
In situ self-reconstructed hierarchical bimetallic oxyhydroxide nanosheets of metallic sulfides for high-efficiency electrochemical water splitting†
Yaning
Fan
a,
Junjun
Zhang
 *a,
Jie
Han
*b,
Mengyuan
Zhang
c,
Weiwei
Bao
b,
Hui
Su
d,
Nailiang
Wang
a,
Pengfei
Zhang
*ac and
Zhenghong
Luo
ac
*a,
Jie
Han
*b,
Mengyuan
Zhang
c,
Weiwei
Bao
b,
Hui
Su
d,
Nailiang
Wang
a,
Pengfei
Zhang
*ac and
Zhenghong
Luo
ac
aState Key Laboratory of High-Efficiency Utilization of Coal and Green Chemical Engineering, College of Chemistry and Chemical Engineering, Ningxia University, Yinchuan, Ningxia 750021, China. E-mail: zhangjj089@nxu.edu.cn; pfzhang@nxu.edu.cn
bNational & Local Joint Engineering Laboratory for Slag Comprehensive Utilization and Environmental Technology, School of Material Science and Engineering, Shaanxi University of Technology, Hanzhong, Shaanxi 723000, P. R. China. E-mail: hanjie@snut.edu.cn
cSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai, 200240, P. R. China. E-mail: chemistryzpf@sjtu.edu.cn
dDepartment of Chemistry, FRQNT Centre for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke Street W., Montreal, QC H3A 0B8, Canada
First published on 29th January 2024
Abstract
The advancement of economically efficient electrocatalysts for alkaline water oxidation based on transition metals is essential for hydrogen production through water electrolysis. In this investigation, a straightforward one-step solvent method was utilized to spontaneously cultivate bimetallic sulfide S-FeCo1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1/NIF on the surface of a nickel–iron foam (NIF). Capitalizing on the synergistic impact between the bimetallic constituents and the highly active species formed through electrochemical restructuring, S-FeCo1:1/NIF exhibited remarkable oxygen evolution reaction (OER) performance, requiring only a 310 mV overpotential based on 500 mA cm−2 current density. Furthermore, it exhibited stable operation at 200 mA cm−2 for 275 h. Simultaneously, the catalyst demonstrated excellent hydrogen evolution reaction (HER) and overall water-splitting capabilities. It only requires an overpotential of 191 mV and a potential of 1.81 V to drive current densities of 100 and 50 mA cm−2. Density functional theory (DFT) calculations were also employed to validate the impact of the bimetallic synergistic effect on the catalytic activity of sulfides. The results indicate that the coupling between bimetallic components effectively reduces the energy barrier required for the rate-determining step in water oxidation, enhancing the stability and activity of bimetallic sulfides. The exploration of bimetallic coupling to improve the OER performance holds theoretical significance in the rational design of advanced electrocatalysts.
:1/NIF on the surface of a nickel–iron foam (NIF). Capitalizing on the synergistic impact between the bimetallic constituents and the highly active species formed through electrochemical restructuring, S-FeCo1:1/NIF exhibited remarkable oxygen evolution reaction (OER) performance, requiring only a 310 mV overpotential based on 500 mA cm−2 current density. Furthermore, it exhibited stable operation at 200 mA cm−2 for 275 h. Simultaneously, the catalyst demonstrated excellent hydrogen evolution reaction (HER) and overall water-splitting capabilities. It only requires an overpotential of 191 mV and a potential of 1.81 V to drive current densities of 100 and 50 mA cm−2. Density functional theory (DFT) calculations were also employed to validate the impact of the bimetallic synergistic effect on the catalytic activity of sulfides. The results indicate that the coupling between bimetallic components effectively reduces the energy barrier required for the rate-determining step in water oxidation, enhancing the stability and activity of bimetallic sulfides. The exploration of bimetallic coupling to improve the OER performance holds theoretical significance in the rational design of advanced electrocatalysts.
New conceptsGreen hydrogen production driven by renewable energy is an important means of reducing carbon emissions. The development of efficient catalysts is crucial for lowering the cost of hydrogen production through the electrolysis of water. Monitoring the evolution of catalysts during actual operation is essential for exploring the catalytic mechanism. In this study, we employed a simple solvothermal method to obtain a single phase of cobalt–iron bimetallic sulfides. These sulfides exhibit high catalytic activity and stability in the water-splitting process. It was observed that the sulfide transformed into bimetallic hydroxyl oxide through various in situ and ex situ tactics, which constituted the real active site in the catalytic process. Theoretical analysis indicates that the center of the d band of bimetallic oxyhydroxide is closer to the Fermi level than that of monometallic hydroxyl oxide. Simultaneously, iron sites in bimetallic oxyhydroxide significantly reduce the energy barrier of the key reaction steps during the catalytic process. This study offers a new perspective on the construction of high-performance transition-metal sulfide electrodes through metal coordination and regulation of the reconstruction process. |
1. Introduction
Hydrogen gas (H2), which has a high energy density and a combustion heat value, is deemed the ultimate energy source of the 21st century.1–4 H2 finds application as an ideal energy carrier in methanol production, catalytic hydrogenation, and various industrial sectors.5,6 Thus, H2 is a central material within the energy resources of modern chemical engineering, with the potential to foster and enhance an economically and environmentally friendly society.7 Additionally, it can contribute to the global mission of achieving “carbon peak and carbon neutrality”.8Water electrolysis is considered an efficient and environmentally friendly method for H2 production. Hydrogen production by water electrolysis involves primarily the OER at the anode and the HER at the cathode.9 The OER process involves a four-electron transfer process, which has problems such as slow kinetics and unfavorable thermodynamic conditions, limiting the overall efficiency of electrolytic water. It is considered to be a key reaction in a variety of conversion-related reactions related to renewable energy,10 such as water splitting, the nitrogen reduction reaction (NRR) and the CO2 reduction reaction (CO2RR) in water.11 Consequently, the development of efficient and stable catalysts to reduce overpotentials and accelerate reaction kinetics is an urgent and challenging task. Presently, RuO2, IrO2, and Pt/C are highly commercialized electrocatalysts. The scarcity of precious metal-based catalysts has kept their commercial prices persistently high. Therefore, the development of cheap and stable metal catalysts is of significant importance to reduce the investment in commercial H2 production.12 In recent years, various compounds such as phosphides, borides, nitrides, hydroxides, selenides and sulfides have been widely reported for the alkaline electrolyte OER and bifunctional water electrolysis.13–19 However, recent studies have shown that these compounds exhibit instability at a high anodic potential required for the OER process. They undergo phase transformation to form the corresponding metal (oxy)hydroxides, which should be considered as a truly active component of the OER process. Therefore, the above electrocatalysts can be called ‘precatalysts’, and the phase transition at the anode potential is called self-reconfiguration. These studies reveal the self-reconfiguration chemistry of precatalysts, including the degree of self-reconfiguration and the mechanism of self-reconfiguration.20–24 In addition, elucidating the nature–structure relationship of self-reconfigurable metal (oxy)hydroxides is of great significance for finding efficient OER electrocatalysts and determining the true OER mechanism and process. CoP nanoparticles have been employed as OER catalysts.23 The research explicitly stated that the catalyst's remarkable OER catalytic activity arises from the generation of CoOOH at high anodic potentials. The conclusions were based on the utilization of extended X-ray absorption fine structure (EXAFS) to track the local structural evolution during the catalyst activation process and X-ray photoelectron spectroscopy to investigate the changes in the oxidation state of cobalt. These findings indicate that CoP undergoes self-reconstruction during the water oxidation process, forming highly active in situ transformed CoOOH catalysts. These catalysts not only exhibit excellent OER efficiency but also demonstrate remarkable stability. Similarly, Liu et al. achieved directed reconstruction of co-doped Fe2O3 and Fe3O4 with Zn and S on iron foam (Zn, S-Fe2O3–Fe3O4/IF) into FeOOH (Zn, S-Fe3O4–FeOOH/IF). Researchers substantiated the restructuring during the electrochemical oxidation process using techniques such as X-ray spectroscopy and in situ Raman spectroscopy. The presence of sulfur facilitated iron dissolution, and the co-deposition of zinc regulated the activity of the obtained FeOOH. Furthermore, the existence of Fe3O4 provided stable sites for FeOOH deposition, resulting in the formation of more active FeOOH components.25
Most nonprecious metal OER bimetallic catalysts require the addition of nonmetal elements, like O, S, P, N, etc., to attain thermodynamic structural stability.26 Transition-metal sulfides, in particular, are cost-effective, abundant and possess unique physical and chemical properties, including excellent electrochemical performance and conductivity.27,28 Their good conductivity stems from their ability to maintain rapid charge transfer. In the case of bimetallic catalysts composed of two nonprecious metals, the interactions between the two metals are expected to enhance their inherent catalytic activity, especially bimetallic sulfides with a pyrite structure that exhibit superior catalytic performance in polysulfide electrocatalysis compared to monometallic sulfides. The introduction of the second metal into a monometallic sulfide as a promoter, known as the synergistic effect, impacts the catalyst's activity and selectivity. Three factors dominate the catalytic activity due to the synergistic effect: site blocking, bifunctionality, and ligand effect.29 Site blocking adversely affects the OER catalytic activity. If one metal is proficient at dissociating a molecule while the other metal possesses a particularly suitable binding energy for a reaction, the favorable binding of the two metals can result in a “whole greater than the sum of its parts” effect, thereby reducing the overpotential of the OER process and enhancing the faradaic efficiency, known as bifunctionality.30 The ligand effect refers to the optimization of the electronic structure of the original metal through the addition of a second metal, leading to changes in the binding and the activation energies, which are reflected in the selectivity of the catalytic activity. Moreover, bimetallic electrocatalysts demonstrate unique properties, and various components, crystal structures, and easily accessible electronic states have improved their electrocatalytic activity.
Research on low-cost transition metal-based alkaline OER electrocatalysts has primarily focused on the composition of transition metal bases with anions (B, S, P, N, etc.), quasi-metals (Si, Ge, As, etc.) and poor metals (Al, Ga, Sn, etc.).31,32 However, these compounds are essentially considered precatalysts. In this regard, transition metal-based compounds transform into active transition metal (oxy)hydroxides during the OER process, while nonmetals mainly leach from the catalyst structure, forming active nanodomains and thus enhancing catalytic activity.33,34 Many experimental results confirm that the (oxy)hydroxides generated during the electrooxidation process are amorphous or poorly crystalline, which is favorable for their intrinsic activity in the OER.35 On the basis of this premise, significant low-crystalline active nanodomains need to be established during the OER through phase reconstruction to achieve high catalytic activity. Since the extent of phase reconstruction highly depends on the particle size of the precatalysts, nanoparticle formation has been proven to be a favorable strategy for achieving deep phase reconstruction.34 Effective phase reconstruction should involve not only the deep conversion of precatalysts but also prevent the aggregation of active nanodomains. Therefore, an ideal precatalyst must exist in nanoscale form, allowing sufficient (oxy)hydroxide ligands to bind to metal sites.36 Additionally, precatalysts should contain nonmetals such as sulfur or phosphorus that exhibit a high thermodynamic destabilization tendency for anions during the OER process, and their leaching will generate pores and promote the deep reconstruction of precatalysts.37 Furthermore, prior research suggests that oxygen anions (SO42−, PO42−) formed during precatalyst reconstruction are expected to adsorb on the surface of transition-metal-based (oxy)hydroxides.38,39 Then, they adjust the electronic structure of the catalytic sites to enhance the intrinsic activity of the catalyst.
