
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discovery of novel SOS1 inhibitors using machine learning†
Lihui
Duo‡
a,
Yi
Chen‡
 ce,
Qiupei
Liu‡
ac,
Zhangyi
Ma
a,
Amin
Farjudian
g,
Wan Yong
Ho
d,
Sze Shin
Low
a,
Jianfeng
Ren
*a,
Jonathan D.
Hirst
*b,
Hua
Xie
*cef and
Bencan
Tang
*a
ce,
Qiupei
Liu‡
ac,
Zhangyi
Ma
a,
Amin
Farjudian
g,
Wan Yong
Ho
d,
Sze Shin
Low
a,
Jianfeng
Ren
*a,
Jonathan D.
Hirst
*b,
Hua
Xie
*cef and
Bencan
Tang
*a
aNottingham Ningbo China Beacons of Excellence Research and Innovation Institute, Key Laboratory for Carbonaceous Waste Processing and Process Intensification Research of Zhejiang Province, Department of Chemical and Environmental Engineering, The University of Nottingham Ningbo China, 199 Taikang East Road, Ningbo 315100, P. R. China. E-mail: Jianfeng.Ren@nottingham.edu.cn; Bencan.Tang@nottingham.edu.cn
bSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: jonathan.hirst@nottingham.ac.uk
cDivision of Antitumor Pharmacology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, 201203 Shanghai, China. E-mail: hxie@simm.ac.cn
dFaculty of Medicine and Health Sciences, University of Nottingham (Malaysia Campus), Semenyih 43500, Malaysia
eUniversity of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing 100049, China
fZhongshan Institute for Drug Discovery, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Zhongshan Tsuihang New District, Zhongshan 528400, China
gSchool of Mathematics, Watson Building, University of Birmingham, Edgbaston, Birmingham, B15 2TT UK
First published on 15th March 2024
Abstract
Overactivation of the rat sarcoma virus (RAS) signaling is responsible for 30% of all human malignancies. Son of sevenless 1 (SOS1), a crucial node in the RAS signaling pathway, could modulate RAS activation, offering a promising therapeutic strategy for RAS-driven cancers. Applying machine learning (ML)-based virtual screening (VS) on small-molecule databases, we selected a random forest (RF) regressor for its robustness and performance. Screening was performed with the L-series and EGFR-related datasets, and was extended to the Chinese National Compound Library (CNCL) with more than 1.4 million compounds. In addition to a series of documented SOS1-related molecules, we uncovered nine compounds that have an unexplored chemical framework and displayed inhibitory activity, with the most potent achieving more than 50% inhibition rate in the KRAS G12C/SOS1 PPI assay and an IC50 value in the proximity of 20 μg mL−1. Compared with the manner that known inhibitory agents bind to the target, hit compounds represented by CL01545365 occupy a unique pocket in molecular docking. An in silico drug-likeness assessment suggested that the compound has moderately favorable drug-like properties and pharmacokinetic characteristics. Altogether, our findings strongly support that, characterized by the distinctive binding modes, the recognition of novel skeletons from the carboxylic acid series could be candidates for developing promising SOS1 inhibitors.
1. Introduction
The rat sarcoma virus (RAS) superfamily members, including HRAS, KRAS, and NRAS in mammals, play significant roles in the pathogenesis of various human cancers.1 Notably, KRAS is commonly mutated, contributing to the activation of this gene in a multitude of cancer cases, including 80% to 90% of pancreatic cancers, 40% to 50% of colorectal cancers, and 30% of non-small cell lung cancers.1 However, the clinical therapeutic options are considerably constrained for individuals harbouring KRAS mutations. There are only two small-molecule inhibitors, sotorasib and adagrasib, currently approved by the FDA for treating the KRAS G12C mutated non-small cell lung cancer, indicating a significant unmet clinical need for KRAS-targeted therapies.2,3 As shown in Fig. 1, the mutation of KRAS is associated with the activation of multiple downstream signalling pathways, notably the RAF–MEK–ERK pathway, within the MAPK family, which are crucial for regulating cell survival and proliferation.1,4 The RAS proteins function as molecular switches, transitioning between the active on-state when bound to guanosine triphosphate (GTP) and the inactive off-state when bound to guanosine diphosphate (GDP).5 This switch is regulated by guanine nucleotide exchange factors, which promote the exchange of GDP for GTP, and GTPase-activating proteins, which enhance the hydrolysis of GTP to GDP.2 Son of sevenless 1 (SOS1), as a major guanine nucleotide exchange factor, plays a crucial role in the RAS signalling pathways by facilitating guanine nucleotide exchange and regulating KRAS switching from “GDP-bound off state” to “GTP-bound on state”.6,7 Hence, inhibiting the interaction between KRAS-GDP and SOS1 effectively reduces the formation of activated KRAS-GTP, thereby suppressing uncontrolled downstream cell proliferation.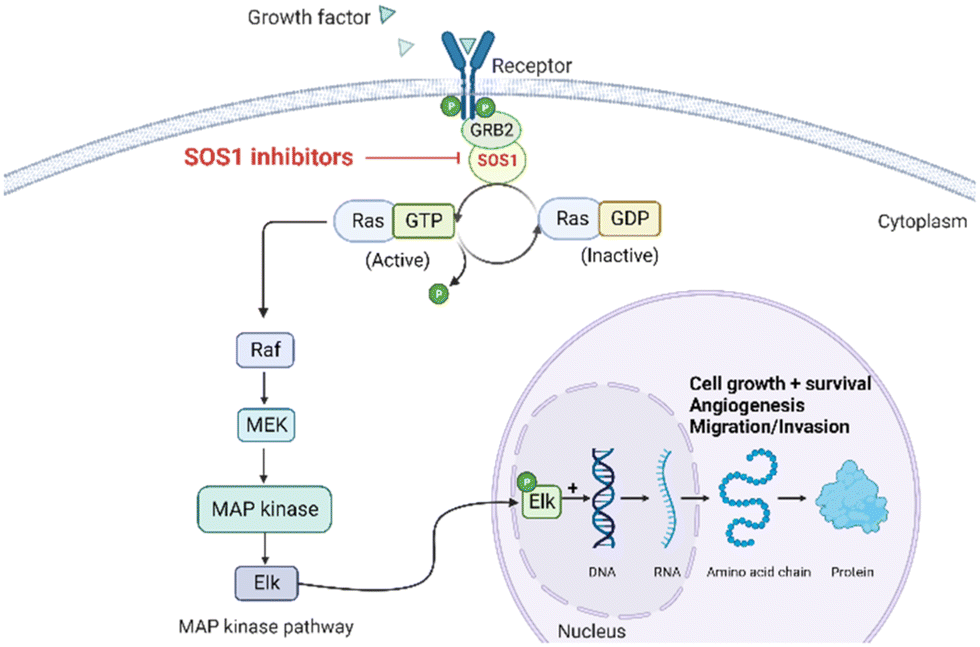 | ||
| Fig. 1 Mechanism of SOS1 inhibitors in RAS-driven malignancies. KRAS can exist in inactive (GDP-bound) or active (GTP-bound) states. SOS1 facilitates the exchange of GDP for GTP, activating RAS. In its activated state, RAS interacts with downstream effectors, initiating vital cellular pathways, such as MAP kinase signalling, critical for cell survival, growth, angiogenesis, cancer cell invasion, and migration. SOS1 inhibitors block GTP loading, maintaining RAS in an inactive state, preventing excessive downstream signalling, and offering a potential therapeutic approach for RAS-driven malignancies. Figure created with https://BioRender.com. | ||
Given the pivotal role of SOS1 in the progression of RAS-driven cancer, inhibiting the binding between SOS1 and RAS has emerged as a promising therapeutic avenue against RAS-driven tumours. Small molecule SOS1 inhibitors modulate RAS activation by binding to the SOS1 protein pocket, affecting the interaction between SOS1 and RAS.8,9Fig. 2 shows some examples of reported SOS1 compounds. For instance, Hillig et al. reported an aminoquinazoline inhibitor, BAY-293, based on fragment and high-throughput screening for the KRAS-SOS1 binding site.10 In 2021, Hofmann et al. discovered another aminoquinazoline compound, BI-3406, as an orally administered, selective, and highly potent SOS1 inhibitory agent.11 Its analogue, BI 1701963 (whose structure has not been disclosed) was introduced into the first clinical study of SOS1 inhibitors but with a disappointing outcome. Another compound, MRTX0902, with a pyridopyridazine core, has just entered clinical trials (NCT05578092), which are designed to elucidate the effectiveness of MRTX0902, either alone or in combination with MRTX849 (adagrasib), in treating solid tumours malignancies among patients harbouring KRAS G12C mutations.12 Similar research has been published by Revolution Medical13 and He et al.14,15 for the SOS1 inhibitors: RMC-0331 with a pyrrolo [3,4-d]pyrimidine scaffold and the tetracyclicquinazoline (37 and 13c) with superior pharmacokinetic properties, respectively (see Fig. 2). Despite these promising advances, there is currently no approved SOS1 inhibitor, and most of these candidates are designed to be combined with anti-cancer drugs targeting the KRAS-MAP kinase pathway. Notably, most current SOS1 inhibitors are derived from BAY-293 and lack structural diversity. Thus, there is an urgent need to identify and explore novel SOS1 inhibitors.
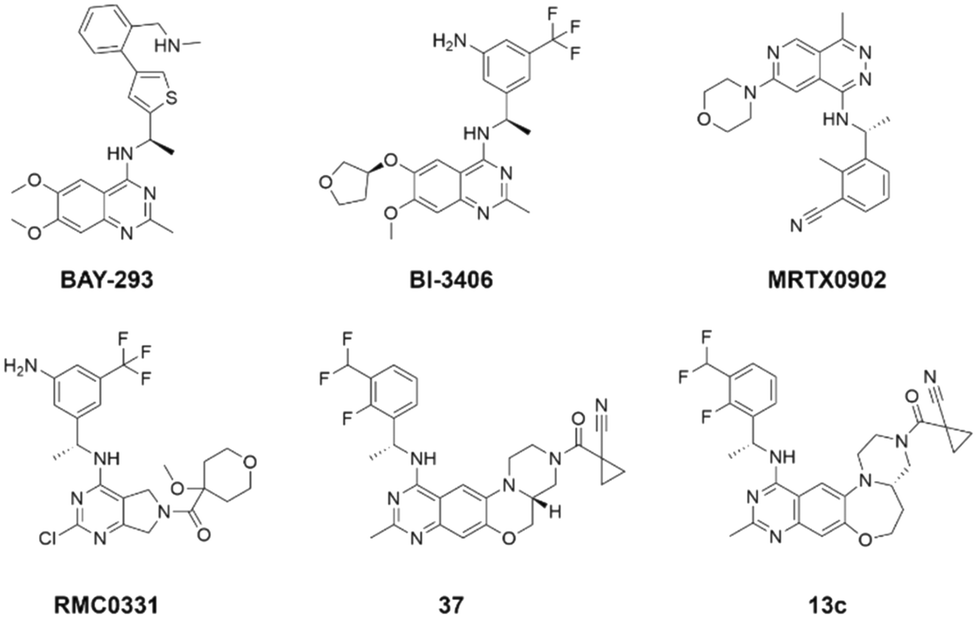 | ||
| Fig. 2 Representative SOS1 inhibitors. | ||
With the increased access to large-scale datasets, ML is revolutionizing drug development endeavours, especially for anti-cancer drug discovery, by offering the unparalleled potential to accelerate this process with higher success rates and lower cost compared with traditional pharmaceuticals.16–18 One common artificial intelligence (AI)-assisted drug discovery technique is virtual screening (VS), which is used to identify prospective leads from large compound libraries.19 Ligand-based virtual screening (LBVS) can be applied to target proteins with unknown structures to discover novel ligands based on the premise that substances with comparable structural properties are likely to exhibit comparable biological activities.20 Quantitative structure–activity relationships (QSAR), an important LBVS strategy, explore the correlation between chemical structures and biological activities of molecules to develop predictive models. Indeed, several such endeavours have been reported recently. Valentini et al. in 2022 utilized a comprehensive QSAR approach, integrating various ML techniques (K-nearest neighbour, gradient bosting, logistic regression, RF, and support vector machine), alongside in vitro and in vivo experimental data, to successfully identify two novel inhibitors of anti-apoptotic proteins with promising efficacy across multiple tumour histocytes.21 In another study, Yang et al. employed a combined approach of ligand-based ML and structural-based molecular docking to screen the latest US Food and Drug Administration approved drug library (∼2600 compounds) for potential inhibitors of adipocyte fatty acid-binding protein, seeking to investigate current medications with established safety characteristics.22 The results demonstrated the efficacy of the naïve Bayesian model in predicting potential inhibitors, leading to the discovery of cobimetinib, which was subsequently confirmed to inhibit A-FABP-activated JNK/C-jun phosphorylation in cellular assays. Other related research efforts are focused on the discovery of lead anti-cancer compounds such as lysine-specific histone demethylase 1,23 and indoleamine 2,3-dioxygenase inhibitors.24 All these initiatives highlight the tremendous promise that ML strategies hold in the field of drug discovery, particularly in the context of LBVS.
In summary, SOS1 is a crucial protein within the RAS pathway. Recently, targeting SOS1 has garnered increasing attention as a promising strategy for treating RAS-driven cancers. However, the development of effective and selective SOS1 inhibitors remains a challenge and an urgent demand. Thus, the aim of this research work is to use ML to predict the bioactivity of small molecules against SOS1 with a view to discovering structurally novel inhibitors.
2. Experimental
Our approach includes seven steps: 1) raw data collection from the ChEMBL dataset, including data curation, data cleaning and the calculation of molecular representations; 2) model development and optimization; 3) evaluation and validation of ten different ML models, selection of the best performing one; 4) VS of molecules from in-house libraries using the robust LBVS model to identify and rank hits; 5) biological experiments focusing on the KRAS G12C/SOS1 PPI assay; 6) molecular docking to analyse hits and receptor interactions; 7) in silico evaluation of drug-likeness properties.2.1 Data collection and pre-processing
To compile the SOS1 training set, the publicly available ChEMBL database (version 31) was queried, focusing on records related to anthropogenic ligands tested against the SOS1 target (ChEMBL ID: CHEMBL2079846). The data were stored in the form of Simplified Molecular Input Line Entry System (SMILES) representations, along with various types of bioactivity measurements such as IC50, EC50, AC50, Fc, Ki, Emax, and Kd, reported in molar concentrations. Data redundancy was assessed based on the SMILES representation. Duplicated molecules were identified and removed, giving 375 unique compounds, and the mean value for each measurement was calculated for each compound. To facilitate further analysis, the bioactivity data were converted to pChEMBL values using the following equation.| pChEMBL = −log10(Effective Value) | (1) |
2.2 Model construction and optimization
In order to construct ML models for regression, the Scikit package is utilized to develop ten regression models,28 including K-nearest-neighbour, Ridge, Lasso, elastic net, decision-tree, RF, extra-tree, adaboost, gradient boosting, and support vector regression (SVR). To ensure an unbiased evaluation of all models, a five-fold cross-validation approach was implemented to assess the performance and the generalization capability of the established models, where the dataset was segmented into five mutually exclusive subsets. In each fold, four subsets are used as the training set to develop the models while the remaining subset is used as the test set to evaluate the predictive performance of models. The use of predetermined random seeds guaranteed that each fold within the cross-validation procedure results in a controlled and reproducible data partition, enabling a rigorous assessment of model stability and consistency. Furthermore, adjusting the hyperparameters can improve the predictive power of a given model. Therefore, to systematically evaluate a range of potential parameter combinations, a grid search is applied on each model to identify the optimal model parameters by incorporating nested cross-validation.2.3 Model evaluation and selection
Two metrics are used to evaluate the developed regressors, i.e., the coefficient of determination R2 and the root mean squared error eRMSE.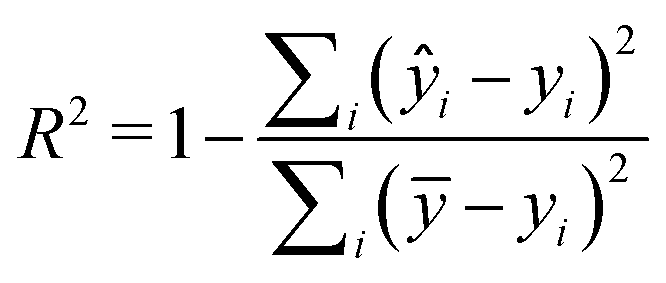 | (2) |
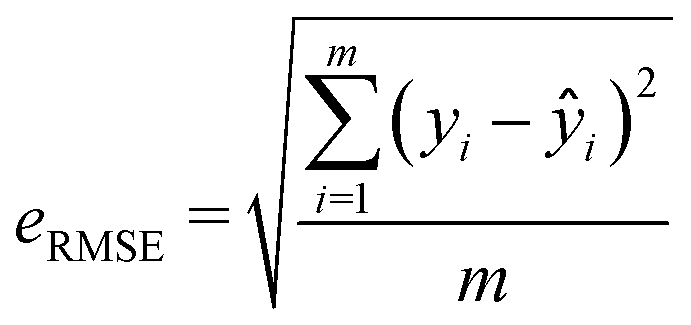 | (3) |
The model validation process here was augmented by using two sets of labelled data, including external testing data comprising an additional 94 molecules derived from the latest literature, as well as a randomly selected 10% of the total dataset. To elaborate further, ten models were reconstructed using 90% of the reshuffled data from the entire dataset, resulting in 337 molecules, while the remaining 10% of data (38 compounds with diverse pChEMBL values) were combined with the aforementioned external data sourced from the literature (94 labelled compounds). Since this external verification set contains molecules of known pChEMBL value and it is never used to construct the models, the above simulated VS process allows a detailed examination of the model's predictions on a specific labelled dataset, evaluates the models on novel unseen molecules and detects the extent of model overfitting by quantifying the extent of the difference between the actual and the predicted activity value for this particular set. Complementing the five-fold cross validation mentioned above, this strategy, resembling a 10-fold cross-validation, providing an opportunity to scrutinize the model's predictions on a specific set of labelled data.
