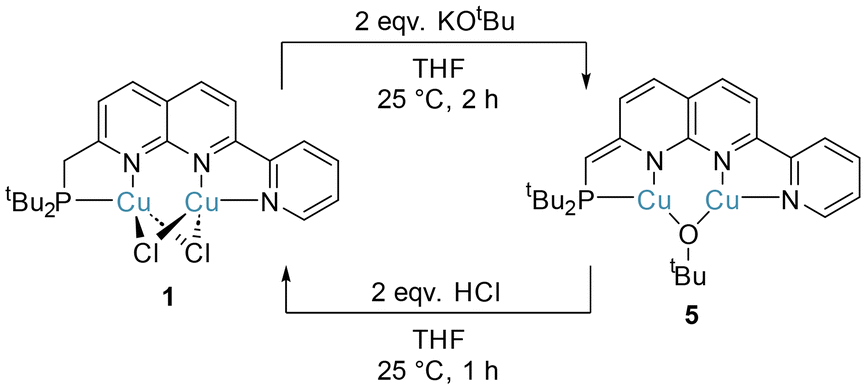 | ||
| Scheme 5 Synthesis of 5 by deprotonation of 1, and reformation of 1 by protonation of 5. | ||
Crystals of 5 were obtained as red prisms from DCM/n-hexane and the structure was determined by single crystal X-ray diffraction (Fig. 5). The solid-state structure of 5 shows that both copper(I) centres sit within the bidentate binding sites, with a bridging tert-butoxide completing the copper coordination sphere. The expected changes are observed upon deprotonation, such as a contraction of the C10–C2 distance (1.376(3) vs. 1.509(4) Å), an elongation of the N1–C2 distance (1.388(3) vs. 1.323(4) Å), an increase in the P1–C10–C2 angle (120.09(16) vs. 112.5(2)°), and increased planarity of the ligand. An examination of the bond lengths throughout the ligand indicate that the dearomatised assignment PNNN* is appropriate (Table S2†). Overall, the metrics largely resemble those of the related dearomatised PNNP* complex [Cu2(μ-OtBu)(PNNP*)].13 A slightly shorter Cu⋯Cu distance is observed in 5 (2.9398(4) Å in 5vs. 3.0468(4) and 3.0220(4) Å in [Cu2(μ-OtBu)(PNNP*)]), likely as a result of the more contracted NN binding pocket compared to PN.
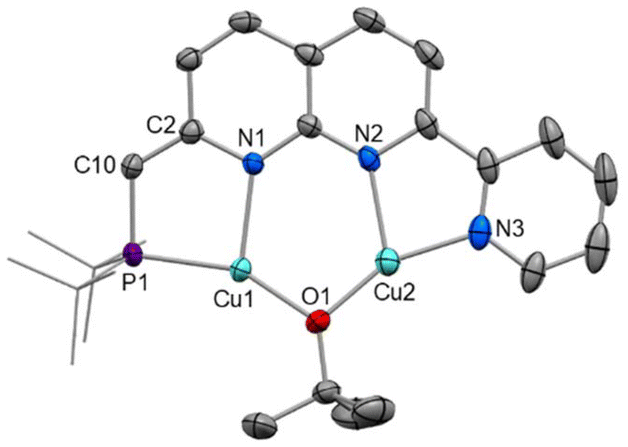 | ||
| Fig. 5 X-ray crystal structure of 5 (50% displacement ellipsoids, H atoms omitted). Selected bond lengths (Å) and angles (°): Cu1⋯Cu2 2.9398(4), Cu1–P1 2.1902(6), Cu1–N1 2.1333(16), Cu2–N2 2.0946(18), Cu2–N3 1.9537(19), P1–C10 1.774(2), C2–C10 1.376(3), Cu1–O1–Cu2 102.61(7), P1–C10–C2 120.09(16). | ||
Attempts to deprotonate the heterobimetallic complex 3 with KOtBu resulted in irreproducible outcomes. However, in the case of [4]2[PF6]2, deprotonation with KOtBu successfully yielded the dearomatised complex [RuCuCl(cymene)(PNNN*)]PF6 ([6]PF6), as shown in Scheme 6. The 31P{1H} NMR spectrum of [6]PF6 is comprised of a single, broad resonance at 20.1 ppm. Similar diagnostic changes were observed in the NMR spectra of [6]PF6 as were seen upon dearomatisation of 1 to 5, including reduction of the P–CH 2JPH coupling constant to 2.2 Hz in [6]PF6 (cf. P–CH22JPH 9.4 Hz in [4]2[PF6]2). Rearomatisation could be effected by treatment of [6]PF6 with one equivalent of HCl (2 M in Et2O) in THF. This led to formation of an orange precipitate, the formulation of which was confirmed as [4]2[PF6]2 by NMR spectroscopy (Scheme 6).
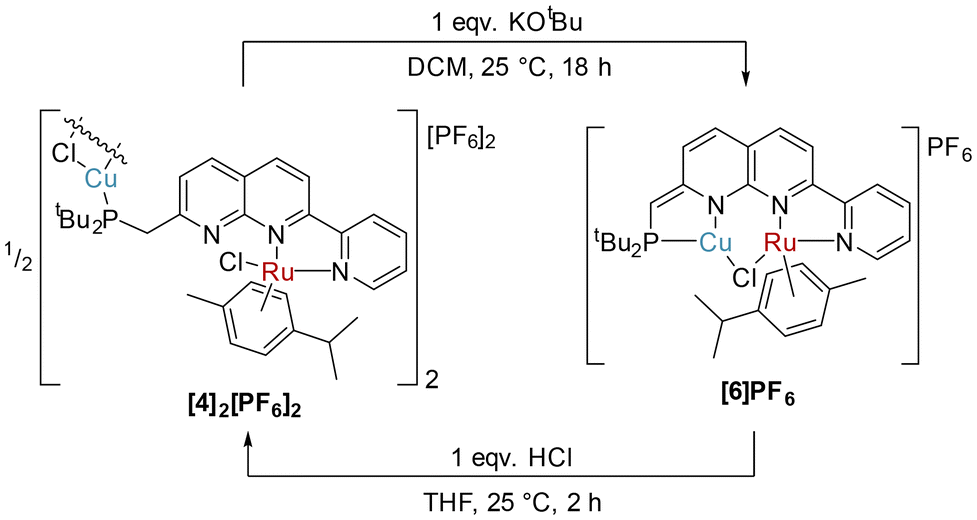 | ||
| Scheme 6 Synthesis of [6]PF6 by deprotonation of [4]2[PF6]2, and reformation of [4]2[PF6]2 by protonation of [6]PF6. | ||
Notably, a change in Cu coordination from κ1-P to κ2-P,N chelation occurs upon dearomatisation, with both metal centres in [6]PF6 now residing in the naphthyridine binding pockets. The ability to control κ1vs. κ2 coordination dependent on the protonation state of the methylene linker has been previously observed upon naphthyridine (de)aromatisation in one other case, namely Khusnutdinova's Pd4 PNNO system.41 Proton-responsive behaviour of related naphthyridinone-type ligands has been successfully implemented in structurally-responsive systems,43 offering control over nuclearity of complexes.19,34,44
The solid-state structure of [6]PF6 (Fig. 6) reveals that both copper(I) and ruthenium(II) centres sit within the bidentate binding sites with a chloride ligand unsymmetrically bridging the two metals (Ru1–Cl1 2.4423(9), Cu1–Cl1 2.2473(9) Å). The ruthenium–copper distance of 2.9770(11) Å is beyond the sum of the covalent radii (2.78 Å),45 and as expected based on the d-electron count there is no formal Ru–Cu bond present. To fit both metals in the bidentate binding pockets, the Cu atom is displaced 0.574 Å from the N2C8 naphthyridine plane, while the Ru atom sits 0.755 Å out from the opposite face of the N2C8 plane to accommodate the bulky cymene ligand. This displacement is more pronounced than in the other Ru complexes reported here in which the second naphthyridine N is unbound, such as 0.278 Å in 3. The variations in bond lengths throughout the PNNN* ligand upon dearomatisation resemble those observed upon dearomatisation of 1 to 5 (Table S2†). This is most notable in the contracted C2–C10 distance ([6]+: 1.360(4), [4]+: 1.497(6) Å) and elongated N1–C2 distance ([6]+: 1.402(4), [4]+: 1.325(6) Å), as well as the change in P1–C10–C2 angle upon the change in hybridisation at C10 from sp3 to sp2 ([6]+: 119.8(2), [4]+: 115.5(3)°).