In addition, the conventional preparation of electrocatalysts involves expensive organic ligands, organic or inorganic templates, intricate ion exchange processes, and subsequent thermal treatments. Consequently, employing a straightforward synthesis method for such catalysts, coupled with the selection of superior conductive substrates and the abandonment of insulating organic binders, has proven effective in enhancing the charge transfer capability of electrocatalysts.40 The utilization of NIF, characterized by its excellent conductivity and high OER activity, makes it an ideal substrate material for the self-growth of catalysts.
On the basis of the aforementioned theoretical considerations, we have developed conventional two- or three-step methods for synthesizing sulfides, instead of a simpler one-step solvothermal approach that facilitates the in situ growth of bimetallic FeCo sulfides on NIF. The S-FeCo1:1/NIF catalyst prepared in this study exhibits outstanding OER, HER, and overall water electrolysis catalytic performances in 1 M KOH. The OER overpotential is measured at 310 mV (j = 500 mA cm−2), the HER overpotential at 197 mV (j = −100 mA cm−2) and the overall water electrolysis potential at 1.81 V (j = 50 mA cm−2). Simultaneously, it demonstrates remarkable electrocatalytic stability and the ability to maintain stability and superior catalytic performance for extended periods under industrial water electrolysis conditions. This excellent catalytic activity arises from the synergistic effect between the bimetallic components that modulates the electronic structure of the catalyst, thereby enhancing the rate of electron transfer and improving the conductivity of the material.
It should be noted that through characterization techniques such as SEM, TEM, XPS, and in situ Raman spectroscopy, we have demonstrated the material surface reconstruction of sulfides during electrooxidation, resulting in the dynamic and stable phase of FeCoOOH as a genuine OER active site. The generation of FeCoOOH provides additional adsorption/desorption sites for active intermediates in the catalytic reaction. DFT calculations were employed to investigate the impact of the synergistic effect between the bimetallic components on the density of states (DOS) and the rate-determining step (RDS) of the OER for the FeCoOOH catalytic site. The results indicate that the synergistic effect between the bimetallic components effectively promotes the d-band center of FeCoOOH toward the Fermi level, accelerating rapid electron transfer, and reducing the reaction energy barrier required for the RDS. This study offers a practical approach for formulating and designing catalysts on a large scale to produce green hydrogen industrially through water splitting employing bimetallic sulfide catalysts.
2. Results and discussion
2.1. Synthesis and characterization
As depicted in Fig. 1a, in contrast to the more intricate two- or three-step methods reported in the literature for sulfide synthesis, the present study employed a straightforward one-step solvothermal approach at 180 °C using Fe(NO3)3·9H2O, Co(NO3)2·6H2O, and thiourea to achieve in situ growth of S-FeCox![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :y/NIF on the surface of NIF. To ascertain the corresponding phases of the synthesized bimetallic sulfides (S-FeCo1:1/NIF, S-FeCo1:2/NIF, and S-FeCo2:1/NIF) and monometallic sulfides (S-Fe/NIF and S-Co/NIF), the powder samples were subjected to X-ray diffraction (XRD) analysis. As illustrated in Fig. 1b, the diffraction peaks in the XRD patterns of S-FeCox:y/NIF, S-Fe/NIF, and S-Co/NIF all exhibited excellent matches with the structures of Co3S4 (PDF#42-1448), Fe3S4 (PDF#16-0713), and Co9S8 (PDF#19-0364), respectively. The diffraction peaks at 2θ = 31.3°, 38.1°, 47.3°, and 55.0° correspond to the (311), (400), (422), and (440) crystal planes of Co3S4 (PDF#42-1448), and similar corresponding crystal planes were observed in the XRD patterns of S-Fe/NIF and S-Co/NIF. Additionally, the exclusive product of monometallic Co–sulfide was identified as Co9S8, while the introduction of equimolar Fe resulted in the transformation of the corresponding sulfide product into Co3S4. This indicates that the addition of Fe significantly alters the coordination environment of Co, thereby favoring the generation of new phases.
:y/NIF on the surface of NIF. To ascertain the corresponding phases of the synthesized bimetallic sulfides (S-FeCo1:1/NIF, S-FeCo1:2/NIF, and S-FeCo2:1/NIF) and monometallic sulfides (S-Fe/NIF and S-Co/NIF), the powder samples were subjected to X-ray diffraction (XRD) analysis. As illustrated in Fig. 1b, the diffraction peaks in the XRD patterns of S-FeCox:y/NIF, S-Fe/NIF, and S-Co/NIF all exhibited excellent matches with the structures of Co3S4 (PDF#42-1448), Fe3S4 (PDF#16-0713), and Co9S8 (PDF#19-0364), respectively. The diffraction peaks at 2θ = 31.3°, 38.1°, 47.3°, and 55.0° correspond to the (311), (400), (422), and (440) crystal planes of Co3S4 (PDF#42-1448), and similar corresponding crystal planes were observed in the XRD patterns of S-Fe/NIF and S-Co/NIF. Additionally, the exclusive product of monometallic Co–sulfide was identified as Co9S8, while the introduction of equimolar Fe resulted in the transformation of the corresponding sulfide product into Co3S4. This indicates that the addition of Fe significantly alters the coordination environment of Co, thereby favoring the generation of new phases.
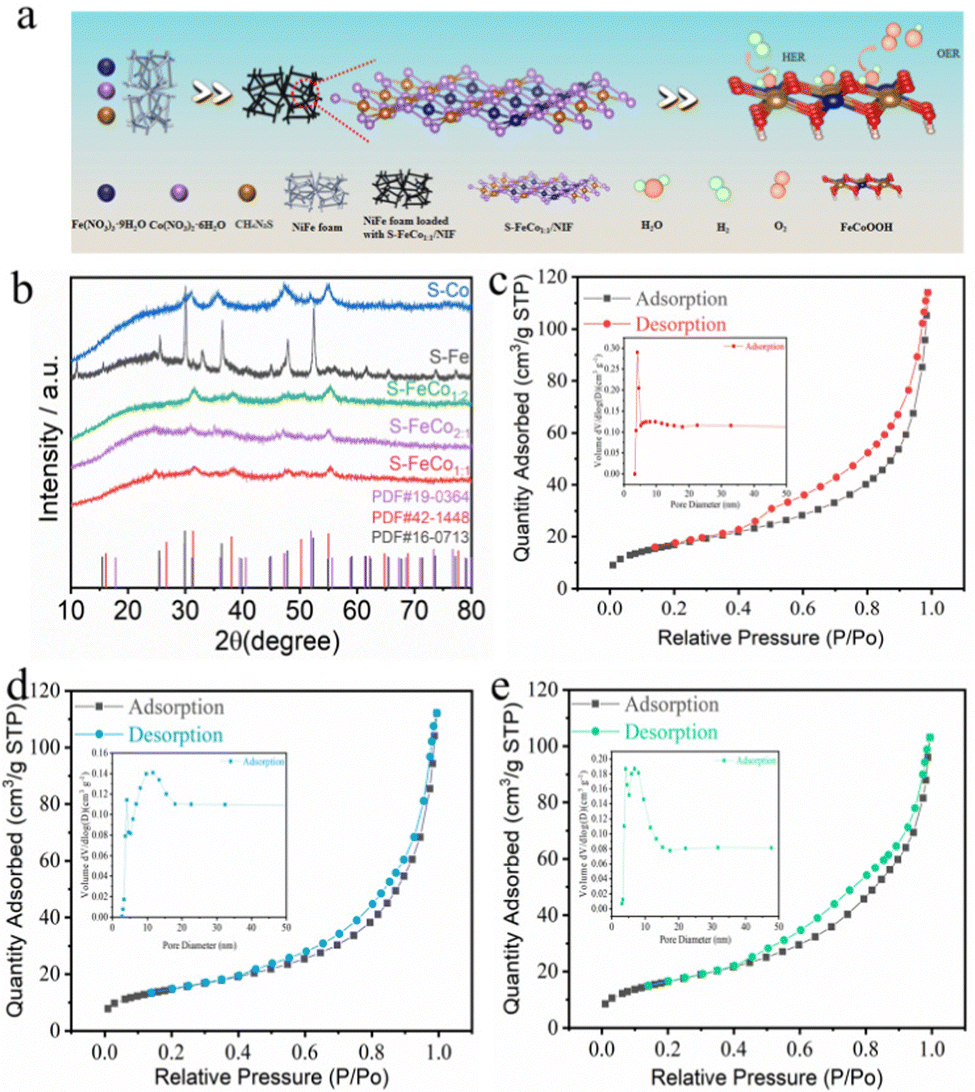 | ||
| Fig. 1 (a) Schematic diagram of the S-FeCo1:1/NIF solvothermal synthesis process. (b) XRD patterns of the S-FeCo1:1, S-FeCo2:1, S-FeCo1:2, S-Fe and S-Co samples. (c)–(e) The N2 adsorption–desorption isotherms and the Barrett–Joyner–Halenda (BJH) pore size distributions for S-FeCo1:1, S-Co and S-Fe samples, respectively. | ||
The surface morphological and microstructural characterization of the S-FeCo1:1/NIF, S-Co/NIF and S-Fe/NIF catalysts were carried out using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The prepared S-FeCo1:1/NIF, S-Co/NIF, and S-Fe/NIF catalysts exhibited a large accumulation of nanospheres on the surface of NIF (Fig. 2a and Fig. S1, ESI†), with the nanospheres assembled from loosely packed nanoparticles (Fig. 2b and Fig. S2, ESI†). The specific surface areas and pore size distributions of the catalyst powders were determined using the Brunauer–Emmett–Teller (BET) method, as illustrated in Fig. 1c–e. The results indicated that the specific surface areas of S-FeCo1:1/NIF, S-Co/NIF and S-Fe/NIF were predominantly 60.40, 59.5, and 52.58 m2 g−1, respectively, with pore size distributions of 11.68, 10.57, and 12.68 nm. This suggests that, under the same conditions, S-FeCo1:1/NIF (60.40 m2 g−1) possessed the maximum specific surface area and a more concentrated mesoporous structure. To demonstrate the hydrophilicity of the bimetallic sulfides, a simplified hydrophilicity test was conducted on the surfaces of the S-FeCo1:1/NIF and bare NIF electrodes. As shown in Fig. S3 (ESI†), when water contacted the S-FeCo1:1/NIF, the solution immediately spread across the catalyst surface. In contrast, when the solution droplets were placed on the NIF surface, they formed water droplets without spreading. This experimental result confirms the good hydrophilicity of the S-FeCo1:1/NIF sample, indicating its strong adsorption capacity for water molecules, which effectively promotes the oxidation efficiency of alkaline solutions.4 The outstanding hydrophilicity and unique microstructure exposed numerous active sites, facilitating the penetration of the electrolyte into the catalyst surface and providing a larger specific surface area and a favorable pore size distribution for the adsorption and desorption of active species. Additionally, it contributed to the deep reconstruction and generation of active species and metal hydroxyoxides, accelerating the OER process.
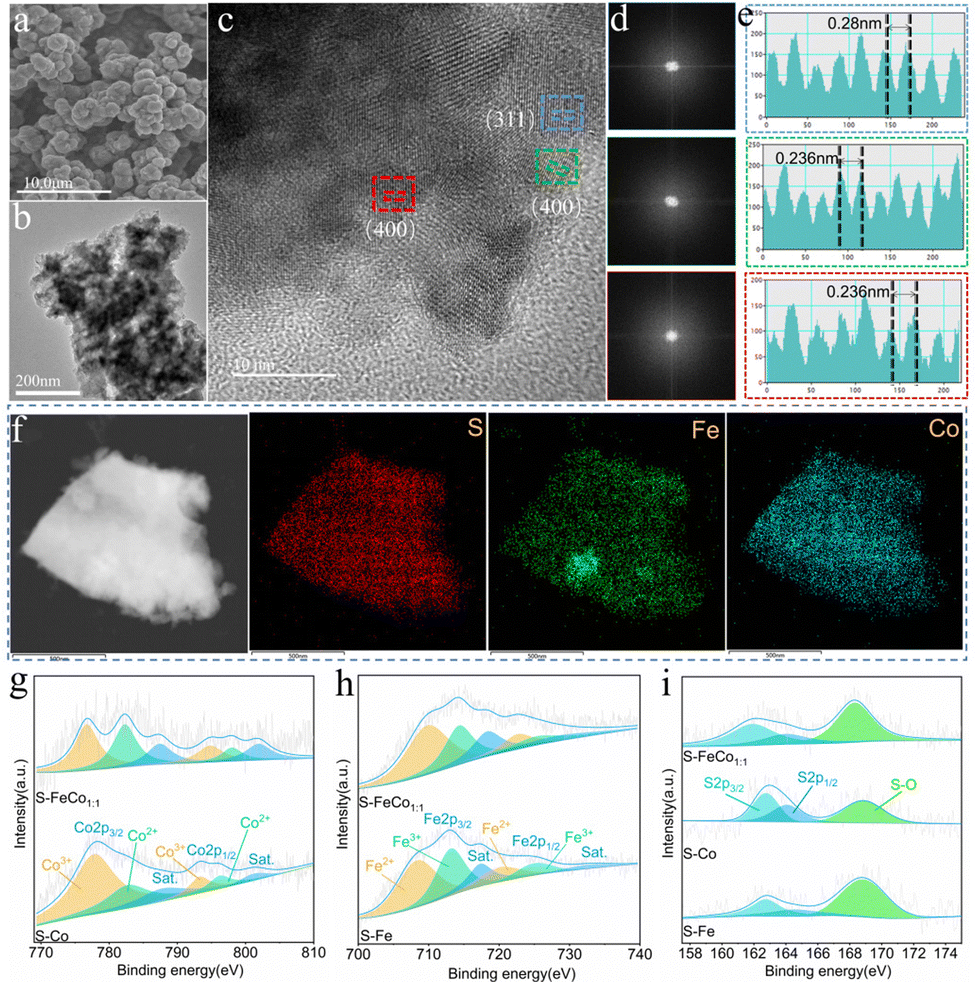 | ||
| Fig. 2 (a) SEM image, (b) TEM image, (c) HRTEM image, (d) fast Fourier transform (FFT) in different regions and (e) corresponding intensity profiles along with the position of S-FeCo1:1/NIF. (f) High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and elemental mapping of S, Fe, and Co. (g) High-resolution Co 2p XPS spectra of S-FeCo1:1/NIF and S-Co/NIF. (h) High-resolution Fe 2p XPS spectra of S-FeCo1:1/NIF and S-Fe/NIF. (i) High-resolution S 2p XPS spectra of S-FeCo1:1/NIF, S-Co/NIF and S-Fe/NIF. | ||
As depicted in Fig. 2c, high-resolution transmission electron microscopy (HRTEM) was used to characterize S-FeCo1:1/NIF. Fast Fourier transform (FFT) analysis of the HRTEM images in Fig. 2d reveals three different FFT patterns extracted from the different regions in Fig. 2c, displaying identifiable diffraction points and indicating their high crystallinity. Moreover, Fig. 2e displays high-resolution lattice fringes measurable at different locations, with interplanar spacings of 0.28 and 0.236 nm, corresponding to the crystal planes (311) and (400) of Co3S4. The lattice distortion of S-FeCo1:1/NIF was further investigated using the inverse fast Fourier transform (IFFT) patterns obtained from the HRTEM image (Fig. S4, ESI†). The corresponding IFFT images showed no significant distortion of the lattice fringes of the (311) and (400) crystal planes. To support this conclusion, the selected area electron diffraction (SAED) patterns also confirmed high crystallinity (Fig. S5, ESI†). The elemental distribution maps in Fig. 2f and Fig. S6 (ESI†) display uniform distributions of S, Fe and Co throughout the sample, indicating a relatively homogeneous distribution of various elements in the catalyst. Furthermore, inductively coupled plasma atomic emission spectroscopy (ICP-AES) was used to analyze the Co element content in the samples (Table S1, ESI†), with a loading of 1.823 mg per unit area for the S-FeCo1:1/NIF catalyst.
HRTEM analyses were conducted on S-Co/NIF and S-Fe/NIF. The HRTEM images of S-Co/NIF and S-Fe/NIF exhibited characteristics similar to those of S-FeCo1:1/NIF (Fig. S7 and S9, ESI†), with identifiable FFT patterns similar to those of S-FeCo1:1/NIF, indicating a similar crystallinity (Fig. S7b and S9b, ESI†). Simultaneously, the lattice fringes in the measurable regions of S-Co/NIF and S-Fe/NIF, with widths of 0.198 nm and 0.298 nm, respectively, were attributed to the (511) crystal plane of Co9S8 (PDF#19-0364) (Fig. S7c, ESI†) and the (311) crystal plane of Fe3S4 (PDF#16-0713) (Fig. S9c, ESI†). The IFFT images further confirmed the absence of significant lattice distortions (Fig. S8 and S10, ESI†), indicating crystalline features that are consistent with the results obtained from XRD.
X-ray photoelectron spectroscopy (XPS) was utilized to analyze the samples for a more thorough assessment of the elemental composition and distribution of valence states on the sample surface. The full XPS spectra of S-FeCo1:1/NIF, S-Co/NIF and S-Fe/NIF confirmed the presence of Fe, Co, O and S elements in the samples (Fig. S11, ESI†), which was consistent with the EDS results. The high-resolution Co 2p spectra exhibit two pairs of spin–orbit doublets (776.76/794.88 eV and 782.3/798.01 eV) along with two satellite peaks (787.52/802.02 eV), corresponding to Co3+ and Co2+ in S-FeCo1:1/NIF, respectively.41 Compared to S-Co/NIF, both spin–orbit doublets in S-FeCo1:1/NIF shifted to lower binding energies, indicative of Co site electron-acceptor behavior (Fig. 2g).19 The high-resolution Fe 2p spectra display six subpeaks, corresponding to Fe2+ (709.84/722.89 eV), Fe3+ (714.44/726.43 eV) and two satellite peaks (718.52/731.06 eV).42 In contrast to S-Fe/NIF, all the peaks in S-FeCo1:1/NIF shifted to higher binding energies (Fig. 2h), suggesting Fe site electron-donor behavior. In S-FeCo1:1/NIF, the electron density around the Co and Fe atoms is balanced, indicating charge synergy between the bimetallic species. Furthermore, according to the results of XPS fitting, the molar ratios of Co2+/Co3+ and Fe2+/Fe3+ in S-FeCo1:1/NIF are 0.83:1 and 1:1.51, respectively, while those in S-Co/NIF and S-Fe/NIF are 0.47:1 and 1:1.34, respectively. The decrease in Co3+ and the increase in Fe2+ can be attributed to the synergistic effect between the bimetals, resulting in a decrease in the oxidation state of Co3+, which, in turn, reduces the electron density around Co, creating more d orbitals and promoting electrocatalytic performance of the catalyst.27,43,44 The aforementioned research elucidates that strong electronic interactions between Co and Fe elements can optimize the adsorption/desorption of intermediates in the OER, enhancing the activity of S-FeCo1:1/NIF, as corroborated by numerous studies.41,45,46 Moreover, as shown in Fig. 2i, the S 2p spectrum of S-FeCo1:1/NIF exhibits two peaks at 161.83 and 164.26 eV, corresponding to S 2p1/2 and S 2p3/2 of S2−, indicating the formation of metal–S bonds in S-FeCo1:1/NIF.47 The peak at 168.27 eV is attributed to the formation of S–O bonds caused by partial oxidation on the surface of S-FeCo1:1/NIF.48 Additionally, similar results were obtained from the S 2p spectra of S-Co/NIF and S-Fe/NIF, but compared to S-FeCo1:1/NIF, all three peaks in S-FeCo1:1/NIF exhibited a leftward shift, suggesting the acceptor role of S, which facilitated the OER/HER processes.
2.2. OER electrocatalytic performance
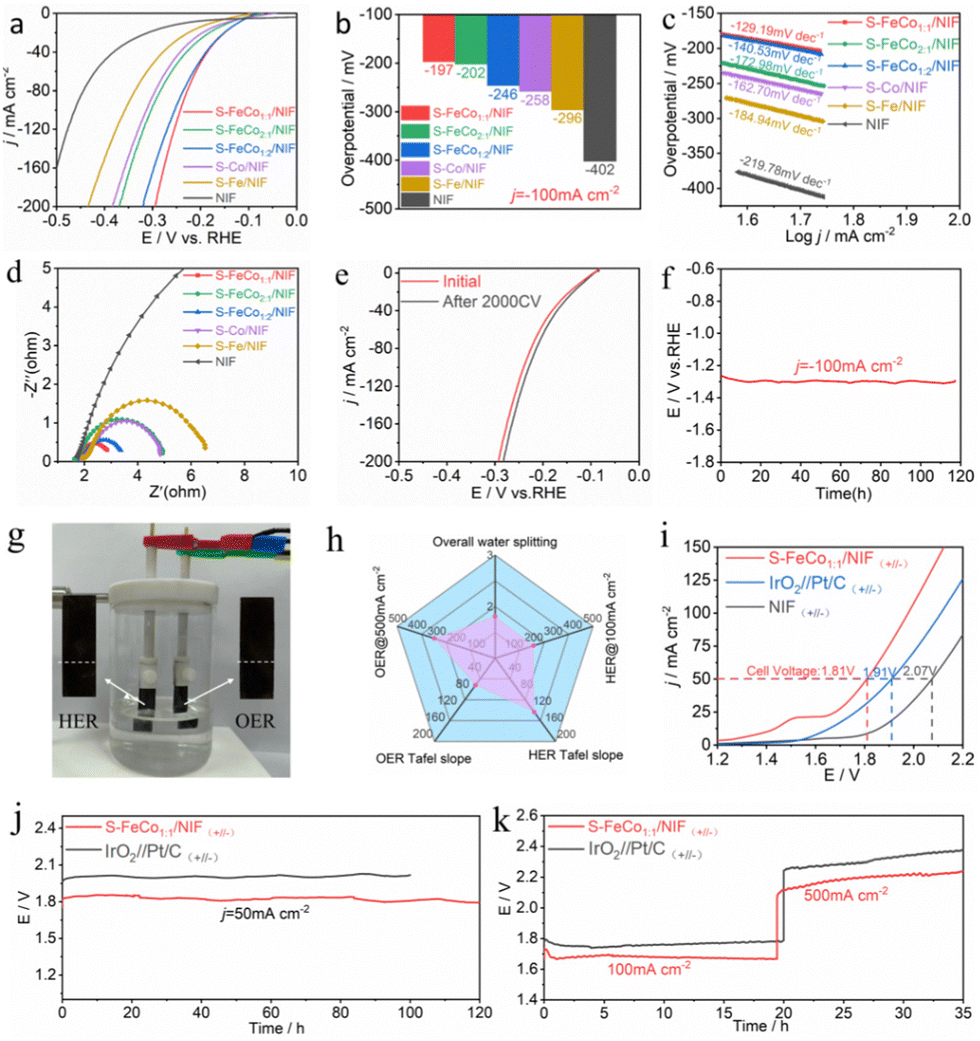
The electrochemical performance of S-FeCo1:1/NIF, S-FeCo2:1/NIF, S-FeCo1:2/NIF, S-Fe/NIF, S-Co/NIF and NIF was assessed in 1.0 M KOH. To prevent interference from the oxidation peaks of Co2+/Co3+ and Fe2+/Fe3+ at around 1.45 V, we recorded the overpotential for the OER using linear sweep voltammetry (LSV) with a negative scan, as illustrated in Fig. 3a. The iR-corrected LSV curves reveal that the S-FeCo1:1/NIF electrode demonstrates superior OER activity in alkaline electrolytes compared to those of the other samples. At a current density of 500 mA cm−2, the overpotential (η) is only 310 mV, which is lower than those of S-FeCo1:2/NIF (η500 = 333 mV), S-FeCo2:1/NIF (η500 = 369 mV), S-Co/NIF (η500 = 392 mV), S-Fe/NIF (η500 = 404 mV), and NIF (η500 = 474 mV) (Fig. 3b). Additionally, as shown in Fig. S12 (ESI†) and described in Table S2 (ESI†), the overpotential of S-FeCo1:1/NIF, produced in this investigation, exceeds those of previously documented sulfide catalysts and alternative nanocatalysts at both 10 mA cm−2 and 100 mA cm−2.
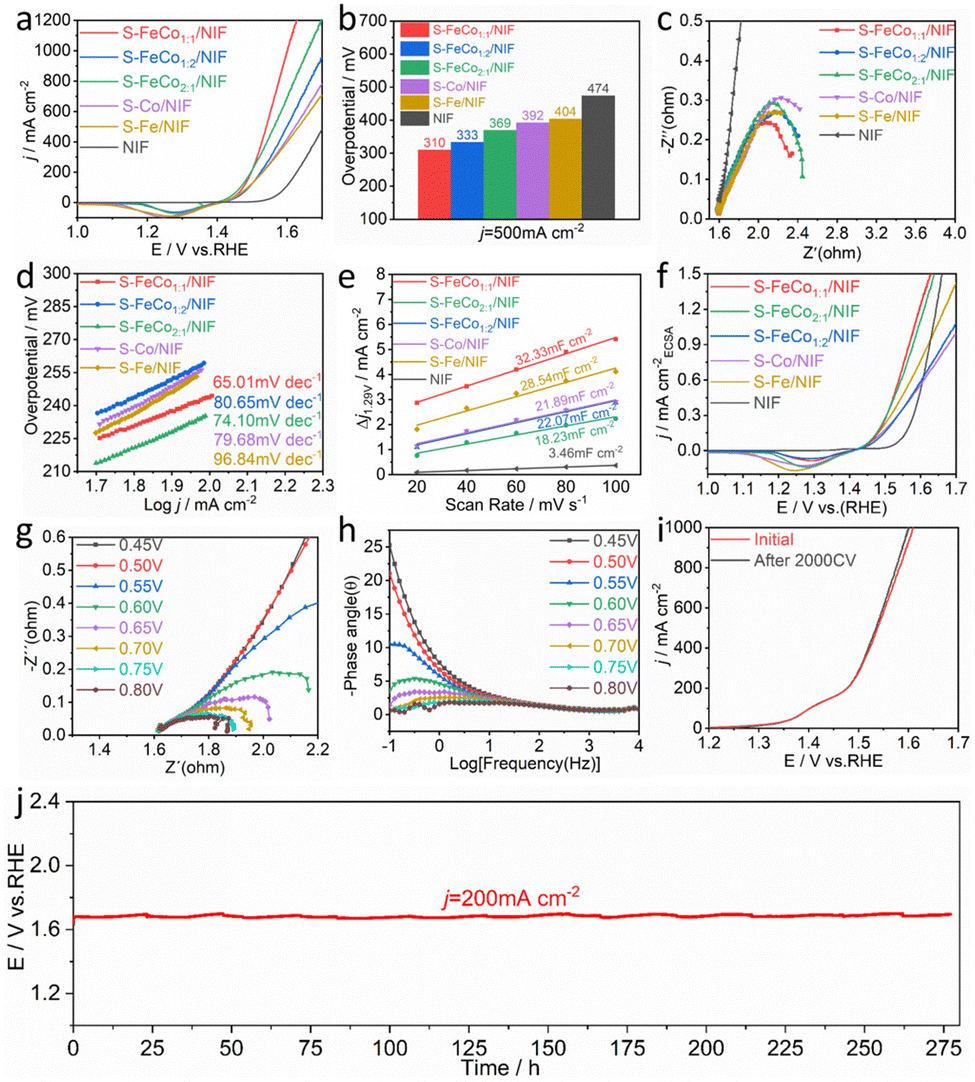 | ||
| Fig. 3 (a) iR-corrected LSV curves, (b) overpotential based on j = 500 mA cm−2, (c) electrochemical impedance spectroscopy, and (d) Tafel slopes for S-FeCo1:1/NIF, S-FeCo2:1/NIF, S-FeCo1:2/NIF, S-Fe/NIF, S-Co/NIF and NIF in 1.0 M KOH electrolyte. (e) Cdl obtained at a specific potential and (f) ECSA normalized linear sweep voltammetry curves of the S-FeCo1:1/NIF, S-FeCo2:1/NIF, S-FeCo1:2/NIF, S-Fe/NIF, S-Co/NIF and NIF electrodes. (g) Operando Nyquist and (h) Bode phase plots of S-FeCo1:1/NIF at various overpotentials in 1.0 M KOH. (i) The LSV curve of S-FeCo1:1/NIF before and after 2000 cycles and (j) the long-term E–t curves of S-FeCo1:1/NIF for the OER under a constant current of 200 mA cm−2. | ||
The electrochemical reaction kinetics of the OER were analyzed using electrochemical impedance spectroscopy (EIS) at η = 300 mV and Tafel slope analysis of the LSV curves, as shown in Fig. 3c and d. The EIS and Tafel slope graphs, obtained by fitting the equivalent circuit diagram (Fig. S13, ESI†), suggest that S-FeCo1:1/NIF has the smallest charge transfer resistance (Rct) and the smallest Tafel slope (65.01 mV dec−1) among all samples. To further investigate the activity of S-FeCo1:1/NIF, the effective electrochemically active surface area (ECSA) of the catalysts was determined by measuring the double-layer capacitance (Cdl). The samples were subjected to multiple cyclic voltammetry (CV) scans at scan rates of 20–100 mV s−1 (Fig. S14, ESI†), and the Cdl values for S-FeCo1:1/NIF, S-FeCo2:1/NIF, S-FeCo1:2/NIF, S-Fe/NIF, S-Co/NIF, and NIF were determined to be 32.33, 18.23, 22.07, 21.89, 28.54, and 3.46 mF cm−2 (Fig. 3e), respectively. The ECSA of the catalysts was then calculated using the formula ECSA = Cdl/Cs, where the value of the specific capacitance (Cs) is typically between 20 and 60 μF cm−2, with 40 μF cm−2 used in this case.
The calculated ECSA values for different electrodes were 808.25, 455.75, 551.75, 547.25, 713.5, and 86.5 cm−2. Using these values, we normalized the LSV curves obtained to compare the intrinsic activityies of the different catalysts. As shown in Fig. 3f, at the same potential, the current density of S-FeCo1:1/NIF remains higher than that of the other samples, indicating its higher intrinsic electrocatalytic activity. The remarkable electrocatalytic activity, fast electron transfer ability, and high intrinsic activity of S-FeCo1:1/NIF are mainly attributed to the nanostructured morphology of the catalyst, which not only provides a larger specific surface area and more active sites, but also enhances the inherent activity of the catalyst. Additionally, the synergistic effects between the bimetallic components result in electronic redistribution within the catalyst, facilitating the adsorption and desorption of the active intermediates and tuning the binding energy of the intermediates.
The intrinsic reaction kinetics of the electrode/electrolyte interface were examined using in situ EIS. The Nyquist plots in Fig. 4g and Fig. S15 (ESI†) depict the variations in catalytic performance among the different catalysts. As the external voltage increases, the resistance gradually diminishes, suggesting that primary charge transfer in the catalytic process predominantly occurs in the low-frequency region. Compared to the catalysts S-FeCo2:1/NIF, S-FeCo1:2/NIF, S-Fe/NIF and S-Co/NIF, catalyst S-FeCo1:1/NIF consistently displays the lowest resistance across all potentials. This indicates that the collaborative effects between the bimetallic components significantly boost the adsorption kinetics of the oxygen-containing active species during the alkaline OER process. The Bode plots depict the relaxation processes of the catalyst samples. A distinct transition peak is observed at a potential of 0.55 V for S-FeCo1:1/NIF and S-FeCo2:1/NIF, which is lower than those for S-FeCo1:2/NIF, S-Fe/NIF and S-Co/NIF, pointing to their more rapid reaction kinetics (Fig. 4h and Fig. S16, ESI†).49 Furthermore, the phase angle of the Bode phase plots for each electrode noticeably decreases with increasing potential, indicating a reduction in faradaic resistance and thereby facilitating electron transfer during intermediate adsorption.17,50–53
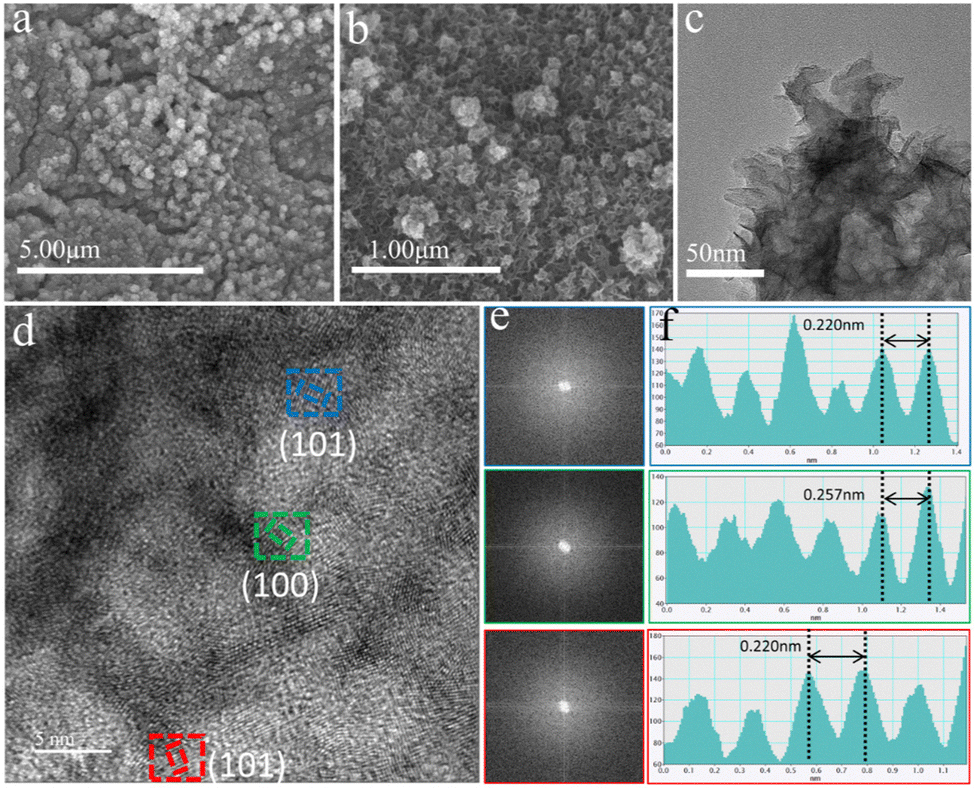 | ||
| Fig. 4 (a) and (b) SEM images of S-FeCo1:1/NIF after OER, (c) TEM image, (d) HRTEM image, (e) fast Fourier transform (FFT) of different regions and (f) corresponding intensity profiles along with the position of S-FeCo1:1/NIF after OER. | ||
The long-term stable operation of an electrocatalyst under higher currents is a critical benchmark for evaluating the catalyst performance. The LSV curves and EIS plots in Fig. 4i and Fig. S17a (ESI†), obtained after 2000 CV cycles of S-FeCo1:1/NIF, indicate a slight enhancement in the activity of S-FeCo1:1/NIF after 2000 CV cycles, accompanied by a minor reduction in charge transfer resistance. This effect may be attributed to incomplete surface restructuring during early activation of the catalyst. Nevertheless, it can still be deduced that the S-FeCo1:1/NIF catalyst maintains relatively good stability under dynamic currents. As depicted in Fig. S17b (ESI†), the S-FeCo1:1/NIF catalyst demonstrates the ability to maintain its stability at various high current densities. Furthermore, the time-dependent potential curve presented in Fig. 4j illustrates that the S-FeCo1:1/NIF catalyst, exhibits no significant potential degradation after 275 h of OER testing at a current density of 200 mA cm−2. This observation suggests the catalyst's outstanding electrocatalytic durability and stability. This exceptional stability may be attributed to the propensity of the self-grown catalyst on the NIF surface to resist detachment from the carrier surface, allowing for sustained, long-term operation without performance deterioration. Additionally, surface self-reconstruction of the precatalyst and leaching of surface elements are also crucial factors that influence stability. Furthermore, using the S-Co/NIF stability test, it was found that the potential of S-Co was significantly higher than that of S-FeCo1:1/NIF after 20 h at the same current density, indicating that the stability of the single metal sulfide was poor (Fig. S18, ESI†). The addition of Fe can significantly improve the stability of bimetallic sulfides due to the synergistic effect between the two metals. When Fe is added, a new surface with high activity and good stability is promoted, which lowers the overpotential required for OER occurrence. Although the Co site still exists, it is no longer the main active site. This conclusion has been proved in many studies to enhance the stability of sulphides.22,54
2.3. Investigation of the OER mechanism
To demonstrate the occurrence of self-reconstruction of the catalyst surface, post-OER characterization of the S-FeCo1:1/NIF catalyst was performed using relevant instrumentation. As depicted in Fig. 4a and b, the SEM and TEM images of the S-FeCo1:1/NIF sample reveal a distinct transformation from relatively thick nanosheets at the edges of the pre-OER nanograins to smaller and thinner nanosheets with a nanoflower-like structure resulting from the reconstruction (Fig. 4c). The corresponding HRTEM images display discernible lattice fringes that are not densely packed (Fig. 4d). Further FFT analysis of the three regions in the HRTEM images (Fig. 4e) reveals two broad and diffuse diffraction rings with no identifiable diffraction spots, indicating their low crystalline nature. At the same time, the lattice fringes belonging to the FeCoOOH (100) and (101) crystal planes can be measured (Fig. 4f), and the lattice width can be observed on the (101) crystal plane, which is reduced by 2.65% compared to the standard (101) crystal plane. From the IFFT diagram (Fig. S19, ESI†), it is found that the lattice has an obvious tensile distortion.55 This distortion may be due to the synergistic effect between the two metals, which changes the corresponding coordination environment, and results in lattice distortion. The results presented here fully prove the formation of low crystalline FeCoOOH. The nature of the nanoflower-like morphology resulting from self-reconstruction provides favorable conditions for efficient electrolyte–catalyst surface contact and the release of bubbles, effectively accelerating the mass transfer rates on the catalyst surface. Furthermore, the presence of low-crystalline species offers additional inherent active sites for oxygen intermediate reactions and promotes electron transfer rates, thus expediting the OER kinetics of S-FeCo1:1/NIF.
For a better understanding of the catalytic reconfiguration process of S-FeCo1:1/NIF, XPS was used for sample analysis to monitor changes in elemental valence states before and after the reaction of S-FeCo1:1/NIF. As depicted in Fig. 5a, the high resolution XPS spectrum of Co 2p reveals two characteristic peaks at 782.33 eV and 798.14 eV,56 corresponding to Co 2p3/2 and Co 2p1/2, respectively. Simultaneously, the high-resolution XPS spectrum of Fe shows two distinctive peaks at 714.42 eV and 725.31 eV, corresponding to Fe 2p3/2 and Fe 2p1/2 (Fig. 5b).57 After a period of OER, the positions of both Co2+ peaks exhibit a partial blueshift, and the Fe2+ peaks are no longer detectable. This signifies the oxidation of Co2+ and Fe2+ to Co3+ and Fe3+, a phenomenon also supported by the peak areas after XPS fitting.58 The abundance of Co3+ and Fe3+ on the surface facilitates the formation of highly active FeCoOOH, which serves as a catalytically active site for OER. Furthermore, the spectrum of O1s before the reaction reveals three peaks at 531.03, 532.60, and 533.48 eV (Fig. S20, ESI†), corresponding to metal–oxygen bonds (M–O), hydroxyl bonds (M–OH) and surface-adsorbed H2O. The ratio of M–O/M–OH is 1:6.9, indicating that the predominant form of oxygen binding in the catalyst is metal–oxygen. After the OER process, the corresponding ratio changes to M–O/M–OH = 5.4:1, signifying the conversion of M–O to M–OH during this process. This transformation is likely attributed to the reconfiguration of the catalyst into highly active hydroxide oxide (FeCoOOH),59,60 which aligns with the results of the corresponding HRTEM analysis. At the same time, the peak area of H2O adsorbed on the surface increased, indicating that the formation of a nanoflower-like material structure is more conducive to the adsorption of water on the surface of the catalyst. It is noteworthy from the high-resolution S 2p XPS spectrum (Fig. 5c) that the intensity of the two diffraction peaks corresponding to S2− decreases noticeably, indicating significant sulfur leaching during the OER process. The peak of S2− moves towards a higher binding energy, indicating that the electron density around the S species decreases and the valence state increases. The decrease in the peak values of S–O (SO42−) and the areas further elucidate that the form of sulfur leaching involves the dissolution of SO42− into the electrolyte, a phenomenon well documented in various studies.57,61
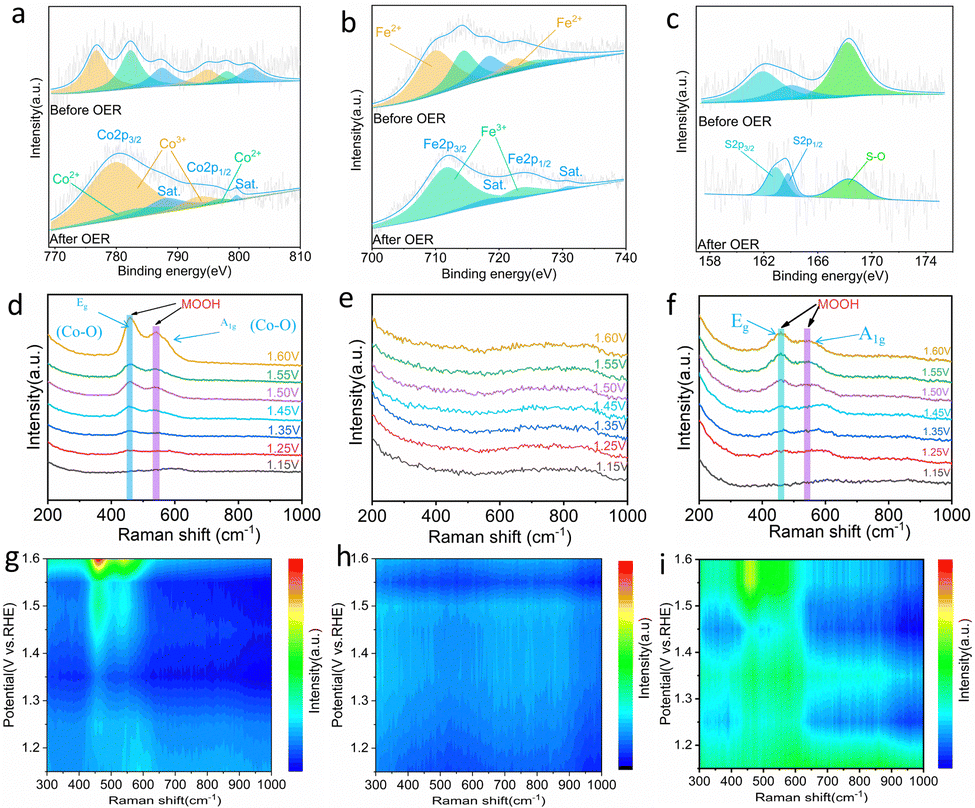 | ||
| Fig. 5 (a) Co 2p, (b) Fe 2p and (c) S 2p XPS spectra of S-FeCo1:1/NIF before and after OER. In situ Raman spectra at actual working conditions of the cycle (applied potential from 1.15 V to 1.6 V) for (d) S-Co, (e) S-Fe and (f) S-FeCo1:1 electrocatalysts and corresponding contour plots of in situ Raman spectra for (g) S-Co, (h) S-Fe and (i) S-FeCo1:1 samples. | ||
To elucidate the electrocatalytic mechanism, in situ Raman spectroscopy was employed to investigate the surface electrooxidation transformation of the catalyst (Fig. S21, ESI†). As depicted in Fig. 5d and g, Raman spectra were acquired for the S-Co sample within the voltage range of 1.15 V to 1.60 V. With a continuous increase in voltage, two characteristic Raman peaks gradually emerged at 458 cm−1 and 546 cm−1. These peaks arise from the bending vibration of the Co–O mode Eg in γ-CoOOH and the stretching vibration of the Co–O mode A1g in β-CoOOH, indicating the complete formation of CoOOH.62–64 In contrast, no conspicuous Raman peaks were observed in the monometallic S-Fe sample (Fig. 5e and h). However, in bimetallic S-FeCo1:1, Raman peaks similar to those in S-Co appeared at 456 cm−1 and 544 cm−1.65 Compared to S-Co, the Raman peaks in S-FeCo1:1 exhibited a slight redshift, which is attributed to the synergistic effect between the bimetallic components that distort the crystal structure of CoOOH (Fig. 5f and i).66,67 Simultaneously, a noticeable decrease in the intensity of the Raman peaks was observed in S-FeCo1:1 as a result of the intermetallic interactions that caused changes in the electronic structure of the catalyst, altering the corresponding coordination environment, and leading to the generation of the new FeCoOOH compound.54
To further prove that the S-Fe catalyst is not reconstructed in the OER, we used SEM, TEM, and HRTEM to characterize the morphology of the catalyst. As shown in Fig. S22 (ESI†), the SEM and TEM images before and after the reaction of S-Co indicate that the morphology of the catalyst changed significantly, and the edge structure of the nanoparticles also changed into a thinner nanosheet structure. On the contrary, S-Fe showed no obvious changes. SEM shows that the catalyst continues to maintain the morphology of its original nanoparticles, but there is still a change in the particle size compared to the initial ones. The TEM characterization further revealed that the edge structure of the nanoparticles did not change much (Fig. S23, ESI†). To clarify whether there is a high activity of hydroxyl oxide formation, the HRTEM images of S-Co and S-Fe were analyzed. The results show that the lattice fringes are blurred and discontinuous in the S-Co image. The visible lattice fringes were measured to be 0.247 nm, which belongs to the lattice width of the (511) crystal plane of typical CoOOH (Fig. S24, ESI†). At the same time, the lattice fringes of the crystal plane (012) belonging to Fe3S4 can be observed in the S-Fe image (Fig. S25, ESI†). These results fully prove that there is no reconstruction of S-Fe during the electrocatalytic OER process, which is consistent with the results obtained by in situ Raman spectroscopy. Existing studies have shown that the introduction of Fe can greatly improve the OER activity and change the natural properties of the active CoOOH sites.68 The partial electron loss at the Co site leads to a higher electron affinity, which promotes the adsorption of the OER by OH− and the charge transfer of the adsorbed OH−. In addition, it is reported that the introduction of Fe can promote the activity and redeposition of the Co element, so that the catalyst has good self-healing ability. Therefore, the excellent OER activity and stability of S-FeCo1:1/NIF are attributed to the synergistic effect of Fe and Co on the self-reconfigurable bimetallic oxyhydroxides.
2.4. DFT calculation
Moreover, for a more profound understanding of the specific function of the collaborative impact between the bimetallic constituents in the catalytic process after electrocatalytic reconstruction and the adsorption proficiency of the active intermediates at distinct metal locations, we conducted DFT calculations of the catalytic process. In this study, the Fe3S4 catalyst did not undergo reconstruction; however, for a comprehensive and accurate comparison, we also constructed FeOOH for energy barrier calculations and analyses. Drawing on the experimental findings presented in Fig. 5 and employing the AEM mechanism for the OER in alkaline conditions, we formulated theoretical models for the FeCoOOH, CoOOH, and FeOOH structures corresponding to various active intermediates (Fig. S26–S29, ESI†). As depicted in Fig. 6a–c, the density of states (DOS) of FeCoOOH, CoOOH, and FeOOH signifies a noteworthy influence of the synergistic effect between the bimetallic constituents on the DOS of FeCoOOH. Initially, compared to CoOOH (−1.287 eV) and FeOOH (−1.882 eV), the center of the d band of FeCoOOH (−1.201 eV) is closer to the Fermi level, facilitating the exposure of active sites and the adjustment of electronic structures, leading to a decrease in the adsorption energy of the active intermediates in contrast to CoOOH and FeOOH.69,70 Furthermore, FeCoOOH displays a heightened DOS at the Fermi level, implying that an increased DOS enables more charge carriers to directly engage in the catalytic reaction, significantly amplifying the OER performance of the catalyst. Lastly, for a more profound understanding of the electronic structure of FeCoOOH, we computed the DOS of the three-dimensional orbitals of each metal constituent (i.e., Fe, Co) in FeCoOOH, as delineated in Fig. 6d. The center of the Fe atom in the d-band is closer to the Fermi level, increasing its capacity to stabilize the *O intermediate and refine the binding energy of the reaction intermediates.71 To summarize, the inherent factors contributing to the enhanced electrocatalytic activity and resilience of FeCoOOH are the electronic coupling between bimetallic atoms within the system and the advantageous adsorption capability of the *O intermediate at the Fe sites.72,73 | ||
| Fig. 6 Calculated DOS of (a) FeCoOOH, (b) CoOOH, (c) FeOOH, and (d) Fe and Co sites of FeCoOOH. (e) Schematic representation of the OER process of FeCoOOH in an alkaline environment. (f) The Gibbs free energies of the different samples in each elementary step of OER at 0 V. | ||
We further examined the OER catalytic activity of FeCoOOH and clarified the water oxidation energy barriers for FeCoOOH, CoOOH, and FeOOH based on ΔG changes along the 4e− pathway.74 The widely acknowledged AEM mechanism for the OER process can be categorized into four stages, encompassing three intermediate states (*OH, *O, *OOH) (Fig. 6e). Table S3 (ESI†) presents the energy requirements for all feasible reaction pathways for FeCoOOH, CoOOH, and FeOOH. In the FeOOH pathway, the highest energy demand is associated with the OH → O step, while for FeCoOOH and CoOOH, it is associated with the O → OOH step. This implies that the rate-determining step (RDS) for FeOOH is OH → O, while those for FeCoOOH and CoOOH is O → OOH. Our initial assessment focused on evaluating the impact of bimetallic synergy on the catalytic activity of FeCoOOH. As depicted in Fig. 6f and Fig. S30 (ESI†), we calculated the influence of bimetallic synergy on the OER process of the active intermediates at distinct Fe and Co sites in FeCoOOH at 0 V and 1.23 V. In the catalytic water oxidation reaction of FeCoOOH, the ΔGmax values for the Fe and Co sites are 1.829 and 1.831 eV, accompanied by η = 0.599 and 0.601 V, respectively. This indicates that during OER catalysis, the Fe sites exhibit superior adsorption performance compared to the Co sites. Subsequent calculations for various substances revealed that ΔGmax and overpotentials for the RDS of FeCoOOH are notably lower than those of CoOOH (ΔGmax = 1.880 eV, η = 0.650 V) and FeOOH (ΔGmax = 2.208 eV, η = 0.979 V). This indicates that the synergistic effect of the metals optimizes the electronic structure of the catalyst, enhances the adsorption capability of the intermediates during the catalytic process, effectively reduces the reaction barrier, and improves the catalytic efficiency and stability.64 In summary, we enhanced the catalytic activity of sulfide catalysts through a straightforward bimetallic synergistic strategy and elevated the activation capability of the rate-limiting step in the water oxidation process through metal coupling, ultimately enhancing the activity and stability of bimetallic sulfides.
2.5. HER and overall water-splitting electrocatalytic performance
The electrochemical HER performances of S-FeCo1:1/NIF, S-FeCo2:1/NIF, S-FeCo1:2/NIF, S-Fe/NIF, S-Co/NIF and NIF were assessed in a 1.0 M KOH electrolyte. As shown in Fig. 7a, compared to the other samples, S-FeCo1:1/NIF exhibited superior HER catalytic activity, requiring only an overpotential of 191 mV to reach a current density of −100 mA cm−2. This was 5 mV lower than that of S-FeCo2:1/NIF (η100 = 202 mV), 49 mV lower than that of S-FeCo1:2/NIF (η100 = 246 mV), 61 mV lower than that of S-Fe/NIF (η100 = 258 mV), 99 mV lower than that of S-Co/NIF (η100 = 246 mV), and 205 mV lower than that of NIF (η100 = 246 mV) (Fig. 7b). We compared the overpotential of the prepared S-FeCo1:1/NIF at a current density of 10 mA cm−2 with the sulfide catalyst and other nanocatalysts reported in the previous literature. The results show that the catalytic performance of the catalysts prepared in this experiment is better than that of some previously reported catalysts (Fig. S31 and Table S4, ESI†). Furthermore, Fig. 7c indicates that S-FeCo1:1/NIF had the smallest Tafel slope, which was 129.19 mV dec−1, while S-FeCo2:1/NIF had a slope of 172.98 mV dec−1, S-FeCo1:2/NIF had a slope of 140.53 mV dec−1, S-Fe/NIF had a slope of 162.71 mV dec−1, S-Co/NIF had a slope of 184.94 mV dec−1, and NIF had a slope of 219.78 mV dec−1, indicating that S-FeCo1:1/NIF possessed faster HER kinetics. The EIS spectra of S-FeCo1:1/NIF displayed a superior Rct compared to those of the other samples (Fig. 7d), suggesting that the synergistic effect between the bimetallic components and low-crystalline species effectively accelerated the reaction rate during water electrolysis. Surprisingly, after 2000 CV cycles, both the LSV curve and the EIS spectra of S-FeCo1:1/NIF exhibited minor enhancements and reductions (Fig. 7e and Fig. S32, ESI†). The improvement in catalyst performance could be attributed to the structural changes on the catalyst's surface and the generation of active species after 2000 CV cycles. In the stability test, at a current density of −100 mA cm−2 for 120 h, the E–t curve remained relatively stable (Fig. 7f), showing no significant upward trend, indicating the stable and excellent catalytic activity of S-FeCo1:1/NIF.
 | ||
| Fig. 7 (a) The HER LSV curves, (b) overpotential, (c) Tafel slope, (d) EIS spectra for the S-FeCo1:1/NIF, S-FeCo2:1/NIF, S-FeCo1:2/NIF, S-Fe/NIF, S-Co/NIF and NIF samples in 1.0 M KOH. (e) The LSV curves of S-FeCo1:1/NIF before and after 2000 cycles. (f) The long-time E–t curves of S-FeCo1:1/NIF for HER under a constant current density of −100 mA cm−2. (g) The two-electrode water- splitting device. (h) Summary of the electrocatalytic performance of S-FeCo1:1/NIF, (i) LSV plots of the S-FeCo1:1/NIF||S-FeCo1:1/NIF, RuO2||Pt/C and NIF||NIF for overall alkaline water splitting, (j) E–t curves of S-FeCo1:1/NIF||S-FeCo1:1/NIF and RuO2||Pt/C for overall water splitting at 50 mA cm−2 in a 1.0 M KOH electrolyte. (k) E–t curves of the S-FeCo1:1/NIF||S-FeCo1:1/NIF and RuO2||Pt/C electrodes for overall water splitting at 100 and 500 mA cm−2 in a 6.0 M KOH electrolyte at 60 °C. | ||
Due to the exceptional OER and HER activities of the S-FeCo1:1/NIF catalyst, it was employed as both the cathode and anode in an electrolyzer for the overall water-splitting reaction (Fig. 7g). A comprehensive evaluation of S-FeCo1:1/NIF for overall water electrolysis performance, reflecting the synergistic effect of the FeCo bimetallic system and the low crystallinity of the water electrolysis, is presented in Fig. 7h. Notably, the S-FeCo1:1/NIF electrolyzer required only 1.81 V and 1.58 V voltage to achieve current densities of 50 mA cm−2 and 10 mA cm−2 (Fig. S33, ESI†), surpassing that of the electrolyzer comprising IrO2||Pt/C (1.91 V) and NIF||NIF (2.07 V) (Fig. 7i). The overpotential of the prepared bifunctional catalyst S-FeCo1:1/NIF at a current density of 10 mA cm−2 is lower than those of some sulfide catalysts and other nanocatalysts reported in the previous literature (Table S5, ESI†). Furthermore, it demonstrated stable operation at a current density of 50 mA cm−2 for 120 h (Fig. 7j), with no significant performance degradation, proving the good stability and durability of the bifunctional S-FeCo1:1/NIF catalyst. More importantly, under extreme water electrolysis conditions of 6.0 M KOH and 60 °C, S-FeCo1:1/NIF maintained long-term operation at high current densities of 100 mA cm−2 and 500 mA cm−2 (Fig. 7k), with almost no change in the overpotential at 100 mA cm−2. At 500 mA cm−2, there was a more noticeable increase in the overpotential, which could be attributed to the evaporation and electrolysis of the solution at higher temperatures and current density during electrolysis.75
3. Conclusion
In summary, we have successfully grown bimetallic sulfide S-FeCo1:1/NIF catalysts in situ on the surface of NIF via a straightforward one-step solvothermal method. S-FeCo1:1/NIF demonstrated remarkable electrocatalytic activity and stability, achieving an overpotential of only 310 mV at a current density of 500 mA cm−2 in 1 M KOH. Furthermore, it exhibited a stable operation for 275 h at a current density of 200 mA cm−2. Additionally, it exhibited excellent performance in both the HER and overall water electrolysis. Characterization by SEM, TEM, XPS, and in situ Raman spectroscopy successfully confirmed the electrochemical restructuring of the precatalyst during the OER process. DFT calculations further validated the impact of bimetallic synergy on the catalytic activity of the sulfides. The results indicate that coupling between bimetallic components effectively reduces the energy barriers required for the rate-determining steps in water electrolysis, enhancing the activity and stability of bimetallic sulfides. The investigation of improving the OER performance through bimetallic coupling provides theoretical guidance for the rational design of advanced electrocatalysts.
Author contributions
Yaning Fan: data acquisition, writing – review & editing. Junjun Zhang: experimental design, funding acquisition, writing – review & editing. Jie Han: theoretical calculation, writing – review & editing. Mengyuan Zhang: visualization and data acquisition. Weiwei Bao: data acquisition, writing – review & editing. Hui Su: writing & editing. Nailiang Wang: data acquisition, funding acquisition and writing & editing. Pengfei Zhang: visualization and acquisition of funds. Zhenghong Luo: supervision and writing-review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21968023, 21902123, 2217821), Inner Mongolia R&D Program Plan (2021ZD0042, 2021EEDSCXSFQZD006), Ordos R&D Program (2121HZ231-8) and Ningxia Key Research and development Program (No. 2021BEE03007). Junjun Zhang thanks the Chinese Academy of Sciences Western Young Scholar Program for the scholarships (2023), the Young Elite Scientist Sponsorship Program of the Ningxia Association for Science and Technology (2023) and the Scientific Research Start-Up Project Program of Ningxia University. We thank eceshi (https://www.eceshi.com/) for the ICP and XPS test.Notes and references
- L. Chen and J. Shi, J. Mater. Chem. A, 2018, 6, 13538–13548 RSC
.
- Q. Fan, J. Su, T. Sun, Z. Bi, H. Wang, S. Zhang, Q. Liu, L. Zhang and G. Hu, Carbon Energy, 2022, 4, 211–236 CrossRef CAS
- Y. He, L. Liu, C. Zhu, S. Guo, P. Golani, B. Koo, P. Tang, Z. Zhao, M. Xu, C. Zhu, P. Yu, X. Zhou, C. Gao, X. Wang, Z. Shi, L. Zheng, J. Yang, B. Shin, J. Arbiol, H. Duan, Y. Du, M. Heggen, R. E. Dunin-Borkowski, W. Guo, Q. J. Wang, Z. Zhang and Z. Liu, Nat. Catal., 2022, 5, 212–221 CrossRef CAS
- W.-Z. Chen, M. Zhang, Y. Liu, X.-M. Yao, P.-Y. Liu, Z. Liu, J. He and Y.-Q. Wang, Appl. Catal. B, 2022, 312, 121432 CrossRef CAS
- S. Atilhan, S. Park, M. M. El-Halwagi, M. Atilhan, M. Moore and R. B. Nielsen, Curr. Opin. Chem. Eng., 2021, 31, 100668 CrossRef
- R. Ramachandran and R. K. Menon, Int. J. Hydrogen Energy, 1998, 23, 593–598 CrossRef CAS
- N. Sazali, Int. J. Hydrogen Energy, 2020, 45, 18753–18771 CrossRef CAS
- S. E. Hosseini and M. A. Wahid, Inter. J. Energy Res., 2020, 44, 4110–4131 CrossRef
- F. Li, H. Wu, S. Lv, Y. Ma, B. Wang, Y. Ren, C. Wang, Y. Shi, H. Ji, J. Gu, S. Tang and X. Meng, Small, 2023, 19, 2309025 CrossRef PubMed
