
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Switchable, chiral aluminium catalysts for ring opening polymerisations†
David T.
Jenkins
a,
Elizabeth C.
Trodden
ab,
John M.
Andresen
b,
Stephen M.
Mansell
 *a and
Ruaraidh D.
McIntosh
*a
*a and
Ruaraidh D.
McIntosh
*a
aInstitute of Chemical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: s.mansell@hw.ac.uk; r.mcintosh@hw.ac.uk; Web: https://www.mansellresearch.org.uk
bResearch Centre for Carbon Solutions (RCCS), Heriot-Watt University, Edinburgh, EH14 4AS, UK
First published on 6th November 2024
Abstract
A switchable, solvent-free catalytic system was developed in which Al methyl aminebis(phenolate) catalysts selectively initiate the formation of a polyether from cyclohexene oxide under CO2 atmosphere or the ring opening copolymerisation (ROCoP) of cyclohexene oxide and CO2 through the addition of a PPNCl (bis(triphenylphosphine)iminium chloride) cocatalyst to form poly(cyclohexene carbonate).
Recent research has focused on the concept of switchable catalysis, defined in a recent themed collection of Royal Society of Chemistry papers as ‘multiple, catalytically active species with different reactivity generated from a single precursor in the presence of external stimuli’.1–4 By switching the activity of the catalyst, a novel polymer forms, including gradient co-polymers and periodic co-block polymers.5 This approach works well with existing reactions such as ring opening polymerisation (ROP) and CO2 co-polymerisations where the polymers are derived from sustainable materials but lack some of the favourable properties found with the more well established oil-derived polymers.6 Thus, utilising a switchable catalyst, functional materials can be produced whilst also harnessing sustainable resources to aid the advancement of renewable materials and the circular economy.7–9 Two classes of polymers which have gained significant interest are polyesters produced from cyclic esters, such as polylactic acid (PLA),10–15 and polycarbonates using CO2 as a co-monomer (Scheme 1).16,17
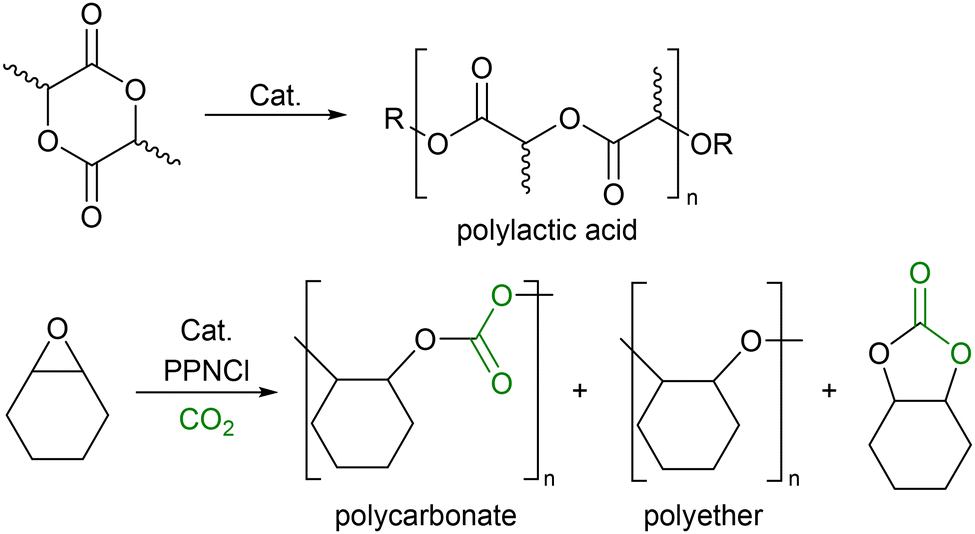 | ||
| Scheme 1 Polylactic acid derived from lactide and the products derived from the reaction of CO2/cyclohexene oxide. | ||
The incorporation of CO2 into the alternating polymer chain with an epoxide is a method of utilising this environmentally deleterious gas, partially replacing carbon in the polymer backbone with a plentiful and freely available C1 source.18,19 While there are many catalysts capable of polymerising one or the other, there are relatively few reported to copolymerising both. Even more scarce are examples where the selectivity of the polymerisation can be influenced by the addition of an external compound.1–4 Within a wide range of catalysts that have been used in the ROP of lactide,20–23 Al complexes have been targeted as low cost, low toxicity, high abundance alternatives to the use of transition metal catalysts, with several groups investigating their activity for ROP.24–26 Although the coordination of amine bis(phenolate) (ABP) ligands to Al has been reported, the effect of an ester-functionalised ligand has not.27–30 Al complexes are also active towards the polymerisation of epoxides and the ROCoP of CO2 and epoxides.31–34 In this paper, we describe the synthesis (Scheme 2) and application of Al-ABP complexes in the polymerisation of rac-lactide, and the switchable ROP/ROCoP catalytic system of CHO (cyclohexene oxide) and CO2.
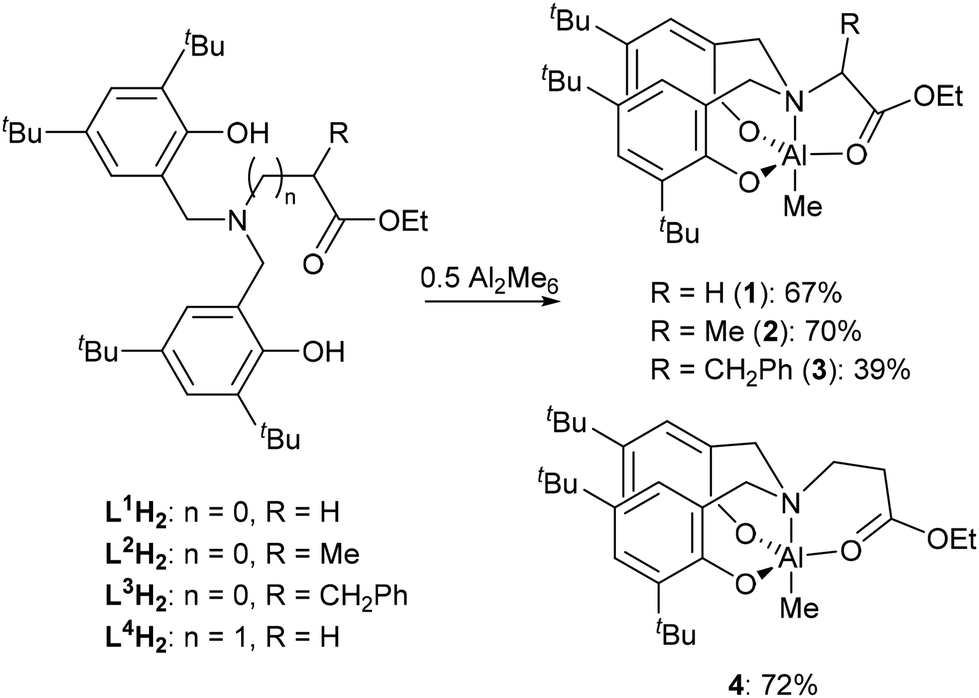 | ||
| Scheme 2 Synthesis of ABP Al–Me complexes. | ||
The proligands35,36 L1H2–L4H2 were reacted with trimethyl aluminium at 90 °C in toluene and, following purification, the corresponding Al complexes were produced in good yields (Scheme 2). Analysis of the 1H NMR spectrum of 1 revealed the absence of the phenolic O–H resonances as well as the presence of doublets in the region of the spectra associated with the protons of the N-methylene bridge, indicating that the atoms had become diastereotopic, which along with a singlet at −0.36 ppm for the Al–Me group confirms the coordination of the ligand to the metal centre.35,36 Similar observations were made in the 1H NMR spectra of 2–4. In each case, the proposed products were found to be consistent with the data obtained from high resolution mass spectrometry (HRMS).
Attempts to grow crystals of 1–4 that were suitable for SCXRD studies proved unsuccessful when using rigorously dry solvents. Previously we have found that adventitious capture of water from bench solvents can result in the formation of oxo-bridged aggregates,36–38 thus crystallisations of 1 and 4 from ‘wet’ solvents revealed either monometallic (5) or bimetallic complexes (Scheme 3, 6 and 7).
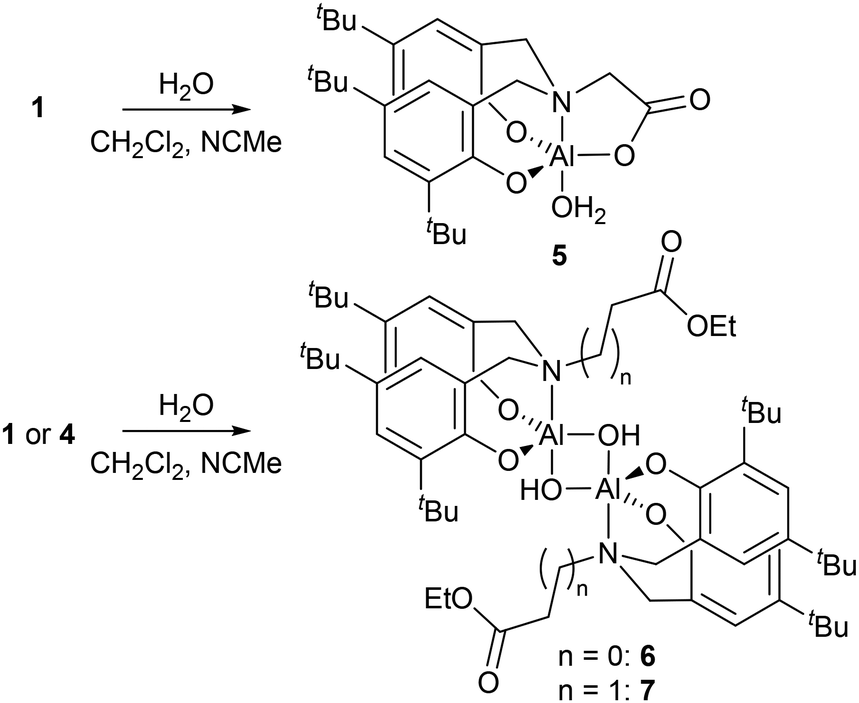 | ||
| Scheme 3 Reactions of 1 and 4 with water. | ||
SCXRD studies of 5 revealed a monometallic complex featuring a carboxylate donor, loss of the Al–Me group and binding of water to the 5-coordinate trigonal bipyramidal Al (Fig. 1). This demonstrates the fate of complex 1 in non-dry reaction and polymerisation conditions.
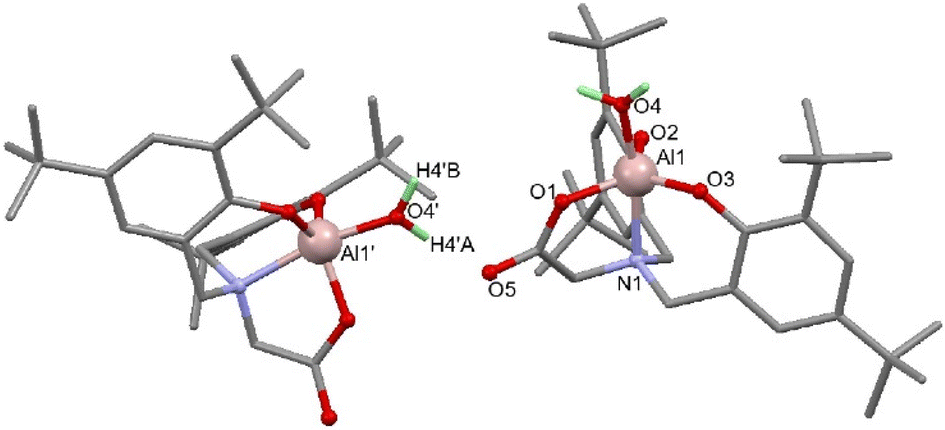 | ||
| Fig. 1 Molecular structure of 5. Disorder and hydrogen atoms (excluding atoms bonded to oxygen) omitted for clarity. Selected bond lengths for (Å): Al1–O1 1.822(6), Al1–O2 1.750(5), Al1–O3 1.767(5), Al1–O4 1.869(5), Al1–N1 2.100(5), O1–C1 1.289(11), C1–O5 1.227(10). Selected bond angles (°): O1–Al1–O2 120.1(3), O1–Al1–O3 118.4(3), O1–Al1–O4 87.6(3), O1–Al1–N1 82.1(2). | ||
With fewer equivalents of water, single crystal X-ray diffraction (SCXRD) experiments revealed hydrolysis of only the Al–Me group in 1 and 4 to give bimetallic Al complexes featuring bridging hydroxide groups and uncoordinated ester functionalities (Fig. 2 for 6, see ESI for 7†)
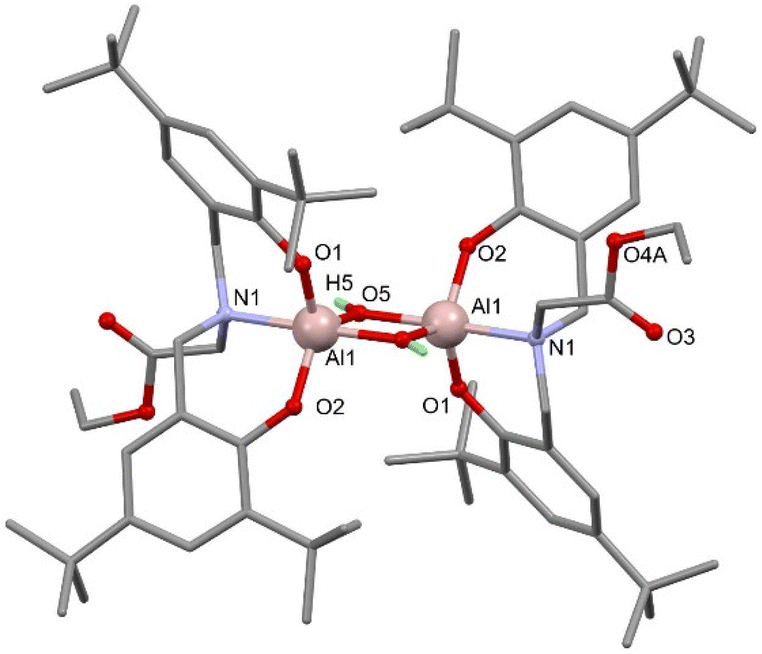 | ||
| Fig. 2 Molecular structure of 6 with hydrogen atoms, except OH, and second position of disordered ester group omitted for clarity. Selected bond lengths (Å): Al1–O1 1.743(2), Al1–O2 1.760(2), Al1–O5 1.792(2), Al1 O5′ 1.879(2), Al1 N1 2.097(2), O3 C32 1.195(3), O4A C32 1.369(4). Selected bond angles (°): N1–Al1–O1 90.5(1), N1–Al1–O2 90.5(1), N1–Al1–O5 93.9(1), N1 Al1 O5′ 172.1(1), Al1–O5–Al1′ 101.9 (1). | ||
Complexes 1–4 were investigated for their ability to initiate the ROP of rac-lactide (catalyst loading = 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with rac-lactide in dry toluene) at 130 °C over 24 hours (Table 1). All complexes showed good activity reaching >90% conversion after 24 hours.
:1 with rac-lactide in dry toluene) at 130 °C over 24 hours (Table 1). All complexes showed good activity reaching >90% conversion after 24 hours.
| Entry | Cat. | Conversionb (%) | P i | M n(calc) (g mol−1) | M n(obs) (g mol−1) | Đ |
|---|---|---|---|---|---|---|
| Conditions [Al]:200[LA], [Al] = 0.01 M, 1 mL of toluene, 130 °C, time = 24 h.a [M]:2[BnOH].b Calculated from 1H NMR spectrum, analysis of the integration of the lactide and poly(lactic acid) resonances in the methylene region.c Determined using homonuclear decoupled 1H NMR spectroscopy.d Calculated as Mn(calc) = ([LA]/[M]) × conversion × MwLA or Mn(calc) = ([LA]/[M]/[BnOH]) × conversion × MwLA for reactions initiating with BnOH.e Determined from GPC trace at 30 °C in THF, using polystyrene standards. |
||||||
| 1 | 1 | 90 | 0.57 | 26000 |
24800 |
1.06 |
| 2a | 1 | 96 | 0.56 | 13800 |
4866 | 1.08 |
| 3 | 2 | 90 | 0.60 | 27200 |
25500 |
1.20 |
| 4a | 2 | 86 | 0.61 | 13900 |
8000 | 1.05 |
| 5 | 3 | 94 | 0.56 | 27200 |
26100 |
1.04 |
| 6a | 3 | 96 | 0.55 | 13900 |
6900 | 1.10 |
| 7 | 4 | 94 | 0.50 | 25800 |
17300 |
1.04 |
| 8a | 4 | 97 | 0.53 | 12400 |
5744 | 1.08 |
Reactions run in the presence/absence of benzyl alcohol proceeded at a similar rate, showing that the Me group does not need to be exchanged for an alkoxide first.27,29 The tacticity of the PLA chains produced was investigated and it was found that complex 2 has an isotactic bias with Pi = 0.61 and 0.60 (Table 1, entries 3 and 4). The other complexes did not show any significant enhancement in the stereoselectivity of the enchainment, suggesting the addition of the chiral methyl group influenced the tacticity of the polymer formed. All complexes showed excellent control over the molecular weight of the polymer chains, with most dispersity values lower than 1.10 and chain lengths close to their calculated values for Mn. Polymerisation in the melt also delivered excellent control (see ESI†).
Complexes 1, 2 and 4 were investigated for their activity as catalysts in the ROCoP of CHO and CO2 using a standard set of reaction conditions (24 hours, 75 °C, 20 bar CO2) and a catalyst loading of 3.5:2000 units of monomer. Initial reactions resulted in formation of poly(cyclohexene oxide) with almost no incorporation of CO2 (Table 2).
| Entry | Cat. | Conv.a (%) | % CO3b | TON/TOFc (h−1) | M w/Mnd (g mol−1) | Đ |
|---|---|---|---|---|---|---|
| Results of polymerisations carried out in neat cyclohexene oxide (2 mL), 20 bar CO2 in absence of initiator (no PPNCl), ratio 3.5 [Al]:2000 [CHO], [cat.]:[monomer]. Conditions 75 °C, 24 hours.a Calculated from 1H NMR spectrum.b % CO3 includes cyclic carbonate and poly carbonate.c Calculated as conversion × (monomer/cat ratio)/time, or TON/time.d Determined from GPC trace at 30 °C in THF, using polystyrene standards. GPC using refractive index detector. |
||||||
| 1 | 1 | 23 | 4 | 135/34 | 25542/17231 |
1.48 |
| 2 | 2 | 14 | 4 | 82/3 | 32624/21149 |
1.54 |
| 3 | 4 | 56 | 5 | 329/14 | 31395/22253 |
1.41 |
Even under a relatively high-pressure of CO2 (20 bar) only trace formation of carbonate was observed (as cyclohexene carbonate). 4 was the most active catalyst with a 56% conversion of polyether observed (Table 3, entry 3). Polyethers with high molecular weight have been synthesised with reasonable Đ values of 1.4 to 1.5 for all the Al-ABP complexes. The Mw values are similar to polymers synthesised in the literature by an aluminium β-ketoamino complex (Mn 1240 g mol−1 to 2500 g mol−1) and in line with a zinc complex [Zn(C6F5)2(toluene)] which generated Mn values in a range of 14600 g mol−1 to 39100 g mol−1.39,40 Plommer and co-workers also found that without an initiator aluminium complexes led to the production of polyether with molecular weights around 180 kg mol−1 (in one minute with a 53% yield) to 500 kg mol−1 (1 h with a 13% yield), both demonstrating narrow polydispersity (obtained using a multi-light scattering detector).32
| Entry | Cat. | T [°C] | Bar | Conv.a (%) | % CO3b | TON/TOFc (h−1) | M w/Mn (g mol−1) | Đ |
|---|---|---|---|---|---|---|---|---|
| Results of polymerisations carried out in neat cyclohexene oxide (2 mL) with PPNCl, ratio 3.5[Al]:3.5[PPNCl]:2000[CHO], [cat.]:[initiator]:[monomer]. Conditions 75 °C.a Calculated from 1H NMR spectrum.b % CO3 includes cyclic carbonate and poly carbonate.c Calculated as conversion × (monomer/cat ratio)/time, or TON/time. |
||||||||
| 1 | 1 | 75 | 5 | 27 | 95 | 158/7 | 2819/2036 | 1.38 |
| 2 | 1 | 75 | 20 | 51 | 94 | 300/13 | 2538/1986 | 1.27 |
| 3 | 2 | 75 | 5 | 32 | 96 | 188/8 | 2641/2152 | 1.22 |
| 4 | 2 | 75 | 20 | 41 | 94 | 241/10 | 3161/2455 | 1.29 |
| 5 | 4 | 75 | 5 | 33 | 96 | 194/8 | 3627/2857 | 1.27 |
| 6 | 4 | 75 | 20 | 56 | 97 | 329/14 | 4658/3757 | 1.24 |
| 7 | 1 | 100 | 20 | 47 | 97 | 276/12 | 4286/3037 | 1.4 |
| 8 | 1 | 75 | 20 | 51 | 94 | 300/13 | 2538/1986 | 1.27 |
| 9 | 1 | 75 | 20 | 37 | 66 | —/— | 3688/2870 | 1.22 |
| 10 | 1 | 60 | 20 | 38 | 95 | 224/9 | 3076/2320 | 1.33 |
| 13 | 2 | 75 | 20 | 41 | 94 | 241/10 | 3161/2455 | 1.29 |
| 14 | 2 | 60 | 20 | 29 | 95 | 171/7 | 1902/1509 | 1.26 |
| 15 | 4 | 75 | 20 | 56 | 97 | 329/14 | 4658/3757 | 1.24 |
| 16 | 4 | 60 | 20 | 56 | 95 | 329/14 | 2074/1986 | 1.20 |
Interestingly, utilising a [1]:[1] ratio of catalyst (1, 2 or 4) to PPNCl cocatalyst under an atmosphere of CO2 resulted in significantly increased rates of CO2 incorporation with poly(cyclohexene carbonate) becoming the major product (Scheme 1 and Table 3), as deduced from analysis of the 1H NMR spectra of the polymers. This demonstrates the potential for a switchable catalytic system in which the Al-ABP complexes can polymerise CHO to a polyether but with the addition of PPNCl, switch to forming a polycarbonate.
The 1H NMR spectra of the crude reaction mixture indicated that the polycarbonate polymer was formed as the major product. Further inspection of the NMR spectra shows only trace quantities of the trans-CHC isomer and there was no observable signal for cis-CHC. This was confirmed with further HSQC experiments were performed on an 800 MHz spectrometer (see ESI†).
At CO2 pressures of 20 bar, conversions to the copolymer were good with very high incorporation of CO2, resulting in almost complete formation of carbonate junctions 94% to 97% for all three catalysts (Table 3 entries 2, 4 and 6) in preference to polyether. Reducing the pressure of CO2 to 5 bar resulted in lower conversion to polymers but these polymers retained their high incorporation of CO2 (Table 3, entries 1, 3 and 5). Increasing the pressure from 5 to 20 bar increased the Mw of the polymers marginally for complexes 2 and 4, however, complex 1 showed a decrease in the Mw of the polymer with the increase of pressure. Complex 4 was the most active catalyst for this reaction, and 4 displayed the same efficiency at the lower temperature 60 °C as the reaction carried out at 75 °C.
There is a clear viscosity limit for this solvent free reaction as conversion did not extend beyond 60%. The addition of 2.2 mL of toluene did not lead to an increase in conversion, however, an increase in the molecular weights was observed alongside a decrease in CO2 incorporation (see ESI, Table S4†). Presumably, the addition of solvent assists with the diffusion of the monomer through the sample, however, the choice of solvent is important as donor solvents such as THF can coordinate to the catalyst and inhibit polymerisation.41 The Mw and Mn values reported here are similar to those found for other related aluminium complexes.31,42 Combined with cocatalysts (such as tetrabutylammonium bromide, TBAB) some aluminium complexes have proven extremely active achieving Mn values of 14.5 kg mol−1.43 Work with aluminium porphyrin complexes has also been successful without the need for a co-catalyst, generating Mn values of 96 kg mol−1.44 With complex 4, the rate of reaction reaches a plateau after 15 hours (see ESI, Table S3†). The conversion of CHO remained roughly constant at ca. 55% over the time period of 15 hours to 48 hours. Again, we anticipate this is viscosity limited due to the decrease in mobility of polymer solutions.
Conclusions
Four novel Al ABP complexes featuring pendent ester arms were synthesised. Complexes 1–4 were active for the ROP of rac-lactide both in the solution phase and melt conditions with low dispersities (<1.10). A switchable catalytic system for the ROCOP of CO2 and cyclohexene oxide was demonstrated by the addition or absence of PPNCl with the aluminium bis(phenolate) complexes. Polycarbonates were successfully synthesised at low pressures of 5 bar CO2 and at temperatures down to 60 °C using PPNCl.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC (EP/N509474/1 and EP/S005781/1) and the EPSRC Centre for Doctoral Training in Critical Resource Catalysis (CRITICAT) for financial support [E.C.T.; EP/L016419/1]. We thank Dr D. Lester at Warwick Analytical Science Centre (WASC), University of Warwick, for Seedcorn Funding to enable some additional polymer analysis on polycarbonate samples, Dr L. Murray and Dr J. Bella at the University of Edinburgh NMR facility for their help with use of the 800 MHz spectrometer and Dr S. Patterson (HWU) for additional guidance with the GPC analysis at HWU.References
- P. L. Diaconescu and C. K. Williams, Themed Collection on Switchable Catalysis, https://rsc.66557.net/en/journals/articlecollectionlanding?sercode=cc&themeid=5eaa255f-7bc6-4283-800b-7c4f4fba65bf.
- Y. Shen, S. M. Shepard, C. J. Reed and P. L. Diaconescu, Chem. Commun., 2019, 55, 5587–5590 RSC.
- S. K. Raman, R. Raja, P. L. Arnold, M. G. Davidson and C. K. Williams, Chem. Commun., 2019, 55, 7315–7318 RSC.
- N. E. Clayman, L. S. Morris, A. M. LaPointe, I. Keresztes, R. M. Waymouth and G. W. Coates, Chem. Commun., 2019, 55, 6914–6917 RSC.
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2016, 116, 1969–1992 CrossRef.
- R. W. F. Kerr and C. K. Williams, J. Am. Chem. Soc., 2022, 144, 6882–6893 CrossRef PubMed.
- G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. Peña Carrodeguas, G. Trott, A. Santmarti, K. Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef PubMed.
- D. C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Fredrick Jr., D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer, T. Tschaplinski and J. P. Hallett, Science, 2006, 311, 484–489 CrossRef PubMed.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef PubMed.
- E. L. Whitelaw, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 10004–10006 RSC.
- M. Vert, Macromol. Symp., 2000, 153, 333–342 CrossRef CAS.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- C.-S. Ha, J. Joseph and A. Gardella, Chem. Rev., 2005, 105, 4205–4232 CrossRef CAS.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS.
- G. A. Bhat and D. J. Darensbourg, Coord. Chem. Rev., 2023, 492, 215277 CrossRef CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS.
- C. M. Kozak, K. Ambrose and T. S. Anderson, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS.
- P. L. Arnold, J. C. Buffet, R. P. Blaudeck, S. Sujecki, A. J. Blake and C. Wilson, Angew. Chem., Int. Ed., 2008, 47, 6033–6036 CrossRef CAS.
- F. Ge, Y. Dan, Y. Al-Khafaji, T. J. Prior, L. Jiang, M. R. J. Elsegood and C. Redshaw, RSC Adv., 2016, 6, 4792–4802 RSC.
- E. Fazekas, G. S. Nichol, J. A. Garden and M. P. Shaver, ACS Omega, 2018, 3, 16945–16953 CrossRef CAS.
- A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC.
- E. Fazekas, P. A. Lowy, M. Abdul Rahman, A. Lykkeberg, Y. Zhou, R. Chambenahalli and J. A. Garden, Chem. Soc. Rev., 2022, 51, 8793–8814 RSC.
- A. J. Chmura, C. J. Chuck, M. G. Davidson, M. D. Jones, M. D. Lunn, S. D. Bull and M. F. Mahon, Angew. Chem., Int. Ed., 2007, 46, 2280–2283 CrossRef CAS.
- A. J. Gaston, G. Navickaite, G. S. Nichol, M. P. Shaver and J. A. Garden, Eur. Polym. J., 2019, 119, 507–513 CrossRef CAS.
- E. D. Cross, G. K. Tennekone, A. Decken and M. P. Shaver, Green Mater., 2013, 1, 79–86 CrossRef.
- Z. Tang and V. C. Gibson, Eur. Polym. J., 2007, 43, 150–155 CrossRef CAS.
- C.-T. Chen, C.-A. Huang and B.-H. Huang, Dalton Trans., 2003, 3799–3803 RSC.
- Y. Li, D. Yu, Z. Dai, J. Zhang, Y. Shao, N. Tang and J. Wu, Dalton Trans., 2015, 44, 5692–5702 RSC.
- H. Plommer, L. Stein, J. N. Murphy, N. Ikpo, N. Mora-Diez and F. M. Kerton, Dalton Trans., 2020, 49, 6884–6895 RSC.
- H. Plommer, I. Reim and F. M. Kerton, Dalton Trans., 2015, 44, 12098–12102 RSC.
- C. Chatterjee and M. H. Chisholm, Chem. Rec., 2013, 13, 549–560 CrossRef.
- G. Chiarioni, M. van Duin and P. P. Pescarmona, Green Chem., 2023, 25, 7612–7626 RSC.
- E. Fazekas, D. T. Jenkins, A. A. Forbes, B. Gallagher, G. M. Rosair and R. D. McIntosh, Dalton Trans., 2021, 50, 17625–17634 RSC.
- D. T. Jenkins, E. Fazekas, S. B. H. Patterson, G. M. Rosair, F. Vilela and R. D. McIntosh, Catalysts, 2021, 11, 551 Search PubMed.
- J. E. Cols, C. E. Taylor, K. J. Gagnon, S. J. Teat and R. D. McIntosh, Dalton Trans., 2016, 45, 17729–17738 RSC.
- J. E. P. Cols, V. G. Hill, S. K. Williams and R. D. McIntosh, Dalton Trans., 2018, 47, 10626–10635 RSC.
- B. Liu, H. Li, C.-S. Ha, I. Kim and W. Yan, Macromol. Res., 2008, 16, 441–445 CrossRef CAS.
- Y. C. A. Sokolovicz, A. Buonerba, C. Capacchione, S. Dagorne and A. Grassi, Catalysts, 2022, 12, 970 CrossRef CAS.
- D. H. Lamparelli and C. Capacchione, Catalysts, 2021, 11, 961 CrossRef CAS.
- K. Nishioka, H. Goto and H. Sugimoto, Macromolecules, 2012, 45, 8172–8192 CrossRef CAS.
- M. A. Fuchs, C. Altesleben, T. A. Zevaco and E. Dinjus, Eur. J. Inorg. Chem., 2013, 2013, 4541–4545 CrossRef CAS.
- X. Sheng, Y. Wang, Y. Qin, X. Wang and F. Wang, RSC Adv., 2014, 4, 54043–54050 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods and analytical data for complexes 1–4; polymerisation conditions and polymer analysis. CCDC 2352910–2352912. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02831g |
| This journal is © The Royal Society of Chemistry 2024 |