2.4 Virtual screening
After evaluating the models developed by the 10 methods, the re-trained regressor with 337 compounds that yielded the optimal performance is utilized to find prospective SOS1 inhibitors from various commercial chemical libraries. Specifically, we initiated the screening of the L-series commercial databases provided by the targetmol company (https://www.tsbiochem.com/), namely L1000, L4000, L6000, L5600, and L9200, which collectively encompass a total of 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 582 compounds. Additionally, we also screened the EGFR-related ChEMBL dataset due to the fact that SOS1 lead compounds originated from the EGFR project and the similarity of molecular structure between EGFR inhibitors and SOS1 inhibitors. During the VS, we employed the predicted pChEMBL value generated by our model to assess the potential inhibitory activity of each compound and molecules with a predicted pChEMBL value of 7 or higher were classified as ‘hits’. To broaden the exploration of chemical diversity, we further conducted screening within the expansive Chinese National Compound Library (Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China), encompassing a substantial collection of 1487140 compounds. During this screening process, the top 200 compounds were selected for subsequent experimental validation without considering an activity threshold.
582 compounds. Additionally, we also screened the EGFR-related ChEMBL dataset due to the fact that SOS1 lead compounds originated from the EGFR project and the similarity of molecular structure between EGFR inhibitors and SOS1 inhibitors. During the VS, we employed the predicted pChEMBL value generated by our model to assess the potential inhibitory activity of each compound and molecules with a predicted pChEMBL value of 7 or higher were classified as ‘hits’. To broaden the exploration of chemical diversity, we further conducted screening within the expansive Chinese National Compound Library (Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China), encompassing a substantial collection of 1487140 compounds. During this screening process, the top 200 compounds were selected for subsequent experimental validation without considering an activity threshold.
2.5 Protein–protein interaction assay
All the test compounds were provided by the CNCL (https://www.cncl.org.cn/). These compounds were supplied as stock solutions at a concentration of 1 or 5 mg mL−1 and dissolved in DMSO. The inhibitory activity was measured using the KRAS G12C/SOS1 PPI homogeneous time-resolved fluorescence (HTRF) assay at a final concentration of 10 μg mL−1, with BI-3406 (MedChemExpress) as a positive control. For the IC50 measure of potentially active molecules, 1 or 5 mg mL−1 compound stock solution was selected to be gradient diluted at a final concentration of 0.5–50 μg mL−1. Briefly, the binding assay was performed in a white 384-shallow well microplate (PerkinElmer) with a final reaction volume of 20 μl in the binding domain detection buffer: 40 nM GST-tagged KRAS G12C protein (AntibodySystem), 20 nM His-tagged SOS1 protein (Cytoskeleton), 10 μM GTP, test compounds, MAb Anti GST-XL665 and MAb Anti-6HIS Tb cryptate Gold (PerkinElmer) were co-incubated at room temperature for 2 h. Subsequently, the HTRF signals were detected using a microplate reader (TECAN Spark®) with an excitation wavelength of 320 nm and emission wavelengths of 620 and 665 nm, respectively. The percentage inhibition was calculated by comparison with the buffer control group. The IC50 value was calculated by GraphPad Prism software in inhibitor versus normalized response (variable slope) mode.2.6 Docking procedure
To identify the binding conformation of potential ligands in the active site, molecular docking was used to predict the interaction mode and binding energy between the selected ligands and SOS1 crystal structure (6SCM) from Protein Data Bank (PDB) using AutoDockVina4.29 Considering docking precision, semi-flexible docking was conducted, based on a stochastic docking procedure within the predefined docking box. The protein and ligand, formatted in PDBQT, were subjected to docking within a cubic box with specific coordinates: center_x = 7.540, center_y = −30.158, center_z = −43.243, and the dimension of 22.5 Å. AutoDockVina4 was used to reproduce nine distinct poses for each compound, but only the lowest energy constructs were considered as the best binding configuration. The analysis of protein-ligand interactions was conducted using protein–ligand interaction profiler (PLIP).302.7 In silico assessment of drug-like characteristics
Regarding the absorption, distribution, metabolism, excretion, toxicity (ADMET) properties, and pharmacochemical characteristics, the SMILES representation of the top molecule was submitted to an integrated online ADMET evaluation platform, ADMETlab2.0.313. Results and discussion
3.1 Dataset
Data collection from the ChEMBL database retrieved a total of 861 related molecules. Rigorous data curation protocols including data cleaning and duplicate removal resulted in 375 distinct molecules, which were used to generate molecular descriptors and construct the regression models. In order to model a QSAR well using regression models, it is desirable for the collection of molecules to encompass a broad range of inhibitory activities. Our data span a range of activities from 2.72 to 9.00. When the activity threshold is set to 7, a total of 214 compounds, representing 57% of the entire dataset, would be regarded as active, and this proportion is slightly higher than that of the inactive compounds.3.2 t-SNE analysis
To explore the distribution of the dataset, t-distributed stochastic neighbour embedding (t-SNE), a data visualization method, is applied to the dataset with the ECFP molecular representation to visualize the spread and clustering of compounds. As shown in Fig. 3, two distinct clusters (orange and blue) are evident, and data points in the same cluster showed a small Euclidean distance between adjacent sets of data points. Such tight clusters with clear boundaries indicated the significant structural difference between active and inactive molecules. By examining the molecular structures in these two groups, we found that different from the orange cluster which contains different core structures, molecules in the blue cluster share similar core structures. As the molecules with similar outputs (pChEMBL values) cluster well in the 2D space obtained using t-SNE here, the analysis suggests that there are some potential patterns in the structures of active molecules that could be potentially recognized through ML models. Motivated by this t-SNE plot, we proceeded to construct regression models to discover the patterns embedded in the distribution of biologically active chemicals against SOS1.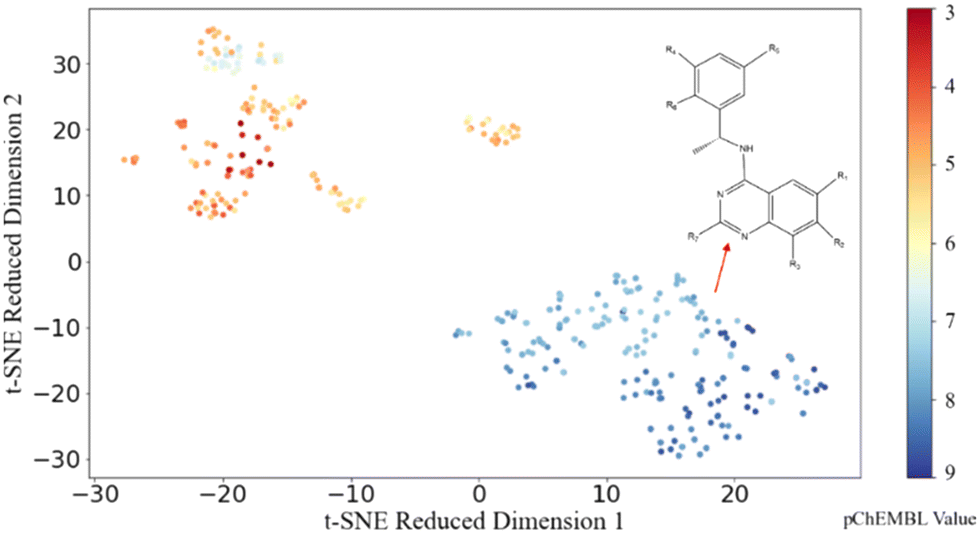 | ||
| Fig. 3 Visualization of the dataset using t-SNE. The colour bar represents pChEMBL values. | ||
3.3 Model validation and model selection
The statistical performance of the ten studied regression models is shown in Table 1. As indicated by their R2 and RMSE values, all these models showed relatively good prediction power (please refer to Table S1 in the ESI† for specific optimization parameters). Specifically, the RF-derived model outperformed others on the test set, achieving the lowest RMSE of 0.441 and the highest R2 of 0.918. Thus, the RF model was taken forward for the VS. Noticing that a relatively high standard deviation of 0.0596 for RF's test RMSE, which could indicate some potential variability in the prediction, to validate the model further, we carried out model reconstruction based on a subset of the whole data and conducted simulated VS using the residual dataset.| Algorithm | Train R2 | Test R2 | Train RMSE | Test RMSE |
|---|---|---|---|---|
| Decision tree | 0.985 (0.0023) | 0.844 (0.0426) | 0.191 (0.0154) | 0.605 (0.0923) |
| Extra trees | 0.994 (0.0012) | 0.847 (0.0365) | 0.119 (0.0134) | 0.600 (0.0765) |
| AdaBoost | 0.937 (0.0050) | 0.899 (0.0176) | 0.392 (0.0161) | 0.489 (0.0548) |
| Ridge | 0.989 (0.0014) | 0.901 (0.0148) | 0.164 (0.0103) | 0.484 (0.0456) |
| SVR | 0.989 (0.0017) | 0.904 (0.0167) | 0.165 (0.0127) | 0.477 (0.0539) |
| K-neighbors | 0.994 (0.0012) | 0.905 (0.0169) | 0.119 (0.0134) | 0.475 (0.0498) |
| Gradient boosting | 0.977 (0.0023) | 0.910 (0.0201) | 0.238 (0.0113) | 0.463 (0.0596) |
| Lasso | 0.943 (0.0033) | 0.912 (0.0154) | 0.373 (0.0109) | 0.457 (0.0546) |
| Elastic net | 0.952 (0.0029) | 0.915 (0.0157) | 0.343 (0.0102) | 0.451 (0.0541) |
| Random Forest | 0.983 (0.0014) | 0.918 (0.0179) | 0.205 (0.0084) | 0.441 (0.0596) |
The performance of ten reconstructed models using 90% of the reshuffled entire data may be found in the ESI† (Table S2), and the evaluation revealed that the predictive power of the RF model remained consistently superior. The simulated screening test on the labelled dataset showed that RF can successfully screen out 27 active molecules from 10% reserved dataset (38 molecules) and accurately identify the 55 out of 64 active molecules from the 94 SOS1-related compounds, utilizing a threshold based on the experimentally determined pChEMBL value of 7. As shown in the supporting information (Table S3†), the difference between predicted and actual activity values of compounds from the 10% reserved data is fairly small, with the maximum difference being 0.86. The reconstitution of models and their evaluation on a validation set provide insights into the model's uncertainty and an understanding of the model's overall performance, assessing the reliability, stability, and generalization capability of the selected model. In addition, when comparing the chemical structure of the external validation dataset molecules with that of the training set molecules, we found that the RF used for the simulated screening identified molecules with slightly different structure from the training set compounds and accurately predicted their activity. For instance, for the novel scaffold inhibitors reported by He et al.15 (as shown in 37 in Fig. 2.), our model could successfully pick out such tetracyclic quinazoline SOS1 inhibitors with a high predicted pChEMBL value of 8.29 against the actual value of 8.33, even though they were not included in the model development process. Therefore, we conclude that the RF regressor has a reliable predictive ability for SOS1 inhibitory activity, and it can also predict the activity of novel molecules with molecular backbones different from those in the training dataset.