- H. Cheng, N. Yang, G. Liu, Y. Ge, J. Huang, Q. Yun, Y. Du, C.-J. Sun, B. Chen, J. Liu and H. Zhang, Adv. Mater., 2020, 32, 1902964 CrossRef CAS PubMed
- S. Zheng, X. Liang, J. Pan, K. Hu, S. Li and F. Pan, ACS Catal., 2023, 13, 2847–2856 CrossRef CAS
- X. Liang, S. Wang, J. Feng, Z. Xu, Z. Guo, H. Luo, F. Zhang, C. Wen, L. Feng, C. Wan and M.-M. Titirici, Inorg. Chem. Front., 2023, 10, 2961–2977 RSC
- Y. Ren, Z. Li, B. Deng, C. Ye, L. Zhang, Y. Wang, T. Li, Q. Liu, G. Cui, A. M. Asiri, Y. Luo and X. Sun, Int. J. Hydrogen Energy, 2022, 47, 3580–3586 CrossRef CAS
- Y. Bai, Y. Wu, X. Zhou, Y. Ye, K. Nie, J. Wang, M. Xie, Z. Zhang, Z. Liu, T. Cheng and C. Gao, Nat. Commun., 2022, 13, 6094 CrossRef CAS PubMed
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2021, 80, 105514 CrossRef CAS
- Y. Li, J. Han, W. Bao, J. Zhang, T. Ai, M. Yang, C. Yang and P. Zhang, J. Energy Chem., 2024, 90, 590–599 CrossRef CAS
- Y. Fan, J. Zhang, K. Luo, X. Zhou, J. Zhao, W. Bao, H. Su, N. Wang, P. Zhang and Z. Luo, Inorg. Chem. Front., 2024, 11, 114–122 RSC
- M. Yang, W. Bao, J. Zhang, T. Ai, J. Han, Y. Li, J. Liu, P. Zhang and L. Feng, J. Colloid Interface Sci., 2024, 658, 32–42 CrossRef CAS PubMed
- R. Yu, C. Wang, D. Liu, Z. Wu, J. Li and Y. Du, Inorg. Chem. Front., 2022, 9, 3130–3137 RSC
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef CAS
- Y. Zhang, B. Ouyang, J. Xu, G. Jia, S. Chen, R. S. Rawat and H. J. Fan, Angew. Chem., Int. Ed., 2016, 55, 8670–8674 CrossRef CAS PubMed
- S. Lee, K. Banjac, M. Lingenfelder and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 10295–10299 CrossRef CAS PubMed
- J. Ryu, N. Jung, J. H. Jang, H.-J. Kim and S. J. Yoo, ACS Catal., 2015, 5, 4066–4074 CrossRef CAS
- Y. Zuo, Y. Liu, J. Li, R. Du, X. Han, T. Zhang, J. Arbiol, N. J. Divins, J. Llorca, N. Guijarro, K. Sivula and A. Cabot, Chem. Mater., 2019, 31, 7732–7743 CrossRef CAS
- H.-J. Liu, S. Zhang, W.-Y. Yang, N. Yu, C.-Y. Liu, Y.-M. Chai and B. Dong, Adv. Funct. Mater., 2023, 33, 2303776 CrossRef CAS
- P. Wang, Y. Luo, G. Zhang, Z. Chen, H. Ranganathan, S. Sun and Z. Shi, Nano-Micro Lett., 2022, 14, 120 CrossRef CAS PubMed
- Y. Huang, D. Lv, Z. Zhang, Y. Ding, F. Lai, Q. Wu, H. Wang, Q. Li, Y. Cai and Z. Ma, Chem. Eng. J., 2020, 387, 124122 CrossRef CAS
- G. Ai, Q. Hu, L. Zhang, K. Dai, J. Wang, Z. Xu, Y. Huang, B. Zhang, D. Li, T. Zhang, G. Liu and W. Mao, ACS Appl. Mater. Inter., 2019, 11, 33987–33999 CrossRef CAS PubMed
- J. Jiang, X.-L. Zhou, H.-G. Lv, H.-Q. Yu and Y. Yu, Adv. Funct. Mater., 2023, 33, 2212160 CrossRef CAS
- J. Liang, F. Ma, S. Hwang, X. Wang, J. Sokolowski, Q. Li, G. Wu and D. Su, Joule, 2019, 3, 956–991 CrossRef CAS
- Q. Che, Q. Li, X. Chen, Y. Tan and X. Xu, Appl. Catal., B, 2020, 263, 118338 CrossRef CAS
- C. Wang, Q. Zhang, B. Yan, B. You, J. Zheng, L. Feng, C. Zhang, S. Jiang, W. Chen and S. He, Nano-Micro Lett., 2023, 15, 52 CrossRef CAS PubMed
- J. N. Hausmann, R. Beltrán-Suito, S. Mebs, V. Hlukhyy, T. F. Fässler, H. Dau, M. Driess and P. W. Menezes, Adv. Mater., 2021, 33, 2008823 CrossRef CAS PubMed
- H. Yang, G. Dai, Z. Chen, J. Wu, H. Huang, Y. Liu, M. Shao and Z. Kang, Small, 2021, 17, 2101727 CrossRef CAS PubMed
- H. Yang, M. Driess and P. W. Menezes, Adv. Energy Mater., 2021, 11, 2102074 CrossRef CAS
- B. Cao, Y. Cheng, M. Hu, P. Jing, Z. Ma, B. Liu, R. Gao and J. Zhang, Adv. Funct. Mater., 2019, 29, 1906316 CrossRef CAS
- Y. Jia, L. Zhang, G. Gao, H. Chen, B. Wang, J. Zhou, M. T. Soo, M. Hong, X. Yan, G. Qian, J. Zou, A. Du and X. Yao, Adv. Mater., 2017, 29, 1700017 CrossRef PubMed
- M. A. McArthur, L. Jorge, S. Coulombe and S. Omanovic, J. Power Sources, 2014, 266, 365–373 CrossRef CAS
- Y. Wu, G.-D. Li, Y. Liu, L. Yang, X. Lian, T. Asefa and X. Zou, Adv. Funct. Mater., 2016, 26, 4839–4847 CrossRef CAS
- T. Zhang, J. Sun and J. Guan, Nano Res., 2023, 16, 8684–8711 CrossRef CAS
- C. Yang, X. Ma, J. Zhou, Y. Zhao, X. Xiang, H. Shang and B. Zhang, Chem. Select, 2023, 8, e202204580 CAS
- F. Xiao, P. He, P. Yang, R. Su, Y. Wang, M. Zhao and B. Jia, J. Alloys Compd., 2023, 938, 168573 CrossRef CAS
- H. Yang, Z. Zhou, H. Yu, H. Wen, R. Yang, S. Peng, M. Sun and L. Yu, J. Colloid Interface Sci., 2023, 636, 11–20 CrossRef CAS PubMed
- D. Yu, Y. Ma, F. Hu, C.-C. Lin, L. Li, H.-Y. Chen, X. Han and S. Peng, Adv. Energy Mater., 2021, 11, 2101242 CrossRef CAS
- Z. Wang, J. Ang, B. Zhang, Y. Zhang, X. Y. D. Ma, T. Yan, J. Liu, B. Che, Y. Huang and X. Lu, Appli. Catal. B, 2019, 254, 26–36 CrossRef CAS
- D. Zhang, H. Zhao, X. Wu, Y. Deng, Z. Wang, Y. Han, H. Li, Y. Shi, X. Chen, S. Li, J. Lai, B. Huang and L. Wang, Adv. Funct. Mater., 2021, 31, 2006939 CrossRef CAS
- J. Yu, C. Lv, L. Zhao, L. Zhang, Z. Wang and Q. Liu, Adv. Mater. Inter., 2018, 5, 1701396 CrossRef
- W. He, C. Wang, H. Li, X. Deng, X. Xu and T. Zhai, Adv. Energy Mater., 2017, 7, 1700983 CrossRef
- T. Wu, S. Xu, Z. Zhang, M. Luo, R. Wang, Y. Tang, J. Wang and F. Huang, Adv. Sci., 2022, 9, 2202750 CrossRef CAS PubMed
- J.-J. Zhang, W.-W. Bao, X.-H. Feng, C.-M. Yang, N.-L. Wang, Y.-J. Qiu, J.-T. Li, P.-F. Zhang and Z.-H. Luo, Int. J. Hydrogen Energy, 2023, 48, 35038–35049 CrossRef CAS
- L. Deng, S.-F. Hung, Z.-Y. Lin, Y. Zhang, C. Zhang, Y. Hao, S. Liu, C.-H. Kuo, H.-Y. Chen, J. Peng, J. Wang and S. Peng, Adv. Mater., 2023, 35, 2305939 CrossRef CAS PubMed
- Y. Li, W. Bao, J. Zhang, T. Ai, D. Wu, H. Wang, C. Yang and L. Feng, J. Ind. Eng. Chem., 2023, 121, 510–518 CrossRef CAS
- C. Yang, C. Wang, L. Zhou, W. Duan, Y. Song, F. Zhang, Y. Zhen, J. Zhang, W. Bao, Y. Lu, D. Wang and F. Fu, Chem. Eng. J., 2021, 422, 130125 CrossRef CAS
- S. Ye, Y. Lei, T. Xu, L. Zheng, Z. Chen, X. Yang, X. Ren, Y. Li, Q. Zhang and J. Liu, Appli. Catal. B, 2022, 304, 120986 CrossRef CAS
- Y. Zhou, Y. Wu, D. Guo, J. Li, Y. Li, X. Yang, S. Fu, G. Sui and D.-F. Chai, ACS Appl. Mater. Inter., 2023, 15, 15797–15809 CrossRef CAS PubMed
- Z. Nie, T. Liu, Y. Chen, P. Liu, Y. Zhang, Z. Fan, H. He, S. Chen and F. Zhang, Electrochim. Acta, 2022, 402, 139558 CrossRef CAS
- A. Wang, X. Zhang, S. Gao, C. Zhao, S. Kuang, S. Lu, J. Niu, G. Wang, W. Li, D. Chen, H. Zhang, X. Zhou, S. Zhang, B. Zhang and W. Wang, Adv. Mater., 2022, 34, 2204247 CrossRef CAS PubMed
- K. Yu, H. Yang, H. Zhang, H. Huang, Z. Wang, Z. Kang, Y. Liu, P. W. Menezes and Z. Chen, Nano-Micro Lett., 2023, 15, 186 CrossRef CAS PubMed
- A. Malek, Y. Xue and X. Lu, Angew. Chem., Int. Ed., 2023, 62, e202309854 CrossRef CAS PubMed
- C. Liu, Y. Han, L. Yao, L. Liang, J. He, Q. Hao, J. Zhang, Y. Li and H. Liu, Small, 2021, 17, 2007334 CrossRef CAS PubMed
- H. Nan, M. Liu, Q. Zhang, M. Wang, S. Liu, L. Qiao, X. Hu and H. Tian, J. Power Sources, 2020, 451, 227822 CrossRef CAS
- Y. Li, J. Cai, J. Zhang, Z. Chen, G. Wang, Q. Chen and M. Chen, Adv. Energy Mater., 2023, 13, 2204114 CrossRef CAS
- H. Wang, Y. Zhou and S. Tao, Appli. Catal. B, 2022, 315, 121588 CrossRef CAS
- H. Chen, J. Chen, H. He, X. Chen, C. Jia, D. Chen, J. Liang, D. Yu, X. Yao, L. Qin, Y. Huang and Z. Wen, Appli. Catal. B, 2023, 323, 122187 CrossRef CAS
- S. H. Ye, Z. X. Shi, J. X. Feng, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2018, 57, 2672–2676 CrossRef CAS PubMed
- W. H. Lee, M. H. Han, Y.-J. Ko, B. K. Min, K. H. Chae and H.-S. Oh, Nat. Commun., 2022, 13, 605 CrossRef CAS PubMed
- X. Han, C. Liu, Y. Tang, Q. Meng, W. Zhou, S. Chen, S. Deng and J. Wang, J. Mater. Chem. A, 2023, 11, 14424–14431 RSC
- M. E. Orazem and B. Tribollet, ChemTexts, 2020, 6, 12 CrossRef CAS
- D. Li, R. Xiang, F. Yu, J. Zeng, Y. Zhang, W. Zhou, L. Liao, Y. Zhang, D. Tang and H. Zhou, Adv. Mater., 2023, 35, 2305685 Search PubMed
- F. Yue, C. Wang, W. Duan, H. Pang, T. Wei, K. Xue, D. Wang, F. Fu and C. Yang, Sci. China: Chem., 2023, 66, 2109–2120 CrossRef CAS
- J. Sun, H. Xue, N. Guo, T. Song, Y.-R. Hao, J. Sun, J. Zhang and Q. Wang, Angew. Chem., Int. Ed., 2021, 60, 19435–19441 CrossRef CAS PubMed
- H. Wu, Q. Lu, Y. Li, J. Wang, Y. Li, R. Jiang, J. Zhang, X. Zheng, X. Han, N. Zhao, J. Li, Y. Deng and W. Hu, Nano Lett., 2022, 22, 6492–6500 CrossRef CAS PubMed
- B. Qiu, L. Cai, Y. Wang, Z. Lin, Y. Zuo, M. Wang and Y. Chai, Adv. Funct. Mater., 2018, 28, 1706008 CrossRef
- E. Wang, M. Guo, J. Zhou and Z. Sun, Mater., 2023, 16, 1457 CrossRef CAS PubMed
- W. Zhang, M. Liu, X. Gu, Y. Shi, Z. Deng and N. Cai, Chem. Rev., 2023, 123, 7119–7192 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh02090h |
| This journal is © The Royal Society of Chemistry 2024 |