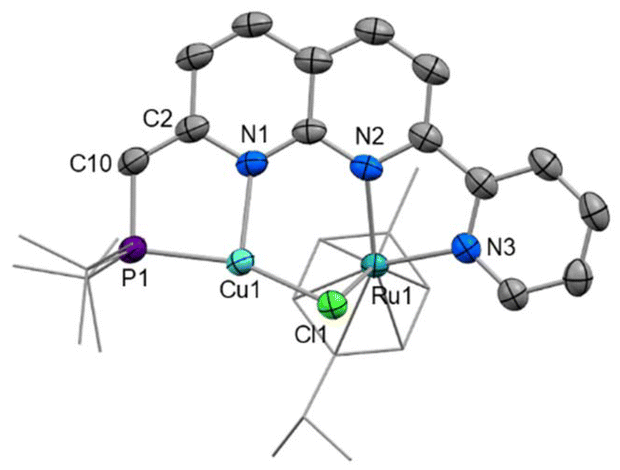 | ||
| Fig. 6 X-ray crystal structure of [6]PF6 (50% displacement ellipsoids, H atoms and PF6− anion omitted). Selected bond lengths (Å) and angles (°): Ru1⋯Cu1 2.9770(11), Ru1–N2 2.112(2), Ru1–N3 2.081(2), Ru1–Cl1 2.4423(9), Cu1–Cl1 2.2473(9), Cu1–P1 2.2103(10), Cu1–N1 2.053(2), P1–C10 1.786(3), C2–C10 1.360(4), Ru1–Cl1–Cu1 78.69(4), P1–C10–C2 119.8(2). | ||
Conclusions
The new PNNN ligand reported herein has been used to prepare a range of dicopper, monoruthenium and ruthenium–copper heterobimetallic complexes. Reaction of PNNN with CuCl forms a bimetallic complex 1, but with the more sterically restricted precursor [RuCl2(cymene)]2 only monometallic N,N-complexation is observed in [2]+. This provides a route to heterobimetallic RuCu complexes 3 and [4]2[PF6]2. In these complexes the PNNN ligand only coordinates κ1-P to the copper centres, rather than in the bidentate P,N pocket, leading to large metal–metal distances. Ruthenium–copper proximity was induced upon deprotonation of [4]2[PF6]2 and formation of the dearomatised complex [6]PF6 with a Ru⋯Cu distance of 2.9770(11) Å. Both the RuCu complex [4]2[PF6]2 and the Cu2 complex 1 exhibit reversible aromatisation–dearomatisation behaviour. The PNNN ligand is not only capable of supporting stable monometallic, homobimetallic and heterobimetallic complexes, it also offers the potential to support metal–ligand, metal–metal and metal–metal–ligand cooperativity.Experimental
General considerations
All experimental work was carried out under a dry, oxygen-free atmosphere of nitrogen or argon using standard Schlenk and glovebox techniques with dried and degassed solvents unless otherwise specified. Once formed, complexes 1, 3, and [4]2[PF6]2 were found to exhibit reasonable air-stability. 2-Chloro-7-methyl-1,8-naphthyridine24,25 and [RuCl2(cymene)]246 were synthesised according to literature procedures. All other reagents were obtained from commercial sources and used as received.
NMR spectra were obtained on a Bruker Avance 400 (1H at 400.1 MHz, 13C{1H} at 100.6 MHz, 31P at 162.0 MHz), a Bruker Avance 600 (1H NMR at 600.0 MHz, 13C{1H} NMR at 150.9 MHz) or a Bruker Avance 700 (1H NMR at 700.0 MHz, 13C{1H} NMR at 176.1 MHz, 31P at 283.4 MHz) spectrometers at 298 K. Chemical shifts (δ) are reported in ppm and are referenced to the residual solvent signal (1H, 13C) or external H3PO4 (31P) with coupling constants given in Hz. The multiplicities of resonances are denoted by the abbreviations s (singlet), d (doublet), t (triplet), sep (septet), m (multiplet), br (broad) and combinations thereof. The numbering system used for NMR assignments is given in Fig. 7.
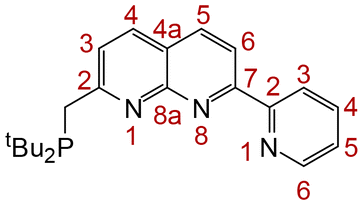 | ||
| Fig. 7 Numbering scheme for NMR spectroscopy assignments. | ||
High resolution electrospray ionisation mass spectrometry (ESI-MS) was performed at the ANU Joint Mass Spectrometry Facility with acetonitrile or methanol as the matrix. For ESI-MS assignments ‘M’ refers to the complex cation. Elemental analysis was conducted by Macquarie University Elemental Microanalysis Service.
X-ray crystallographic data were collected with an Agilent SuperNova diffractometer using Cu-Kα radiation (λ = 1.54184 Å) or on the MX1 beamline at the ANSTO research facilities at the Australian Synchrotron.47 Data obtained in-house was reduced and finalised using the CrysAlis PRO software.48 Raw frame data for synchrotron data (including data reduction, interframe scaling and unit cell refinement) were processed using XDS.49 The structures were solved by direct or Patterson methods and refined by full-matrix least-squares on F2 using the SHELXT and SHELXL programs with the Olex2 interface.50–52 Hydrogen atoms were placed at calculated positions and refined using a riding model.
Synthesis of 2-(2-pyridyl)-7-methyl-1,8-naphthyridine
The synthesis was adapted from literature syntheses of 2,7-dipyridyl-1,8-naphthyridines.10,26 A suspension of 2-chloro-7-methyl-1,8-naphthyridine (2.95 g, 16.6 mmol), 2-tributylstannylpyridine (7.33 g, 19.9 mmol) and tetrakis(triphenylphosphine)palladium (960 mg, 0.831 mmol, 5 mol%) in toluene (100 mL) was heated to reflux for 48 hours, during which time the solution turned dark purple. After this time, the mixture was allowed to cool to room temperature. The reaction mixture was filtered through a Celite plug and washed through with dichloromethane. The solvent was removed under reduced pressure and the residue was triturated in diethyl ether (100 mL) for 5 minutes. The purple solid was collected by filtration and washed with hexane (10 mL) then diethyl ether (10 mL) to give pure 2-(2-pyridyl)-7-methyl-1,8-naphthyridine (2.57 g, 11.62 mmol, 70% yield). 1H NMR (400 MHz, CDCl3, 298 K) δH = 8.93 (d, 3JHH = 7.9 Hz, 1H, Py–H), 8.78–8.71 (m, 2H), 8.30 (d, 3JHH = 8.6 Hz, 1H, Naph–H), 8.17 (d, 3JHH = 8.3 Hz, 1H, Naph–H), 7.90 (ddd, 3JHH = 7.8, 1.6 Hz, 1H, Py–H), 7.49–7.36 (m, 2H), 2.90 (s, 3H, Naph–Me). The spectroscopic data were in agreement with those reported in the literature.53Synthesis of 2-((di-tert-butylphosphino)methyl)-7-(2-pyridyl)-1,8-naphthyridine (PNNN)
2-(2-Pyridyl)-7-methyl-1,8-naphthyridine (303 mg, 1.37 mmol) was dissolved in dry THF (10 mL) at room temperature. LiNiPr2 (2.0 M in THF, 1.37 mL, 2.74 mmol) was added dropwise to the reaction mixture, during which time the solution turned dark blue–purple. The mixture was cooled to −78 °C and PCltBu2 (0.32 mL, 1.68 mmol) was added dropwise. The resulting mixture was stirred at −78 °C for a further four hours, then allowed to gradually warm to room temperature overnight while stirring. Dried and degassed ethanol (1.0 mL) was added and the mixture turned dark green. The volatiles were removed in vacuo, to give crude PNNN as a green solid. The resulting solid was suspended in toluene, stirred overnight, filtered and the toluene was removed under vacuum. The residue was washed with hexane and dried under vacuum to give PNNN as a green solid (250 mg, 0.684 mmol, 50% yield). Crystals of PNNN suitable for X-ray diffraction were obtained from DCM/n-pentane vapour diffusion at −30 °C for two months, granting colourless needles. 1H NMR (400 MHz, CDCl3, 298 K) δH = 8.86 (d, 3JHH = 8.1 Hz, 1H, Py–H3), 8.71 (d, 3JHH = 7.5 Hz, 1H, Py–H6), 8.64 (d, 3JHH = 8.4 Hz, 1H, Naph–H6), 8.23 (d, 3JHH = 8.4 Hz, 1H, Naph–H5), 8.08 (d, 3JHH = 8.4 Hz, 1H, Naph–H4), 7.85 (ddd, 3JHH = 7.7, 1.8 Hz, 1H, Py–H4), 7.76 (d, 3JHH = 8.3 Hz, 1H, Naph–H3), 7.35 (ddd, 3JHH = 6.1, 1.3 Hz, 1H, Py–H5), 3.36 (d, 2JPH = 3.9 Hz, 2H, CH2), 1.18 (d, 3JPH = 11.4 Hz, 18H, tBu2). 13C{1H} NMR (101 MHz, CDCl3, 298 K) δC = 167.1 (d, 2JPC = 14.7 Hz, Naph–C2), 159.0 (s, Naph–C7), 155.8 (s, Py–C2), 155.6 (s, Naph–C8a), 149.2 (s, Py–C6), 137.6 (s, Naph–C5), 137.0 (s, Py–C4), 136.5 (s, Naph–C4), 124.6 (s, Py–C5), 123.6 (d, 3JPC = 11.0 Hz, Naph–C3), 122.9 (s, Py–C3), 121.1 (s, Naph–C4a), 119.4 (s, Naph–C6), 33.5 (d, 1JPC = 24.6 Hz, CH2), 32.4 (d, 1JPC = 20.9 Hz, PC(CH3)2), 29.8 (d, 2JPC = 13.2 Hz, PC(CH3)2). 31P{1H} NMR (162 MHz, CDCl3, 298 K) δP = 35.2 (s, PtBu2). Accurate mass: found 366.2104 [M + H]+. Calcd for C15H13N3P: 366.2099.Synthesis of [Cu2Cl2(PNNN)] (1)
PNNN (300 mg, 0.821 mmol) and CuCl (162.5 mg, 1.64 mmol) were suspended in dry THF (10 mL), and the mixture was stirred for 2 h at room temperature. The reaction mixture was filtered and the resulting solid was washed with THF and dried in vacuo to give 1 as a red solid (370 mg, 0.657 mmol, 80% yield). Crystals of 1 suitable for X-ray diffraction were obtain from DCM/n-hexane vapour diffusion at −20 °C, granting red blocks. 1H NMR (400 MHz, CD2Cl2, 298 K) δH = 9.10 (br, 1H, Py–H6), 8.53 (d, 3JHH = 8.5 Hz, 1H, Naph–H5), 8.32 (br, 3H, Naph–H4, Py–H3, Naph–H6), 8.04 (t, 3JHH = 7.6 Hz, 1H, Py–H4), 7.68 (br, 1H, Py–H5), 7.63 (d, 3JHH = 8.3 Hz, 1H, Naph–H3), 3.46 (d, 2JPH = 7.3 Hz, 2H, CH2), 1.33 (d, 3JPH = 13.4 Hz, 18H, tBu2). 13C{1H} NMR (201 MHz, CD2Cl2, 298 K): δC = 165.0 (s, Naph–C2), 154.6 (s, Naph–C8a), 151.4 (s, Py–C2), 151.3 (s, Naph–C7), 150.1 (s, Py–C6), 139.1 (s, Naph–C5), 138.4 (s, Naph–C4), 137.7 (s, Py–C4), 127.7 (s, Py–C5), 125.2 (s, Naph–C3), 123.7 (s, Naph–C4a), 123.4 (s, Py–C3), 119.3 (s, Naph–C6), 33.2 (d, 1JPC = 8.4 Hz, PC(CH3)2), 32.8 (d, 1JPC = 12.5 Hz, CH2), 29.4 (d, 2JPC = 8.3 Hz, PC(CH3)2). 31P{1H} NMR (162 MHz, CD2Cl2, 298 K) δP = 27.9 (br, PtBu2). Accurate mass: found 528.0273 [M]+. Calcd for C22H28Cl2N3PCu2: 528.0278.Synthesis of [RuCl(cymene)(PNNN)]Cl ([2]Cl)
PNNN (486 mg, 1.330 mmol) and [RuCl2(cymene)]2 (370 mg, 0.604 mmol) were dissolved in DCM (10 mL). The resulting mixture was stirred at room temperature for two hours, during which time the solution turned black–orange. The resulting solution was concentrated in vacuo and toluene was added to precipitate a yellow solid, which was isolated by filtration and dried under vacuum to give [2]Cl as a yellow solid (600 mg, 0.893 mmol, 74% yield). Crystals of [2]Cl suitable for X-ray diffraction were obtained from MeCN/n-hexane vapour diffusion at −30 °C, granting orange blocks. 1H NMR (400 MHz, CD3CN, 298 K) δH = 9.55 (d, 3JHH = 5.6 Hz, 1H, Py–H6), 8.69 (d, 3JHH = 8.6 Hz, 1H, Naph–H5), 8.61 (d, 3JHH = 8.1 Hz, 1H, Py–H3), 8.48 (d, 3JHH = 8.6 Hz, 1H, Naph–H6), 8.45 (d, 3JHH = 8.4 Hz, 1H, Naph–H4), 8.24 (td, 3JHH = 7.8, 1.5 Hz, 1H, Py–H4), 7.98 (d, 3JHH = 8.3 Hz, 1H, Naph–H3), 7.83–7.73 (m, 1H, Py–H5), 6.48 (d, 3JHH = 5.7 Hz, 1H, cymene–Ar), 6.24 (d, 3JHH = 6.2 Hz, 1H, cymene–Ar), 6.15 (d, 3JHH = 6.1 Hz, 1H, cymene–Ar), 5.86 (d, 3JHH = 6.4 Hz, 1H, cymene–Ar), 3.57 (dd, 2JHH = 14.24, 2JPH = 3.24 Hz, 1H, CH2), 3.49 (dd, 2JHH = 14.23, 2JPH = 2.63 Hz, 1H, CH2), 2.44 (sep, 3JHH = 6.9 Hz, 1H, cymene–CH), 2.31 (s, 3H, cymene–CH3), 1.27 (d, 3JPH = 11.1 Hz, 9H, PtBu2), 1.17 (d, 3JPH = 11.1 Hz, 9H, PtBu2), 0.87 (d, 3JHH = 6.9 Hz, 3H, cymene–CH(CH3)2), 0.84 (d, 3JHH = 6.9 Hz, 3H, cymene–CH(CH3)2). 13C NMR (101 MHz, CD3CN, 298 K) δC = 171.1 (s, Naph–C2), 158.9 (s, Naph–C7), 156.9 (s, Py–C6), 156.3 (s, Py–C2), 155.3 (s, Naph–C8a), 142.5 (s, Naph–C5), 140.6 (s, Py–C4), 138.9 (s, Naph–C4), 129.1 (s, Py–C5), 127.6 (d, 3JPC = 7.7 Hz, Naph–C3), 126.8 (s, Py–C3), 123.9 (s, Naph–C4a), 120.7 (s, Naph–C6), 110.1 (br, cymene–Ar), 108.8 (br, cymene–Ar), 90.0 (br, cymene–Ar), 88.4 (br, cymene–Ar), 84.9 (br, cymene–Ar), 82.2 (br, cymene–Ar), 34.5 (d, 1JPC = 27.1 Hz, CH2), 33.2 (d, 1JPC = 22.7 Hz, PC(CH3)2), 32.8 (dd, 1JPC = 22.4 Hz, PC(CH3)2), 31.8 (s, cymene–CH), 30.2 (d, 2JPC = 13.5 Hz, PC(CH3)2), 30.0 (d, 2JPC = 13.5 Hz, PC(CH3)2), 22.4 (s, cymene–CH(CH3)2), 19.7 (s, cymene–CH3). 31P{1H} NMR (162 MHz, CD3CN, 298 K) δP = 39.1 (s, PtBu2). Accurate mass: found 636.1840 [M]+. Calcd for C32H42ClN3PRu: 636.1854.Synthesis of [RuCl(cymene)(PNNN)]PF6 ([2]PF6)
PNNN (211 mg, 0.577 mmol), [RuCl2(cymene)]2 (176.8 mg, 0.289 mmol) and NaPF6 (97 mg, 0.577 mmol) were dissolved in DCM (5 mL). The resulting mixture was stirred at room temperature for two hours, during which time the solution turned black–orange. The solvent was removed in vacuo, to give crude [2]PF6 as a dark orange oily solid. The resulting solid was crystallised from acetonitrile/diethyl ether at −20 °C, the crystals were isolated by filtration and dried under vacuum to give [2]PF6 as a yellow solid (270 mg, 0.346 mmol, 60% yield). 1H NMR (400 MHz, CD3CN, 298 K) δH = 9.58 (d, 3JHH = 5.6 Hz, 1H, Py–H6), 8.69 (d, 3JHH = 8.4 Hz, 1H, Naph–H5), 8.63 (d, 3JHH = 8.1 Hz, 1H, Py–H3), 8.50 (d, 3JHH = 8.6 Hz, 1H, Naph–H6), 8.45 (d, 3JHH = 8.4 Hz, 1H, Naph–H4), 8.24 (td, 3JHH = 7.8, 1.5 Hz, 1H, Py–H4), 7.98 (d, 3JHH = 8.4 Hz, 1H, Naph–H3), 7.83–7.73 (m, 1H, Py–H5), 6.49 (d, 3JHH = 6.1 Hz, 1H, cymene–Ar), 6.24 (d, 3JHH = 6.1 Hz, 1H, cymene–Ar), 6.17 (d, 3JHH = 6.1 Hz, 1H, cymene–Ar), 5.88 (d, 3JHH = 6.7 Hz, 1H, cymene–Ar), 3.57 (dd, 2JHH = 14.24, 2JPH = 3.24 Hz, 1H, CH2), 3.49 (dd, 2JHH = 14.23, 2JPH = 2.63 Hz, 1H, CH2), 2.44 (sep, 3JHH = 6.3 Hz, 1H, cymene–CH), 2.31 (s, 3H, cymene–CH3), 1.27 (d, 3JPH = 11.1 Hz, 9H, PtBu2), 1.16 (d, 3JPH = 11.1 Hz, 9H, PtBu2), 0.87 (d, 3JHH = 6.9 Hz, 3H, cymene–CH(CH3)2), 0.84 (d, 3JHH = 6.9 Hz, 3H, cymene–CH(CH3)2). 13C NMR (101 MHz, CD3CN, 298 K) δC = 171.2 (d, 2JPC = 12.7 Hz, Naph–C2), 158.9 (s, Naph–C7), 157.0 (s, Py–C6), 156.3 (s, Py–C2), 155.3 (s, Naph–C8a), 142.5 (s, Naph–C5), 140.6 (s, Py–C4), 138.9 (s, Naph–C4), 129.1 (s, Py–C5), 127.6 (d, 3JPC = 7.6 Hz, Naph–C3), 126.8 (s, Py–C3), 123.9 (s, Naph–C4a), 120.7 (s, Naph–C6), 90.0 (br, cymene–Ar), 88.4 (br, cymene–Ar), 84.9 (br, cymene–Ar), 82.2 (br, cymene–Ar), 34.5 (d, 1JPC = 27.5 Hz, CH2), 33.2 (d, 1JPC = 23.1 Hz, PC(CH3)2), 32.8 (dd, 1JPC = 22.3 Hz, PC(CH3)2), 31.8 (s, cymene–CH), 30.2 (d, 2JPC = 13.5 Hz, PC(CH3)2), 30.0 (d, 2JPC = 13.5 Hz, PC(CH3)2), 22.4 (s, cymene–CH(CH3)2), 19.7 (s, cymene–CH3). 31P{1H} NMR (162 MHz, CD2Cl2, 298 K) δP = 38.8 (s, PtBu2). 31P{1H} NMR (162 MHz, CD3CN, 298 K) δP = 39.1 (s, PtBu2), −144.6 (sep, 1JPF = 711.1 Hz, PF6). Accurate mass: found 636.1853 [M]+. Calcd for C32H42ClN3PRu: 636.1854.Synthesis of [RuCuCl3(cymene)(PNNN)] (3)
[2]Cl (292 mg, 0.435 mmol) and CuCl (43 mg, 0.435 mmol) were dissolved in DCM (8 mL). The resulting mixture was stirred at room temperature for two hours, during which time the solution turned red and orange solid precipitated out of solution. The resulting mixture was filtered and the solid was dried under vacuum to give 3 as an orange solid (165 mg, 0.205 mmol, 47% yield). Crystals of 3 suitable for X-ray diffraction were obtained from slow evaporation of an MeCN solution, granting orange blocks. 1H NMR (400 MHz, (CD3)2SO, 298 K) δH = 9.65 (d, 3JHH = 5.6 Hz, 1H, Py–H6), 8.94 (d, 3JHH = 8.5 Hz, 1H, Naph–H4), 8.89 (d, 3JHH = 8.1 Hz, 1H, Py–H3), 8.81 (d, 3JHH = 8.6 Hz, 1H, Naph–H3), 8.69 (d, 3JHH = 8.6 Hz, 1H, Naph–H5), 8.64 (d, 3JHH = 8.4 Hz, 1H, Naph–H6), 8.37 (dd, 3JHH = 7.8, 7.8 Hz, 1H, Py–H4), 7.88 (dd, 3JHH = 6.7, 6.7 Hz, 1H, Py–H5), 6.44 (d, 3JHH = 6.1 Hz, 1H, cymene–Ar), 6.27 (d, 3JHH = 6.1 Hz, 1H, cymene–Ar), 6.04 (d, 3JHH = 6.4 Hz, 1H, cymene–Ar), 6.01 (d, 3JHH = 6.2 Hz, 1H, cymene–Ar), 3.71 (dd, 2JHH = 13.8, 2JPH = 7.5 Hz, 1H, CH2), 3.68 (dd, 2JHH = 13.8, 2JPH = 9.1 Hz, 1H, CH2), 2.38–2.31 (m, 1H, cymene–CH), 2.29 (s, 3H, cymene–CH3), 1.40 (d, 3JPH = 12.8 Hz, 9H, PtBu2), 1.14 (d, 3JPH = 13.2 Hz, 9H, PtBu2), 0.76 (d, 3JHH = 6.9 Hz, 3H, cymene–CH(CH3)2), 0.73 (d, 3JHH = 6.8 Hz, 3H, cymene–CH(CH3)2). 13C NMR (201 MHz, (CD3)2SO, 298 K) δC = 167.0 (s, Naph–C8a), 157.5 (s, Naph–C7), 156.3 (d, 2JPC = 21.8 Hz, Py–C6), 154.9 (s, Py–C2), 153.7 (s, Naph–C2), 141.7 (s, Naph–C4), 139.9 (s, Py–C4), 137.9 (s, Naph–C6), 128.2 (s, Py–C5), 127.0 (s, Naph–C5), 125.8 (s, Py–C3), 122.9 (s, Naph–C4a), 120.0 (s, Naph–C3), 90.0 (br, cymene–Ar), 87.8 (br, cymene–Ar), 83.2 (br, cymene–Ar), 80.2 (br, cymene–Ar), 34.0 (d, 1JPC = 6.0 Hz, PC(CH3)2), 33.8 (d, 1JPC = 7.3 Hz, PC(CH3)2), 32.2 (s, CH2), 30.4 (s, cymene–CH), 29.3 (d, 2JPC = 8.2 Hz, PC(CH3)2), 29.2 (d, 2JPC = 8.1 Hz, PC(CH3)2), 21.9 (s, cymene–CH(CH3)2), 21.7 (s, cymene–CH(CH3)2), 19.2 (s, cymene–CH3). 31P{1H} NMR (162 MHz, (CD3)2SO, 298 K) δP = 38.6 (s, PtBu2). Accurate mass: found 736.0818 [M]+. Calcd for C32H42Cl2N3PRuCu: 736.0825.Synthesis of [RuCuCl2(cymene)(PNNN)]2[PF6]2 ([4]2[PF6]2)
[2]Cl (348 mg, 0.518 mmol) and [Cu(NCMe)4]PF6 (194 mg, 0.518 mmol) were dissolved in MeCN (10 mL). The resulting mixture was stirred at room temperature for two hours, during which time the solution turned red. The reaction mixture was concentrated under vacuum, and subjected to layering recrystallisation in acetonitrile/diethyl ether at −20 °C. The crystals were isolated by filtration and dried under vacuum to give [4]2[PF6]2 as an orange solid (249 mg, 0.141 mmol, 55% yield). Crystals of [4]2[PF6]2 suitable for X-ray diffraction were obtain from slow evaporation of an MeCN solution, granting orange blocks. 1H NMR (400 MHz, CD3CN, 298 K) δH = 9.43 (d, 3JHH = 7.3 Hz, 1H, Py–H6), 8.52 (d, 3JHH = 8.5 Hz, 1H, Naph–H5), 8.47 (d, 3JHH = 8.4 Hz, 1H, Naph–H4), 8.43 (d, 3JHH = 5.1 Hz, 1H, Py–H3), 8.41 (d, 3JHH = 5.2 Hz, 1H, Naph–H3), 8.27 (d, 3JHH = 8.5 Hz, 1H, Naph–H6), 8.14 (dd, 3JHH = 7.2, 7.2 Hz, 1H, Py–H4), 7.76–7.71 (m, 1H, Py–H5), 6.22 (d, 3JHH = 6.1 Hz, 1H, cymene–Ar), 5.99 (d, 3JHH = 6.1 Hz, 1H, cymene–Ar), 5.95 (d, 3JHH = 6.1 Hz, 1H, cymene–Ar), 5.78 (d, 3JHH = 6.1 Hz, 1H, cymene–Ar), 3.72 (d, 2JPH = 9.4 Hz, 2H, CH2), 2.36 (sep, 3JHH = 6.9 Hz, 1H, cymene–CH), 2.32 (s, 3H, cymene–CH3), 1.47 (d, 3JPH = 13.6 Hz, 9H, PtBu2), 1.30 (d, 3JPH = 13.8 Hz, 9H, PtBu2), 0.79 (d, 3JHH = 6.9 Hz, 3H, cymene–CH(CH3)2). 13C NMR (101 MHz, CD3CN, 298 K) δC = 167.9 (d, 2JPC = 4.1 Hz, Naph–C2), 158.6 (s, Naph–C7), 156.8 (s, Py–C2), 155.9 (s, Py–C6), 155.1 (s, Naph–C8a), 142.3 (s, Naph–C5), 140.6 (s, Py–C4), 139.1 (s, Naph–C4), 129.2 (s, Py–C5), 128.2 (d, 3JPC = 4.6 Hz, Naph–C3), 126.7 (s, Py–C3), 124.2 (s, Naph–C4a), 120.7 (s, Naph–C6), 108.9 (br, cymene–Ar), 104.5 (br, cymene–Ar), 89.7 (br, cymene–Ar), 88.4 (br, cymene–Ar), 84.8 (br, cymene–Ar), 82.3 (br, cymene–Ar), 35.8 (d, 1JPC = 10.3 Hz, PC(CH3)2), 35.1 (d, 1JPC = 11.7 Hz, PC(CH3)2), 33.1 (d, 1JPC = 6.2 Hz, CH2), 31.7 (s, cymene–CH), 30.1 (d, 2JPC = 7.4 Hz, PC(CH3)2), 29.7 (d, 2JPC = 7.4 Hz, PC(CH3)2), 22.3 (s, cymene–CH(CH3)2), 22.2 (s, cymene–CH(CH3)2), 19.7 (s, cymene–CH3). 31P{1H} NMR (162 MHz, CD3CN, 298 K) δP = 39.8 (s, PtBu2), −144.6 (sep, 1JPF = 711.1 Hz, PF6). Accurate mass: found 736.0836 [M]+. Calcd for C32H42Cl2N3PRuCu: 736.0825. Anal. found: C, 43.69; H, 4.80; N, 4.78. Calcd for C64H84N6P4Cl4F12Ru2Cu2: C, 43.67; H, 4.81; N, 4.77.Synthesis of [Cu2(OtBu)(PNNN*)] (5)
A suspension of KOtBu (125 mg, 1.13 mmol) in THF (5 mL) was added dropwise to a vigorously stirred suspension of 1 (290 mg, 0.515 mmol) in THF (10 mL). The resulting mixture was stirred at room temperature for two hours. The reaction mixture was filtered and the solvent was removed in vacuo to give 5 as a deep purple solid (124 mg, 0.228 mmol, 44% yield). Crystals of 5 suitable for X-ray diffraction were obtain from DCM/n-hexane vapour diffusion at −30 °C for seven days, granting red prisms. 1H NMR (400 MHz, CD2Cl2, 298 K) δH = 8.72 (d, 3JHH = 5.6 Hz, 1H, Py–H6), 7.97 (d, 3JHH = 8.3 Hz, 1H, Py–H3), 7.85 (t, 3JHH = 8.0 Hz, 1H, Py–H4), 7.39 (t, 3JHH = 6.5 Hz, 1H, Py–H5), 7.08 (d, 3JHH = 7.5 Hz, 1H, Naph–H5), 7.00 (d, 3JHH = 7.5 Hz, 1H, Naph–H6), 6.58 (d, 3JHH = 9.2 Hz, 1H, Naph–H4), 6.46 (d, 3JHH = 9.0 Hz, 1H, Naph–H3), 4.14 (d, 2JPH = 7.3 Hz, 1H, CH), 1.47 (s, 9H, OtBu), 1.25 (d, 2JPH = 13.3 Hz, 18H, PtBu2). 13C{1H} NMR (101 MHz, CD2Cl2, 298 K): δC = 165.1 (d, 2JPC = 13.5 Hz, Naph–C2), 158.4 (s, Naph–C8a), 155.9 (s, Py–C2), 149.6 (s, Naph–C7), 149.2 (s, Py–C6), 137.7 (s, Py–C4), 132.9 (s, Naph–C5), 129.3 (d, 3JPC = 9.9 Hz, Naph–C3), 128.2 (d, 4JPC = 2.2 Hz, Naph–C4), 124.8 (s, Py–C5), 123.2 (d, 5JPC = 2.2 Hz, Naph–C4a), 121.5 (s, Py–C3), 108.2 (s, Naph–C6), 82.9 (d, 1JPC = 41.4 Hz, CH), 71.4 (s, OC(CH3)), 35.8 (s, OC(CH3)), 33.2 (d, 1JPC = 16.4 Hz, PC(CH3)2), 29.9 (d, 2JPC = 8.7 Hz, PC(CH3)2). 31P{1H} NMR (162 MHz, CD2Cl2, 298 K) δP = 11.0 (br, PtBu2). Accurate mass data for 5 could not be obtained due to the air and moisture sensitivity of the complex.Synthesis of [RuCuCl(cymene)(PNNN*)]PF6 ([6]PF6)
[4]2[PF6]2 (318 mg, 0.181 mmol) and KOtBu (45 mg, 0.391 mmol) were dissolved in DCM (10 mL). The reaction mixture immediately turned black–green. The resulting mixture was stirred at room temperature overnight, during which time the solution turned black–blue. The reaction mixture was filtered and the solvent was removed in vacuo to give crude [6]PF6 as a deep blue solid. The resulting solid was subjected to layering recrystallisation with THF/hexane at −20 °C. The crystals were isolated by filtration and dried under vacuum to give [6]PF6 as a deep blue solid (150 mg, 0.178 mmol, 49% yield). Crystals of [6]PF6 suitable for X-ray diffraction were obtain from THF/hexane vapour diffusion at −30 °C, granting blue blocks. 1H NMR (400 MHz, CD2Cl2, 298 K) δH = 9.13 (d, 3JHH = 5.6 Hz, 1H, Py–H6), 8.01 (t, 3JHH = 7.7 Hz, 1H, Py–H4), 7.88 (d, 3JHH = 8.2 Hz, 1H, Py–H3), 7.59 (t, 3JHH = 6.6 Hz, 1H, Py–H5), 7.18 (d, 3JHH = 7.2 Hz, 1H, Naph–H5), 7.08 (d, 3JHH = 7.3 Hz, 1H, Naph–H6), 6.74 (d, 3JHH = 9.2 Hz, 1H, Naph–H3), 6.69 (d, 3JHH = 9.2 Hz, 1H, Naph–H4), 5.91 (d, 3JHH = 6.0 Hz, 1H, cymene–Ar), 5.83 (d, 3JHH = 6.0 Hz, 1H, cymene–Ar), 5.78 (dd, 3JHH = 5.3, 5.3 Hz, 2H, cymene–Ar), 4.59 (d, 2JPH = 2.2 Hz, 1H, P–CH), 2.60 (sep, 3JHH = 6.8 Hz, 1H, cymene–CH), 2.09 (s, 3H cymene–CH3), 1.38 (d, 3JPH = 14.1 Hz, 9H, PtBu2), 1.22 (d, 3JPH = 6.8 Hz, 3H, cymene–CH(CH3)2), 1.18 (d, 3JHH = 14.3 Hz, 9H, PtBu2), 1.08 (d, 3JHH = 6.8 Hz, 3H, cymene–CH(CH3)2). 13C NMR (101 MHz, CD3CN, 298 K) δC = 161.7 (d, 2JPC = 11.4 Hz, Naph–C2), 161.1 (s, Naph–C8a), 157.9 (s, Py–C6), 155.4 (s, Py–C2), 151.9 (s, Naph–C7), 139.8 (s, Py–C4), 132.1 (s, Naph–C5), 130.3 (d, 3JPC = 8.8 Hz, Naph–C3), 128.0 (d, 4JPC = 1.7 Hz, Naph–C4), 126.7 (s, Py–C5), 123.1 (s, Naph–C4a), 122.4 (s, Py–C3), 111.8 (s, Naph–C6), 106.9 (s, cymene–Ar), 101.1 (s, cymene–Ar), 90.2 (d, 1JPC = 40.7 Hz, CH), 88.1 (s, cymene–Ar), 85.9 (s, cymene–Ar), 81.7 (s, cymene–Ar), 81.6 (s, cymene–Ar), 34.0 (d, 1JPC = 16.9 Hz, PC(CH3)2), 32.8 (d, 1JPC = 17.2 Hz, PC(CH3)2), 31.8 (s, cymene–CH), 30.4 (d, 2JPC = 7.7 Hz, PC(CH3)2), 29.7 (d, 2JPC = 8.8 Hz, PC(CH3)2), 22.3 (s, cymene–CH(CH3)2), 21.8 (s, cymene–CH(CH3)2), 18.8 (s, cymene–CH3). 31P{1H} NMR (162 MHz, CD2Cl2, 298 K) δP = 20.1 (s, PtBu2), −144.6 (sep, 1JPF = 711.1 Hz, PF6). Accurate mass: found 698.1105 [M]+. Calcd for C32H41ClN3PRuCu: 698.1070.Author contributions
Synthesis and characterisation were performed by JW. X-ray crystallographic analysis was performed by JW, MAS and MGG. Data analysis was conducted by JW, MAS and ALC. Supervision was performed by MAS and ALC. Project conceptualisation, oversight and funding acquisition was conducted by ALC. The manuscript was written by JW and ALC with contributions from all authors.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for PNNN, 1, [2]Cl, 3, [4]2[PF6]2, 5 and [6]PF6 have been deposited in the CCDC 2386870–2386876 and can be obtained from https://www.ccdc.cam.ac.uk/structures/.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Technical support from Dr Doug Lawes, and financial support from the Australian Research Council (DE200100450), is gratefully acknowledged. This research was undertaken in part using the MX1 beamline at the Australian Synchrotron, part of ANSTO.References
- Z. Fickenscher and E. Hey-Hawkins, Molecules, 2023, 28, 4233 CrossRef CAS PubMed.
- R. Maity, B. S. Birenheide, F. Breher and B. Sarkar, ChemCatChem, 2021, 13, 2337–2370 CrossRef CAS.
- R. Govindarajan, S. Deolka and J. R. Khusnutdinova, Chem. Sci., 2022, 13, 14008–14031 RSC.
- Q. Wang, S. H. Brooks, T. Liu and N. C. Tomson, Chem. Commun., 2021, 57, 2839–2853 RSC.
- B. Chatterjee, W.-C. Chang, S. Jena and C. Werlé, ACS Catal., 2020, 10, 14024–14055 CrossRef CAS.
- M. A. Stevens and A. L. Colebatch, Chem. Soc. Rev., 2022, 51, 1881–1898 RSC.
- Q. Wang, S. Zhang, P. Cui, A. B. Weberg, L. M. Thierer, B. C. Manor, M. R. Gau, P. J. Carroll and N. C. Tomson, Inorg. Chem., 2020, 59, 4200–4214 CrossRef CAS PubMed.
- P. C. Duan, R. A. Schulz, A. Römer, B. E. Van Kuiken, S. Dechert, S. Demeshko, G. E. Cutsail, S. DeBeer, R. A. Mata and F. Meyer, Angew. Chem., Int. Ed., 2021, 60, 1891–1896 CrossRef CAS PubMed.
- W. R. Tikkanen, E. Binamira-Soriaga, W. C. Kaska and P. C. Ford, Inorg. Chem., 1983, 22, 1147–1148 CrossRef CAS.
- P. D. Hall, M. A. Stevens, J. Y. J. Wang, L. N. Pham, M. L. Coote and A. L. Colebatch, Inorg. Chem., 2022, 61, 19333–19343 CrossRef CAS PubMed.
- I. Dutta, A. Sarbajna, P. Pandey, S. M. W. Rahaman, K. Singh, J. K. Bera, S. M. Wahidur Rahaman, K. Singh, J. K. Bera, S. M. W. Rahaman, K. Singh and J. K. Bera, Organometallics, 2016, 35, 1505–1513 CrossRef CAS.
- Y. Y. Zhou, D. R. Hartline, T. J. Steiman, P. E. Fanwick and C. Uyeda, Inorg. Chem., 2014, 53, 11770–11777 CrossRef CAS PubMed.
- E. Kounalis, M. Lutz and D. L. J. Broere, Chem. – Eur. J., 2019, 25, 13280–13284 CrossRef CAS PubMed.
- A. R. Scheerder, M. Lutz and D. L. J. Broere, Chem. Commun., 2020, 56, 8198–8201 RSC.
- A. R. Delaney, L.-J. Yu, M. L. Coote and A. L. Colebatch, Dalton Trans., 2021, 50, 11909–11917 RSC.
- B.-C. Tsai, Y.-H. Liu, S.-M. Peng and S.-T. Liu, Eur. J. Inorg. Chem., 2016, 2016, 2783–2790 CrossRef CAS.
- B. Saha, S. M. Rahaman, P. Daw, G. Sengupta, J. K. Bera, S. M. Wahidur Rahaman, P. Daw, G. Sengupta and J. K. Bera, Chem. – Eur. J., 2014, 20, 6542–6551 CrossRef CAS PubMed.
- M. A. Stevens, L. F. Lim, L. N. Pham, N. Cox, M. L. Coote and A. L. Colebatch, Dalton Trans., 2023, 53, 1284–1294 RSC.
- A. J. H. Multem, A. R. Delaney, A. A. Kroeger, M. L. Coote and A. L. Colebatch, Chem. – Asian J., 2024, 19, e202301071 CrossRef CAS PubMed.
- A. R. Delaney, L.-J. Yu, V. Doan, M. L. Coote and A. L. Colebatch, Chem. – Eur. J., 2023, 29, e202203940 CrossRef CAS PubMed.
- A. R. Delaney, A. A. Kroeger, M. L. Coote and A. L. Colebatch, Chem. – Eur. J., 2023, 29, e202302366 CrossRef CAS PubMed.
- E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 16756–16758 CrossRef CAS PubMed.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed.
- C.-Y. Huang, K.-Y. Kuan, Y.-H. Liu, S.-M. Peng and S.-T. Liu, Organometallics, 2014, 33, 2831–2836 CrossRef CAS.
- L. Meng, Y. Wu and T. Yi, Chem. Commun., 2014, 50, 4843–4845 RSC.
- W. Lu, L. H. Zhang, X. S. Ye, J. Su and Z. Yu, Tetrahedron, 2006, 62, 1806–1816 CrossRef CAS.
- A. Nicolay and T. D. Tilley, Chem. – Eur. J., 2018, 24, 10329–10333 CrossRef CAS PubMed.
- M.-U. Hung, B.-S. Liao, Y.-H. Liu, S.-M. Peng and S.-T. Liu, Appl. Organomet. Chem., 2014, 28, 661–665 CrossRef CAS.
- I. Dutta, S. De, S. Yadav, R. Mondol and J. K. Bera, J. Organomet. Chem., 2017, 849–850, 117–124 CrossRef CAS.
- K. T. Prasad, B. Therrien and K. M. Rao, J. Organomet. Chem., 2008, 693, 3049–3056 CrossRef CAS.
- I. Kusuma, T. Komuro and H. Tobita, Chem. Lett., 2018, 47, 400–403 CrossRef CAS.
- E. Binamira-Soriaga, N. L. Keder and W. C. Kaska, Inorg. Chem., 1990, 29, 3167–3171 CrossRef CAS.
- C. B. van Beek, L. Killian, M. Lutz, M. Weingarth, A. S. Asundi, R. Sarangi, R. J. M. Klein Gebbink and D. L. J. Broere, Chem. – Eur. J., 2022, 28, e202202527 CrossRef CAS PubMed.
- M. Gallardo-Villagrán, O. Rivada-Wheelaghan, S. M. W. Rahaman, R. R. Fayzullin and J. R. Khusnutdinova, Dalton Trans., 2020, 49, 12756–12766 RSC.
- A. E. M. Boelrijk, T. X. Neenan and J. Reedijk, J. Chem. Soc., Dalton Trans., 1997, 4561–4570 RSC.
- M. A. Stevens, P. D. Hall and A. L. Colebatch, Aust. J. Chem., 2021, 75, 135–141 CrossRef.
- T.-C. Su, Y.-H. Liu, S.-M. Peng and S.-T. Liu, Eur. J. Inorg. Chem., 2013, 2362–2367 CrossRef CAS.
- W. Tikkanen, W. Kaska, S. Moya, T. Layman, R. Kane and C. Krüger, Inorg. Chim. Acta, 1983, 76, L29–L30 CrossRef CAS.
- S. Deolka, O. Rivada-Wheelaghan, S. L. Aristizábal, R. R. Fayzullin, S. Pal, K. Nozaki, E. Khaskin and J. R. Khusnutdinova, Chem. Sci., 2020, 11, 5494–5502 RSC.
- O. Rivada-Wheelaghan, A. Comas-Vives, R. R. Fayzullin, A. Lledós and J. R. Khusnutdinova, Chem. – Eur. J., 2020, 26, 12168–12179 CrossRef CAS PubMed.
- O. Rivada-Wheelaghan, S. Deolka, R. Govindarajan, E. Khaskin, R. R. Fayzullin, S. Pal and J. R. Khusnutdinova, Chem. Commun., 2021, 57, 10206–10209 RSC.
- T. Shimbayashi and K. Fujita, Catalysts, 2020, 10, 635 CrossRef CAS.
- J. M. Blacquiere, ACS Catal., 2021, 11, 5416–5437 CrossRef CAS.
- O. Rivada-Wheelaghan, S. L. Aristizábal, J. López-Serrano, R. R. Fayzullin and J. R. Khusnutdinova, Angew. Chem., Int. Ed., 2017, 56, 16267–16271 CrossRef CAS PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- M. A. Bennett, T.-N. Huang, T. W. Matheson, A. K. Smith, S. Ittel and W. Nickerson, Inorg. Synth., 1982, 21, 74–78 CrossRef CAS.
- N. P. Cowieson, D. Aragao, M. Clift, D. J. Ericsson, C. Gee, S. J. Harrop, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Williamson and T. Caradoc-Davies, J. Synchrotron Radiat., 2015, 22, 187–190 CrossRef CAS PubMed.
- CrysAlisPRO, Oxford Diffraction/Agilent Technologies UK Ltd., Yarnton, England, 2020 Search PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- S. Jiang, Z. Yang, Z. Guo, Y. Li, L. Chen, Z. Zhu and X. Chen, Org. Biomol. Chem., 2019, 17, 7416–7424 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental spectra and crystallographic data. CCDC 2386870–2386876. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02755h |
| This journal is © The Royal Society of Chemistry 2024 |