3.4 Virtual screening of chemical libraries
The RF model was used as a filtering tool to predict promising candidates as potential SOS1 inhibitors from commercially available molecule databases. Employing a pChEMBL value 7 as a cut-off, a total of four hits from the L4000 small database and nine hits from EGFR ChEMBL were predicted to be active. A summary, including detailed information on these molecules resulting from the initial VS process, is presented in Tables 2 and 3.
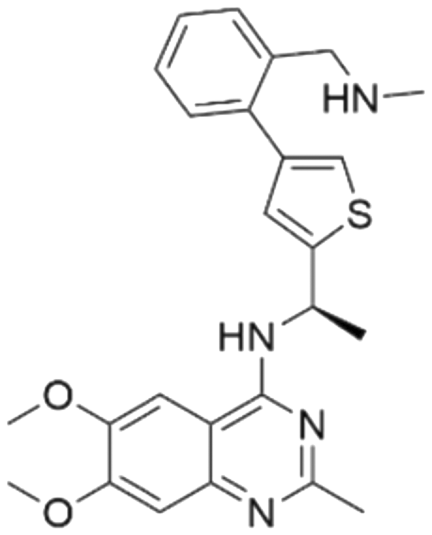
| Series | Compound ID | Structure | Predicted pChEMBL | Actual pChEMBL | Actual IC50 (nM) | Ref. |
|---|---|---|---|---|---|---|
| 1 | CHEMBL5084571 |
|
7.73 | 6.97 | 106 | 33 |
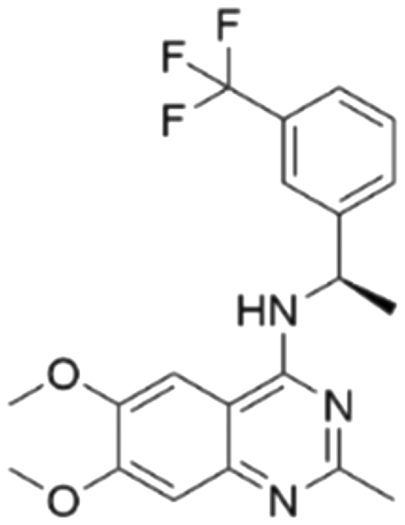
| 2 | CHEMBL4436731 |
|
7.71 | 6.04 | 901 | 34 |
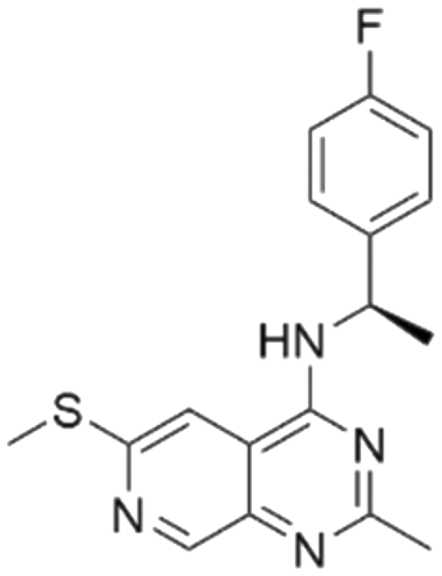
| 3 | CHEMBL5087955 |
|
7.63 | 6.34 | 453 | 33 |
| 4 | CHEMBL4532624 |
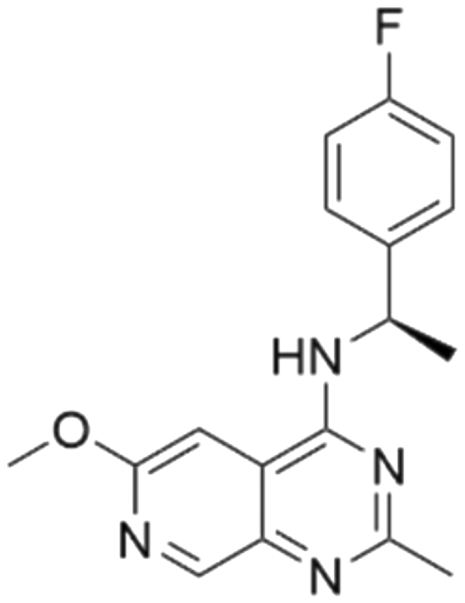
|
7.60 | 5.60 | 2510 | 34 |
| 5 | CHEMBL4445586 |
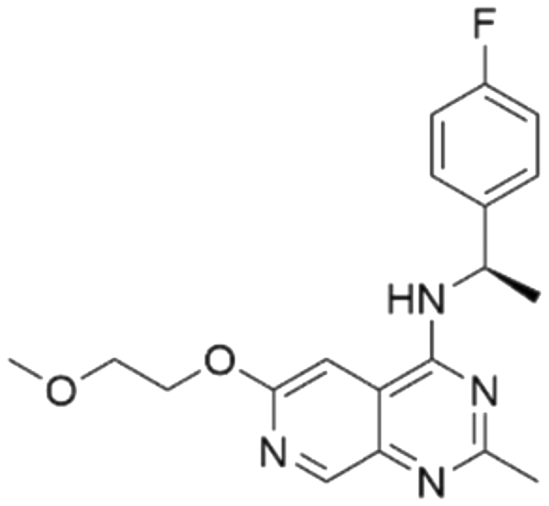
|
7.58 | 6.03 | 934 | 34 |
| 6 | CHEMBL4545109 |
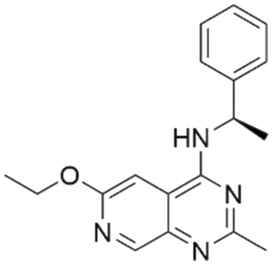
|
7.53 | 5.69 | 2060 | 34 |
| 7 | CHEMBL5085531 |
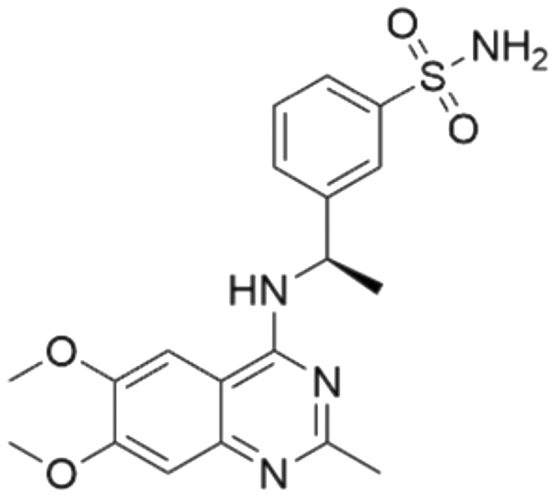
|
7.50 | 7.25 | 56 | 33 |
| 8 | CHEMBL4456281 |
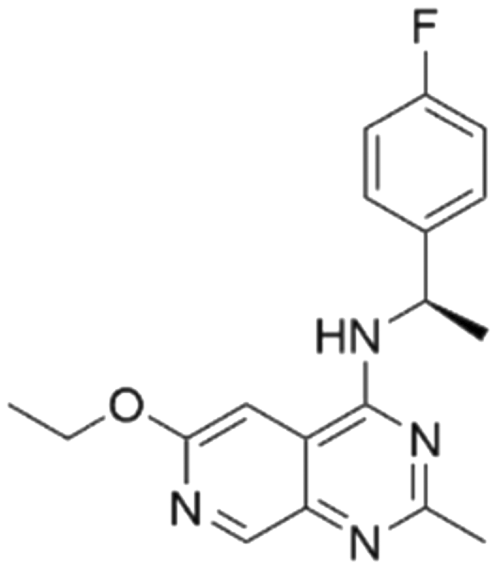
|
7.47 | 5.03 | 9300 | 34 |
| 9 | CHEMBL4463000 |
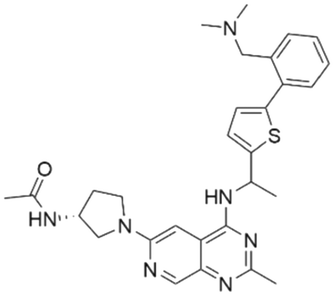
|
7.15 | 7.82 | 15 | 34 |
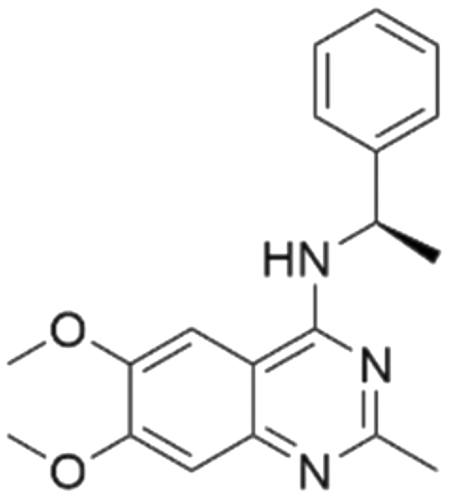
Among those screened molecules, BI-3406 and BAY-293 were reported to be SOS1 inhibitors.10,11 We found that four compounds' predicted pChEMBL values were very close to their actual ones, i.e., 8.23 and 8.30 (5 nM) for BI-3406, 7.39 and 7.68 (21 nM) for BAY-293, 7.50 and 7.25 (56 nM) for CHEMBL5085531, and 7.15 and 7.82 (15 nM) for CHEMBL4463000, respectively. Noteworthily, we also identified eight compounds with novel inhibitor skeletons from those hits, with six of them sourced from the EGFR database (CHEMBL4436731, CHEMBL4532624, CHEMBL4445586, CHEMBL4545109, CHEMBL4456281, and CHEMBL4463000), exhibiting moderate activity as SOS1 inhibitors, and two of them, I-37 and I-49, derived from an SOS1-related patent, although the patent does not provide quantitative activity information.32 Further structural analysis revealed that in contrast to known amino quinazoline inhibitors, those eight hits all have a pyrido pyrimidine scaffold, which did not appear in the training set. This structure has been designed and synthesized recently by Liu et al.35 as a novel potent SOS1 inhibitor pharmacophore. Furthermore, we compare the structural similarity of I-37 and I-49 with the training set based on the Tanimoto coefficient. The above two molecules did not share a high structural similarity, with Tanimoto coefficients of 0.439 for I-37 and 0.433 for I-49. The limited similarity may be attributed to the absence of a quinazoline core, unlike the parent structure in the active compounds of the training set. In general, regression models are most likely to find highly analogous compounds or “me-too” hits, but using Tanimoto scores as a reference, the identified molecules were found to be structurally distinct from known SOS1 inhibitors, which revealed that the ML model in our study is capable of mining SOS1 inhibitors with novel chemical structures. Based on the insights from earlier investigations and exploring a broader chemical space, screening the large CNCL database helps us complement our VS efforts and identify novel SOS1 inhibitors. The top-ranked 200 promising compounds from the CNCL were subsequently submitted for further biological evaluation.
3.5 Biological activity test analysis
To validate our predictions, we measured experimentally the biochemical activity. During this screening process, we employed the HTRF PPI assay to evaluate the inhibitory effects of the compounds on the interaction between KRAS G12C and SOS1. We selected BI-3406 as the positive control, and an IC50 of 31 nM was detected, which closely aligns with literature reports. It is worth noting that the Z′-score for this assay method is 0.95, exceeding the threshold of 0.5, indicating the reliability and accuracy of this assay. Out of the ranked top 200 compounds, we found nine of them exhibited modest inhibition at the final concentration of 10 μg mL−1 primary screening. Subsequently, we conducted activity confirmation assays at 25 μg mL−1 for these nine candidates (Table 4). The resulting four compounds with carboxylic acid backbone showed slightly stronger inhibitory activity against SOS1-KRAS PPI. Dose–response testing was performed on such potential hits, among which CL01545365 was the most potent with an IC50 value close to 20 μg mL−1. Collectively, we identified a novel structural series capable of disrupting the SOS1-KRAS interaction.| Series | Compound ID | Structure | Inhibition rate at 25 μg mL−1 (%) | IC50 (μg mL−1) | Predicted pChEMBL | Docking energy (kcal mol−1) |
|---|---|---|---|---|---|---|
| a The half-inhibitory concentration is calculated using the molecular weight of each compound. b Not detected due to low activity. | ||||||
| 1 | CL01545444 |
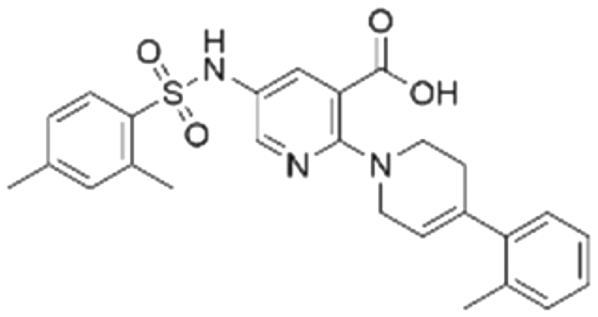
|
31.1 ± 1.4 | 49.2 (103 μMa) | 5.83 | −8.5 |
| 2 | CL01545464 |
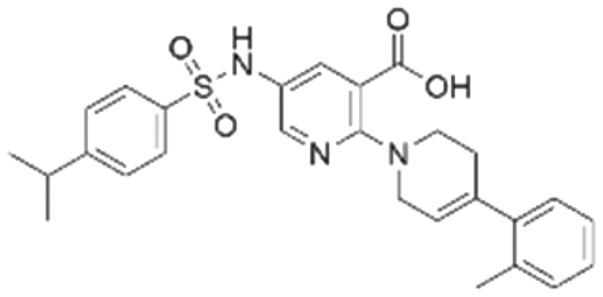
|
42.9 ± 3.4 | 32.6 (66.3 μMa) | 5.96 | −8.4 |
| 3 | CL01545365 |
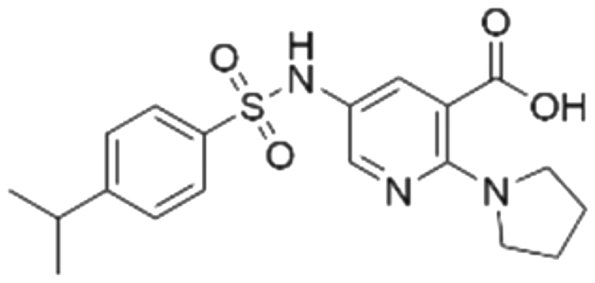
|
53.8 ± 1.6 | 20.9 (53.7 μMa) | 5.81 | −7.1 |
| 4 | CL01545355 |
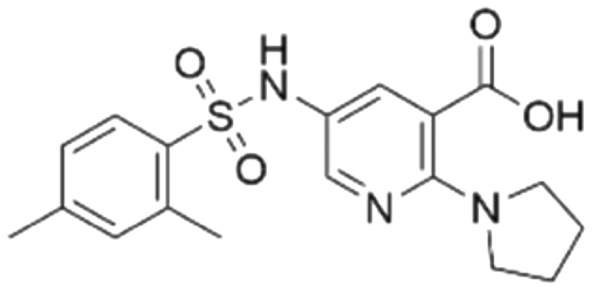
|
38.0 ± 3.3 | 35.5 (94.6 μMa) | 5.80 | −8.3 |
| 5 | CL00838284 |
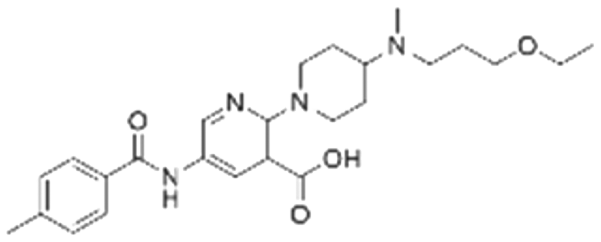
|
25.3 ± 0.3 | NDb | 5.83 | −7.7 |
| 6 | CL01132463 |
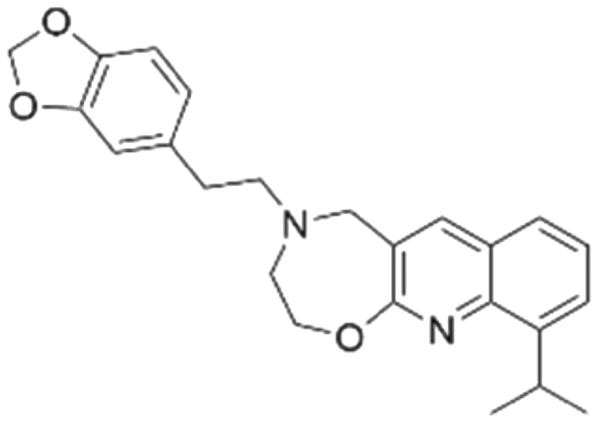
|
−6.5 ± 2.5 | NDb | 6.05 | −8.8 |
| 7 | CL00838287 |
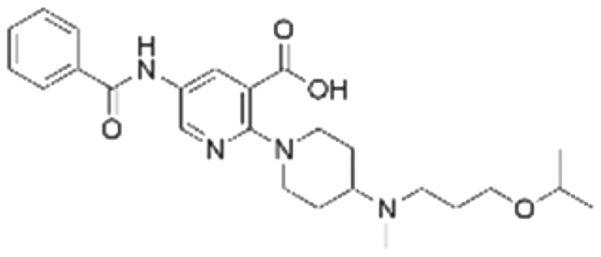
|
6.3 ± 6.6 | NDb | 5.85 | −7.0 |
| 8 | CL00817024 |
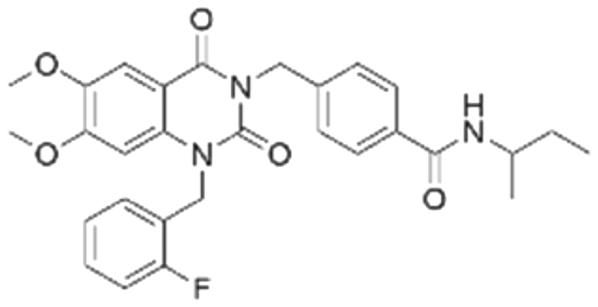
|
2.0 ± 0.8 | NDb | 5.82 | −7.9 |
| 9 | CL01027021 |
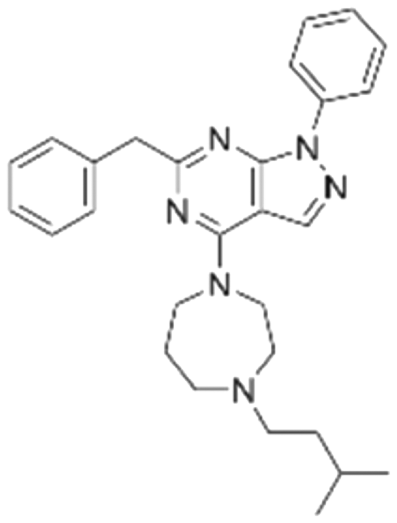
|
2.1 ± 1.4 | NDb | 6.07 | −7.8 |
3.6 Binding mode analysis of selected molecules
The binding mode analysis of hit compounds and the target is carried out with respect to the known inhibitor, BI-3406. We observed that among the nine compounds selected from biological tests, two were confined within the same binding groove of the SOS1 crystal structure (CL01132463 and CL01545355), while the remaining seven were found in binding pockets that are either identical to or in close proximity to the binding site of molecule CL01545365 (See Table 2 for structural information, Fig. 4 for a comparison of docking modes, and Fig. S1† for the 2D receptor–ligand interaction).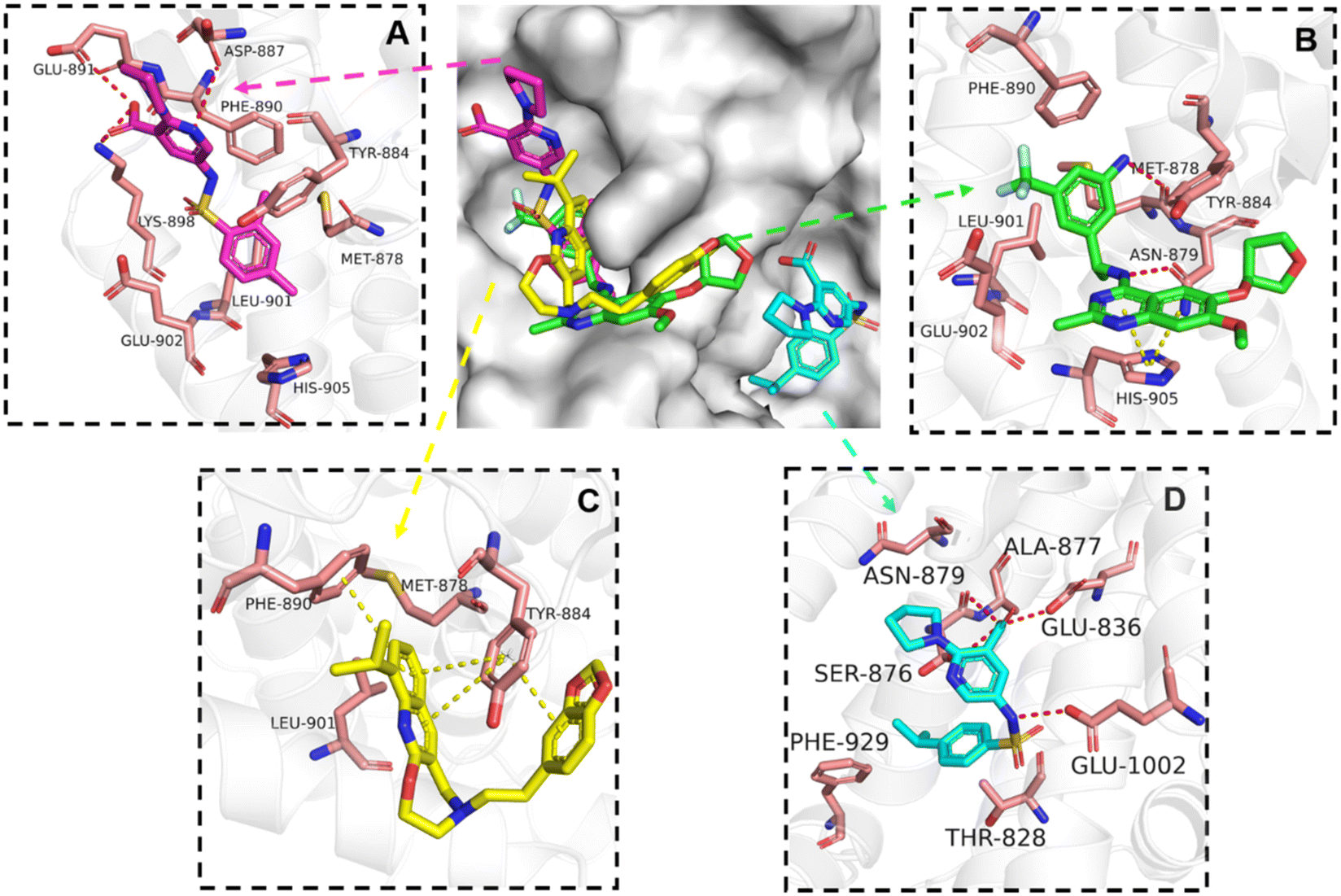 | ||
| Fig. 4 Interaction mode comparison of the hit compound and the known inhibitor against SOS1 protein (PDB:6SCM). (A) CL01545355; (B) BI-3406; (C) CL01132463; (D) CL01545365; the red dashed line represents the hydrogen bond interaction, and the yellow dashed line represents pi stacking; the protein-ligand interactions were analysed by PLIP (protein–ligand interaction profiler).31 | ||
The docking studies demonstrated that the predicted binding energy (−8.8 kcal mol−1) for compound CL01132463 against 6SCM was higher than other hits. Such strong binding is mainly attributed to a series of pi stacking interactions between the ligand and residues Tyr884 and Phe890, which anchor it at the ligand-binding pocket of SOS1. Although those two residues form similar hydrophobic interactions, BI-3406 employs a different anchoring method by establishing two pairs of key hydrogen bonds, including a key polar anchor between aniline N–H and Asn879, and interactions between amino substituent at the phenyl moiety and Met878. Moreover, the quinoline group of CL01132463 protrudes into the hydrophobic pocket, forming another pi-stacking interaction with Phe890, while the oxazepino in the middle bends, allowing the ethyl group to interact well with Tyr884, and disrupt the R73 KRAS–Tyr884 binding like BI-3406, which was confirmed to be responsible for inhibitory function. Additionally, a series of hydrophobic interactions also contribute to the binding of CL01132463.
CL01545355, with the fourth-highest estimated binding affinity (−8.3 kcal mol−1), occupies the same binding site as BI-3406 but exhibits a slightly different binding mode. Specifically, by engaging in hydrophobic interactions with Phe890, Tyr884, His905, and Leu901, the dimethylphenyl group is positioned in this hydrophobic area, and three hydrogen bonds are formed between the polar atoms on the niacin group and the surrounding polar protein residues, Asp887, Lys898, and Glu891. Notably, tetramine on pyrrolidine interacts with Asp887, and the carboxylate interacts with Lys898, forming two salt bridges. The hydrogen of the sulfonamide sulfur atom forms a pi-donor hydrogen bond with Phe890.
Interestingly, although CL01545365 with the greatest potency has a similar scaffold structure to CL01545355, molecular docking suggested that it does not bind with the residues known to be involved in binding with known SOS1 inhibitors. Instead, by establishing three hydrogen bonds, docking predicts it to bind to the neighbouring pocket of BI-3406 with a predicted binding energy of −7.7 kcal mol−1, as depicted in Fig. 4. The different binding energies (−7.7 kcal mol−1versus −8.3 kcal mol−1) observed between the two molecules may suggest distinct modes of interaction with the target, potentially indicating their accommodation within different pockets of 6SCM. The hydrogen atom on the carboxylic acid hydroxyl acts as a donor, forming hydrogen bond interactions with Glu836, Ser876 and Ala877, respectively, and a similar strong binding pattern can also be observed between the sulfonyl amide group and Glu1002 (1.79 Å). The dimethyl phenyl substituent forms two pi-alkyl interactions with Phe929, and other dominant nonpolar contributions including benzene ring binding to Thr828 and Ser876, were also identified as a factor for the anchoring of CL01545365. In comparison, the critical hydrogen bond formed by BI-3406 with Asn879 was replaced by a hydrophobic interaction in CL01545365. Importantly, although compound CL01545365 showed no interaction with Tyr884, which is believed to be crucial for the previous SOS1 inhibitory activity, biological activity testing revealed its ability to disrupt the interaction between SOS1 and KRAS at a micro-molar level. This implies that the binding site associated with this compound is likely to serve as a promising target for developing novel SOS1 inhibitors. Moreover, the docking suggested that CL01545365 fills a shallow and wide pocket, which may reflect its relative low affinities in the predicted VINA scoring function. With the substantial structural difference from BI-3406, the CL01545365 binding mode does not fully overlap with the BI-3406 docked conformation. The comparable affinity reinforces the potential of such novel chemical structures for further development.
3.7 In silico evaluation of drug-like properties
The in silico ADMET of the top compound (CL01545365) was estimated to predict its pharmacochemical characteristics. As shown in Table S5,† all its properties fall within acceptable limits, except for the logP of 4.128, which is slightly larger than the maximum recommended value of 3. In addition, other drug-likeness parameters, including the MDCK permeability of 1.1 × 10−5 cm s−1 and volume of distribution of 0.245 L kg−1, are situated within the ideal range of 2 to 20 × 10−6 cm s−1 and 0.04 to 20 respectively. These predictions collectively suggest that molecule CL01545365 possesses a relatively good pharmacokinetic profile and basically satisfies Lipinski's rule of five with only one violation, demonstrating its potential as a foundational candidate for the development of an SOS1 inhibitors. However, it is worth noting that while several ADMET properties of this molecule may not currently align with established drug-likeness criteria, further implementation of assays for physicochemical property evaluation (such as the Caco-2 cells assay for intrinsic permeability assessment, cardiotoxicity assay, etc.) should be undertaken to provide more accurate and pertinent information on this compound.
SOS1, a crucial protein within the RAS pathway, has recently garnered increasing attention as a promising strategy for treating RAS-driven cancers. However, the development of effective and selective SOS1 inhibitors remains a challenge. In our study, we employed LBVS combined with ML models to identify novel SOS1 inhibitors. We utilized the ChEMBL database for SOS1 as the target, which encompassed a diverse set of molecules with broad-ranging activity. Through the application of ML algorithms, such as SVR, RF Regressor, and Ridge Regressor, we evaluated and compared their performance in predicting the activity of SOS1 inhibitors. Among these algorithms, the RF model with the highest R2 and lowest RMSE in the test set demonstrated robustness and accuracy, making it the optimal choice for VS. Using the RF model, we screened commercially available compound databases. From the L4000 database and EGFR relevant dataset, we identified several inhibitory compounds reported in the literature, such as documented inhibitors BAY-293 and BI-3406, and molecules previously reported in a patent, including I-37 and I-49. Our findings also revealed that a subset of the above-screened molecules exhibited similar pharmacophore structures to the training set and positive controls but with distinct scaffolds, which highlights the potential of RF in discovering SOS1 inhibitors with different core structures. Building on the insights gained from the aforementioned results, we extended our screening efforts to the broader chemical space CNCL, encompassing more than 1.4 million compounds. From the pool of the 200 top-ranked molecules, we successfully identified nine candidate compounds exhibiting entirely distinct scaffolds from acknowledged SOS1 inhibitors, illustrating the novelty of the discovered inhibitors in this work. Represented by CL01545365, which has carboxylic acid as the skeleton, this type of molecule displayed moderate potency in the subsequent biological inhibitory activity assay: SOS1-mediated protein–protein interaction. Molecular docking indicated the presence of advantageous binding configurations, relatively strong binding energies and an unprecedented docking conformation, while an assessment of their suitability as drug candidates indicated their comparatively good drug-like properties, further supporting its potential as a SOS1 inhibitor. These compounds represent a prospective reservoir of novel molecular frameworks demonstrating efficacy against SOS1, thereby contributing to the design and refinement of related SOS1 inhibitors.
Several ongoing clinical trials are assessing the effectiveness of SOS1 inhibitors in cancer treatment. Specifically, these trials involve investigating BI 1701963 as monotherapy and in combination with adagrasib (NCT04975256), or BI 1823911 (NCT04973163), or MEK inhibitor trametinib (NCT04111458)36 or irinotecan (NCT0462742)37 for treating patients with KRAS mutated solid tumours. Thus, the utilization of CL01545365 in conjunction with other chemotherapeutic drugs targeting the RAS oncogenic driver pathway aims to achieve enhanced therapeutic efficacy. Of course, there are some the limitations to our study. Data-driven AI models rely on a substantial volume of high-quality data as input for effective model training. Thus, a more extensive and diverse dataset of SOS1-related compounds would likely improve the predictive accuracy of the model. To mitigate the issue of data scarcity, a pre-trained model or transfer learning paradigm could be explored, and might enhance the model performance and reducing training time. We considered a single type of molecular representation for model construction; future work may incorporate other representations and fusion of the resultant composite features into deep learning models. Other future computational investigation will employ atomistic molecular dynamics simulation, to consider the pertinent dynamic interactions and conformational changes and go beyond the docking insights based on the static binding. Finally, experimental verification for the predicted drug-likeness properties of the lead compound is needed.
4. Conclusions
Overall, our research has revealed the potential of AI-driven LBVS as a valuable tool in discovering effective SOS1 inhibitors. By leveraging computational approaches, we have successfully identified and characterized lead compounds with new chemotype from databases, making a positive contribution to expanding the SOS1 inhibitor repertoire. These findings offer valuable insights for subsequent investigations and underscore new opportunities for therapeutic intervention in RAS-driven cancers.Abbreviations
The following abbreviations are used in this manuscript:| ADMET | Absorption, distribution, metabolism, excretion, and toxicity |
| AI | Artificial intelligence |
| CNCL | Chinese National Compound Library |
| ECFP | Extended-connectivity fingerprints |
| GDP | Guanosine diphosphate |
| GTP | Guanosine triphosphate |
| HTRF | Homogeneous time-resolved fluorescence |
| LBVS | Ligand-based virtual screening |
| ML | Machine learning |
| PDB | Protein Data Bank |
| PLIP | Protein–ligand interaction profiler |
| QSAR | Quantitative structure–activity relationship |
| RAS | Rat sarcoma virus |
| RF | Random forest |
| SMILES | Simplified molecular input line entry system |
| SOS1 | Son of sevenless 1 |
| SVR | Support vector regressor |
| t-SNE | t-distributed stochastic neighbour embedding |
| VS | Virtual screening |
Software and data availability
The data and code used in this study are available online at: https://github.com/cristinaduo/ML-for-SOS1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ningbo Science and Technology Bureau under CM2025 programme (Grant No. 2020Z092), Ningbo Natural Science Foundation Programme (Grant No. 2022 J171), National Natural Science Foundation of China (Grant No. 22171153 & 21502101), Ministry of Science and Technology of the People's Republic of China under funding scheme National Key R&D Program of Intergovernmental Kay Projects (Grant No. 2018YFE0101700), and Ningbo Municipal Key Laboratory on Clean Energy Conversion Technologies (Grant No. 2014A22010) as well as the Zhejiang Provincial Key Laboratory for Carbonaceous Waste Processing and Process Intensification Research funded by the Zhejiang Provincial Department of Science and Technology (Grant No. 2020E10018). Professor Hirst acknowledges support from the Department of Science, Innovation and Technology (DSIT) and the Royal Academy of Engineering under the Chairs in Emerging Technologies scheme. The authors also acknowledge The University of Nottingham, England, and the University of Nottingham Ningbo China for the use of their High-Performance Computing facilities.References
- A. R. Moore, S. C. Rosenberg, F. McCormick and S. Malek, Nat. Rev. Drug Discovery, 2020, 19, 533–552 CrossRef CAS PubMed.
- S. R. Punekar, V. Velcheti, B. G. Neel and K.-K. Wong, Nat. Rev. Clin. Oncol., 2022, 19, 637–655 CrossRef CAS PubMed.
- A. D. Cox, S. W. Fesik, A. C. Kimmelman, J. Luo and C. J. Der, Nat. Rev. Drug Discovery, 2014, 13, 828–851 CrossRef CAS PubMed.
- B. Papke and C. J. Der, Science, 2017, 355, 1158–1163 CrossRef CAS PubMed.
- F. McCormick, Clin. Cancer Res., 2015, 21, 1797–1801 CrossRef CAS PubMed.
- J. Gureasko, W. J. Galush, S. Boykevisch, H. Sondermann, D. Bar-Sagi, J. T. Groves and J. Kuriyan, Nat. Struct. Mol. Biol., 2008, 15, 452–461 CrossRef CAS PubMed.
- H. Sondermann, S. M. Soisson, S. Boykevisch, S.-S. Yang, D. Bar-Sagi and J. Kuriyan, Cell, 2004, 119, 393–405 CrossRef CAS PubMed.
- D. Kessler, D. Gerlach, N. Kraut and D. B. McConnell, Curr. Opin. Chem. Biol., 2021, 62, 109–118 CrossRef CAS PubMed.
- G. Luo, B. Wang, Q. Hou and X. Wu, J. Med. Chem., 2023, 66, 4324–4341 CrossRef CAS PubMed.
- R. C. Hillig, B. Sautier, J. Schroeder, D. Moosmayer, A. Hilpmann, C. M. Stegmann, N. D. Werbeck, H. Briem, U. Boemer, J. Weiske, V. Badock, J. Mastouri, K. Petersen, G. Siemeister, J. D. Kahmann, D. Wegener, N. Böhnke, K. Eis, K. Graham, L. Wortmann, F. von Nussbaum and B. Bader, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2551–2560 CrossRef CAS PubMed.
- M. H. Hofmann, M. Gmachl, J. Ramharter, F. Savarese, D. Gerlach, J. R. Marszalek, M. P. Sanderson, D. Kessler, F. Trapani and H. Arnhof, others, Cancer Discovery, 2021, 11, 142–157 CrossRef CAS PubMed.
- J. M. Ketcham, J. Haling, S. Khare, V. Bowcut, D. M. Briere, A. C. Burns, R. J. Gunn, A. Ivetac, J. Kuehler, S. Kulyk, J. Laguer, J. D. Lawson, K. Moya, N. Nguyen, L. Rahbaek, B. Saechao, C. R. Smith, N. Sudhakar, N. C. Thomas, L. Vegar, D. Vanderpool, X. Wang, L. Yan, P. Olson, J. G. Christensen and M. A. Marx, J. Med. Chem., 2022, 65, 9678–9690 CrossRef CAS PubMed.
- A. Buckl, E. Quintana, G. J. Lee, N. Shifrin, M. Zhong, L. S. Garrenton, D. C. Montgomery, C. Stahlhut, F. Zhao and D. M. Whalen, others, Cancer Res., 2021, 81, 1273–1273 CrossRef.
- S. Zhang, Y. Zhang, X. Chen, J. Xu, H. Fang, Y. Li, Y. Liu and H. He, J. Med. Chem., 2022, 65, 15856–15877 CrossRef CAS PubMed.
- H. He, Y. Zhang, J. Xu, Y. Li, H. Fang, Y. Liu and S. Zhang, J. Med. Chem., 2022, 65, 13158–13171 CrossRef CAS PubMed.
- A. S. Rifaioglu, H. Atas, M. J. Martin, R. Cetin-Atalay, V. Atalay and T. Doğan, Briefings Bioinf., 2019, 20, 1878–1912 CrossRef CAS PubMed.
- J. Vamathevan, D. Clark, P. Czodrowski, I. Dunham, E. Ferran, G. Lee, B. Li, A. Madabhushi, P. Shah, M. Spitzer and S. Zhao, Nat. Rev. Drug Discovery, 2019, 18, 463–477 CrossRef CAS PubMed.
- S. Pandiyan and L. Wang, Comput. Biol. Med., 2022, 150, 106140 CrossRef CAS.
- J. Bajorath, Nat. Rev. Drug Discovery, 2002, 1, 882–894 CrossRef CAS PubMed.
- M. A. Johnson and G. M. Maggiora, Concepts and Applications in Molecular Similarity, Wiley, New York, 1990 Search PubMed.
- E. Valentini, S. D'Aguanno, M. Di Martile, C. Montesano, V. Ferraresi, A. Patsilinakos, M. Sabatino, L. Antonini, M. Chiacchiarini, S. Valente, A. Mai, G. Colotti, R. Ragno, D. Trisciuoglio and D. D. Bufalo, Theranostics, 2022, 12, 2427–2444 CrossRef CAS PubMed.
- S. Yang, S. Li and J. Chang, RSC Adv., 2022, 12, 13500–13510 RSC.
- J. Zhou, S. Wu, B. G. Lee, T. Chen, Z. He, Y. Lei, B. Tang and J. D. Hirst, Molecules, 2021, 26, 7492 CrossRef CAS PubMed.
- H. Zhang, W. Liu, Z. Liu, Y. Ju, M. Xu, Y. Zhang, X. Wu, Q. Gu, Z. Wang and J. Xu, MedChemComm, 2018, 9, 937–945 RSC.
- P. Carracedo-Reboredo, J. Liñares-Blanco, N. Rodríguez-Fernández, F. Cedrón, F. J. Novoa, A. Carballal, V. Maojo, A. Pazos and C. Fernandez-Lozano, Comput. Struct. Biotechnol. J., 2021, 19, 4538–4558 CrossRef CAS.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS.
- G. Landrum, RDKit: Open-Source Cheminformatics Software Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
- M. F. Adasme, K. L. Linnemann, S. N. Bolz, F. Kaiser, S. Salentin, V. J. Haupt and M. Schroeder, Nucleic Acids Res., 2021, 49, W530–W534 CrossRef CAS PubMed.
- G. Xiong, Z. Wu, J. Yi, L. Fu, Z. Yang, C. Hsieh, M. Yin, X. Zeng, C. Wu and A. Lu, others, Nucleic Acids Res., 2021, 49, W5–W14 CrossRef CAS PubMed.
- J. Ramharter, C. Kofink, H. Stadtmueller, T. Wunberg, M. H. Hofmann, A. Baum, M. Gmachl, D. L. Rudolph, F. Savarese, M. Ostermeier, M. Frank, A. Gille, S. Goepper, M. Santagostino and J. Wippoch, Novel benzylamino substituted pyridopyrimidinones and derivatives as Sos1 inhibitors, WO/2019122129, 2019.
- J. Ramharter, D. Kessler, P. Ettmayer, M. H. Hofmann, T. Gerstberger, M. Gmachl, T. Wunberg, C. Kofink, M. Sanderson, H. Arnhof, G. Bader, K. Rumpel, A. Zöphel, R. Schnitzer, J. Böttcher, J. C. O'Connell, R. L. Mendes, D. Richard, N. Pototschnig, I. Weiner, W. Hela, K. Hauer, D. Haering, L. Lamarre, B. Wolkerstorfer, C. Salamon, P. Werni, S. Munico-Martinez, R. Meyer, M. D. Kennedy, N. Kraut and D. B. McConnell, J. Med. Chem., 2021, 64, 6569–6580 CrossRef CAS PubMed.
- G. Rosse, ACS Med. Chem. Lett., 2020, 11, 2–3 CrossRef CAS PubMed.
- M. Liu, G. Zhou, W. Su, Y. Gu, M. Gao, K. Wang, R. Huo, Y. Li, Z. Zhou and K. Chen, others, ACS Med. Chem. Lett., 2023, 14, 183–190 CrossRef CAS PubMed.
- M. L. Johnson, E. Gort, S. Pant, M. P. Lolkema, M. Sebastian, M. Scheffler, J. Hwang, U. Dünzinger, K. Riemann and T. Kitzing, Ann. Oncol., 2021, 32, S591–S592 CrossRef.
- D. Gerlach, M. Gmachl, J. Ramharter, J. Teh, S.-C. Fu, F. Trapani, D. Kessler, K. Rumpel, D.-A. Botesteanu and P. Ettmayer, Cancer Res., 2020, 80, 1091–1091 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00063c |
| ‡ Lihui Duo, Yi Chen, and Qiupei Liu contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |