
Metal ion interference therapy: metal-based nanomaterial-mediated mechanisms and strategies to boost intracellular “ion overload” for cancer treatment
Yutang
Li
a,
Yandong
Wang
a,
Li
Zhao
a,
Martina H.
Stenzel
 *b and
Yanyan
Jiang
*a
*b and
Yanyan
Jiang
*a
aKey Laboratory for Liquid–Solid Structural Evolution & Processing of Materials (Ministry of Education), School of Materials Science and Engineering, Shandong University, Jinan, Shandong 250061, P. R. China. E-mail: yanyan.jiang@sdu.edu.cn
bSchool of Chemistry, University of New South Wales (UNSW), Sydney, NSW 2052, Australia. E-mail: m.stenzel@unsw.edu.au
First published on 29th June 2024
Abstract
Metal ion interference therapy (MIIT) has emerged as a promising approach in the field of nanomedicine for combatting cancer. With advancements in nanotechnology and tumor targeting-related strategies, sophisticated nanoplatforms have emerged to facilitate efficient MIIT in xenografted mouse models. However, the diverse range of metal ions and the intricacies of cellular metabolism have presented challenges in fully understanding this therapeutic approach, thereby impeding its progress. Thus, to address these issues, various amplification strategies focusing on ionic homeostasis and cancer cell metabolism have been devised to enhance MIIT efficacy. In this review, the remarkable progress in Fe, Cu, Ca, and Zn ion interference nanomedicines and understanding their intrinsic mechanism is summarized with particular emphasis on the types of amplification strategies employed to strengthen MIIT. The aim is to inspire an in-depth understanding of MIIT and provide guidance and ideas for the construction of more powerful nanoplatforms. Finally, the related challenges and prospects of this emerging treatment are discussed to pave the way for the next generation of cancer treatments and achieve the desired efficacy in patients.
Wider impactThis review will significantly impact the advancement of innovative MIIT, personalized medicine, and interdisciplinary research. It has the potential to inspire researchers to develop more groundbreaking MIIT approaches, including novel treatment regimens, advanced drug delivery systems, and enhanced strategies for tailoring treatments based on patient genotypes, molecular characteristics, and pathology. Consequently, this will improve treatment accuracy and success rates. Additionally, the advancement of MIIT involves multiple fields such as biochemistry, nanotechnology, materials science and medicine, fostering interdisciplinary collaboration and cutting-edge research to tackle intricate scientific challenges. |
1. Introduction
Malignant tumors pose a severe threat to human life and health as they are highly aggressive and metastatic.1 Research indicates that tumor metastasis is accountable for approximately 90% of all cancer-related deaths.2 Once a primary tumor infiltrates nearby tissues, it gains access to the lymphatic and blood systems, utilizing them as conduits for metastasis and the formation of secondary tumors.3 Over the past decade, scientists have endeavored to develop diverse cancer treatments with enhanced efficacy against malignant tumors.4,5 Traditional cancer treatments, including surgical resection, chemotherapy, radiation, and immunotherapy, have been optimized to eradicate primary tumors.6 However, the clinical treatment of metastatic tumors remains extremely challenging and is associated with serious side effects such as surgical trauma, immune system damage, and neurotoxicity.7With the continuous advancement of nanotechnology, there is growing interest in utilizing nanomaterials for the detection and treatment of metastatic tumors.8–10 Compared to traditional small molecule drugs, biodegradable metal-based nanomaterials exhibit advantages, including enhanced permeability and retention (EPR) effect, diverse functionalities, and responsiveness to the tumor microenvironment (TME), making them more selective in tumor treatment while minimizing the potential adverse effects on normal tissues.11–13 Degradation-released metal ions disrupt homeostasis in cancer cells, leading to irreversible damage or even death.14–16 Based on this, biodegradable metal-based nanomaterials largely achieve the localized accumulation of metal ions at tumor sites, which is a difficult task to accomplish solely through the administration of metal ions. Therefore, extensive research on metal ion interference therapy (MIIT) has led to the development of a novel treatment strategy.
As research progresses, various roles of metal ions in human physiological activities are being increasingly elucidated. Certain metal ions are closely related to protein stability and biological activity. For example, Fe3+ and Cu2+ can disrupt redox homeostasis and induce ferroptosis in tumor cells by inhibiting the activity of glutathione peroxidase 4 (GPX4).17–19 Moreover, excess Cu+ can trigger the aggregation of lipoylated proteins and destabilize iron–sulfur cluster proteins, resulting in mitochondrial dysfunction and cuproptosis.20 Similarly, Zn2+, Ca2+, and Mg2+ can also act on mitochondria, resulting in cellular metabolic disorders.21–23 Specifically, Zn2+ and Mg2+ can inhibit the activity of glycolysis-related enzymes, cut off the energy supply, and cause a starvation effect in cancer cells, thereby inducing apoptosis of cancer cells.24,25 In the case of Ca2+, it can induce the release of cysteine aspartate-specific proteases (caspase) to initiate apoptosis by destroying the mitochondrial morphology. Furthermore, the released caspase 3 can activate the formation of transmembrane pores, leading to the occurrence of pyroptosis,26,27 which is similar to the caspase 1-mediated pyroptosis initiated by Na+/K+ overload.28
Additionally, metal ions have been found to serve as immune activators, stimulating the body's immune response to overcome clinical barriers associated with low tumor immunogenicity and immune-related adverse events linked to immunotherapy. For instance, Zn2+ and Mn2+ can stimulate the cyclic GMP-AMP synthase/stimulator of interferon gene (cGAS/STING) immune signaling pathway in tumor cells, thereby suppressing immune escape effects and enhancing the body's immune response to tumors.29,30 Therefore, MIIT is a powerful therapeutic tool with strong tumor cell-killing capacity and abundant immunological effects. However, it is important to recognize that research on MIIT is still in its infancy. To date, numerous studies examining various types of MIIT have investigated their different molecular mechanisms and amplification strategies, thereby enhancing our understanding of this emerging therapeutic modality. A critical aspect in the advancement of MIIT is the design of amplification strategies that leverage interference mechanisms to optimize its therapeutic efficacy. These strategies aim to augment the treatment outcomes through synergistic treatment, disruption of ion homeostasis, and intensification of metabolic damage across diverse pathways. However, disparities exist in the research progress in different types of MIIT, necessitating a meticulous and comprehensive review to offer valuable insights for more recent advancements in the development of MIIT. By comparing differences and similarities, our perception of MIIT can be reshaped, moving beyond the limited focus on individual MIIT types to a holistic understanding of MIIT as a collective entity.
In this comprehensive review, we focus on the intrinsic mechanism of different metal ion (such as Fe3+/Fe2+, Cu2+/Cu+, Ca2+, Zn2+, Mn2+, Na+/K+, and Mg2+)-mediated interference therapies and their research progress in cancer treatment. Moreover, we shed light on the amplification strategies of MIIT, such as modulation of relevant metabolic pathways to sensitize cancer cells to MIIT, inhibition of channel protein activity to disrupt original homeostasis, and synergy with other therapeutics. Moreover, the current challenges encountered in this field and its prospects for clinical translation are also discussed. It is worth noting that this review is the first comprehensive report on the progress of emerging MIIT and its corresponding amplification strategies. We anticipate that this review will offer valuable guidance for the future development of efficient metal-based nanomaterials for effective MIIT and lay a solid foundation for the early clinical translation of MIIT.
2. Conception of metal ion interference therapy
2.1. Accurate delineation of metal ion interference therapy
Endogenous metal ions are crucial for various physiological processes within cells, such as signaling pathway regulation, enzyme catalysis, and protein synthesis.31,32 Accordingly, to maintain these metal ions at levels within acceptable ranges, cells have established sophisticated and highly collaborative ion homeostatic regulatory systems for commonly found metal ions.33,34 However, when the dynamic balance is severely disrupted, intracellular original metabolisms are blocked and the process of programmed cell death (e.g. apoptosis, pyroptosis, ferroptosis, and cuproptosis) is initiated.35,36 Additionally, some nutritional metal ions can also stimulate the body's immune response to tumor cells, giving access to a novel form of immunotherapy.37 Therefore, supplementation of exogenous metal ions or disruption of intracellular regulatory systems has been adopted to restrain the progression of tumors. In this case, given the huge potential of metal ions in combating tumors, the method of manipulating the level of metal ions at the tumor site has been defined as metal ion interference therapy.2.2. Nanomaterial-facilitated metal ion interference anticancer therapy
The initial prerequisite to achieve efficient MIIT is to precisely elevate the levels of metal ions at the tumor sites. However, the short blood half-life and non-specificity of metal ions increase the risk of serious whole-body toxicity and undesirable therapeutic effects. In this case, with the rapid advancement of nanotechnology, biodegradable metal-based nanomaterials (e.g. metal oxides, metal peroxides, metal salts, metal organic frameworks (MOF), and metal complexes) have been adopted as carriers for metal ions to address the challenge in their delivery.38,39 Most of these metal-based nanomaterials typically demonstrate TME-responsive properties, where they are particularly sensitive to the weak acidity and elevated levels of glutathione (GSH) in the TME.40,41 Consequently, at tumor sites, these nanomaterials degrade rapidly, leading to the specific release of metal ions. This targeted degradation reduces the risk of metal ion leakage into adjacent tissues, thus minimizing the potential adverse effects of MIIT. Furthermore, the versatility of metal-based nanomaterials allows the integration of amplification strategies such as synergistic therapy and disruption of homeostatic regulatory systems, which augment the therapeutic efficacy of MIIT.423. Progress in metal ion interference therapy
The underlying mechanisms of various types of MIIT are intricate and diverse, making it suitable for addressing a wide range of tumors. As researchers strive to optimize MIIT, several amplification strategies have been proposed and explored. These strategies are based on the disruption of the ion homeostasis regulation systems in cancer cells to induce a more pronounced ion imbalance. In this section, we provide a detailed summary of the intrinsic mechanisms of different MIIT approaches, including iron ion interference, copper ion interference, Ca2+ interference, Zn2+ interference, and some other metal ion interference therapy (Scheme 1). By virtue of their Fenton-catalytic property, iron ions generate intracellular reactive oxygen species (ROS), disrupting the redox balance, and thereby triggering ferroptosis mediated by the accumulation of toxic lipid peroxides (LPO). The aggregation of lipoylated proteins and destabilization of Fe–S cluster proteins caused by copper ions induce cuproptosis, a novel form of cell death resulting from proteotoxic stress. Ca2+ overload can cause mitochondrial dysfunction to initiate caspase 3-mediated apoptosis and degrade the cell skeleton to restrict the metastasis of cancer cells. Zn2+ interference blocks the development of tumors in multiple ways through the inhibition of glycolysis, cGAS/STING activation, and mutant p53 protein (Mutp53) degradation. However, the understanding of the effect if other metal ions on metabolic pathways by is currently rudimentary and lacks systematicity.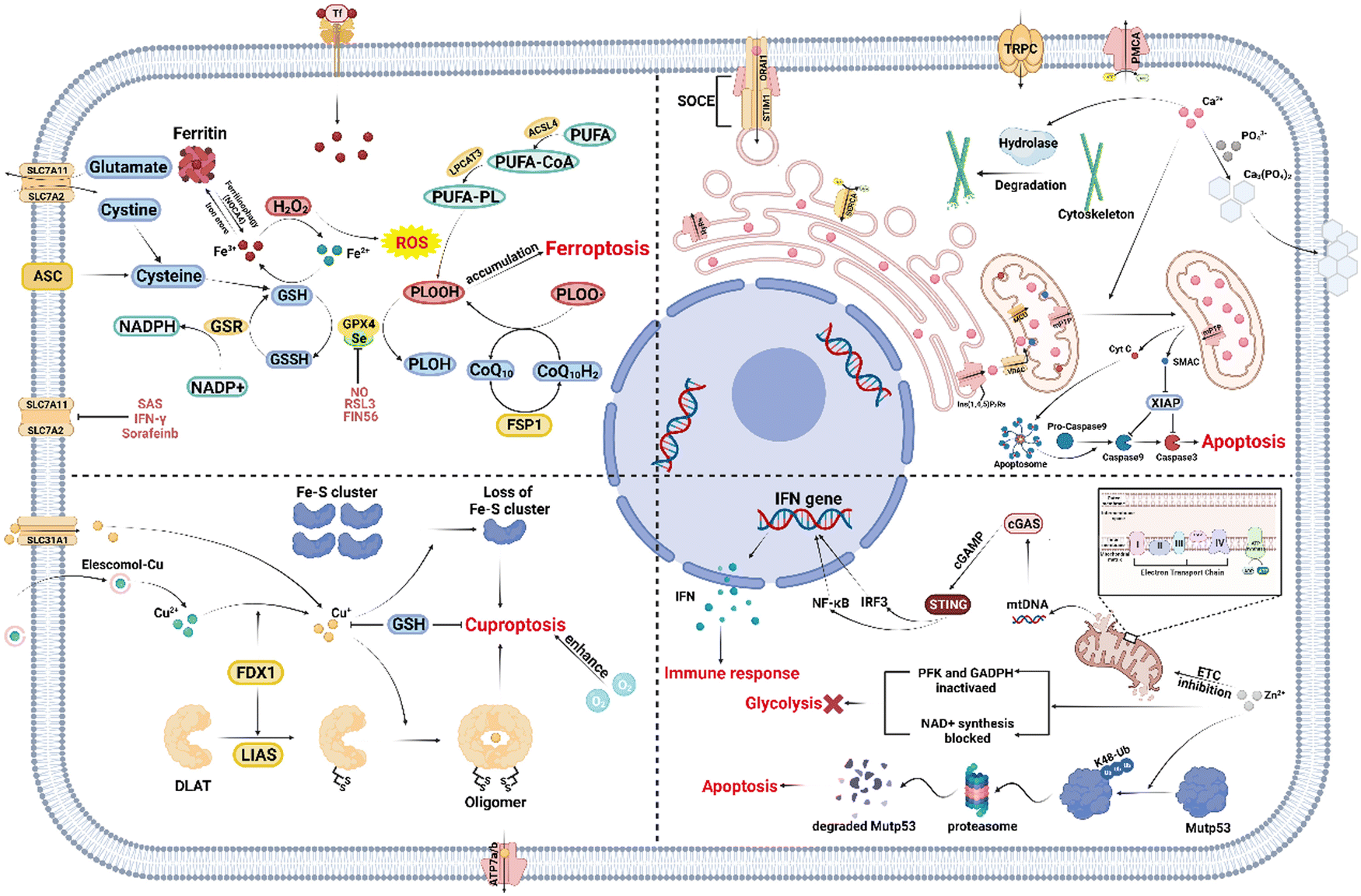 | ||
| Scheme 1 Schematic illustration of the mechanism pathways of various metal ion interference therapies, mainly including ferroptosis, cuproptosis, Ca2+ interference therapy, and Zn2+ interference therapy. This scheme was created with Biorender.com. | ||
3.1. Iron ion interference therapy
 | ||
| Fig. 1 (A) Mechanism diagram of the transformation between Cro-Fe and Cro.66 (B) Iron ion release profile of Cro-Fe at different pH values.66 (C) Photothermal conversion capacity of Cro-Fe at different pH values.66 Reprinted with permission from ref. 66. Copyright 2021, Wiley-VCH GmbH. (D) Schematic illustration of the pathways of PdPtAu@TF to ferroptosis.70 (E)–(G) GSH level (E), expression of GPX4 (F) and LPO level (G) in 4T1 cells treated with different concentrations of PdPtAu@TF.70 Reprinted with permission from ref. 70. Copyright 2023, Wiley-VCH GmbH. (H) Synthetic process and TEM image of Ce6-PEG-HNK15.71 (I) Degree of co-localization of ferritin and Ce6-PEG-HNK15.71 (J) Schematic diagram of Ce6-PEG-HNK15 destroying ferritin.71 (K) Cytosolic iron levels in 4T1 cells treated with different formulations.71 (L) LPO levels in different groups.71 Reprinted with permission from ref. 71. Copyright 2022, Wiley-VCH GmbH. (M) Mechanism diagram of COS@MOF.72 (N) Bio-TEM of autophagosomes and mitochondrial changes in CT26 cell.72 (Yellow box: autophagosomes and red box: mitochondrial changes). (O) Autophagy indicator LC3II/LC3I ratio.72 (P) Expression of p62, NCOA4 and GPX4. Reprinted with permission from ref. 71. Copyright 2023, Wiley-VCH GmbH. | ||
Ferritin, the intracellular “Fe pool”, has a powerful buffering effect on Fe ions, thus weakening exogenous ferroptosis. Additionally, ferritin has been found to be overexpressed in various malignant tumors, causing them to have greater anti-interference ability for Fe ions than normal cells.73 Therefore, target-attacking ferritin can potentially be a strategy to provoke Fe ion disturbance, thus improving the therapeutic effect of ferroptosis. The ferritin-homing peptide HKN15 provides a precise target to attack ferritin to achieve endogenous Fe ion interference.74 Ferritin-hijacking Ce6-PEG-HKN15 NPs prepared by self-assembly were validated to destroy ferritin under laser irradiation (Fig. 1H).71 Specifically, Ce6-PEG-HKN15 NPs preferentially gathered around ferritin due to the targeting of HKN15, which was supported by the higher overlap of ferritins and Ce6-PEG-HKN15 (Fig. 1I). The ROS generated by laser-activated Ce6 could destroy ferritin to induce the release of Fe3+ (Fig. 1J). Furthermore, the disruption of the “Fe pool” effectively enhanced ferroptosis, as confirmed by the significantly elevated cytoplasmic level of Fe ions and LPO content (Fig. 1K and L). Moreover, activated autophagy is another promising pathway to induce the degradation of ferritin.75 Du's group72 connected the autophagy accelerator chitosan oligosaccharides (COS) to an etched MOF to fabricate an autophagy-amplifying nanocomposite, COS@MOF (Fig. 1M). COS@MOF produced an increased number of autophagosomes, and the elevated LC3II/LC3I further confirmed the occurrence of the cellular autophagy process (Fig. 1N and O). Furthermore, the increased presence of NCOA4 and decreased levels of GPX4 suggested that autophagy facilitated the degradation of ferritin to enhance ferroptosis (Fig. 1P). Overall, specifically target-attacking ferritin or enhancing the autophagy of cells can facilitate the degradation of ferritin, thereby increasing the Fe ion interference in cancer cells.
GSH is critical in maintaining the redox balance within cells, and thus disrupting the GSH-centered antioxidant defense mechanism is an effective strategy to enhance the sensitivity of cancer cells to ferroptosis. Firstly, the content of intracellular GSH can be reduced by limiting the cellular uptake of Cys. Science reported that Cys starvation, achieved through the deletion of subunit SLC7A11, is a translatable method to induce tumor-selective ferroptosis.76 The clinical drug sulfasalazine (SAS) can delete SLC7A11 to inhibit the synthesis of GSH, thereby strengthening the ferroptosis of cancer cells.77 Cai's team78 designed a hypoxia-responsive dual-drug delivery system, PAD@MS, by simply coordinating Fe3+ with mitoxantrone (MTO), sulfasalazine (SAS), and dopamine derivative of polyethylene glycol (Fig. 2A). Under hypoxia TME, PAD@MS obviously degraded and released SAS, which significantly down-regulated the expression of SLC7A11 to strengthen immunogenic ferroptosis therapy with ROS burst (Fig. 2B and C). The immunogenic ferroptosis could lead to the up-regulation of interferon γ (IFN-γ), which further contributed to the down-regulation of SLC7A11, ultimately resulting in a significant decrease in GSH generation (Fig. 2D). Moreover, several Cys transporter system Xc-inhibitors such as erastin and sorafenib have also been identified to strengthen ferroptosis.45,79 In addition to small molecule drugs, the corresponding siRNA can silence the expression of system Xc- to achieve the same effect of limiting Cys uptake and GSH synthesis.80 Besides cutting off the external supply of Cys, the Cys-depleting strategy realized based on the strong nucleophilic nature of the mercapto moiety has also received extensive attention.81 In the current work, researchers utilized the acrylate group of Aza-BDY to selectively consume intracellular Cys through Michael addition and subsequent cyclization reactions (Fig. 2E and F).82 Meanwhile, Aza-BDY converted into the sonosensitizer Azb-BDY, which was confirmed by the shift in its absorption peak (Fig. 2G). Consequently, there was a significant increase in the level of LPO after Aza-BDY + US treatment (Fig. 2H). In conclusion, strategically blocking the Cys supply by down-regulating SLC7A11 and inhibiting system Xc-activity can effectively inhibit intracellular GSH synthesis to enhance ferroptosis.
 | ||
| Fig. 2 (A) Synthetic process of PAD@MS.78 (B) Degradation of PAD@MS under hypoxia condition.78 (C) Release profile of SAS.78 (D) SLC7A11 content in tumor cells with different treatments.78 Reprinted with permission from ref. 78. Copyright 2023, Wiley-VCH GmbH. (E) Mechanism diagram of the transformation between Aza-BDY and Azb-BDY.82 (F) UV/vis absorption of Aza-BDY treated with different amino acids. (G) Change in UV/vis absorption of Aza-BDY before and after reaction with Cys.82 (H) Confocal image of LPO in tumor cells.82 Reprinted with permission from ref. 82. Copyright 2022, Wiley-VCH GmbH. | ||
Some types of cancer cells can bypass system Xc-, leading to system Xc-inhibitors that cannot achieve the desired effect.83 Fortunately, there are alternative strategies to promote the sensitization of tumor cells to ferroptosis. One of these strategies is the suppression of GPX4, which is the central regulator of ferroptosis. As mentioned above, by accelerating the ferroptosis cycle or introducing additional free radicals, the depletion of GSH and subsequent inactivation of GPX4 can be achieved. Nitric oxide (NO), an endogenous signaling gasotransmitter, has the ability to generate the highly reactive free radical ˙NO to induce oxidative stress and inactivate GPX4. Additionally, certain small molecule drugs, including RSL3, RSL5, and FIN56, can trigger ferroptosis by directly acting on GPX4. Among them, RSL can specifically target the nucleophilic site of GPX4 and effectively deactivate it through the alkylation of selenocysteine. Thus, nano-delivery strategies for RSL have been developed to strengthen ferroptosis.84 Differently, FIN56 selectively enhances the lysosomal degradation of GPX4, and concurrently activates squalene synthase to deplete CoQ10. The GDY-FIN56-RAP nanoplatform containing FIN56 was successfully employed to induce ferroptosis in glioblastoma, demonstrating great potential for clinical therapy.85 In summary, the direct or indirect inactivation of GPX4 has gradually become a favored option to improve the therapeutic effect of ferroptosis.
Most ferroptosis-related amplification strategies are based on targeting the redox balance within tumor cells. Strategies such as attacking intracellular ferritin, supplying H2O2, and enhancing the temperature aim to increase ROS production, leading to the accumulation of toxic LPO. Simultaneously, disrupting the GSH-centered antioxidant defense system can be achieved by either blocking system Xc- or inactivating GPX4.
Iron ion interference therapy, one of the most extensively researched forms of MIIT, has shown synergistic benefits when combined with innovative therapeutic methods such as PDT/SDT and PTT. High temperature can enhance the cell membrane permeability and speed up the Fenton reaction within tumor cells, hence amplifying intracellular oxidative stress. Besides, iron ion interference alleviates the drawback of tumor thermoresistance to PTT by suppressing the expression of HSP. Subsequently, PDT and SDT function as an additional pathway to boost ROS, thus elevating the LPO levels and strengthening ferroptosis. Belonging to ICD, ferroptosis plays a significant role in modulating the immunosuppressive TME. After recognizing DAMPs such as ATP and HMGB1 released through ferroptosis, dendritic cells (DCs) undergo maturation and antigen presentation to activate cytotoxic T lymphocytes (CTLs) for tumor cell elimination. In turn, IFNγ secreted by CTLs downregulates the expression of SLC7A11 in tumor cells to accelerate ferroptosis, creating a positive feedback loop to enhance the immune response. Additionally, recent evidence indicates that the immune cells within the TME also experience ferroptosis, affecting their immune and anti-tumor characteristics. For instance, stimuli-promoting ferroptosis can modulate multiple metabolic pathways to polarize pro-tumor M2 macrophages towards anti-tumor M1 phenotypes, awakening potent phagocytic killing abilities and migration inhibition in tumor-associated macrophages.
3.2. Copper ion interference therapy
Cuproptosis, a form of proteotoxic stress-induced cell death, is closely associated with the aggregation of lipoylated proteins and the instability of iron–sulfur cluster proteins. Protein lipoylation involves the attachment of lipoic acid to specific mitochondrial proteins through amide bonds. Several key mitochondrial proteins participate in this process, including dihydrolipoamide S-acetyltransferase (DLAT), dihydrolipoamide branched chain transacylase E2 (DBT), glycine cleavage system protein H (GCSH), and dihydrolipoamide S-succinyltransferase (DLST), which are all vital for regulating carbon entry into the TCA cycle. When Cu+ binds to lipoylated proteins, it leads to the formation of oligomers, which severely disrupt mitochondrial respiration. Alternatively, proteins that are not lipoylated cannot bind to Cu+. The key genes associated with this process were identified by screening to be ferredoxin 1 (FDX1) and lipoyl synthase (LIAS). The FDX1 gene is responsible for encoding a small iron–sulfur protein that converts Cu2+ to the more toxic Cu+, while LIAS, belonging to the family of lipoic acid synthases, catalyzes the final step in the de novo synthesis of lipoic acid. After the knockdown of FDX1, the complete loss of protein lipoylation and a significant decrease in respiration were observed, confirming that FDX1 is an upstream regulator. In addition, genes that encode the component of the lipoic acid pathway, such as lipolytransferase1 (LIPT1), lipoyl synthase (LIAS), and dihydrolipoamide dehydrogenase (DLD), undoubtedly also participate in the cuproptosis process. It is important to note that tumor cells predominantly rely on glycolysis for energy metabolism, which is known as the Warburg effect, making them insensitive to cuproptosis compared to normal cells. Most Fe–S cluster proteins are important coenzymes involved in electron transfer chains (ETC) and other biochemical processes. Copper ion overload triggers a remarkable loss of Fe–S cluster proteins in an FDX1-dependent manner. However, due to its recent discovery, only the basic principles of cuproptosis are clearly understood. Therefore, the associated metabolic processes still need to be further explored to help fully recognize this new type of cell death.
Among the Cu ionophores, traditional small molecule Cu ionophores cannot effectively deliver sufficient Cu to tumor sites due to their limitations such as short blood half-life and poor specificity.91 Thus, to address these issues, intelligent nanoplatforms have been developed as Cu ionophores to induce cuproptosis, leveraging the advancements in nanotechnology. A novel ROS-responsive NP@ESCu nanoparticle was created by encapsulating the amphiphilic biodegradable polymer PHPM on the surface of elesclomol-Cu92 (Fig. 3A). The thioketal bonds of PHPM were broken by the intertumoral excessive ROS, leading to the release of elesclomol-Cu (Fig. 3B). Upon targeted transportation to the mitochondria, Cu2+ was released from the elesclomol-Cu complex and reduced to Cu+ by FDX1. Simultaneously, elesclomol rapidly effluxed and chelated with extracellular Cu2+ before being transported into tumor cells. This shuttle mechanism facilitated the continuous influx of Cu2+, thereby reinforcing cuproptosis. The shuttle mechanism was validated by the observed decrease in the IC50 of NP@ESCu to BIU-87, reaching 6 nM in the presence of preincubated CuCl2 (Fig. 3C). Notably, elevated Cu+ concentrations consistently upregulated PD-L1 on the surface of cancer cells, thereby enhancing the efficacy of αPD-L1 (Fig. 3D).
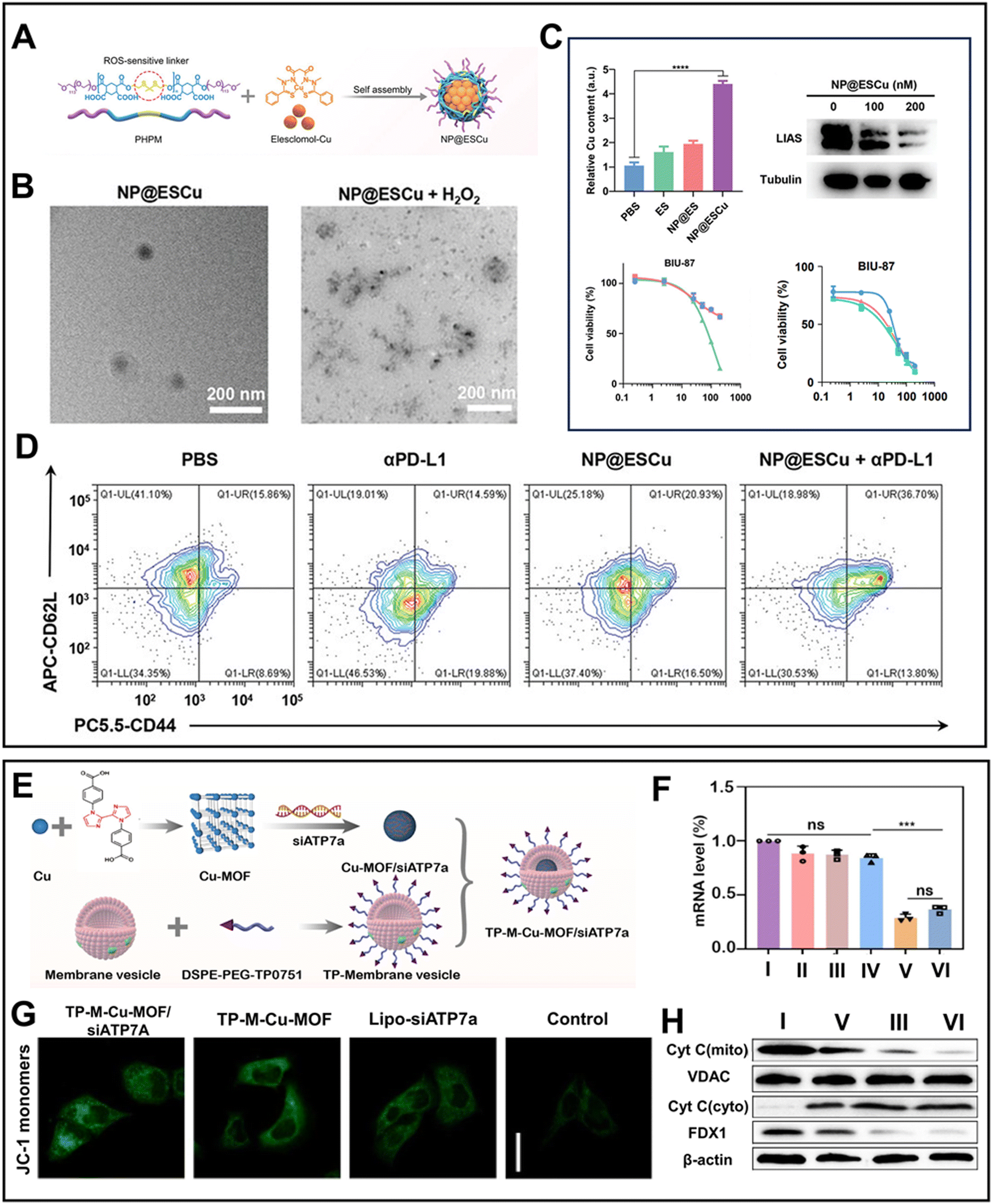 | ||
| Fig. 3 (A) Synthetic process of NP@ESCu.92 (B) Degradation of NP@ESCu in response to ROS.92 (C) Relative Cu content in tumor, expression of LIAS, and cell viability of BIU-87 cells treated with CuCl2 and NP@ESCu.92 (D) Flow cytometry maps of DCs with various treatments.92 Reprinted with permission from ref. 92. Copyright 2023, Wiley-VCH GmbH. (E) Synthetic process of TP-M-Cu-MOF/siATP7a.93 (F) mRNA level and expression of ATP7a in H69 cells with different treatments.93 (G) Mitochondrial membrane potential in H69 cells treated with various formulations.93 (H) Expression of Cyt C and FDX1.93 All groups: I Control; II siATP7a; III TP-M-Cu-MOF; IV TP-M-Cu-MOF/siRNANC; V lipo-siATP7a; and VI TP-M-Cu-MOF/siATP7a. Reprinted with permission from ref. 93. Copyright 2023, Elsevier B.V. | ||
Unfortunately, the long-term use of the Cu supplementation strategy in chronic diseases may cause tolerance to cuproptosis. Copper chaperone proteins and membrane transporters play crucial roles in maintaining intracellular Cu+ homeostasis. Among these proteins, the p-type ATPase copper-transporting ATPase (ATP7a/b), which can modulate the efflux of Cu+, is a target for disrupting intracellular Cu homeostasis.94 Inspired by this, a pH-responsive Cu-MOF loaded with siATP7a, named TP-M-Cu-MOF/siATP7a, was designed to strengthen cuproptosis for cancer therapy93 (Fig. 3E). The transcription of the ATP7a gene was significantly restricted due to the robust gene silencing, which further reduced the Cu+ efflux and elevated the intracellular Cu+ level (Fig. 3F). Furthermore, the Fenton-like catalytic property of Cu+ disrupted the mitochondrial metabolism to activate the related cuproptosis pathways (Fig. 3G and H). Moreover, recently, H2S-activitated Cu2(PO4)(OH) NPs, which could generate ultra-small copper sulfide (Cu9S8) nanoparticles through an in situ sulfidation reaction, were synthesized (Fig. 4A).95 The ultra-small Cu9S8 nanoparticles with an average size of 15 nm could quickly enter tumor cells through endocytosis and be degraded (Fig. 4B). Specifically, the higher endocytosis efficiency of Cu9S8 resulted in an intracellular ROS storm and restricted the energy supply, leading to a noticeable decrease in the activity of ATP7a (Fig. 4C). Then, nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasomes were activated and the activity of FDX1 was inhibited to induce a highly effective combination of pyroptosis and cuproptosis (Fig. 4D).
 | ||
| Fig. 4 (A) Mechanism diagram of Cu2(PO4)(OH)-mediated cuproptosis and pyroptosis.95 (B) Change in particle size and Cu2+ release profile of Cu9S8 at different pH values.95 (C) Content of cellular copper and expression of ATP7A in HCT116 cells treated with different formulations.95 (D) FDX1 and NLRP3 staining in HCT116 cells (scale bar = 100 μm).95 Reprinted with permission from ref. 95. Copyright 2023, Wiley-VCH GmbH. (E) Synthetic process of BCMD NPs.64 (F) pH-responsive-degradation of BCMD.64 (G) Cu2+ and BSO release profiles of BCMD.64 (H) Immunofluorescence image of DLAT in tumor cells with different treatments.64 (I) Western blot analysis of POLD1, FDX1, LIAS, ACO-2 and HSP70.64 Reprinted with permission from ref. 64. Copyright 2023, Elsevier Ltd. | ||
With further exploration, glycolytic inhibition has been confirmed by several studies as an innovative way to strengthen cuproptosis. Improving the hypoxic microenvironment of tumors can significantly inhibit tumor glycolysis, and thus augment cuproptosis. Based on this mechanism, a Cu-based nanoreactor CCJD-FA, which is mainly composed of CaO2, Cu-based shell, and JQ-1 (a bromodomain-containing protein 4 inhibitor), was fabricated to facilitate immunogenic cuproptosis.96 In this nanosystem, the Cu-based shell responded to overexpressed GSH to release Cu2+, and subsequently the exposed CaO2 decomposed into H2O2, which in turn produced O2 through Cu2+-mediated catalase-like properties, thus relieving the hypoxia TME. Furthermore, JQ-1 restricted the expression of glycolysis-related enzymes. Consequently, O2 combined with JQ-1 to effective suppress glycolysis, thereby blocking ATP generation and inactivating ATP7b. As previously stated, GOx can consume energy substrates of tumor cells and disrupt the intracellular energy metabolism. Recently, employing a GOx-engineered nonporous copper(I) 1,2,4-triazolate coordination polymer nanoplatform (GOx@[Cu(tz)]), it was confirmed that starvation therapy indeed improved cuproptosis.97 Regrettably, the above-mentioned studies did not elaborate on the intrinsic mechanism of glycolysis inhibition-enhanced cuproptosis. Given the metabolic plasticity of cancer cells, glycolysis inhibition may trigger a metabolic shift to aerobic respiration, thus sensitizing them to cuproptosis. Furthermore, a high endogenous level of GSH can function as a thiol-containing copper chelator to prevent the occurrence of cuproptosis.98 Thus, to address this, Huang et al.64 devised a bidirectional amplification strategy for cuproptosis by constructing a Cu-based nanoplatform, BSO-CAT@MOF-199@DDM (BCMD) (Fig. 4E). The pH-responsive degradation of BCMD destroyed its structure, releasing Cu2+, buthionine-sulfoximine (BSO), and CAT (Fig. 4F and G). The goal of this strategy was to increase the O2 concentration and decrease the GSH levels in tumors. Specifically, the secreted INF-γ combined with the released BSO to suppress the intracellular GSH content by inhibiting its synthesis. Meanwhile, the relief of hypoxia was aided by the catalytic capacity of CAT. Compared to the BMD or CMD groups, different types of lipoylated proteins and iron–sulfur cluster proteins in tumors were elevated and decreased in the BCMD group, respectively, indicating the occurrence of stronger cuproptosis (Fig. 4H and I).
Cuproptosis alone may not meet the needs of tumor treatment, and thus some work has been conducted to achieve a “1 + 1 > 2” effect by combining it with other therapies. Disulfiram (DSF) has been explored for its potential in combination with cuproptosis.99 DSF is rapidly converted to diethyldithiocarbamate (DTC) in the cell, which can chelate with Cu2+ to form the more toxic and stable bis(diethyldithiocarbamate)-Cu (CuET).100 Zhou et al.101 constructed an Au@MSN-Cu/PEG/DSF nanoplatform, as depicted in Fig. 5A and B. Under laser irradiation, Au@MSN-Cu/PEG/DSF exhibited an excellent photothermal conversion capacity (Fig. 5C). Subsequently, the Cu-doped silicone frameworks underwent temperature- and pH-dependent decomposition, leading to the release of Cu2+ and DSF (Fig. 5D and E). One part of Cu2+ was reduced to Cu+ by FDX1 to participate in the protein-lipid acylation process, while the other part was directly bound to DTC to form CuET (Fig. 5F). The aggregation of the NPL4 protein mediated by CuET resulted in the abnormal metabolism of ubiquitinated proteins, ultimately leading to apoptosis (Fig. 5G). The synergy of Cu2+ and DSF exhibited a remarkable enhancement in tumor inhibition (Fig. 5H). Another study demonstrated that CuET possessed better reduction resistance to the traditional anticancer agent cisplatin, inert reactivity with GSH, and tumor-selectivity toxicity.102 Therefore, CuET can be used as a robust and cuproptosis-dependent anticancer drug. Alternatively, Chan's group103 synthesized a programmed responsive nanosystem, DMMA@Cu2−xSe, to achieve the mutual promotion of thermotherapy and cuproptosis (Fig. 5I). Compared to other transition metal ions, selenium has a stronger binding capacity to copper ions, especially Cu+ (Fig. 5J). In an aqueous solution, Cu2−xSe NPs gradually oxidized and released Cu2+/Cu+, exhibiting the bio-responsive release of Cu2+/Cu+ (Fig. 5K). Besides, Cu2−xSe exhibited strong absorption and photothermal conversion efficiency in the far infrared region. Thermotherapy causes damage to cancer cells by increasing the temperature and causing mitochondrial disruption, which result in the upregulation of ROS. More ROS promoted the degradation of Cu2−xSe NPs, which contributed to the release of Cu2+/Cu+. Finally, in synergy with thermotherapy, the degree of cuproptosis substantially increased (Fig. 5L and M).
 | ||
| Fig. 5 (A) Synthetic process and (B) TEM image of Au@MSN-Cu/PEG/DSF.101 (C) Photothermal conversion capacity of Au@MSN-Cu/PEG/DSF.101 Release profile of (D) Cu and (E) DSF at different temperatures and pH values.101 (F) Mechanism diagram of Au@MSN-Cu/PEG/DSF-mediated cuproptosis and apoptosis.101 (G) Expression of DLAT, LIAS and NPL4 in 4T1 cells.101 (H) Tumor volume change.101 All groups: I control; II Au@MSN-Cu/PEG/DSF; and III Au@MSN-Cu/PEG/DSF + laser. Reprinted with permission from ref. 101. Copyright 2022, Wiley-VCH GmbH. (I) Schematic illustration of DMMA@Cu2-xSe-mediated synergy of cuproptosis and thermotherapy.103 (J) Binding capacity of selenium to different metal ions.103 (K) Copper concentration in tumors treated with different formulations.103 (L) Western blot analysis of FDX1, LIAS, DLST and DBT.103 (M) Mechanism diagram of cuproptosis-mediated mitochondrial dysfunction and ROS burst.103 Reprinted with permission from ref. 103. Copyright 2023, Wiley-VCH GmbH. | ||
Due to the recent discovery of cuproptosis, there are limited therapeutic platforms and amplification strategies available. Among them, the most commonly utilized methods include inhibiting Cu ion efflux and enhancing the synergy with other therapeutic tools. One major challenge in copper ion interference therapy is the high resistance exhibited by cancer cells towards cuproptosis, which can be attributed to the metabolic shifts towards glycolysis and the high glutathione (GSH) content. Various approaches have been proposed in previous studies to address this issue of resistance, such as glycolysis inhibition and GSH depletion, with the intention of restoring the sensitivity of cancer cells to cuproptosis. Notably, gene p53, which is closely linked to elevated GSH levels and metabolic alterations in cancer cells, stands out as a potential target for enhancing cuproptosis. Nevertheless, the precise role of p53 in regulating cuproptosis still needs to be conclusively proven. Therefore, further investigations should be directed towards exploring this aspect in future studies.
Previous studies demonstrated the synergistic enhancement of treatments such as PTT, SDT, and PDT with copper ion-mediated Fenton-like catalysis, similar to iron ion interference. Nevertheless, although these treatments have exhibited enhanced macroscopic therapeutic effects in proteotoxic stress-induced cuproptosis, to date, research has not clearly elucidated the underlying intrinsic mechanisms. Therefore, further efforts are needed to understand these mechanisms at the molecular level. Currently, there is limited research on the immunological effects of copper ion interference, besides its induction of ICD. Extensive studies have shown that copper-mediated cell death processes can reduce the surface PD-L1 levels in cancer cells, potentially enhancing the treatment effectiveness by combining with immune checkpoint inhibitors to overcome immune evasion. However, the regulatory effects of copper ions on various immune cells remain poorly understood, necessitating further research to explore the immunological behavior influenced by copper ion interference.
3.3. Calcium ion interference therapy
Cytoplasmic Ca2+ overload disrupts osmotic pressure balance, making it difficult for cells to maintain normal physiological processes. Excessive Ca2+ combines with phosphate to form calcium phosphate (CaP), which deposits on the cell membrane and results in cell calcification.119 This calcification affects the binding capacity of membrane proteins and interferes with signal transduction, ultimately disrupting cellular metabolism. Besides, excessive cytoplasmic calcium activates hydrolases, which break down the cytoskeleton, repressing the migration and invasion of tumor cells. More importantly, Ca2+ overload can cause mitochondrial dysfunction to kill tumor cells. High levels of mitochondrial Ca2+ have been shown to regulate the metabolic shift from oxidative phosphorylation (OXPHOS) to glycolysis (Warburg effect). This shift typically occurs in tumor cells to support their rapid proliferation.120 Therefore, higher Ca2+ uptake capacity is considered a hallmark of tumorigenesis but it also augments the tumor vulnerability to mitochondrial Ca2+ overload-induced apoptosis.33 Mitochondrial Ca2+ overload leads to the opening of mPTP, allowing more molecules to enter the mitochondrial matrix and resulting in the remodel of IMM.121 Consequently, the second mitochondria-derived activator of caspase (SMAC) and Cyt C are released from the cristae of IMM into the cytoplasm.122 Subsequently, SMAC activates the conversion of pro-caspase 9 to caspase 9 by binding to the inhibitor of apoptosis proteins, while Cyt C, together with cytoplasmic protein APAF1 and caspase 9, forms apoptosomes, which can induce apoptosis.123 Meanwhile, the increase in mPTP permeability depolarizes the mitochondrial membrane, leading to mitochondrial dysfunction and the release of ROS into the cytoplasm through mPTP. Overall, Ca2+ overload induces oxidative stress and apoptosis via mitochondrial dysfunction to kill cancer cells.
P-glycoprotein (PgP), an ATP-dependent drug pump overexpressed in tumor cells, is the major cause of the failure of clinical chemotherapy. Some studies have shown that the upregulation of HIF-1α can enhance the expression of PgP under hypoxic TME by regulating the MDR gene.126 Ca2+ overload-induced mitochondrial dysfunction can decrease the oxygen (O2) need of cancer cells, thus alleviating the hypoxia dilemma of chemotherapy. Thus, to achieve Ca2+ overload-augmented chemotherapy, a tumor-targeting “Ca2+ nanogenerator” (TCaNG) mainly containing CaP and doxorubicin (DOX) was prepared127 (Fig. 6A). In response to the acidic TME, TCaNG specifically degraded and released Ca2+ and DOX in tumor cells, resulting in mitochondrial dysfunction and the significant reduction in PgP content on the cell membrane (Fig. 6B and C). Moreover, Ca2+ interference-mediated ATP reduction also limited the efficiency of PgP and further reduced DOX pumping out (Fig. 6D). Consequently, TCaNG effectively reversed the drug resistance in cancer cells and enhanced the treatment of the chemotherapy drug DOX. Additionally, disrupting the Ca2+ buffering capacity of tumor cells through the modulation of channel proteins is an effective means of achieving Ca2+ accumulation. The reversal of drug resistance caused by Ca2+ overload can prevent the efflux of modulators, facilitating their full utilization. Kaempferol-3-O-rutinoside (KAE) exhibits excellent anti-cancer properties by suppressing SERCA1 and PMCA to disrupt Ca2+ homeostasis and induce apoptosis.128 Based on this, the M@CaCO3@KAE nanocomposite, incorporating CaCO3 and KAE, was fabricated for the successful delivery of drugs and amplification of Ca2+ overload129 (Fig. 6E and F). Encapsulation of the cancer cell membrane enabled M@CaCO3@KAE to specifically target the tumor sites through innate properties of immune escape and homologous aggregation. In this case, these two channel proteins were potently suppressed to block Ca2+ in the cytoplasm and mitochondria, thus amplifying the Ca2+ interference (Fig. 6G). Subsequently, hydrolase calpain1 activated by excessive Ca2+ degraded the cytoskeleton to limit the ability of tumor cells to migrate and invade (Fig. 6H). Besides, the Ca2+ overload-induced mitochondrial dysfunction effectively suppressed the polymerization of F-actin, which is indispensable in tumor metastasis (Fig. 6I).130 The degradation of F-actin caused the plasma membrane to fold, resulting in vague borders and unclear silhouettes. Therefore, the metastasis of the tumor was significantly impaired (Fig. 6J).
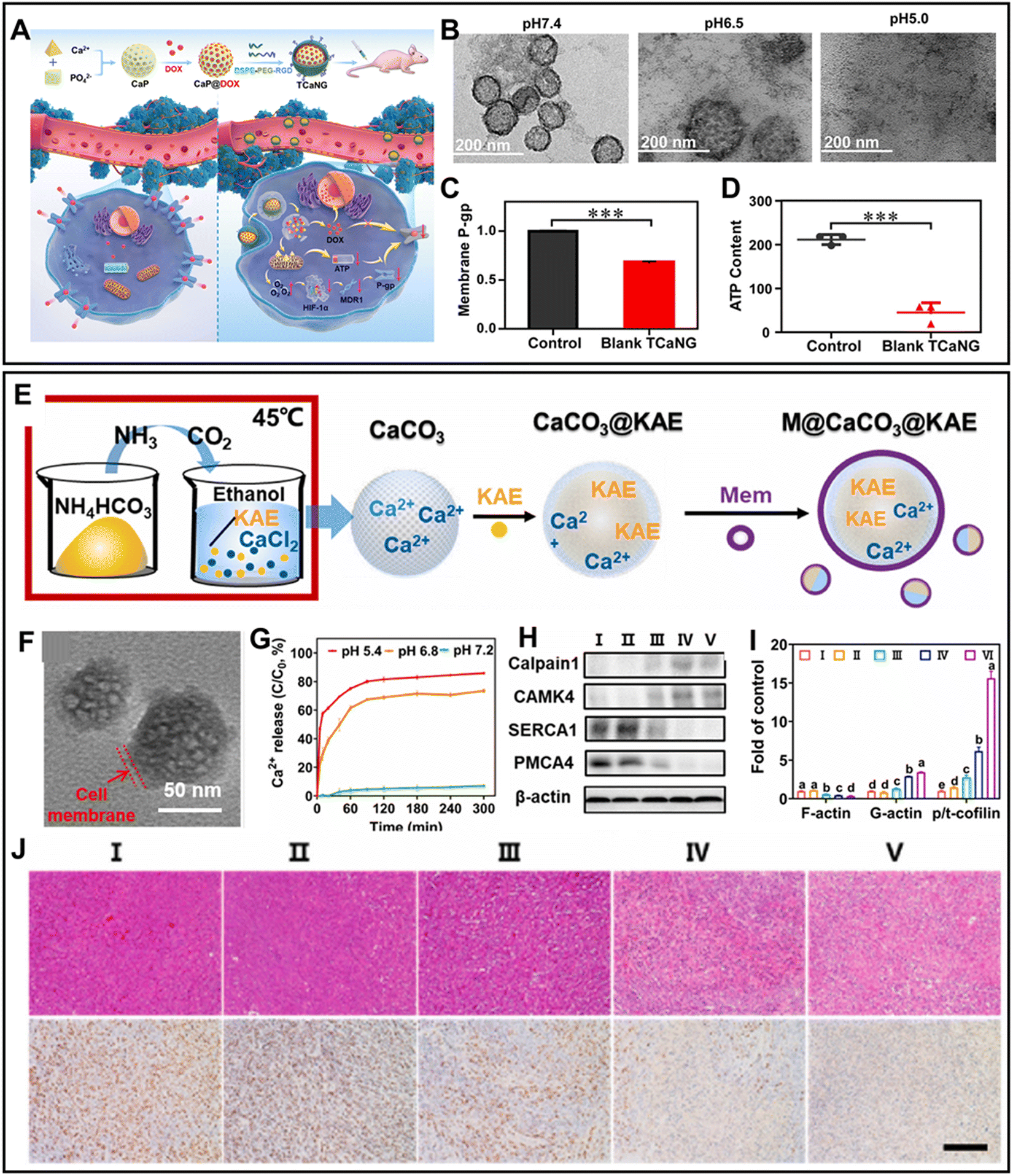 | ||
| Fig. 6 (A) Schematic illustration of TCaNG-mediated reversal of drug resistance.127 (B) Degradation of TCaNG at different pH values.127 (C) and (D) Expression of membrane P-gp and ATP content in tumor tissues treated with blank TCaNG for 24 h.127 Reprinted with permission from ref. 127. Copyright 2020, the American Chemical Society. (E) Synthetic process of M@CaCO3@KAE.129 (F) TEM image of M@CaCO3@KAE.129 (G) Ca2+ release profile of M@CaCO3@KAE at different pH.129 (H) Western blot analysis of Calpain1 and CAMK4.129 (I) Intensity of p/t-coffin, G-actin and F-actin.129 (J) Apoptosis and ki67 expression within tumor tissues after different treatments.129 (I) Control; (II) CaCO3 NPs; (III) KAE; (IV) CaCO3@KAE NPs; and (V) M@CaCO3@KAE NPs. Reprinted with permission from ref. 129. Copyright 2021, Elsevier Ltd. | ||
Similarly, curcumin (CUR) has the same effect as KAE in enhancing Ca2+ interference therapy.131 For example, Zheng's group26 prepared biodegradable Ca2+ nanomodulators (CaNMs) composed of CUR and CaCO3 to activate pyroptosis and robust antitumor immunity (Fig. 7A). CaNMs triggered a much lower MMP compared to CUR or CaCO3, indicating more severe mitochondria dysfunction. The activation of caspase 3 by Cyt C led to the cleavage of gasdermin E (GSDME), resulting in the formation of an N-terminal, which bind to the cell membrane phospholipids, constructing transmembrane pores (Fig. 7B). These pores facilitated the release of inflammatory molecules and cellular contents, such as lactate dehydrogenase (LDH) and ATP, thus promoting the maturation of DCs and enhancing T cell activation (Fig. 7C). Therefore, combining Ca2+ regulators with Ca-based nanomaterials can be expected to become a new type of immunomodulator to activate the body's immune response. However, the antigen presentation efficiency of DCs is still limited by autophagy inhibition and low viability in the complex TME.132 Antigens require digestion and processing through autophagy before being presented.133 Thus, to address these obstacles, a simple and versatile Ca2+ nanogenerator (OVA@CaCO3) was constructed.134 This nanosystem could disrupt a series of obstacles in antigen presentation according to the following mechanisms: (i) neutralizing the acidic TME to restore the viability of DCs; (ii) alleviating autophagy inhibition to promote the processing of antigens; and (iii) triggering DAMPs to stimulate DCs (Fig. 7D). In detail, as intracellular Ca2+ increased, more vesicles and LC3II (an important autophagy marker) appeared in DCs, indicating a higher level of autophagy (Fig. 7E and F). Furthermore, the higher colocalization rate of antigen and autophagosome confirmed that OVA@CaCO3 could strengthen the autophagy ability of DCs, thus enhancing Ca2+ interference-mediated immunotherapy (Fig. 7F).
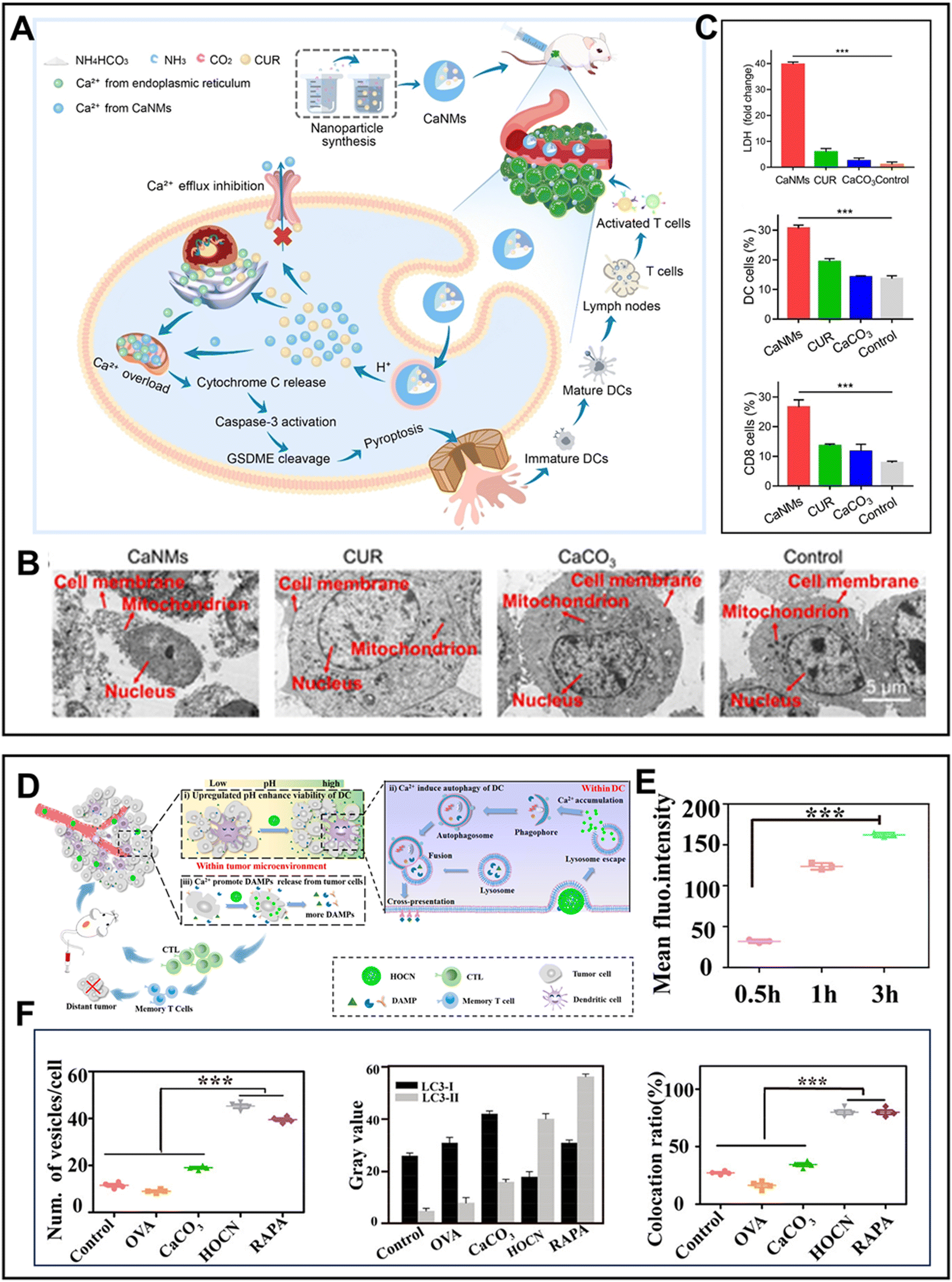 | ||
| Fig. 7 (A) Schematic illustration of mitochondrial Ca2+ overload-mediated pyroptosis and immunogenic cell death.26 (B) Bio-TEM images of cancer cells.26 (C) Released LDH content and the populations of DCs and CD8+ T cells in the spleen.26 Reprinted with permission from ref. 26. Copyright 2022, Wiley-VCH GmbH. (D) Mechanism diagram of HOCN-mediated disruption of three barriers in antigen presentation of DCs.134 (E) Ca2+ level in DCs.134 (F) Number of vesicles, LC3-I/II protein, and colocation ratio of LC3-II and antigen in DCs.134 Reprinted with permission from ref. 134. Copyright 2020, the American Chemical Society. | ||
Moreover, as chemical messengers for the transfer of biological information, some gaseous molecules (e.g. H2S and NO) actively participate in maintaining the homeostasis and physiological processes of cells.135 H2S plays a crucial role in regulating the channel proteins responsible for the flow of Ca2+.136 Based on this, poly(acrylic acid)-stabilized CaS nanoparticles loaded with zinc protoporphyrin (ZnPP@PAA-CaS) were developed to implement H2S gas therapy to enhance the Ca2+ interference and signal the cascade of Ca2+-induced ICD137 (Fig. 8A). Under acidic lysosome conditions, the nano messenger broke into Ca2+ and H2S, and subsequently ruptured the lysosomes via the “proton sponge effect” to release them (Fig. 8B). Fig. 8C displays that cytoplasmic Ca2+ stress was sharply elevated in the presence of PAA-CaS, whereas it was barely detectable with PAA-Ca. Moreover, the heme oxygenase-1 (HO-1) protection pathway for the tumor to escape death was blocked by the amplifier ZnPP, which strengthened Ca2+-induced apoptosis.138 Consequently, tumor-related antigens were released and acted as an in situ vaccine to activate a powerful immune response and restrict tumor metastasis (Fig. 8D and E). Similarly, NO-induced S-nitrosylation of cysteine residues on RyRs bends its structure and keeps it open, resulting in the leakage of Ca2+ from the ER store.139 Based on this mechanism, Chu's group140 prepared a Ca2+ store-regulating system UC-ZIF/BER by loading berbamine (BER) on upconversion nanoparticles (UCNPs) coated with ZIF-82 (Fig. 8F and G). The UV emission of UCNPs triggered by NIR light isomerized the nitro–nitrite in ZIF-82, resulting in NO production and structure collapse of ZIF-82 (Fig. 8H). UC-ZIF/BER exhibited stronger cytotoxicity in 4T1 and MB-231 cells with overexpressed RyRs, confirming the key role of RyRs in NO-induced cell death (Fig. 8I and J). Meanwhile, BER was released and bound to Ca2+-excreted pumps to inhibit Ca2+ efflux. Through the synergy of BER-mediated efflux protein inhibition and NO-induced RyRs deformation, UC-ZIF/BER showed outstanding in vivo antitumor efficiency in a 4T1 subcutaneous tumor model. Additionally, Chen et al.141 designed a multichannel nanomodulator, Lipo CaO2-TA-Fe3+/PArg, using polyarginine (PArg) as a precursor of NO to achieve a similar effect. Moreover, the high oxidative stress environment induced by ˙OH simultaneously down-regulated the Ca2+ efflux pump PMCA and up-regulated the Ca2+ influx channel on the plasma membrane.142
 | ||
| Fig. 8 (A) Synthetic process of ZnPP@PAA-CaS nanomessenger.137 (B) Release profiles of Ca2+ and H2S at different pH values.137 (C) Intracellular H2S and Ca2+ content after different treatments.137 (D) Released CRT and HMGB1. (E) Number of metastatic nodules in the lung after various treatments.137 (C) and (D) (1) Blank; (2) PAA-Ca; (3) ZnPP; (4) PAA-CaS; and (5) ZnPP@PAA-CaS. (E) Number of metastatic nodules in the lungs after various treatments.137 All groups: (1) blank; (2) PAA-Ca; (3) ZnPP; (4) αPD-1; (5) PAA-CaS; (6) ZnPP@PAA-CaS; (7) PAA-CaS + αPD-1; and (8) ZnPP@PAA-CaS + αPD-1. Reprinted with permission from ref. 137. Copyright 2021, the American Chemical Society. (F) Synthetic process and (G) TEM image of UC-ZIF/BER NPs.140 (H) NO release capacity of UC-ZIF/BER under 980 nm NIR irradiation.140 (I) Mechanism diagram of the role of NO on RyRs.140 (J) Expression of RyRs in various tumor cell models.140 Reprinted with permission from ref. 140. Copyright 2021, Wiley-VCH GmbH. | ||
As previously stated, the combination of Fenton agents with Ca-based nanomaterials can effectively enhance the level of cellular oxidative stress, thereby promoting Ca2+ inflow and causing Ca2+ disorder in cancer cells. Interestingly, Ca-based nanomaterials can be effectively endowed with Fenton-like properties by changing their crystal structure, synthesizing nano-reagents with high efficiency in inducing intracellular oxidative stress and Ca2+ disturbance through a one-step method. For the first time, a recent study reported that a type of valency-invariable CaF2 crystal obtained by the direct precipitation method possesses peroxidase (POD)-like activity143 (Fig. 9A). The activity of CaF2, unlike the valence change-dependent POD-like activity of transition metal ions, was derived from the CaF2(110) facet. Specifically, H2O2 was absorbed on the (110) facet with an energy drop of 0.92 eV, and then decomposed into OH*, which formed a bond with CaF2via Ca–O binds. In addition, free ˙OH was generated with an energy of 1.67 eV. Subsequently, OH* combined with nearby H+ to produce H2O, which desorbed from the facet, accompanied with a low endothermic energy of 0.78 eV (Fig. 9B). The cavitation effect of US irradiation could accelerate the rate and promote the degree of this process (Fig. 9C). Consequently, the generated ˙OH inhibited the expression of Ca2+ pump PMCA4 ATPase (ATP2B4) (Fig. 9D). Moreover, the mitochondrial dysfunction-induced decrease in ATP content further impaired the efficiency of ATP2B4, leading to an intensified Ca2+ overload and creating a “vicious cycle” (Fig. 9E). Therefore, the powerful Ca2+ interference capacity of CaF2 could effectively inhibit the growth of tumors.
 | ||
| Fig. 9 (A) Schematic illustration of CaF2-mediated ultrasound-augmented calcium interference therapy.143 (B) POD-like catalytic pathway and the free energy variation of CaF2(110) facet.143 (C) ESR spectra of ˙OH trapped by DMPO in different groups.143 (D) Expression of Calpain-1 and ATP2B4 in 4T1 cells.143 (E) Relative ATP content in 4T1 cells with various treatment.143 Reprinted with permission from ref. 143. Copyright 2022, Wiley-VCH GmbH. (F) Schematic illustration of plasma membrane damage mediated amplification of calcium interference therapy.144 (G) Cell membrane-anchoring capacity of CMA-nPS.144 (H) H1299 cell killing capacity of CMA-nPS.144 (I) Digital photos of tumors with different treatments.144 Reprinted with permission from ref. 144. Copyright 2022, Wiley-VCH GmbH. | ||
Different from supplementing exogenous Ca2+ and regulating channel proteins to disrupt Ca2+ homeostasis, inducing cell membrane damage is a novel approach to trigger endogenous Ca2+ overload. Inspired by this, Gao et al.144 proposed a strategy for achieving endogenous Ca2+ interference therapy by damaging tumor plasma membranes through the photodamage caused by photocatalysis. They constructed a cell-membrane-anchoring nano-photosensitizer (CMA-nPS) by embedding a dibenzocyclooctyne (DBCO)-decorated photosensitizer into a nanovesicle (Fig. 9F). In the acidic TME, the fusion of CMA-nPS and the tumor plasma membrane was triggered due to the transformation of the VSV-G structure into an unfolded fusion state. With an increase in the co-incubation time, CMA-nPS was observed to colocalize well with the plasma membrane (Fig. 9G). Subsequently, under NIR laser irradiation, the ROS in situ generated by the anchored photosensitizer significantly oxidized the phospholipids and increased the membrane permeability. The integrity of the plasma membrane was assessed using propidium iodide, and the results showed that CMA-nPS + L caused the most severe damage to the cell membrane, enabling extracellular Ca2+ influx and leading to tumor cell death, exhibiting an outstanding antitumor effect (Fig. 9H and I).
One of the most remarkable features of Ca2+ interference therapy is its ability to limit malignant tumor metastasis and invasion through cell calcification and cytoskeleton degradation. Given the clear understanding of the homeostatic regulatory system of Ca2+, the inhibition of channel proteins to control intracellular Ca2+ distribution has become the mainstream strategy for amplifying Ca2+ interference therapy. With the help of various channel protein inhibitors (e.g. small molecule drugs, ROS, and gas molecules), Ca2+ interference therapy has demonstrated powerful anti-tumor effects and considerable clinical translational potential. Moving forward, it is crucial to focus on the modulation of relevant immune cells by Ca2+ as a second messenger. This will allow the utilization of Ca2+ interference as a means of enhancing immunotherapy.
The effects of synergistic therapeutics can disrupt intracellular Ca2+ homeostasis by altering the state of channel proteins and amplifying the interference of Ca2+ on metabolism. The photothermal effect activates TRPV channels, leading to the promotion of Ca2+ influx and exacerbation of ion overload in cancer cells. In addition, ROS generated by PDT and SDT regulate the endogenous Ca2+ distribution by changing the conformation of channel proteins, resulting in localized Ca2+ overload. Besides, Ca2+ overload-mediated mitochondrial dysfunction further exacerbates the intracellular oxidative stress, thereby intensifying the therapeutic effects of ROS-dependent treatment. Ca2+ interference therapy can also trigger different forms of ICD, such as apoptosis and pyroptosis, to evoke the body's immune response to tumor cells. Besides elevating the abundance of tumor-associated antigens and DAMPs, enhancing the antigen processing and presentation efficiency of DCs is also an effective way to improve the cytotoxic T cell-mediated killing of tumor cells. High Ca2+ levels can enhance autophagy in DCs to accelerate antigen processing and promote immunological synapse formation to facilitate antigen presentation from DCs to CTLs. The latest research has shown that high calcium levels can polarize macrophages towards the M1 phenotype, thereby triggering the innate immune system's recognition and elimination of cancer cells. Furthermore, the NF-kB/IRF3 pathway activated by Ca2+ within tumor cells can also reprogram macrophage polarization to the M1 phenotype via the upregulation of IFN-1 expression. Therefore, Ca2+ interference can comprehensively coordinate the innate and adaptive immune systems for efficient immunotherapy.
3.4. Zinc ion interference therapy
 | ||
| Fig. 10 (A) Mechanism diagram of HZ@GD-activated gene silencing therapy.25 (B) Degradability of HZ@GD treated with HAase at different pH values.25 (C) Zn2+ and DNAzyme release profile of HZ@GD with different treatments.25 (D) Immunofluorescence image of GLUT1 protein.25 (Blue: DAPI; red: GLUT1; scale bar: 100 μm.) (E) Expression of GLUT1 mRNA and relative LA and ATP content in tumor tissues with different treatments.25 Reprinted with permission from ref. 25. Copyright 2021, The Authors. (F) Synthetic process of Zn-Car NMs.163 (G) Mechanism diagram and release profile of Zn2+ from Zn-Car.163 (H) Confocal fluorescent images of free Cu in 4T1 cells treated with different formulations.163 (I) COX activity, ratio of JCagg/JCmono, ratio of NAD+/NADH and ATP content in 4T1 cells.163 (J) Relative 4T1 tumor volume with i.t. injection.163 Reprinted with permission from ref. 163. Copyright 2023, the American Chemical Society. | ||
Mutp53 is another star target for Zn2+ interference therapy.164 For instance, Zhang's group156 synthesized ZnFe NPs, which exhibited the strongest Mutp53 degradation ability with a Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe ratio of 1:2 (denoted as ZnFe-4) (Fig. 11A and B). ZnFe-4 selectively degraded several types of translated Mutp53 rather than disrupting their transcription and translation processes (Fig. 11C and D). Furthermore, the degradation of Mutp53 can up-regulate the expression of p21, another tumor suppressor. The treatment with ZnFe-NPs significantly induced cell cycle arrest, decreased migratory and invasive capacity, and reduced colony formation (Fig. 11E). In another study, Mn-ZnO2 NPs were found to exhibit the ability to promote the degradation of Mutp53 and accumulation of WTp53. This co-regulation was achieved through the synergistic effects among Mn2+, Zn2+, and ROS165 (Fig. 11F and G). Specifically, a high concentration of Mn2+ and ROS activated the ataxia-telangiectasia mutated (ATM)-p53 signaling pathway to stabilize and accumulate WTp53.166 The activated WTp53 induced the transcription of proapoptotic genes such as Bax, thereby suppressing tumor growth (Fig. 11H). Zn2+-mediated Mutp53 degradation and Mn2+-mediated WTp53 accumulation effectively impaired GOF and initiated the death process in tumor cells. Consequently, Mn-ZnO2 exhibited better tumor inhibition than ZnO2 alone (Fig. 11I).
:Fe ratio of 1:2 (denoted as ZnFe-4) (Fig. 11A and B). ZnFe-4 selectively degraded several types of translated Mutp53 rather than disrupting their transcription and translation processes (Fig. 11C and D). Furthermore, the degradation of Mutp53 can up-regulate the expression of p21, another tumor suppressor. The treatment with ZnFe-NPs significantly induced cell cycle arrest, decreased migratory and invasive capacity, and reduced colony formation (Fig. 11E). In another study, Mn-ZnO2 NPs were found to exhibit the ability to promote the degradation of Mutp53 and accumulation of WTp53. This co-regulation was achieved through the synergistic effects among Mn2+, Zn2+, and ROS165 (Fig. 11F and G). Specifically, a high concentration of Mn2+ and ROS activated the ataxia-telangiectasia mutated (ATM)-p53 signaling pathway to stabilize and accumulate WTp53.166 The activated WTp53 induced the transcription of proapoptotic genes such as Bax, thereby suppressing tumor growth (Fig. 11H). Zn2+-mediated Mutp53 degradation and Mn2+-mediated WTp53 accumulation effectively impaired GOF and initiated the death process in tumor cells. Consequently, Mn-ZnO2 exhibited better tumor inhibition than ZnO2 alone (Fig. 11I).
 | ||
| Fig. 11 (A) Preparation and treatment mechanism of ZnFe-4 nanoparticles.156 (B) Morphological change in ZnFe-4 at different pH values.156 (C) Capacity of ZnFe-4 to degrade different types of Mutp53.156 (D) Relative level of p53 mRNA in H1299 cells.156 (E) Cell cycle arrest and clonal formation of ES-2 cells treated with PBS or ZnFe-4 for 12h.156 Reprinted with permission from ref. 156. Copyright 2020, Wiley-VCH GmbH. (F) Schematic illustration of the preparation and treatment mechanism of Mn-ZnO2 NP.165 (G) Zn2+ release profile of Mn-ZnO2.165 (H) Expression of Mutp53, ATM, p-ATM, p-WTp53, WTp53 and Bax in MDA-MB-231 cells.165 (I) TUNEL staining analysis to detect the apoptotic MDA-MB-231 cells.165 Reprinted with permission from ref. 165. Copyright 2022, The Authors. | ||
Zn-based nanomaterials also play a crucial role in immune activation and regulation, which is helpful in achieving a synergistic enhancement in interference therapy with metal ion immunotherapy. Zhang's team synthesized a Zn2+-doped LDH (Zn-LDH)-based immunomodulating adjuvant167 (Fig. 12A). In the acidic TME, Zn–OH is susceptible to hydrolytic fracture to elevate the pH of TME and release a certain amount of Zn2+, thereby effectively disrupting endo-/lysosomes to block autophagy and triggering mitochondrial damage. Moreover, Zn-LDHs can activate the cGAS/STING pathway, as confirmed by the significant up-regulation of IFNB1 and ISG56 (Fig. 12B). Consequently, the treatment of Zn-LDHs allowed up-regulation of the “eat me” signal (CRT and p53) and down-regulation of the “do not eat me” signal (PD-L1 and CD47) (Fig. 12C and D). Meanwhile, these variations promoted the conversion of M2-type tumor-associated macrophages (TAMs) to M1-type TAMs and the maturation of DCs, which can highly increase the efficiency of antigen presentation (Fig. 12E). In general, Zn-LDHs are simple but robust immunomodulating adjuvants that not only induce ICD but also reduces immune escape. In addition to enhancing the infiltration of immune cells to tumor by inducing ICD, Zn-based nanomaterials can also directly act on DCs and activate the matrix metalloproteinase pathway to stimulate the immune response of the body and alleviate the tumor suppressive microenvironment. Matrix metalloproteinase is an endogenous Zn2+-dependent proteolytic enzyme, which is responsible for the degradation of the tumor extracellular matrix (ECM) and matrix membranes.168 The dense structure of the tumor ECM is a physical barrier that prevents the deep penetration of T cells and antibodies, impairing their killing ability. Also, an increase in the stiffness of the tumor ECM hampers the ability of DCs to re-recognize and internalize tumor antigens.169 Thus, to overcome these tissues, Ding's group designed a Zn-MOF vaccine that directly targets DCs to enhance antigen uptake and presentation.170 This vaccine, when taken up by DCs, enhanced antigen cross-presentation through lysosomal swelling-released OVA and cGAS/STING pathway-activated expression of interferon. Furthermore, in the acidic TME, the vaccine released Zn2+ to activate matrix metalloproteinase-2, thereby enhancing the T cell invasion and tumor-killing ability. However, despite the successful immune response elicited by these immune adjuvants, the low enrichment efficiency at the tumor site remains a significant obstacle in achieving the desired therapeutic effect. Recently, it has been shown that disruption of epithelial cells at the tumor vasculature can effectively enhance the EPR effect. Yang et al.157 constructed a STING agonist, ZnCDA, which significantly reduced the blood vessel density at the tumor. Knocking out the STING gene in epithelial cells significantly attenuated this effect. This study implied that the targeting of nanomedicines can be enhanced through the STING pathway, and thus greatly increase their potential for clinical application.
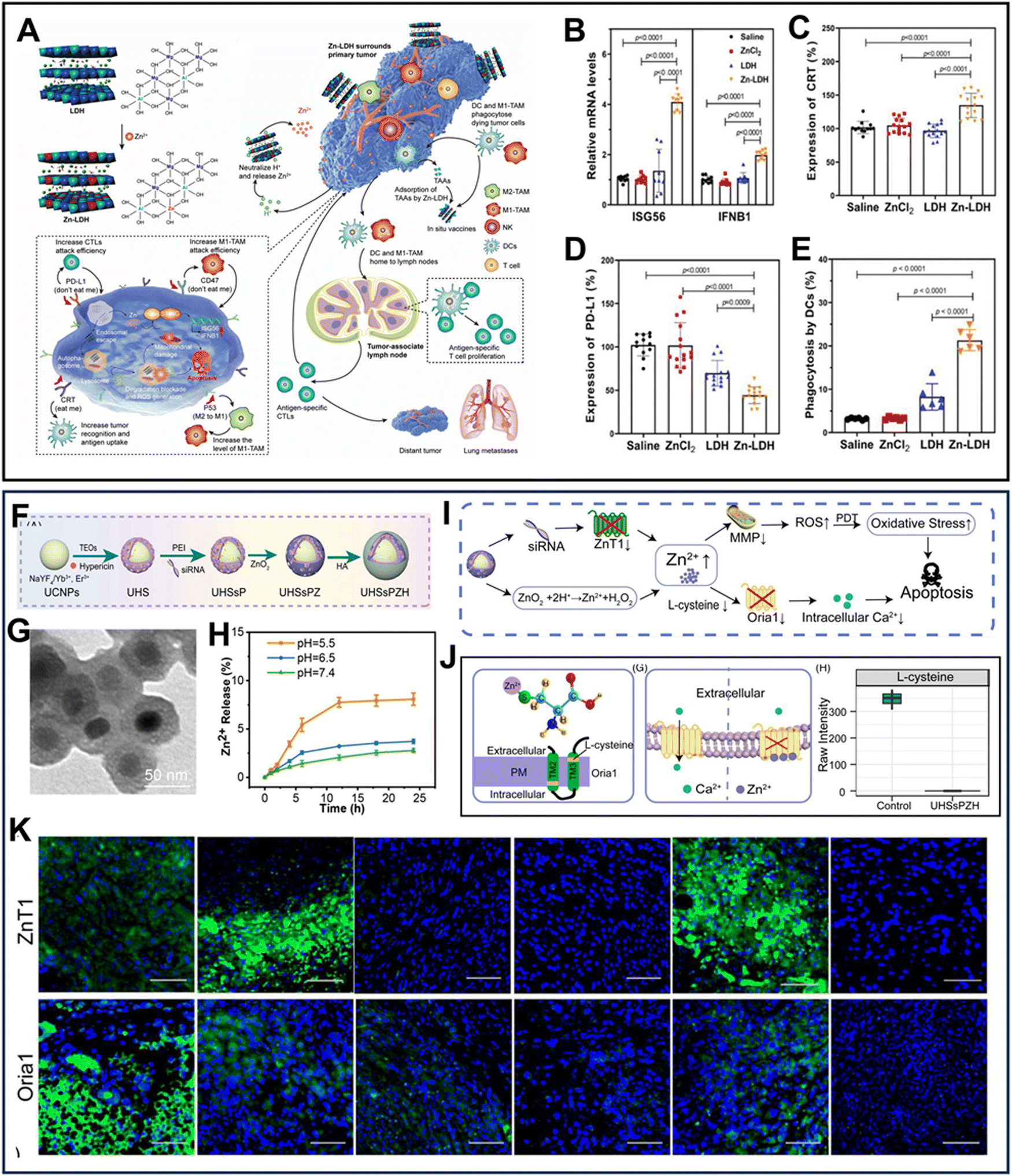 | ||
| Fig. 12 (A) Schematic illustration of the preparation of Zn-LDHs and mechanism of Zn-LDH-mediated robust and safe metalloimmunotherapy.167 (B)–(D) Relative levels of ISG56 and IFNB1 mRNA (B), CRT (C), and PD-L1(D) in B16F10 cells.167 (E) Phagocytosis of dying B16F10 cells by DCs.167 Reprinted with permission from ref. 167. Copyright 2022, Wiley-VCH GmbH. (F) Synthetic process and (G) TEM image of UHSsPZH NPs.171 (H) Zn2+ release profile of UHSsPZH NPs at different pH values. (I) Mechanism diagram of Zn2+-mediated Ca2+ channel protein destruction in vivo.171 (J) Two pathways of Zn2+-induced apoptosis.171 (H) Expression of Oria1 and ZnT1.171 Reprinted with permission from ref. 171. Copyright 2023, The Authors. | ||
Most of the above-mentioned studies focused on the use of Zn-based nanomaterials to supplement exogenous Zn2+ and achieve Zn2+ interference therapy. To maintain the intracellular Zn2+ levels, the body strictly regulates the activities of the Zrt/Irt-related protein (ZIP) and Zn transporter (ZnT1).172 ZIP is responsible for the influx of Zn2+ from extracellular organelles into the cytoplasm, while ZnT1 plays the opposite role.173 Jiang et al.171 proposed a “block and attack” strategy, which is achieved by combining gene silencing using siRNA with Zn2+ supplementation. To implement this strategy, they created a nanocomposite called UHSsPZH, which stands for UCNPs-Hy@mSiO2@PEI-siRNA@ZnO2@HA (Fig. 12F and G). “Attack” is the typical Zn2+ supplementation strategy, whereas “block” is to limit the Zn2+ efflux via gene silencing of ZnT1. Compared to “block” or “attack” alone, “block and attack” triggered a higher intracellular Zn2+ concentration (Fig. 12H and I). Furthermore, Zn2+ can chelate with the abundant cysteine residues on the Ca2+ channel protein ORAI1 to form a Zn2+-cysteine complex, which distorts the conformational structure of ORAI1 (Fig. 12J). In vivo studies showed that USsPZH could inhibit both the ZnT1 and ORAI1 channels, and thus trigger dual-ion disorder (Fig. 12K). Therefore, the normal physiological function of ORAI1 was compromised, resulting in the down-regulation of the intracellular Ca2+ concentration. Ultimately, the oxidative stress generated by Zn2+-interference worked synergistically with the disturbances in Ca2+ homeostasis to accelerate apoptosis.
Zn2+ interference with the multidimensional containment of tumor cell development can be summarized as Mutp53 degradation, cGAS/STING activation, and glycolysis inhibition. Among these mechanisms, Zn2+ uniquely possesses the ability to degrade Mutp53 compared to other metal ions. Mutp53 is a crucial regulator of tumor cell metabolic remodeling, rapid multiplication, and apoptosis inhibition. Therefore, Zn2+ interference can reverse the GOF properties of cancer cells, thereby enhancing the effectiveness of other therapeutic treatments. Current research on Zn2+ interference therapy has primarily focused on understanding the underlying mechanisms and has only explored the inhibition of Zn2+ efflux proteins in isolation. Moving forward, it is necessary to devote greater attention to regulating the metabolism of Zn2+ and improving the efficiency of Zn2+-activated antitumor pathways.
Zn2+ interference therapy in combination with other therapeutic modalities, specifically ROS-dependent treatments such as PDT and SDT, has not been extensively studied. These therapies can induce mitochondrial damage and subsequent release of mitochondrial DNA, which amplifies Zn2+-mediated cGAS/STING activation. Additionally, photothermal effects have been found to enhance the glycolysis-inhibiting properties of Zn2+. More importantly, Zn2+ interference has the potential to improve the immunosuppressive TME and reverse the immune evasion of tumor cells from multiple dimensions. Basically, Zn2+ activates the cGAS/STING pathway by promoting the separation of the cGAS-DNA phase to restore the body's immune surveillance of the tumors. Recently, the physical nature of the TME has gained significant attention as a crucial factor affecting the effectiveness of immunotherapy, complementing the traditional focus on its biochemical aspects. At the tumor site, the ECM is notably stiffer than in normal tissue, creating a formidable physical barrier that hinders the antigen presentation capacity of DCs and the infiltration of T cells. Matrix metalloproteinases, when activated by Zn2+ acting as cofactors, play a key role in ECM modification by promoting its softening, thereby facilitating the functional activities of immune cells within the TME. Notably, Zn2+ exhibits a further significant impact by selectively degrading Mutp53, disrupting the immune evasive strategies employed by tumor cells. This mechanism involves the down-regulation of PD-L1 and the release of inflammatory cytokines, ultimately enhancing the immune response against tumor cells.
3.5. Other metal ion interference therapies
Recently, a pH-responsive MRI theranostic nanoplatform was constructed, which was comprised of Se nanoparticles, manganese carbonyl (MnCO), and Fe3O4.180 The Se nanoparticles facilitated O2− production and rapidly activated superoxide dismutase, triggering cascade reactions that produced H2O2. The presence of Mn2+ facilitated the Fenton-like reaction, leading to the generation of ˙OH, which induced quick protein degradation and oxidation. Additionally, Mn2+ played a role in inhibiting the generation of ATP, thereby effectively starving cancer cells. Moreover, similar to Zn2+, Mn2+ also participates in activating the cGAS/STING pathway in humans to stimulate the body's immune response and facilitate Mn2+ interference-mediated immunotherapy.181,182 To enhance the Mn2+ activation of the cGAS/STING pathway, a gas nano-adjuvant based on gas-induced mitochondria dysfunction was designed (Fig. 13A). Once inside cancer cells, the gas nano-adjuvant disassembled in response to the overexpressed GSH and low pH, leading to the release of MnCO and H2S (Fig. 13B). Under NIR-irradiated AIEgen-based PDT/PTT, the prodrug MnCO in situ decomposed to Mn2+ and CO. Consequently, the MMP was depolarized due to the induction of H2S and CO, leading to the subsequent release of mitochondrial DNA (Fig. 13C). Moreover, mitochondrial DNA and Mn2+ can trigger the activation of the cGAS/STING pathway to promote the synthesis of related cytokines through phosphorylation of TBK1 and IRF3, thereby arousing the immune response to tumors (Fig. 13C–E). It should be noted that Mn2+ exhibits a distinct feature from Zn2+ given that it can bypass STING to directly or indirectly enhance the phosphorylation of two downstream transcription factors, p65 and IRF3.30 Notably, recent studies have established the relationship between IFN gene transcription and the formation of enhancesomes, which include phosphorylated IRF3 and p65.183 In this context, the research by Sun's group's showed that Mn2+ has the ability to trigger the STING-independent phosphorylation of p65 and TBK130 (Fig. 13F). Through thermal shift tests, it was found that Mn2+ could not enhance the affinity between STING and STING agonists (Fig. 13G). After knocking out STING, Mn2+ could still initiate the phosphorylation process of TBK1 and p65, but not IRF3. Therefore, Mn2+ exhibited robust immune activation potential by strengthening the phosphorylation of IRF3 in the presence of STING agonists. Inspired by this, Mn2+ was combined with cyclic dinucleotide (CDN), a conventional STING agonist, to achieve the amplification of the STING signaling cascade and up-regulation of IFN-β (Fig. 13H). Moreover, Mn2+ promotes the survival and proliferation of memory T cells and significantly enhances the killing effect of natural killer cells (NKs), thereby increasing the host's immune surveillance of the tumor. In summary, Mn2+-mediated immunotherapy has shown great potential for clinical application, and thus received widespread attention.
 | ||
| Fig. 13 (A) Synthetic process of gas nanoadjuvant.184 (B) Degradability and gas release profiles of gas nanoadjuvants.184 (C) Cytosolic mitochondrial DNA and released cytokines IFN-β, CXCL10, and IL-6.184 (D) Expression of STING, TBK1, IRF3 and their phosphorylation in tumors.184 (E) Flow cytometric assay of mature DCs and tumor-infiltrating CD8+ T cells.184 Reprinted with permission from ref. 184. Copyright 2023, The Author(s). (F) Mechanism diagram of Mn2+-mediated elevation of IFN-β expression, phosphorylation of p65, TBK1, and IRF3 in STING knock-out cells, and inhibition of Mn2+-mediated IFN-β by p65 nucleus translocation.30 (G) Affinity between STING and STING agonists.30 (H) Synthetic process of CDN-Mn nanoparticles.30 Reprinted with permission from ref. 30. Copyright 2021, The Author(s). | ||
 | ||
| Fig. 14 (A) Synthetic process and corresponding TEM images of NaCl@ssss-VHMS.186 (B) CLSM images of Na+ and Cl− level.186 (C) Morphology changes in HepG2 cells treated with NaCl@ssss-VHMS.186 (D) Caspase-1 activation and Capase-3 activation in HepG2 cells with different formulations.186 Reprinted with permission from ref. 186. Copyright 2022, the American Chemical Society. (E) Schematic illustration of Zr NP-mediated pyroptosis-enhanced immunotherapy.187 (F) Level of K+ in 4T1 cells.187 (G) Relative c-GSDMD content and profiles of released IL-1β in 4T1 cells, mature rate of DCs, population of CD8+ T cells, and CD8+/CD4+ ratio in the lymph node.187 Reprinted with permission from ref. 187. Copyright 2021, the American Chemical Society. | ||
Mg-based nanomaterials have various applications in clinical settings, such as being used as screws and pins in orthopedics and as biodegradable scaffolds in the cardiovascular field.196,197 Additionally, Mg-based nanomaterials exhibit increased cytotoxicity towards cancer cells. The biocompatibility and degradability of magnesium-based nanomaterials lead to the generation of surface effects, including increase in pH, increase in osmotic pressure, and hydrogen evolution, which have the potential to directly target cancer cells and reduce tumor growth.198 Moreover, the Mg2+ released by biodegradable Mg-based nanomaterials can serve as a coenzyme factor, which effectively collaborates with DNA enzymes to inhibit the expression of MDR mRNA and further reverse the drug resistance of tumors to chemotherapy. Inspired by this, a versatile DNA nanodrug (MCD@TMPyP4@DOX) was fabricated to achieve effective MDR silencing via Mg2+-activated DNAzymes.23 Triggered by photodynamic-induced ROS burst, the nanodrug self-disassembled to release DNAzymes and Mg2+. Subsequently, DNAzymes cleaved the MDR mRNA under the action of cofactor Mg2+, resulting in a significant decrease in PgP expression. Meanwhile, ROS-mediated mitochondrial dysfunction impaired ATP production, thus limiting the DOX pumping efficiency of PgP. This research presented a new way that Mg2+ effectively improves the effect of gene therapy as a cofactor.
Mg2+ can also actively participate in cellular energy metabolism. Mg2+-dependent enzymes such as HK,199 PFK,200 and pyruvate kinase (PK),201 which are the three key enzymes involved in glycolysis, make Mg2+ deficiency an effective strategy for blocking glycolysis in tumor cells. Recently, a GSH-responsive nanoplatform MSN/EDTA&rotenone@PAASH was subtly designed to achieve Mg2+ interference-mediated glycolysis inhibition.202 Within this nanoplatform, EDTA chelated high intracellular concentrations of Mg2+ to form EDTA-Mg complexes, inactivating HK, PFK, and PK. Simultaneously, rotenone diffused into the intermembrane space and blocked the electron transfer during OXPHOS, thereby disrupting ATP synthesis. In addition to participating in energy generation, Mg2+ has a close relationship with DNA replication and stability, which participates in various DNA repair patterns, such as nucleotide excision repair, base excision repair, and mismatch repair, and stabilizes the natural DNA conformation through hydrogen bonding. Therefore, when there is an intracellular deficiency of Mg2+, DNA becomes more vulnerable to damage from ROS. Several studies have shown that a moderate Mg2+ level is strongly associated with mitochondrial health and intracellular redox homeostasis. Insufficient Mg2+ disrupts the ETC, which leads to a burst of ROS, subsequently disrupting the existing antioxidant defense mechanisms of cells. This process includes the down-regulation of intracellular GSH and vitamin E levels, which accelerates the oxidation of lipids.
Metal-based nanomaterials, including various types such as metal oxides, metal peroxides, metal salts, MOFs, and complexes, have been extensively utilized for targeted metal ion interference therapy in the context of cancer treatment. Here, we comparatively analyze the types, sizes, and responsive properties of the different metal-based nanomaterials that have been developed thus far and the treated tumor models (Table 1). Most of these nanomaterials demonstrated a propensity to actively interact with the acidic tumor microenvironment and exploit overexpressed GSH to modulate the metal ion levels within the ECM. Consequently, the use of degradable metal-based nanomaterials offers heightened tumor selectivity, while minimizing the adverse impacts on healthy tissues. Concomitantly, nano-sized drugs are capable of being internalized by cancer cells through endocytosis, subsequently undergoing degradation within lysosomes, which in turn leads to elevated intracellular metal ion concentrations. Additionally, we summarize the biosafety, injection modalities, dosages, and corresponding therapeutic effects of various nanomaterial-mediated MIIT in Table 2. Furthermore, the use of complex functionalized nanomedicines for achieving tumor ablation through multimodal synergistic therapy adds another layer of complexity, making it difficult to compare the therapeutic effects of metal ion interference therapy alone. Fortunately, the current nanomedicine being studied has demonstrated excellent biocompatibility and therapeutic outcomes in xenograft mice. By incorporating diverse amplification strategies and metal-based nanomaterials through a rational design approach, the interference of metal ions can be maximized for enhanced therapeutic efficacy. Consequently, it is evident that the utilization of nanotechnology-mediated metal ion interference therapy holds promise as a potent strategy for advancing cancer treatment methodologies.
| Type | Formulation | Metal-based nanomaterials | Metal ion content (%) | Particle size | Responsive degradation | Tumor model | Ref. |
|---|---|---|---|---|---|---|---|
| Iron ion interference | LDHP | Fe/Al-LDH | 38.52 | 433 | pH | 4T1 cells | 203 |
| Cro-Fe@BSA | Cro-Fe complex | 17.0 ± 3.6 | 91.3 ± 10.4 | pH | 4T1 cells | 66 | |
| sSFP | FePt | 7.57 | 325 | pH | 4T1 cells | 204 | |
| Fe-MOF-RP | Fe-TCPP | 1.03 | ∼150 | GSH | B16F10 cells | 205 | |
| PFP@Au-Fe2C-SRF | Fe2C | 22.7 | 320.2 | pH and laser | 4T1 cells | 206 | |
| PAD@MS | Fe complex | 10.2 | ∼130 | Hypoxia and pH | CT26 and MC38 cells | 78 | |
| PCSFG | Fe-GA coordinate | 13.5 | ∼45 | Temperature and pH | 4T1 cells | 207 | |
| Calcium ion interference | M@CaCO3@KAE | CaCO3 | — | ∼100 | pH | A549 cells | 129 |
| CSSG | CaS | — | 80 | pH | 4T1 cells | 208 | |
| ZnPP@PAA-CaS | CaS | 51 | 122 | pH | 4T1 cells | 137 | |
| CaO2-N770@MSNs | CaO2 | 8.1 | 464.1 | pH | CT26 cells | 140 | |
| Lipo CaO2-TA-Fe3+/PArg | CaO2 | 4.11 | 43.8 | pH | 4T1 cells | 141 | |
| CCMH | CaO2 | 20.21 | 180 | pH | 4T1 cells | 209 | |
| CEL/ACC-LP NPs | CaCO3 | 17.61 ± 0.41 | ∼120 | pH | CT26 cells | 210 | |
| Copper ion interference | NP@ESCu | Elescomol-Cu | — | 87.5 | ROS | MB49 cells | 92 |
| RBC-Zr@APC/C | CuO/Cu-MOF | 8.61 | 184 ± 10 | — | 4T1 cells | 211 | |
| CuO2-MSN@TA-Cu2+ | CuO2/TA-Cu2+ | 2.46 | 167 ± 0.344 | pH | 4T1 cells | 212 | |
| T-HCN@CuMS | Cu2+ | 0.44 | — | pH | 143B cells | 213 | |
| Cu-GA | Cu-gallic acid | 20 | 120 | pH and GSH | 4T1 cells | 214 | |
| CAT-ecSNA-Cu | Cu2+ | 80.8 | 415 | — | CT26 cells | 215 | |
| DMMA@Cu2−xSe | Cu2−xSe | — | 130 | H2O2 and pH | A375 cells | 103 | |
| Cu-MCGH | Cu-MCN | 0.47 | ∼250 | — | 4T1 cells | ||
| BSO-CAT@MOF-199 @DDM | MOF-199 | — | ∼50 | pH | GL261 cells | 64 | |
| Au@MSN-Cu/PEG/DSF | Cu-MSN | 11.5 | 221.81 | Temperature and pH | 4T1 cells | 101 | |
| Zinc ion interference | Zn-LDHs | Zn-LDH | 6.7 | ∼100 | pH | B16F10 cells | 167 |
| ZnS@BSA | ZnS | — | ∼100 | pH | HCC cells | 161 | |
| ALA&Dz@ZIF-PEG | ZIF-8 | 15.5 | ∼78 | pH | 4T1 cells | 216 | |
| HZ@GD | ZIF-8 | — | 117 | HAase and pH | B16F10 cells | 25 | |
| DSF@Zn-DMSNs | Zn-DMSNs | 26.4 | 230.10 ± 0.40 | pH | CT26 cells | 217 | |
| UHSsPZH NPs | ZnO2 | — | 60.67 ± 8.57 | pH | 4T1 cells | 171 | |
| ZnFe-4 NPs | Zn oleate | 25 | 32 | pH | ES-2 cells | 156 | |
| Manganese ion interference | IFNγ/uMn-LDHs | Mn-LDHs | — | 58 ± 1 | GSH and Ph | 4T1 cells | 218 |
| CDN-Mn particle | Mn complex | — | 117.9 ± 41.42 | — | CT26 and B16F10 cells | 30 | |
| BPNS@Mn2+/CpG | BPNS@Mn2+ | — | ∼310 | Temperature and pH | 4T1 cells | 219 | |
| MTHMS | MnCO | — | 128.1 ± 5.9 | Temperature and pH | 4T1 cells | 184 | |
| Sodium/potassium ion interference | K3ZrF7:Yb/Er UCNPs | K3ZrF7 | — | ∼20 | pH | 4T1 cells | 187 |
| NaCl@ssss-VHMS | NaCl | ∼39.5 | ∼150 | GSH and pH | HepG2 cells | 186 | |
| Formulation | Dose | Synergy therapy | Injection modality | Tumor inhibition (%) | Biosafety | Ref. |
|---|---|---|---|---|---|---|
| A@P/uLDHs | 5 mg kg−1 | — | i.v. injected at day 0, 4, 8, 12, 16 | 95 | Good | 220 |
| LDHP | 10 mg kg−1 | — | i.t. injected at day 0, 3, 6 | ∼76 | Good | 203 |
| Fe-RA | 12 mg kg−1 | — | i.t. injected at day 0, 2, 4, 6, 8 | ∼63 | Good | 221 |
| AuSi@FePB | 5 mg | PTT | i.v. injected at day −1, 1, 3 | ∼87 | Good | 222 |
| PDA-DTC/Cu | 5 mg kg−1 | — | i.v. injected every 2 days | 66.3 | Good | 223 |
| CuP/Er | Cu: 3.5 mg kg−1 | — | i.v. injection every 3 days | 86.5 | Good | 224 |
| PTC | 5 mg kg−1 | PDT | i.v. injection at day 0, 3, 6, 9, 12, 15, 18 | 90 | Good | 225 |
| ECPCP | ES concentration: 5 mg kg−1 | — | i.v. injected at day 0, 2, 4, 6 | 74.8 | Good | 226 |
| CD@CaCO3P | CaCO3 dose at 15 mg kg−1 | — | i.v. injected at day 1, 4, 7 | 85 | Good | 227 |
| PEG/CaO2@p-780 | CaO2 dose at 2 mg kg−1 | PTT | i.v. injected at day 0, 2, 4, 6, 8, 10, 12 | 80 | Good | 228 |
| CaGlu NPs | 62.5 mg kg−1 | — | i.v. injected at day 0, 2, 4, 6, 8, 10, 12, 14 | 71.4 | Good | 229 |
| ZPM@OVA-CpG | OVA: 50 μg | — | i.v. injected at day 7, 14, 21 | 76.2 | Good | 230 |
| ZMCH | 10 mg kg−1 | PDT | i.v. injected every two days | 90 | Good | 231 |
| Zn-LDH | 1mg | — | i.t. injected at day 0, 6 | 88.2 | Good | 232 |
| PZnO@DOX | 1 mg | SDT and chemotherapy | i.v. injected at day 0, 2, 4 | 70.6 | Good | 233 |
| MnPt | Mn: 2.5 mg kg−1 | — | i.v. injected at day 0, 2, 4, 6, 8, 10, 12 | 72.1 | Good | 234 |
| MMP | 20 mg kg−1 | — | i.v. injected at day 0, 2, 4 | ∼88 | Good | 235 |
4. Summary and outlook
4.1. Summary
In this review, we systematically summarized the intrinsic mechanisms of different types of MIIT and provided an overview of the recent advancements in the corresponding amplification strategies. Based on the concept of MIIT, the most basic amplification strategy is to disrupt the ion homeostatic regulatory mechanisms in cancer cells, thus exacerbating the abnormal accumulation of metal ions. With a deeper understanding of metal ion-related metabolism, the modulation of these metabolic pathways, in conjunction with metal ion overloading, can sensitize cancer cells to MIIT. Additionally, the pursuit of further therapeutic efficacy has led to the exploration and utilization of various amplification strategies. Metal-based nanoplatforms with sono/photo-catalytic properties or photothermal conversion capacity have been also investigated to combine SDT/PDT/PTT with MIIT, resulting in enhanced therapeutic outcomes compared to single treatments. Furthermore, taking advantage of TME-responsive nanoplatforms, the spatial-temporal precision and efficacy of MIIT have been highly improved. Next, we discuss the challenges encountered by MIIT in the clinical treatment of cancers and some perspectives on its future development.4.2. Challenges
(i) Currently, some understanding of mechanisms is still in its infancy and lacks systematicity, often attributing the therapeutic effects to a single pathway and ignoring the impact of metal ions on full cellular metabolism. In fact, tumor cell metabolism encompasses a complex interplay among various pathways, wherein perturbations in one pathway can cascade into others.(ii) The uncertain fate of nanoparticles in the body following systemic or topical administration is a hurdle in clinical translation. Without proper surface engineering, nanomedicines are rapidly captured by the mononuclear phagocytic system (MPS), and subsequently cleared from the circulation. Also, despite evading MPS clearance, traversing the blood–brain barrier remains a formidable challenge for efficient nanoparticle accumulation at tumor sites.
(iii) Assessing the in vivo safety of degradable metal-based nanomedicines also poses a significant challenge. Given their status as trace elements in the human body, metal ions have the potential to disrupt homeostatic equilibrium. Consequently, the inevitable leakage of these ions into the body's fluid circulation poses potential risks, affecting the body's homeostatic regulatory system. For instance, the excessive leakage of calcium ions may precipitate blood clot formation in blood vessels. Hence, the judicious assessment of the introduced metal ion burden on the body represents a critical challenge that cannot be overlooked.
4.3. Perspectives
(i) The current focus of MIIT research predominantly focuses on single metal ion interference, overlooking the potential of bi-metal ion interference therapy. However, understanding the underlying mechanism of MIIT suggests the possibility of a synergistic effect from multi-metal ion interference. Compared to single MIIT, bi-MIIT exhibits mutual reinforcement in disrupting intracellular ion homeostasis, amplifying metabolic damage, and modulating the immunosuppressive TME, which is expected to improve the therapeutic efficiency of MIIT. For instance, the mitochondrial disorder induced by Ca2+ overload could impair ATP synthesis, and then block the copper ion efflux mediated by Cu-ATP, resulting in more severe copper ion overload and aggregation of lipoylated proteins.236 Moreover, the challenge of inefficient immunotherapy caused by the immunosuppressive TME can be overcome by the synergistic enhancement of the immune response through multi-MIIT. ICD induced by ferroptosis and the cGAS/STING pathway activated by Mn2+ can collectively promote DC maturation, which addresses the issue of poor therapeutic efficacy due to insufficient antigen presentation resulting from single stimulation.237 The interference of genes or proteins crossing over in metabolic pathways by different metal ions shows the potential feasibility of amplified metabolic disruption by multi-metal ion interference therapy. The latest research has shown that the degradation of Mutp53 by Zn2+ has the potential to reverse the high GSH content in tumor cells, consequently sensitizing them to cuproptosis and ferroptosis.238 Hence, it is imperative to recognize the potential of bi-metal ion interference in MIIT and prioritize its exploration in future developments, given that it exhibits potential to achieve more significant therapeutic effects with greater efficiency.(ii) Emerging research has demonstrated that metal ions play an important role in immune modulation, pioneering a new form of immunotherapy known as metalloimmunotherapy. However, the current studies on metalloimmunotherapy are mainly focused on the activation and regulation of antigen presenting cells (APCs) and CTLs, while neglecting the effect NKs and B lymphocyte cells. Additionally, metal ions have the potential to down-regulate the expression of immunosuppressive cytokines and remodel the TME to reverse tumor immune escape. Therefore, future research efforts should aim to elucidate the underlying mechanisms of metalloimmunotherapy to achieve the desired anti-tumor effects comprehensively.
(iii) The surface modification of nanomaterials is essential to enhance the precision of metal ion delivery given that it can improve their internalization and targeting capabilities. Encapsulating nanomedicines with erythrocyte membranes and homologous cancer cell membranes enables them to effectively evade the immune system, leading to increased accumulation at the tumor site. Additionally, incorporating targeting molecules such as hyaluronic acid and folic acid facilitates the binding of nanomedicines to specific receptors on the membrane of cancer cells, thereby enhancing their cellular uptake. Moreover, the internalization efficiency of nanomedicines is also influenced by factors such as their size, shape, and surface charge. Thus, considering a combination of these factors can optimize the delivery of metal ions.
(iv) In xenograft mouse models, numerous metal-based nanomaterials have been designed and confirmed to possess exceptional therapeutic effects. However, the in vivo fate and ultimate therapeutic efficacy of nanomedicines are significantly influenced by critical properties such as their size, shape, and surface properties. Therefore, relying on incomplete or one-sided data from individual cases may result in overlooking certain elusive biological phenomena. By integrating detailed information from these cases, the creation of a field known as nanomedomics can enhance our comprehension of nanomedicine.239 Within nanomedomics, examining crucial factors affecting barriers such as the blood circulation, accumulation at the tumor site, intratumor penetration, and endocytosis can substantially optimize the design of nanomedicines, thereby increasing the likelihood of their successful clinical translation. Establishing a comprehensive database opens up opportunities for employing artificial intelligence and machine learning tools to design nanomedicines from diverse perspectives, challenging the conventional therapeutic effect-centered design approach. In addition to adopting a more rational design of nanomedicines, innovative techniques, strategies, and tools have been employed to address the limitations of xenograft mouse models in nanomedicine research. Researchers have turned to microfluidic tumor microarrays and 3D in vitro models to simulate the human TME and vasculature system.240 These models aim to provide a more comprehensive understanding of the nanoparticle enrichment at the tumor site by addressing the shortcomings of EPR-mediated passive or active targeting in xenograft models. It is acknowledged that the EPR effect in xenograft models does not accurately represent real tumors found in the human body due to the inherent tumor heterogeneity and individual factors, leading to uncertainty. As a response, patients are now being graded based on the strength of the EPR effect, increasing the likelihood of benefiting from nanomedicine therapies.
In summary, an overview of the development and strategies of MIIT can expedite the clinical application of nanomedicine. However, as a nascent therapeutic approach, MIIT still faces several challenges and opportunities for improvement. Addressing the aforementioned issues is crucial for realizing the full potential of MIIT and facilitating a qualitative leap in nanomedicine development.
Abbreviations
| EPR | Enhanced permeability and retention |
| TME | Tumor microenvironment |
| GPX4 | Glutathione peroxidase 4 |
| Caspase | Cysteine aspartate-specific proteases |
| cGAS | Cyclic GMP-AMP synthase |
| STING | Stimulator of interferon genes |
| MIIT | Metal ion interference therapy |
| TCA | Tricarboxylic acid |
| RSL | RAS-selective lethal |
| ROS | Reactive oxygen species |
| PLOOHs | Phospholipid hydroperoxides |
| PUFA-PL | Phospholipid-containing polyunsaturated fatty acid chain |
| ACSL4 | Acyl-CoA synthetase long-chain family member 4 |
| PUFA | Polyunsaturated fatty acyl |
| LPCAT3 | Lysophosphatidylcholine acyltransferase 3 |
| TF | Transferrin |
| TfR | Transferrin receptor |
| NCOA4 | Nuclear receptor coactivator 4 |
| GSH | Glutathione |
| Cys | Cystine |
| BAP1 | BRCA1-associated protein 1 |
| ASC | Alanine-serine-cysteine |
| GSR | Glutathione–disulfide reductase |
| GSSG | Oxidized glutathione |
| FSP1 | Ferroptosis inhibitor protein 1 |
| CoQ10 | Ubiquinone |
| CoQ10H2 | Panthenol |
| PgP | P-glycoprotein |
| MDR | Multi drug resistance |
| POD | Peroxidase |
| LDHs | Layered double hydroxides |
| Art | Artemisinin |
| HSP | Heat shock protein |
| PTT | Photothermal therapy |
| LPO | Lipid hydroperoxides |
| GOx | Glucose oxidase |
| ST | Starvation therapy |
| CAT | Catalase |
| Ce6 | Chlorin e6 |
| COS | Chitosan oligosaccharides |
| MOF | Metal organic frame |
| SAS | Sulfasalazine |
| MTO | Mitoxantrone |
| IFN | Interferon |
| NO | Nitric oxide |
| FGF | Fibroblast growth factor |
| FDX1 | Ferredoxin 1 |
| IL | Interleukin |
| ETC | Electron transport chain |
| LIAS | Lipoyl synthase |
| DLAT | Dihydrolipoamide S-acetyltransferase |
| DBT | Dihydrolipoamide branched chain transacylase E2 |
| GCSH | Glycine cleavage system protein H |
| DLST | Dihydrolipoamide S-succinyltransferase |
| PDH | Pyruvate dehydrogenase |
| α-KDH | α-Ketoglutarate dehydrogenase |
| ATP7a/b | Copper-transporting ATPase |
| Cyt C | Cytochrome C |
| NLRP3 | Nucleotide-binding oligomerization domain-like receptor protein 3 |
| BSO | Butythione sulfoxideimine |
| DDM | Dodecyl-beta-D-maltoside |
| DSF | Disulfiram |
| DTC | Diethyldithiocarbamate |
| CuET | Bis(diethyldithiocarbamate)–copper |
| NIR | Near infrared |
| ORAI1 | Ca2+ release-activated Ca2+ channel protein 1 |
| TRPCs | Transient receptor potential channels |
| VGCCs | Voltage-gated calcium channels |
| STIM1 | Ca2+ sensor protein stromal interaction molecule 1 |
| ER | Endoplasmic reticulum |
| SOCE | Store-operated calcium entry |
| PMCAs | Ca2+ ATPases |
| mPTP | Mitochondrial permeability transition pore |
| VDACs | Voltage-dependent anion-selective channel proteins |
| OMM | Outer mitochondrial membrane |
| MCU | Mitochondrial Ca2+ uniporter |
| RyRs | Ryanodine receptors |
| Ins (1,4,5) P3Rs | Inositol 1,4,5-triphosphate receptors |
| SERCAs | Sarcoplasmic/endoplasmic reticulum Ca2+ ATPases |
| CaP | Calcium phosphate |
| OXPHOS | Oxidative phosphorylation |
| SMAC | Second mitochondria-derived activator of caspase |
| HIF-1α | Hypoxia-inducible factor-1α |
| DOX | Doxorubicin |
| KAE | Kaempferol-3-O-rutinoside |
| CUR | Curcumin |
| GSDME | Gasdermin E |
| LDH | Lactate dehydrogenase |
| BER | Berbamine |
| UCNPs | Upconversion nanoparticles |
| PArg | Polyarginin |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| PFK | Phosphofructokinase |
| NAD+ | Nicotinamide adenine dinucleotide |
| Mutp53 | Mutant p53 proteins |
| WTp53 | Wild-type p53 protein |
| GOF | Gain-of-function |
| UPS | Ubiquitination-mediated proteasomal |
| LND | Lonidamine |
| HK | Hexokinase |
| GLUT1 | Glucose transporter |
| Car | Carnosine |
| ATM | Ataxia telangiectasia mutated |
| ECM | Extracellular matrix |
| ZIP | Zrt/Irt-related protein |
| ZnT1 | Zn transporter |
| NKs | Natural killer cells |
| PK | Pyruvate kinase |
| APCs | Antigen presenting cells |
| CTLs | Cytotoxic T lymphocytes |
| MPS | Mononuclear phagocytic system |
Data availability
Data availability is not applicable to this article as no new data were created or analyzed in this study.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors gratefully acknowledge the financial support from National Natural Science Foundation of China (Grant No. 52101287).References
- Z. Megyesfalvi, C. M. Gay, H. Popper, R. Pirker, G. Ostoros, S. Heeke, C. Lang, K. Hoetzenecker, A. Schwendenwein, K. Boettiger, P. A. Bunn, F. Renyi-Vamos, K. Schelch, H. Prosch, L. A. Byers, F. R. Hirsch and B. Dome, Ca-Cancer J. Clin., 2023, 73(6), 620–652 CrossRef PubMed.
- C. L. Chaffer and R. A. Weinberg, Science, 2011, 331, 1559–1564 CrossRef CAS PubMed.
- M. Brown, F. P. Assen, A. Leithner, J. Abe, H. Schachner, G. Asfour, Z. Bago-Horvath, J. V. Stein, P. Uhrin, M. Sixt and D. Kerjaschki, Science, 2018, 359, 1408–1411 CrossRef CAS.
- H. Zhou, D. Tang, Y. Yu, L. Zhang, B. Wang, J. Karges and H. Xiao, Nat. Commun., 2023, 14, 5350 CrossRef CAS PubMed.
- D. A. Jaffray, F. Knaul, M. Baumann and M. Gospodarowicz, Nat. Cancer, 2023, 4, 1228–1238 CrossRef.
- S. Gao, T. Li, Y. Guo, C. Sun, B. Xianyu and H. Xu, Adv. Mater., 2020, 32, 1907568 CrossRef CAS.
- J. Chen, C. Ning, Z. Zhou, P. Yu, Y. Zhu, G. Tan and C. Mao, Prog. Mater. Sci., 2019, 99, 1–26 CrossRef CAS PubMed.
- D. J. Irvine and E. L. Dane, Nat. Rev. Immunol., 2020, 20, 321–334 CrossRef CAS.
- S. Wang, K. Cheng, K. Chen, C. Xu, P. Ma, G. Dang, Y. Yang, Q. Lei, H. Huang, Y. Yu, Y. Fang, Q. Tang, N. Jiang, H. Miao, F. Liu, X. Zhao and N. Li, Nano Today, 2022, 45, 101512 CrossRef CAS.
- P. Sun, Z. Li, D. Zhang, W. Zeng, Y. Zheng, L. Mei, H. Chen, N. Gao and X. Zeng, Chin. Chem. Lett., 2024, 35, 108346 CrossRef CAS.
- Q. Wu, Z. He, X. Wang, Q. Zhang, Q. Wei, S. Ma, C. Ma, J. Li and Q. Wang, Nat. Commun., 2019, 10, 240 CrossRef.
- H. Zhou, D. Tang, Y. Yu, L. Zhang, B. Wang, J. Karges and H. Xiao, Nat. Commun., 2023, 14, 5350 CrossRef CAS PubMed.
- H. Chen, W. Zhang, G. Zhu, J. Xie and X. Chen, Nat. Rev. Mater., 2017, 2, 17024 CrossRef CAS.
- A. Fromain, J. E. Perez, A. Van de Walle, Y. Lalatonne and C. Wilhelm, Nat. Commun., 2023, 14, 4637 CrossRef CAS PubMed.
- F. Fang, S. Wang, Y. Song, M. Sun, W.-C. Chen, D. Zhao and J. Zhang, Nat. Commun., 2023, 14, 1660 CrossRef CAS.
- B. Yang, H. Yao, H. Tian, Z. Yu, Y. Guo, Y. Wang, J. Yang, C. Chen and J. Shi, Nat. Commun., 2021, 12, 3393 CrossRef CAS.
- T. Song, G. Yang, H. Zhang, M. Li, W. Zhou, C. Zheng, F. You, C. Wu, Y. Liu and H. Yang, Nano Today, 2023, 51, 101896 CrossRef CAS.
- W. Xie, Z. Guo, L. Zhao and Y. Wei, Prog. Mater. Sci., 2023, 138, 101145 CrossRef CAS.
- X. Dai, Y. Xie, W. Feng and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202309160 CrossRef CAS.
- W. Xu, J. Qian, G. Hou, T. Wang, J. Wang, Y. Wang, L. Yang, X. Cui and A. Suo, Adv. Funct. Mater., 2022, 32, 2205013 CrossRef CAS.
- Y. Huang, G. Qin, T. Cui, C. Zhao, J. Ren and X. Qu, Nat. Commun., 2023, 14, 4647 CrossRef CAS PubMed.
- K. Zhang, J. Wu, X. Zhao, J. Qin, Y. Xue, W. Zheng, L. Wang, H. Wang, H. Shen, T. Niu, Y. Luo, R. Tang and B. Wang, ACS Nano, 2021, 15, 19838–19852 CrossRef CAS.
- D. Wang, H. Yi, S. Geng, C. Jiang, J. Liu, J. Duan, Z. Zhang, J. Shi, H. Song, Z. Guo and K. Zhang, ACS Nano, 2023, 17, 16923–16934 CrossRef CAS PubMed.
- C. C. Daw, K. Ramachandran, B. T. Enslow, S. Maity, B. Bursic, M. J. Novello, C. S. Rubannelsonkumar, A. H. Mashal, J. Ravichandran, T. M. Bakewell, W. Wang, K. Li, T. R. Madaris, C. E. Shannon, L. Norton, S. Kandala, J. Caplan, S. Srikantan, P. B. Stathopulos, W. B. Reeves and M. Madesh, Cell, 2020, 183, 474–489.e417 CrossRef CAS.
- S. Wu, K. Zhang, Y. Liang, Y. Wei, J. An, Y. Wang, J. Yang, H. Zhang, Z. Zhang, J. Liu and J. Shi, Adv. Sci., 2022, 9, e2103534 CrossRef.
- P. Zheng, B. Ding, G. Zhu, C. Li and J. Lin, Angew. Chem., Int. Ed., 2022, 61, e202204904 Search PubMed.
- Y. Deng, F. Jia, P. Jiang, L. Chen, L. Xing, X. Shen, L. Li and Y. Huang, Biomaterials, 2023, 301, 122293 CrossRef CAS.
- B. Ding, P. Zheng, J. Tan, H. Chen, Q. Meng, J. Li, X. Li, D. Han, Z. Li, X. Ma, P. Ma and J. Lin, Angew. Chem., Int. Ed., 2023, 62, e202307706 Search PubMed.
- D. Cen, Q. Ge, C. Xie, Q. Zheng, J. Guo, Y. Zhang, Y. Wang, X. Li, Z. Gu and X. Cai, Adv. Mater., 2021, 33, 2104037 CrossRef CAS.
- X. Sun, Y. Zhang, J. Li, K. S. Park, K. Han, X. Zhou, Y. Xu, J. Nam, J. Xu, X. Shi, L. Wei, Y. L. Lei and J. J. Moon, Nat. Nanotechnol., 2021, 16, 1260–1270 CrossRef CAS.
- D. Tang, G. Kroemer and R. Kang, Nat. Rev. Clin. Oncol., 2024, 21, 370–388 CrossRef CAS.
- B. Chen, P. Yu, W. N. Chan, F. Xie, Y. Zhang, L. Liang, K. T. Leung, K. W. Lo, J. Yu, G. M. K. Tse, W. Kang and K. F. To, Signal Transduction Targeted Ther., 2024, 9, 6 CrossRef CAS.
- C. Giorgi, S. Marchi and P. Pinton, Nat. Rev. Mol. Cell Biol., 2018, 19, 713–730 CrossRef CAS.
- L. Chen, J. Min and F. Wang, Signal Transduction Targeted Ther., 2022, 7, 378 CrossRef CAS.
- P. Tsvetkov, S. Coy, B. Petrova, M. Dreishpoon, A. Verma, M. Abdusamad, J. Rossen, L. Joesch-Cohen, R. Humeidi, R. D. Spangler, J. K. Eaton, E. Frenkel, M. Kocak, S. M. Corsello, S. Lutsenko, N. Kanarek, S. Santagata and T. R. Golub, Science, 2022, 375, 1254–1261 CrossRef CAS PubMed.
- X. Jiang, B. R. Stockwell and M. Conrad, Nat. Rev. Mol. Cell Biol., 2021, 22, 266–282 CrossRef.
- H. Lei, G. Hou, M. Chen, J. Ji and L. Cheng, Nano Today, 2023, 53, 102033 CrossRef CAS.
- Y. Wang, F. Gao, L. Zhao, Y. Wu, C. Li, H. Li and Y. Jiang, Coord. Chem. Rev., 2024, 500, 215535 CrossRef CAS.
- S. Bai, Y. Lan, S. Fu, H. Cheng, Z. Lu and G. Liu, Nano-Micro Lett., 2022, 14, 145 CrossRef CAS.
- H. Fan and Z. Guo, Coord. Chem. Rev., 2023, 480, 215027 CrossRef CAS.
- H. Lei, Z. Pei, C. Jiang and L. Cheng, Exploration, 2023, 3, 20220001 CrossRef CAS PubMed.
- X. Song, Q. Zhang, M. Chang, L. Ding, H. Huang, W. Feng, T. Xu and Y. Chen, Adv. Mater., 2023, 35, 2212259 CrossRef CAS.
- X. Zhao, J. Zhou, G. Du and J. Chen, Trends Biotechnol., 2021, 39, 286–297 CrossRef CAS PubMed.
- C. Wincup, G. Robinson, T. Mcdonnell, A. Radziszewska, F. Farinha and A. Rahman, Ann. Rheum. Dis., 2020, 79, 5 CrossRef.
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison and B. R. Stockwell, Cell, 2012, 149, 1060–1072 CrossRef CAS.
- Y. Cheng, O. Zak, P. Aisen, S. C. Harrison and T. Walz, Cell, 2004, 116, 565–576 CrossRef CAS.
- M. Gao, P. Monian, Q. Pan, W. Zhang, J. Xiang and X. Jiang, Cell Res., 2016, 26, 1021–1032 CrossRef CAS PubMed.
- J. D. Mancias, X. Wang, S. P. Gygi, J. W. Harper and A. C. Kimmelman, Nature, 2014, 509, 105–109 CrossRef CAS PubMed.
- S. Doll, F. P. Freitas, R. Shah, M. Aldrovandi, M. C. da Silva, I. Ingold, A. Goya Grocin, T. N. Xavier da Silva, E. Panzilius, C. H. Scheel, A. Mourão, K. Buday, M. Sato, J. Wanninger, T. Vignane, V. Mohana, M. Rehberg, A. Flatley, A. Schepers, A. Kurz, D. White, M. Sauer, M. Sattler, E. W. Tate, W. Schmitz, A. Schulze, V. O’Donnell, B. Proneth, G. M. Popowicz, D. A. Pratt, J. P. F. Angeli and M. Conrad, Nature, 2019, 575, 693–698 CrossRef CAS PubMed.
- P. Koppula, L. Zhuang and B. Gan, Protein Cell, 2020, 12, 599–620 CrossRef PubMed.
- Y. Yan, H. Teng, Q. Hang, L. Kondiparthi, G. Lei, A. Horbath, X. Liu, C. Mao, S. Wu, L. Zhuang, M. James You, M. V. Poyurovsky, L. Ma, K. Olszewski and B. Gan, Nat. Commun., 2023, 14, 3673 CrossRef CAS PubMed.
- S.-J. Wang, D. Li, Y. Ou, L. Jiang, Y. Chen, Y. Zhao and W. Gu, Cell Rep., 2016, 17, 366–373 CrossRef CAS PubMed.
- Y. Zhang, J. Shi, X. Liu, L. Feng, Z. Gong, P. Koppula, K. Sirohi, X. Li, Y. Wei, H. Lee, L. Zhuang, G. Chen, Z.-D. Xiao, M.-C. Hung, J. Chen, P. Huang, W. Li and B. Gan, Nat. Cell Biol., 2018, 20, 1181–1192 CrossRef CAS PubMed.
- Z. Shi, N. Naowarojna, Z. Pan and Y. Zou, Nat. Commun., 2021, 12, 4792 CrossRef CAS.
- A. Mikou, A. Cabayé, A. Goupil, H.-O. Bertrand, J.-P. Mothet and F. C. Acher, Sci. Rep., 2020, 10, 3731 CrossRef CAS.
- M. Jia, D. Qin, C. Zhao, L. Chai, Z. Yu, W. Wang, L. Tong, L. Lv, Y. Wang, J. Rehwinkel, J. Yu and W. Zhao, Nat. Immunol., 2020, 21, 727–735 CrossRef CAS.
- C. Wu, D. Xu, M. Ge, J. Luo, L. Chen, Z. Chen, Y. You, Y.-X. Zhu, H. Lin and J. Shi, Nano Today, 2022, 46, 101574 CrossRef CAS.
- K. Bersuker, J. M. Hendricks, Z. Li, L. Magtanong, B. Ford, P. H. Tang, M. A. Roberts, B. Tong, T. J. Maimone, R. Zoncu, M. C. Bassik, D. K. Nomura, S. J. Dixon and J. A. Olzmann, Nature, 2019, 575, 688–692 CrossRef CAS.
- S. Zhang, S. Gou, Q. Zhang, X. Yong, B. Gan and D. Jia, Cell Res., 2023, 33, 967–970 CrossRef CAS.
- J. Y. Cao, A. Poddar, L. Magtanong, J. H. Lumb, T. R. Mileur, M. A. Reid, C. M. Dovey, J. Wang, J. W. Locasale, E. Stone, S. P. C. Cole, J. E. Carette and S. J. Dixon, Cell Rep., 2019, 26, 1544–1556.e1548 CrossRef CAS.
- X. Yang, L. Wang, S. Guo, R. Li, F. Tian, S. Guan, S. Zhou and J. Lu, Adv. Healthcare Mater., 2021, 10, e2100539 CrossRef.
- W. Li, S. Yin, Y. Shen, H. Li, L. Yuan and X.-B. Zhang, J. Am. Chem. Soc., 2023, 145, 3736–3747 CrossRef CAS.
- L. Zhang, A. Song, Q.-C. Yang, S.-J. Li, S. Wang, S.-C. Wan, J. Sun, R. T. K. Kwok, J. W. Y. Lam, H. Deng, B. Z. Tang and Z.-J. Sun, Nat. Commun., 2023, 14, 5355 CrossRef CAS.
- P. Huang, Y. Yang, W. Wang, Z. Li, N. Gao, H. Chen and X. Zeng, Biomaterials, 2023, 299, 122157 CrossRef CAS PubMed.
- K. Li, K. Xu, S. Liu, Y. He, M. Tan, Y. Mao, Y. Yang, J. Wu, Q. Feng, Z. Luo and K. Cai, ACS Nano, 2023, 17, 20218–20236 CrossRef CAS PubMed.
- F. Zeng, L. Tang, Q. Zhang, C. Shi, Z. Huang, S. Nijiati, X. Chen and Z. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202112925 Search PubMed.
- W. Guo, Y. Ren, Z. Chen, G. Shen, Y. Lu, H. Zhou, Z. Li, Z. Li, X. Lu, G. Li, Z. Shen and Y. Hu, Adv. Funct. Mater., 2023, 33, 2213921 CrossRef CAS.
- X. Wan, L. Song, W. Pan, H. Zhong, N. Li and B. Tang, ACS Nano, 2020, 14, 11017–11028 CrossRef CAS.
- J. J. Kamphorst, J. R. Cross, J. Fan, E. de Stanchina, R. Mathew, E. P. White, C. B. Thompson and J. D. Rabinowitz, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8882–8887 CrossRef CAS.
- M. Hou, H. Su, Q. Wu, W. Sun, P. Zhang, Y. Jiang, K. Qian and C. Zhang, Adv. Funct. Mater., 2023, 33, 2215510 Search PubMed.
- L. Zhu, Y. You, M. Zhu, Y. Song, J. Zhang, J. Hu, X. Xu, X. Xu, Y. Du and J. Ji, Adv. Mater., 2022, 34, e2207174 CrossRef PubMed.
- J. Du, M. Zhou, Q. Chen, Y. Tao, J. Ren, Y. Zhang and H. Qin, Adv. Funct. Mater., 2023, 33, 2215244 CrossRef CAS.
- Z. Zhang, M. Guo, Y. Li, M. Shen, D. Kong, J. Shao, H. Ding, S. Tan, A. Chen, F. Zhang and S. Zheng, Autophagy, 2020, 16, 1482–1505 CrossRef CAS PubMed.
- A. J. Huhn, D. Parsonage, D. A. Horita, F. M. Torti, S. V. Torti and T. Hollis, Protein Sci., 2014, 23, 1013–1022 CrossRef CAS PubMed.
- J. Yang, L. Ding, L. Yu, Y. Wang, M. Ge, Q. Jiang and Y. Chen, Sci. Bull., 2021, 66, 464–477 CrossRef CAS PubMed.
- M. A. Badgley, D. M. Kremer, H. C. Maurer, K. E. DelGiorno, H. J. Lee, V. Purohit, I. R. Sagalovskiy, A. Ma, J. Kapilian, C. E. M. Firl, A. R. Decker, S. A. Sastra, C. F. Palermo, L. R. Andrade, P. Sajjakulnukit, L. Zhang, Z. P. Tolstyka, T. Hirschhorn, C. Lamb, T. Liu, W. Gu, E. S. Seeley, E. Stone, G. Georgiou, U. Manor, A. Iuga, G. M. Wahl, B. R. Stockwell, C. A. Lyssiotis and K. P. Olive, Science, 2020, 368, 85–89 CrossRef CAS.
- C. Liang, X. Zhang, M. Yang and X. Dong, Adv. Mater., 2019, 31, e1904197 CrossRef PubMed.
- C. Cai, J. Zhu, X. Huang, C. Xu, Z. Wang, T. You, X. Wang, J. Xiao and X. Duan, Adv. Funct. Mater., 2023, 33, 2214998 CrossRef CAS.
- X. Chen, R. Kang, G. Kroemer and D. Tang, Nat. Rev. Clin. Oncol., 2021, 18, 280–296 CrossRef CAS PubMed.
- Z.-H. Li, Y. Chen, X. Zeng and X.-Z. Zhang, Nano Today, 2021, 38, 101150 CrossRef CAS.
- Y. Shi and K. S. Carroll, Acc. Chem. Res., 2020, 53, 20–31 CrossRef CAS PubMed.
- C. You, X. Li, D. Wang, H. Chen, L. Liang, Y. Chen, Y. Zhao and H. Xiang, Angew. Chem., Int. Ed., 2022, 61, e202210174 Search PubMed.
- B. R. Stockwell, J. P. Friedmann Angeli, H. Bayir, A. I. Bush, M. Conrad, S. J. Dixon, S. Fulda, S. Gascón, S. K. Hatzios, V. E. Kagan, K. Noel, X. Jiang, A. Linkermann, M. E. Murphy, M. Overholtzer, A. Oyagi, G. C. Pagnussat, J. Park, Q. Ran, C. S. Rosenfeld, K. Salnikow, D. Tang, F. M. Torti, S. V. Torti, S. Toyokuni, K. A. Woerpel and D. D. Zhang, Cell, 2017, 171, 273–285 CrossRef CAS.
- K. Li, C. Lin, M. Li, K. Xu, Y. He, Y. Mao, L. Lu, W. Geng, X. Li, Z. Luo and K. Cai, ACS Nano, 2022, 16, 2381–2398 CrossRef CAS.
- L. X. Zhao, Z. Q. Gong, Q. Zhang, D. L. He, R. L. Ge, J. Meng, H. Ren, Y. G. Fan and Z. Y. Wang, J. Controlled Release, 2023, 359, 12–25 CrossRef CAS.
- R. A. Festa and D. J. Thiele, Curr. Biol., 2011, 21, R877–883 CrossRef CAS.
- D. C. Brady, M. S. Crowe, D. N. Greenberg and C. M. Counter, Cancer Res., 2017, 77, 6240–6252 CrossRef CAS.
- R. Hu, Y. Fang, M. Huo, H. Yao, C. Wang, Y. Chen and R. Wu, Biomaterials, 2019, 206, 101–114 CrossRef CAS PubMed.
- Y. Wang, F. Gao, X. Li, G. Niu, Y. Yang, H. Li and Y. Jiang, J. Nanobiotechnol., 2022, 20, 69 CrossRef CAS PubMed.
- K. Wang, W. Mao, X. Song, M. Chen, W. Feng, B. Peng and Y. Chen, Chem. Soc. Rev., 2023, 52, 6957–7035 RSC.
- P. Zheng, C. Zhou, L. Lu, B. Liu and Y. Ding, J. Exp. Clin. Cancer Res., 2022, 41, 271 CrossRef CAS PubMed.
- B. Guo, F. Yang, L. Zhang, Q. Zhao, W. Wang, L. Yin, D. Chen, M. Wang, S. Han, H. Xiao and N. Xing, Adv. Mater., 2023, 35, 2370152 CrossRef.
- J. Zhang, M. Han, J. Zhang, M. Abdalla, P. Sun, Z. Yang, C. Zhang, Y. Liu, C. Chen and X. Jiang, Int. J. Pharm., 2023, 640, 123025 CrossRef CAS.
- S. Lutsenko, N. L. Barnes, M. Y. Bartee and O. Y. Dmitriev, Physiol. Rev., 2007, 87, 1011–1046 CrossRef CAS PubMed.
- F. Zhao, L. Liang, H. Wang, C. Wang, D. Su, Y. Ying, W. Li, J. Li, J. Zheng, L. Qiao, X. Mou, S. Che and J. Yu, Adv. Funct. Mater., 2023, 33, 2300941 CrossRef CAS.
- X.-K. Jin, J.-L. Liang, S.-M. Zhang, Q.-X. Huang, S.-K. Zhang, C.-J. Liu and X.-Z. Zhang, Mater. Today, 2023, 68, 108–124 CrossRef CAS.
- Y. Xu, S. Y. Liu, L. Zeng, H. Ma, Y. Zhang, H. Yang, Y. Liu, S. Fang, J. Zhao, Y. Xu, C. R. Ashby, Jr., Y. He, Z. Dai and Y. Pan, Adv. Mater., 2022, 34, e2204733 CrossRef.
- C. Y. Chung, J. M. Posimo, S. Lee, T. Tsang, J. M. Davis, D. C. Brady and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18285–18294 CrossRef.
- R. Safi, E. R. Nelson, S. K. Chitneni, K. J. Franz, D. J. George, M. R. Zalutsky and D. P. McDonnell, Cancer Res., 2014, 74, 5819–5831 CrossRef CAS.
- Z. Skrott, M. Mistrik, K. K. Andersen, S. Friis, D. Majera, J. Gursky, T. Ozdian, J. Bartkova, Z. Turi, P. Moudry, M. Kraus, M. Michalova, J. Vaclavkova, P. Dzubak, I. Vrobel, P. Pouckova, J. Sedlacek, A. Miklovicova, A. Kutt, J. Li, J. Mattova, C. Driessen, Q. P. Dou, J. Olsen, M. Hajduch, B. Cvek, R. J. Deshaies and J. Bartek, Nature, 2017, 552, 194–199 CrossRef CAS PubMed.
- J. Zhou, Q. Yu, J. Song, S. Li, X. L. Li, B. K. Kang, H. Y. Chen and J. J. Xu, Angew. Chem., Int. Ed., 2023, 62, e202213922 Search PubMed.
- Y. Lu, Q. Pan, W. Gao, Y. Pu and B. He, J. Mater. Chem. B, 2022, 10, 6296–6306 RSC.
- L. Chan, Y. Liu, M. Chen, Y. Su, J. Guo, L. Zhu, M. Zhan, T. Chen and L. Lu, Adv. Funct. Mater., 2023, 33, 2302054 CrossRef CAS.
- J. C. Liu, J. Liu, K. M. Holmström, S. Menazza, R. J. Parks, M. M. Fergusson, Z.-X. Yu, D. A. Springer, C. Halsey, C. Liu, E. Murphy and T. Finkel, Cell Rep., 2016, 16, 1561–1573 CrossRef CAS PubMed.
- G. Santulli and A. Marks, Curr. Mol. Pharmacol., 2015, 8, 206–222 CrossRef CAS.
- C. Giorgi, A. Danese, S. Missiroli, S. Patergnani and P. Pinton, Trends Cell Biol., 2018, 28, 258–273 CrossRef CAS PubMed.
- M. Zhang, R. Song, Y. Liu, Z. Yi, X. Meng, J. Zhang, Z. Tang, Z. Yao, Y. Liu, X. Liu and W. Bu, Chem, 2019, 5, 2171–2182 CAS.
- C. Giorgi, S. Marchi and P. Pinton, Nat. Rev. Mol. Cell Biol., 2018, 19, 713–730 CrossRef CAS.
- M. Prakriya, S. Feske, Y. Gwack, S. Srikanth, A. Rao and P. G. Hogan, Nature, 2006, 443, 230–233 CrossRef CAS PubMed.
- N. Siri-Angkul, R. Gordan, S. Wongjaikam, N. Fefelova, J. Gwathmey, S. Chattipakorn, N. Chattipakorn and L.-H. Xie, Circ. Res., 2019, 125, A507–A507 Search PubMed.
- P. Gilon, H. Y. Chae, G. A. Rutter and M. A. Ravier, Cell Calcium, 2014, 56, 340–361 CrossRef CAS PubMed.
- R. S. Lewis, Cold Spring Harbor Perspect. Biol., 2020, 12, a035055 CrossRef CAS.
- L. S. Jouaville, P. Pinton, C. Bastianutto, G. A. Rutter and R. Rizzuto, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13807–13812 CrossRef CAS.
- S. Marchi, S. Patergnani, S. Missiroli, G. Morciano, A. Rimessi, M. R. Wieckowski, C. Giorgi and P. Pinton, Cell Calcium, 2018, 69, 62–72 CrossRef CAS PubMed.
- V. Shoshan-Barmatz, Y. Krelin and A. Shteinfer-Kuzmine, Cell Calcium, 2018, 69, 81–100 CrossRef CAS.
- M. Fan, J. Zhang, C.-W. Tsai, B. J. Orlando, M. Rodriguez, Y. Xu, M. Liao, M.-F. Tsai and L. Feng, Nature, 2020, 582, 129–133 CrossRef CAS.
- M. Prakriya and R. S. Lewis, Physiol. Rev., 2015, 95, 1383–1436 CrossRef CAS.
- D. Kusumoto, S. Yuasa and K. Fukuda, Circ. Res., 2019, 124, 668–670 CrossRef CAS.
- B. Liu, S. Liang, Z. Wang, Q. Sun, F. He, S. Gai, P. Yang, Z. Cheng and J. Lin, Adv. Mater., 2021, 33, 2101223 CrossRef CAS.
- P. E. Porporato, N. Filigheddu, J. M. B.-S. Pedro, G. Kroemer and L. Galluzzi, Cell Res., 2018, 28, 265–280 CrossRef CAS.
- J. F. Garbincius, T. S. Luongo, J. P. Lambert, A. S. Mangold, E. K. Murray, A. N. Hildebrand, P. Jadiya and J. W. Elrod, Circulation, 2020, 142, A16592–A16592 Search PubMed.
- E. Norberg, V. Gogvadze, M. Ott, M. Horn, P. Uhlén, S. Orrenius and B. Zhivotovsky, Cell Death Differ., 2008, 15, 1857–1864 CrossRef CAS.
- D. C. Wallace, Nat. Rev. Cancer, 2012, 12, 685–698 CrossRef CAS.
- M. Chang, Z. Hou, D. Jin, J. Zhou, M. Wang, M. Wang, M. Shu, B. Ding, C. Li and J. Lin, Adv. Mater., 2020, 32, 2004647 CrossRef CAS.
- G. Li, Y. Chen, L. Zhang, M. Zhang, S. Li, L. Li, T. Wang and C. Wang, Nano-Micro Lett., 2017, 10, 7 CrossRef PubMed.
- D. Samanta, D. M. Gilkes, P. Chaturvedi, L. Xiang and G. L. Semenza, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E5429–E5438 CrossRef CAS.
- J. Liu, C. Zhu, L. Xu, D. Wang, W. Liu, K. Zhang, Z. Zhang and J. Shi, Nano Lett., 2020, 20, 8102–8111 CrossRef CAS.
- Y. Li, X. Yu, Y. Wang, X. Zheng and Q. Chu, Food Funct., 2021, 12, 8351–8365 RSC.
- Y. Li, S. Zhou, H. Song, T. Yu, X. Zheng and Q. Chu, Biomaterials, 2021, 277, 121080 CrossRef CAS.
- C. Yang and T. M. Svitkina, Nat. Cell Biol., 2019, 21, 603–613 CrossRef CAS PubMed.
- L. Yin, Z. Meng, Y. Zhang, K. Hu, W. Chen, K. Han, B.-Y. Wu, R. You, C.-H. Li, Y. Jin and Y.-Q. Guan, J. Controlled Release, 2018, 271, 31–44 CrossRef CAS.
- T. Alissafi, A. Banos, L. Boon, T. Sparwasser, A. Ghigo, K. Wing, D. Vassilopoulos, D. Boumpas, T. Chavakis, K. Cadwell and P. Verginis, J. Clin. Invest., 2017, 127, 2789–2804 CrossRef PubMed.
- K. Cadwell, Nat. Rev. Immunol., 2016, 16, 661–675 CrossRef CAS PubMed.
- J. An, K. Zhang, B. Wang, S. Wu, Y. Wang, H. Zhang, Z. Zhang, J. Liu and J. Shi, ACS Nano, 2020, 14, 7639–7650 CrossRef CAS PubMed.
- C. Szabo, Nat. Rev. Drug Discovery, 2016, 15, 185–203 CrossRef CAS PubMed.
- C. Peers, C. C. Bauer, J. P. Boyle, J. L. Scragg and M. L. Dallas, Antioxid. Redox Signaling, 2011, 17, 95–105 CrossRef PubMed.
- H. Zhao, L. Wang, K. Zeng, J. Li, W. Chen and Y. N. Liu, ACS Nano, 2021, 15, 13188–13199 CrossRef CAS.
- M. Mayerhofer, K. V. Gleixner, J. Mayerhofer, G. Hoermann, E. Jaeger, K. J. Aichberger, R. G. Ott, K. Greish, H. Nakamura, S. Derdak, P. Samorapoompichit, W. F. Pickl, V. Sexl, H. Esterbauer, I. Schwarzinger, C. Sillaber, H. Maeda and P. Valent, Blood, 2008, 111, 2200–2210 CrossRef CAS.
- S. Kakizawa, T. Yamazawa, Y. Chen, A. Ito, T. Murayama, H. Oyamada, N. Kurebayashi, O. Sato, M. Watanabe, N. Mori, K. Oguchi, T. Sakurai, H. Takeshima, N. Saito and M. Iino, EMBO J., 2012, 31, 417–428 CrossRef CAS PubMed.
- X. Chu, X. Jiang, Y. Liu, S. Zhai, Y. Jiang, Y. Chen, J. Wu, Y. Wang, Y. Wu, X. Tao, X. He and W. Bu, Adv. Funct. Mater., 2021, 31, 2008507 CrossRef CAS.
- X. Chen, C. Xu, P. Zhao, Y. Zhang, J. Guo, X. Hu, H. Gao, C. Zhang, X. Qu and J. Zhang, Chem. Eng. J., 2023, 463, 142478 CrossRef CAS.
- N. Ogawa, T. Kurokawa and Y. Mori, Cell Calcium, 2016, 60, 115–122 CrossRef CAS.
- C. Dong, X. Dai, X. Wang, Q. Lu, L. Chen, X. Song, L. Ding, H. Huang, W. Feng, Y. Chen and M. Chang, Adv. Mater., 2022, 34, e2205680 CrossRef.
- M. Gao, T. Yang, W. Qin, Q. Wang, M. Huang, H. Peng, M. Shao, W. Yao, X. Yi, G. Sun and X. He, Small, 2022, 18, e2204689 CrossRef.
- G. Faa, V. M. Nurchi, A. Ravarino, D. Fanni, S. Nemolato, C. Gerosa, P. Van Eyken and K. Geboes, Coord. Chem. Rev., 2008, 252, 1257–1269 CrossRef CAS.
- Y. Liu, Y. Wang, S. Song and H. Zhang, Natl. Sci. Rev., 2022, 9, nwab139 CrossRef CAS.
- M. A. Sirover, Cancer Metastasis Rev., 2018, 37, 665–676 CrossRef CAS PubMed.
- I. G. Gazaryan, I. P. Krasinskaya, B. S. Kristal and A. M. Brown, J. Biol. Chem., 2007, 282, 24373–24380 CrossRef CAS PubMed.
- K. T. Bieging, S. S. Mello and L. D. Attardi, Nat. Rev. Cancer, 2014, 14, 359–370 CrossRef CAS PubMed.
- P. A. Muller and K. H. Vousden, Cancer Cell, 2014, 25, 304–317 CrossRef CAS PubMed.
- P. A. J. Muller and K. H. Vousden, Nat. Cell Biol., 2013, 15, 2–8 CrossRef CAS.
- K. P. Olive, D. A. Tuveson, Z. C. Ruhe, B. Yin, N. A. Willis, R. T. Bronson, D. Crowley and T. Jacks, Cell, 2004, 119, 847–860 CrossRef CAS PubMed.
- S. Di Agostino, S. Strano, V. Emiliozzi, V. Zerbini, M. Mottolese, A. Sacchi, G. Blandino and G. Piaggio, Cancer Cell, 2006, 10, 191–202 CrossRef CAS PubMed.
- A. Padmanabhan, N. Candelaria, K.-K. Wong, B. C. Nikolai, D. M. Lonard, B. W. O’Malley and J. S. Richards, Nat. Commun., 2018, 9, 1270 CrossRef PubMed.
- G. Foggetti, L. Ottaggio, D. Russo, C. Mazzitelli, P. Monti, P. Degan, M. Miele, G. Fronza and P. Menichini, Biosci. Rep., 2019, 39, BSR20181345 CrossRef CAS PubMed.
- J. Qian, W. Zhang, P. Wei, G. Yao, T. Yi, H. Zhang, H. Ding, X. Huang, M. Wang, Y. Song, S. Zhong, L. Yang, J. Gao, Z. Zhou, L. P. Wen and Y. Zhang, Adv. Funct. Mater., 2020, 30, 2001994 CrossRef CAS.
- K. Yang, W. Han, X. Jiang, A. Piffko, J. Bugno, C. Han, S. Li, H. Liang, Z. Xu, W. Zheng, L. Wang, J. Wang, X. Huang, J. P. Y. Ting, Y. X. Fu, W. Lin and R. R. Weichselbaum, Nat. Nanotechnol., 2022, 17, 1322–1331 CrossRef CAS.
- L. Sun, J. Wu, F. Du, X. Chen and Z. J. Chen, Science, 2013, 339, 786–791 CrossRef CAS.
- K. M. MacDonald, S. Nicholson-Puthenveedu, M. M. Tageldein, S. Khasnis, C. H. Arrowsmith and S. M. Harding, Nat. Commun., 2023, 14, 556 CrossRef CAS.
- M. Du and Z. J. Chen, Science, 2018, 361, 704–709 CrossRef CAS PubMed.
- D. Cen, Q. Ge, C. Xie, Q. Zheng, J. Guo, Y. Zhang, Y. Wang, X. Li, Z. Gu and X. Cai, Adv. Mater., 2021, 33, 2104037 CrossRef CAS.
- Z. Meng, X. Zhang, H. Tan and H. Lian, Chem. Eng. J., 2022, 435, 134781 CrossRef CAS.
- L. Lei, B. Nan, F. Yang, L. Xu, G. Guan, J. Xu, R. Yue, Y. Wang, S. Huan, X. Yin, X. B. Zhang and G. Song, Nano Lett., 2023, 23, 2659–2668 CrossRef CAS.
- M. J. Duffy, N. C. Synnott and J. Crown, Eur. J. Cancer, 2017, 83, 258–265 CrossRef CAS PubMed.
- J. Wang, C. Qu, X. Shao, G. Song, J. Sun, D. Shi, R. Jia, H. An and H. Wang, Bioact. Mater., 2023, 20, 404–417 CAS.
- A. M. Tidball, M. R. Bryan, M. A. Uhouse, K. K. Kumar, A. A. Aboud, J. E. Feist, K. C. Ess, M. D. Neely, M. Aschner and A. B. Bowman, Hum. Mol. Genet., 2015, 24, 1929–1944 CrossRef CAS.
- L. Zhang, J. Zhao, X. Hu, C. Wang, Y. Jia, C. Zhu, S. Xie, J. Lee, F. Li and D. Ling, Adv. Mater., 2022, 34, e2206915 CrossRef.
- G. A. Cabral-Pacheco, I. Garza-Veloz, C. Castruita-De la Rosa, J. M. Ramirez-Acuña, B. A. Perez-Romero, J. F. Guerrero-Rodriguez, N. Martinez-Avila and M. L. Martinez-Fierro, Int. J. Mol. Sci., 2020, 21, 9739 CrossRef CAS.
- A. Chakravarthy, L. Khan, N. P. Bensler, P. Bose and D. D. De Carvalho, Nat. Commun., 2018, 9, 4692 CrossRef.
- L. Ding, M. Liang, Y. Li, M. Zeng, M. Liu, W. Ma, F. Chen, C. Li, R. L. Reis, F. R. Li and Y. Wang, Adv. Sci., 2023, e2302967, DOI:10.1002/advs.202302967.
- Y. Jiang, K. Shao, F. Zhang, T. Wang, L. Han, X. Kong and J. Shi, Aggregate, 2023, 4, e321 CrossRef CAS.
- E. Bafaro, Y. Liu, Y. Xu and R. E. Dempski, Signal Transduction Targeted Ther., 2017, 2, 17029 CrossRef.
- B. H. Bin, J. Seo and S. T. Kim, J. Immunol. Res., 2018, 2018, 9365747 Search PubMed.
- Q. Wu, Q. Mu, Z. Xia, J. Min and F. Wang, Semin. Cell Dev. Biol., 2021, 115, 45–53 CrossRef CAS.
- J. Azadmanesh and G. E. O. Borgstahl, Antioxidants, 2018, 7, 25 CrossRef.
- X. Sun, Y. Zhang, J. Li, K. S. Park, K. Han, X. Zhou, Y. Xu, J. Nam, J. Xu, X. Shi, L. Wei, Y. L. Lei and J. J. Moon, Nat. Nanotechnol., 2021, 16, 1260–1270 CrossRef CAS.
- T. Taguchi, K. Mukai, E. Takaya and R. Shindo, Front. Immunol., 2021, 12, 646304 CrossRef CAS.
- X. Wang, W. Zhang, Y. Wang, X. Zhu, Z. Liu, M. Liu, Z. Wu, B. Li, S. Liu, S. Liao, P. Zhu, B. Liu, C. Li, Y. Wang and Z. Chen, ACS Nano, 2024, 18, 6946–6962 CrossRef CAS PubMed.
- S. Huang, Y. Gao, H. Li, R. Wang, X. Zhang, X. Wang, D. Huang, L. Zhang, H. A. Santos, Z. Yin and B. Xia, Adv. Mater., 2024, 36, 2310979 CrossRef CAS.
- J. Xiao, G. Zhang, R. Xu, H. Chen, H. Wang, G. Tian, B. Wang, C. Yang, G. Bai, Z. Zhang, H. Yang, K. Zhong, D. Zou and Z. Wu, Biomaterials, 2019, 216, 119254 CrossRef CAS.
- M. Lv, M. Chen, R. Zhang, W. Zhang, C. Wang, Y. Zhang, X. Wei, Y. Guan, J. Liu, K. Feng, M. Jing, X. Wang, Y.-C. Liu, Q. Mei, W. Han and Z. Jiang, Cell Res., 2020, 30, 966–979 CrossRef CAS PubMed.
- W. Zeng, Z. Li, Q. Huang, C. Ding, L. Yang, W. Wang, Z. Shi, Y. Yang, H. Chen, L. Mei and X. Zeng, Adv. Funct. Mater., 2024, 34, 2307241 CrossRef CAS.
- J. P. Ting, J. A. Duncan and Y. Lei, Science, 2010, 327, 286–290 CrossRef CAS PubMed.
- K. Wang, Y. Li, X. Wang, Z. Zhang, L. Cao, X. Fan, B. Wan, F. Liu, X. Zhang, Z. He, Y. Zhou, D. Wang, J. Sun and X. Chen, Nat. Commun., 2023, 14, 2950 CrossRef CAS.
- P. T. Nguyen, C. Deisl, M. Fine, T. S. Tippetts, E. Uchikawa, X.-C. Bai and B. Levine, Nat. Commun., 2022, 13, 5293 CrossRef CAS.
- Y. Li, J. Lin, P. Wang, F. Zhu, M. Wu, Q. Luo, Y. Zhang and X. Liu, ACS Nano, 2022, 16, 7380–7397 CrossRef CAS.
- B. Ding, J. Sheng, P. Zheng, C. Li, D. Li, Z. Cheng, P. Ma and J. Lin, Nano Lett., 2021, 21, 8281–8289 CrossRef CAS.
- L. Cassetta and J. W. Pollard, Nat. Rev. Drug Discovery, 2018, 17, 887–904 CrossRef CAS.
- D. G. DeNardo and B. Ruffell, Nat. Rev. Immunol., 2019, 19, 369–382 CrossRef CAS PubMed.
- H. Xu, S. Xiong, Y. Chen, Q. Ye, N. Guan, Y. Hu and J. Wu, Adv. Mater., 2023, 35, 2303357 CrossRef CAS.
- H. M. Nguyen, E. M. Grössinger, M. Horiuchi, K. W. Davis, L. W. Jin, I. Maezawa and H. Wulff, Glia, 2017, 65, 106–121 CrossRef.
- S. Chen, W. Cui, Z. Chi, Q. Xiao, T. Hu, Q. Ye, K. Zhu, W. Yu, Z. Wang, C. Yu, X. Pan, S. Dai, Q. Yang, J. Jin, J. Zhang, M. Li, D. Yang, Q. Yu, Q. Wang, X. Yu, W. Yang, X. Zhang, J. Qian, K. Ding and D. Wang, Cell Metab., 2022, 34, 1843–1859.e1811 CrossRef CAS PubMed.
- S. Ashique, S. Kumar, A. Hussain, N. Mishra, A. Garg, B. H. J. Gowda, A. Farid, G. Gupta, K. Dua and F. Taghizadeh-Hesary, J. Health, Popul. Nutr., 2023, 42, 74 CrossRef.
- D. W. Killilea and A. N. Killilea, Free Radical Biol. Med., 2022, 182, 182–191 CrossRef CAS.
- J. Lötscher, I. L. A. A. Martí, N. Kirchhammer, E. Cribioli, G. M. P. Giordano Attianese, M. P. Trefny, M. Lenz, S. I. Rothschild, P. Strati, M. Künzli, C. Lotter, S. H. Schenk, P. Dehio, J. Löliger, L. Litzler, D. Schreiner, V. Koch, N. Page, D. Lee, J. Grählert, D. Kuzmin, A. V. Burgener, D. Merkler, M. Pless, M. L. Balmer, W. Reith, J. Huwyler, M. Irving, C. G. King, A. Zippelius and C. Hess, Cell, 2022, 185, 585–602.e529 CrossRef.
- S. Bruns, D. Krüger, S. Galli, D. C. F. Wieland, J. U. Hammel, F. Beckmann, A. Wennerberg, R. Willumeit-Römer, B. Zeller-Plumhoff and J. Moosmann, Bioact. Mater., 2023, 28, 155–166 CAS.
- R. Menze, B. Hesse, M. Kusmierczuk, D. Chen, T. Weitkamp, S. I. Bettink and B. Scheller, Bioact. Mater., 2023, 32, 1–11 Search PubMed.
- N. Yang, F. Gong, B. Liu, Y. Hao, Y. Chao, H. Lei, X. Yang, Y. Gong, X. Wang, Z. Liu and L. Cheng, Nat. Commun., 2022, 13, 2336 CrossRef CAS.
- S. Golshani-Hebroni, Gene, 2016, 581, 1–13 CrossRef CAS PubMed.
- S.-G. Kim, M. Cavalier, M. R. El-Maghrabi and Y.-H. Lee, J. Mol. Biol., 2007, 370, 14–26 CrossRef CAS PubMed.
- H. P. Morgan, F. J. O’Reilly, M. A. Wear, J. R. O’Neill, L. A. Fothergill-Gilmore, T. Hupp and M. D. Walkinshaw, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5881–5886 CrossRef CAS.
- Y. Xu, Y. Guo, L. Chen, D. Ni, P. Hu and J. Shi, Chem. Sci., 2021, 12, 7763–7769 RSC.
- W. Fang, Z. Yu, P. Hu and J. Shi, Adv. Funct. Mater., 2024, 2405483 CrossRef.
- Z.-H. Li, Y. Chen, X. Zeng and X.-Z. Zhang, Nano Today, 2021, 38, 101150 CrossRef CAS.
- Z. Fan, S. Wu, H. Deng, G. Li, L. Huang and H. Liu, ACS Nano, 2024, 18, 12261–12275 CrossRef CAS PubMed.
- J. Wang, Z. Fang, C. Zhao, Z. Sun, S. Gao, B. Zhang, D. Qiu, M. Yang, F. Sheng, S. Gao and Y. Hou, Adv. Mater., 2024, 36, 2307006 CrossRef CAS.
- D. Xie, C. Hu, C. Jiang, J. Xia, L. Ye, Y. Jin, S. Jiang, Y. Ji, Z. Zhang, H. Song, Y. Zhu, P. Tang, Z. Hu, Y. Xiao, J. Dai and Z. Hu, Chem. Eng. J., 2023, 476, 146788 CrossRef CAS.
- Q. Yu, Q. Li, L. Tu, Y. Zhou, H. Zhu, Q. Zhang, M. Liu and Y. Sun, Chem. Eng. J., 2023, 477, 147085 CrossRef CAS.
- C. Huang, B. Lin, C. Chen, H. Wang, X. Lin, J. Liu, Q. Ren, J. Tao, P. Zhao and Y. Xu, Adv. Mater., 2022, 34, 2207593 CrossRef CAS.
- W. Chen, Y. Lu, Y. Xu, Y. Chen, S. Lin, X. He, C. Zhang and C. Yuan, Chem. Eng. J., 2024, 490, 151838 CrossRef CAS.
- Z. Chen, Q. Wu, W. Guo, M. Niu, L. Tan, N. Wen, L. Zhao, C. Fu, J. Yu, X. Ren, P. Liang and X. Meng, Biomaterials, 2021, 276, 121016 CrossRef CAS PubMed.
- X. Tian, H. Xu, F. Zhou, X. Gong, S. Tan and Y. He, Chem. Mater., 2024, 36, 815–828 CrossRef CAS.
- J. Xia, C. Hu, Y. Ji, M. Wang, Y. Jin, L. Ye, D. Xie, S. Jiang, R. Li, Z. Hu and J. Dai, ACS Nano, 2023, 17, 21134–21152 CrossRef.
- F. Zhao, H. Yu, L. Liang, C. Wang, D. Shi, X. Zhang, Y. Ying, W. Cai, W. Li, J. Li, J. Zheng, L. Qiao, S. Che and J. Yu, Adv. Healthcare Mater., 2023, 12, 2301346 CrossRef CAS PubMed.
- Y. Huang, X. Liu, J. Zhu, Z. Chen, L. Yu, X. Huang, C. Dong, J. Li, H. Zhou, Y. Yang and W. Tan, J. Am. Chem. Soc., 2024, 146, 13805–13816 CrossRef CAS PubMed.
- X. Zhao, H. Cheng, Q. Wang, W. Nie, Y. Yang, X. Yang, K. Zhang, J. Shi and J. Liu, ACS Nano, 2023, 17, 13746–13759 CrossRef CAS PubMed.
- Y. Yang, Y. Zhu, K. Wang, Y. Miao, Y. Zhang, J. Gao, H. Qin and Y. Zhang, Bioact. Mater., 2023, 29, 116–131 CAS.
- J. Liu, J. Zhan, Y. Zhang, L. Huang, J. Yang, J. Feng, L. Ding, Z. Shen and X. Chen, Adv. Mater., 2024, 36, 2309562 CrossRef CAS.
- K. Ling, J. Zheng, X. Jiang, W. Huang, Y. Mai, C. Liao, S. Fan, J. Bu, R. Li, B. Zeng, Q. Zheng, R. Huang, Z. Li, N.-K. Wong and H. Jiang, ACS Nano, 2024, 18, 2841–2860 CrossRef CAS.
- X. Yang, L. Wang, S. Guo, R. Li, F. Tian, S. Guan, S. Zhou and J. Lu, Adv. Healthcare Mater., 2021, 10, 2100539 CrossRef CAS PubMed.
- Y. Chen, X. Yang, H. Li, X. Wu, W. Wu, J. Chen, A. Wu and X. Wang, Small, 2024, 2402073 CrossRef PubMed.
- T. Sun, L. Zhang, S. Xiao, Q. Xie, M. Wang, Y. Zhao, C. Zhou, M. Gong and D. Zhang, ACS Mater. Lett., 2024, 6, 985–998 CrossRef CAS.
- J. Chang, W. Yin, H. Zhi, S. Chen, J. Sun, Y. Zhao, L. Huang, L. Xue, X. Zhang, T. Zhang, H. Dong and Y. Li, Small, 2024, 20, 2308565 CrossRef CAS.
- Y. Li, J. Liu, Y. Chen, R. R. Weichselbaum and W. Lin, Adv. Sci., 2024, 11, 2310309 CrossRef CAS.
- S. Ning, M. Lyu, D. Zhu, J. W. Y. Lam, Q. Huang, T. Zhang and B. Z. Tang, ACS Nano, 2023, 17, 10206–10217 CrossRef CAS PubMed.
- H. Wu, Z. Zhang, Y. Cao, Y. Hu, Y. Li, L. Zhang, X. Cao, H. Wen, Y. Zhang, H. Lv and X. Jin, Adv. Sci., 2024, 11, 2401047 CrossRef CAS PubMed.
- S. Qin, L. Ma, R. Li, P. Yuan, Y. Shi, X. Ji, W. Xue, Y. Li and W. Liu, Chem. Eng. J., 2023, 477, 147118 CrossRef CAS.
- T. Zhang, H. Tian, S. Qin, Y. Gao, X. Zhang, E. C. Nice, Z. Du and C. Huang, Adv. Funct. Mater., 2024, 34, 2313384 CrossRef CAS.
- B. Fu, J. Hu, A. Yu and Y. Wang, Adv. Funct. Mater., 2024, 34, 2307823 CrossRef CAS.
- L. Ding, M. Liang, Y. Li, M. Zeng, M. Liu, W. Ma, F. Chen, C. Li, R. L. Reis, F.-R. Li and Y. Wang, Adv. Sci., 2023, 10, 2302967 CrossRef CAS.
- H. Zhu, F. Gao, Y. Li, M. Jiang, Y. Zhang, C. Kan, L. Han, S. Xue, K. Wang, Q. Fan, H. Hu, F. Sun and Z. Ming, Nano Today, 2024, 56, 102308 CrossRef CAS.
- L. Zhang, J. Zhao, X. Hu, C. Wang, Y. Jia, C. Zhu, S. Xie, J. Lee, F. Li and D. Ling, Adv. Mater., 2022, 34, 2206915 CrossRef CAS PubMed.
- Y. Tian, H. Tian, B. Li, C. Feng and Y. Dai, Small, 2024, 20, 2309850 CrossRef CAS.
- L. Huo, S. Zhu, J. Zeng, R. Lin, W. Chen, M. Li, X. Sun, M. Tan, G. Huang, K. Xu and Z. Zhao, ACS Mater. Lett., 2024, 6, 885–895 CrossRef CAS.
- H. Lei, Q. Li, G. Li, T. Wang, X. Lv, Z. Pei, X. Gao, N. Yang, F. Gong, Y. Yang, G. Hou, M. Chen, J. Ji, Z. Liu and L. Cheng, Bioact. Mater., 2024, 31, 53–62 CAS.
- C. Du, X. Guo, X. Qiu, W. Jiang, X. Wang, H. An, J. Wang, Y. Luo, Q. Du, R. Wang, C. Cheng, Y. Guo, H. Teng, H. Ran, Z. Wang, P. Li, Z. Zhou and J. Ren, Adv. Sci., 2024, 11, 2306031 CrossRef CAS.
- Q. Du, Y. Luo, L. Xu, C. Du, W. Zhang, J. Xu, Y. Liu, B. Liu, S. Chen, Y. Wang, Z. Wang, H. Ran, J. Wang and D. Guo, J. Nanobiotechnol., 2024, 22, 95 CrossRef CAS.
- Y. Lu, Y. Chen, G. Hou, H. Lei, L. Liu, X. Huang, S. Sun, L. Liu, X. Liu, J. Na, Y. Zhao, L. Cheng and L. Zhong, ACS Nano, 2024, 18, 10542–10556 CrossRef CAS.
- G. Liang, W. Cao, D. Tang, H. Zhang, Y. Yu, J. Ding, J. Karges and H. Xiao, ACS Nano, 2024, 18, 10979–11024 CrossRef CAS.
- K. E. de Roode, K. Hashemi, W. P. R. Verdurmen and R. Brock, Small, 2024, 2402311 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |