
A mitochondria-targeted anticancer copper dithiocarbamate amplifies immunogenic cuproptosis and macrophage polarization†
Yao
Lu
 a,
Xi
Fan
a,
Qingqing
Pan
*b,
Bin
He
*a and
Yuji
Pu
*a
a,
Xi
Fan
a,
Qingqing
Pan
*b,
Bin
He
*a and
Yuji
Pu
*a
aNational Engineering Research Center for Biomaterials, College of Biomedical Engineering, Sichuan University, Chengdu 610064, China. E-mail: bhe@scu.edu.cn; yjpu@scu.edu.cn
bSchool of Preclinical Medicine, Chengdu University, Chengdu 610106, China. E-mail: panqingqing@cdu.edu.cn
First published on 23rd January 2024
Abstract
The way that cancer cells die inspires treatment regimens and cytolytic cuproptosis induced by copper complexes, like copper(II) bis(diethyldithiocarbamate) (CuET), has emerged as a novel therapeutic target. Herein, a triphenylphosphonium-modified CuET (TPP-CuET) is designed to target mitochondrial metabolism, triggering intense immunogenic cuproptosis in breast cancer cells and remodeling tumor-associated macrophages. TPP-CuET enables an enhanced mitochondrial copper accumulation in comparison to CuET (29.0% vs. 19.4%), and severely disrupts the morphology and functions of mitochondria, encompassing the tricarboxylic acid cycle, ATP synthesis, and electron transfer chain. Importantly, it triggers amplified immunogenic death of cancer cells, and the released damage-associated molecular patterns effectively induce M1 polarization and migration of macrophages. Transcriptome analysis further reveals that TPP-CuET promotes antigen processing and presentation in cancer cells through the MHC I pathway, activating the immune response of CD8 T cells and natural killer cells. To the best of our knowledge, TPP-CuET is the first mitochondrial targeted immunogenic cuproptosis inducer and is expected to flourish in antitumor immunotherapy.
Introduction
Cancer is a worldwide life-threatening disease. While current treatments including chemotherapy and radiotherapy can suppress tumor growth, they are plagued by pronounced systemic toxicity and a lack of immunogenicity. This dearth of immunogenicity hampers their ability to incite a robust immune response, thereby diminishing the immune system's ability to identify and eliminate malignant cells. The primary culprit for this reduced immunogenicity lies in the predominant mode of cell death induced by chemotherapeutic agents and radiation, which is apoptosis—a form of programmed cell death. Apoptotic cancer cells maintain their membrane integrity, concealing a wealth of latent immunogenic damage-associated molecular patterns (DAMPs) within their structure. This concealment allows them to evade the vigilant scrutiny of neighboring immune cells.1,2 Consequently, there arises a pressing need to induce a highly immunogenic form of cell death in cancer cells, both as a standalone therapeutic approach and as a complementary strategy alongside immunotherapies. Fortunately, the crosstalk among necroptosis, ferroptosis, pyroptosis, and antitumor immunity has been emphasized and applied in immunotherapy.3–5 For example, Shao and coworkers reported that pyroptosis is a cytolytic death mode, in which released inflammatory cytokines trigger robust antitumor immunity.6,7Mitochondria were once misunderstood as bystanders of the oncogenic process, and it was not until recent years that the crosstalk between mitochondrial metabolism and tumorigenesis was gradually emphasized.8,9 Increasing evidence suggests that mitochondrial dysfunction augments cancer cellular emission of DAMPs.10,11 The latest study by Mangalhara et al. shows that enhanced tumor antigen presentation and T cell-mediated killing can be achieved by manipulating mitochondrial electron flow.12 Therefore, targeting mitochondrial vulnerabilities emerges as a promising therapy model for cancer therapy. Tsvetkov et al. discovered that some copper ionophores, like diethyldithiocarbamate, could bind lipoylated proteins of the (tricarboxylic acid) TCA cycle at mitochondria and induce cuproptosis in cancer cells.13 Our and other studies revealed that cuproptosis induced by copper(II) bis(diethyldithiocarbamate) (CuET), a complex produced by cupric ions and disulfiram, is a pro-inflammatory cytolytic death.14–17 However, the immunogenicity of cuproptosis has not been thoroughly studied yet. Therefore, we envision that mitochondrial copper delivery could enhance the degree of cuproptosis and induce a robust immune response for cancer therapy.
Herein, we developed a triphenylphosphonium (TPP)-modified CuET (TPP-CuET) for enhanced mitochondrial exposure to copper, thereby promoting robust cuproptosis (Scheme 1). Notably, the complex demonstrates significantly augmented mitochondrial copper distribution and induces substantial mitochondrial damage in 4T1 breast cancer cells when compared to the unmodified CuET. Importantly, the intense cuproptosis is highly immunogenic and capable of eliciting M1 polarization of macrophages. The underlying mechanisms of robust immunogenic cell death (ICD) and antitumor immune activity by TPP-CuET are further investigated by transcriptome analysis. This study highlights the potential of mitochondrial targeted cuproptosis inducers as a new approach for enhancing ICD in cancer treatment.
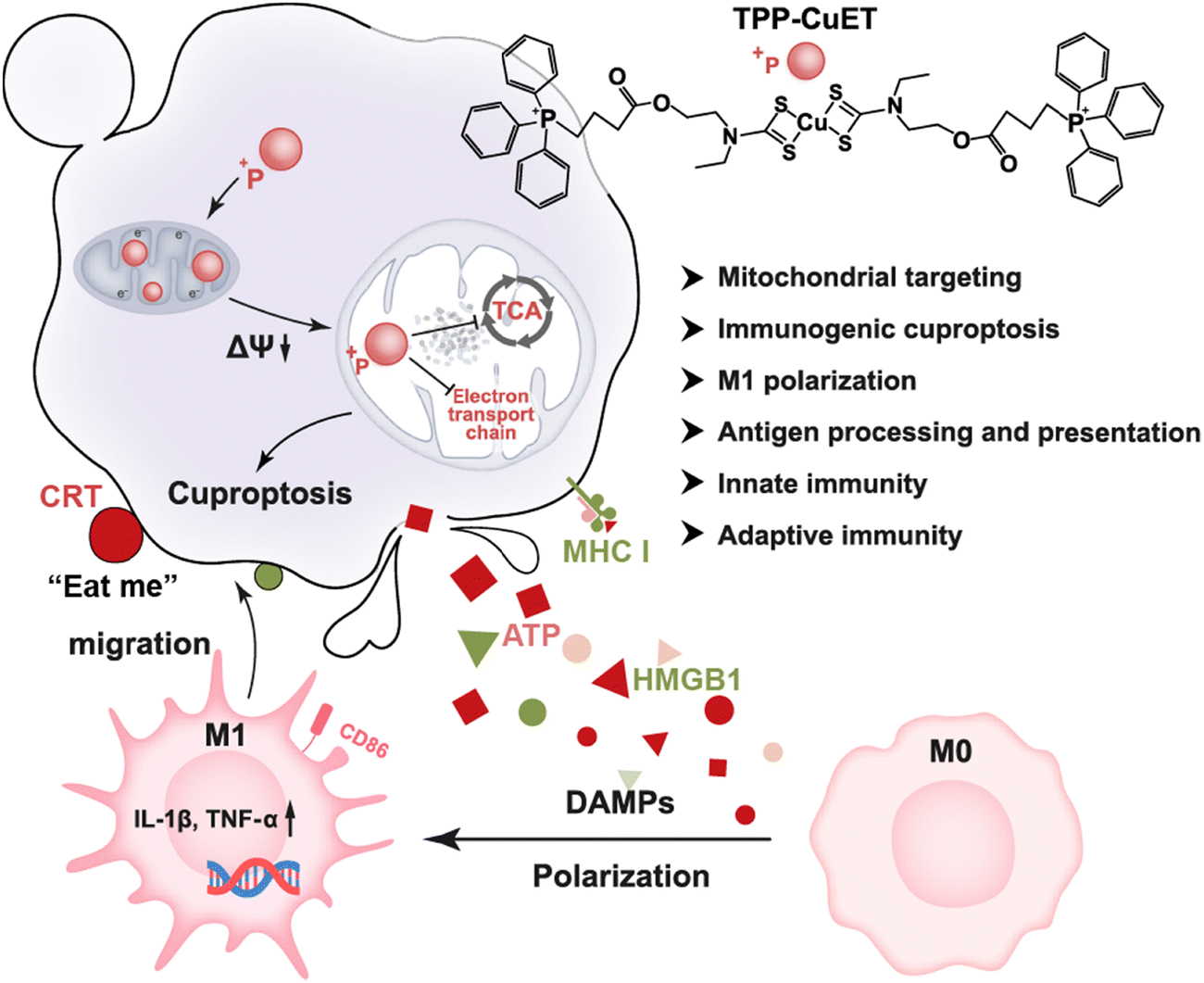 | ||
| Scheme 1 Schematic illustration of amplified immunogenic cuproptosis and macrophage M1 polarization by mitochondrial targeting TPP-CuET. | ||
Results and discussion
Design and synthesis of cuproptosis inducer TPP-CuET
Before designing a mitochondria-targeted copper complex, we verified that CuET could elicit cuproptosis, a distinct cellular response, as opposed to pyroptosis induced by metal ion overload18,19 or the more common apoptotic pathway, within 4T1 breast cancer cells owing to our interest in breast cancer, which is the number one malignant tumor in the world.20 We first studied the viability of cells treated with dimethyl fumarate, 3,4-methylenedioxy-β-nitrostyrene (MNS)/Tranilast, VX-765, and lenalidomide, which are pyroptosis inhibitors targeting GSDMD, NLRP3 protein, caspase-1, and pyroptosis regulator molecules, respectively.21 Our findings indicated that none of these inhibitors were capable of rescuing cells from the lethality induced by CuET, effectively ruling out the possibility of CuET-triggered pyroptosis (Fig. 1A). Similar outcomes were observed when employing inhibitors of necrosis (Nec-1) and ferroptosis (Fer-1). In contrast, the introduction of the mitochondrial pyruvate uptake inhibitor UK-509922 demonstrated a mitigating effect on cellular toxicity. Collectively, these results provide compelling evidence for the induction of mitochondrial respiration-dependent cuproptosis by CuET in cancer cells.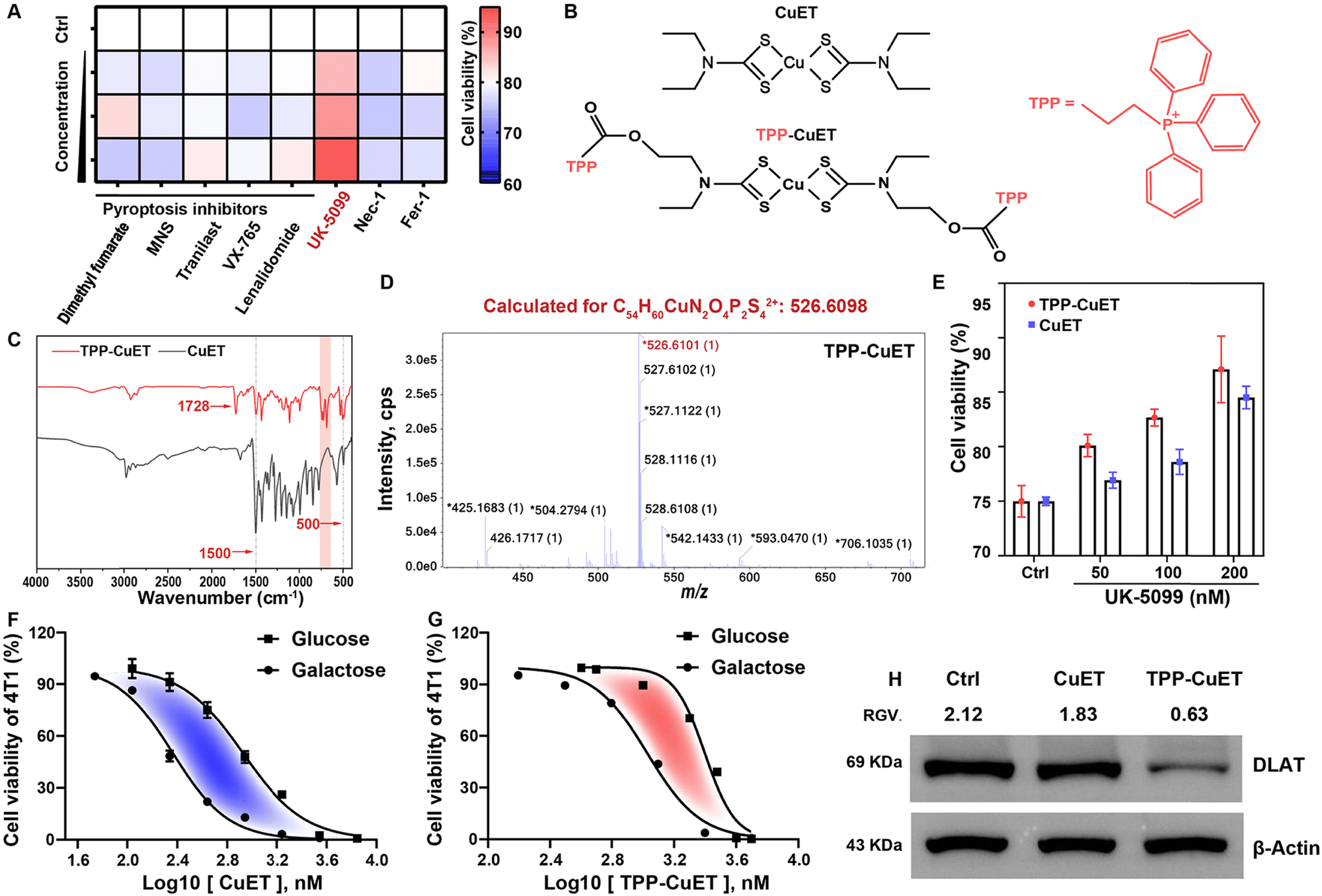 | ||
| Fig. 1 Design of mitochondrial targeted TPP-CuET as a robust cuproptosis inducer. (A) Heatmap of viability of 4T1 breast cancer cells pretreated with various death inhibitors of different concentrations and then treated with 250 nM CuET. (B) Chemical structures of CuET and TPP-CuET. (C) FT-IR spectrum and (D) high-resolution mass spectrum of TPP-CuET. (E) Viability of 4T1 cells subsequently treated with UK-5099 and 2 μM TPP-CuET. Anticancer efficacy of (F) CuET and (G) TPP-CuET against 4T1 cells in media containing glucose or galactose (n = 4, mean ± S.D.). (H) Immunoblotting and relative gray values (RGVs) of DLAT in 4T1 cells treated with TPP-CuET, wherein β-Actin was selected as the loading control. | ||
Next, we designed a mitochondrial-targeting CuET, TPP-CuET (Fig. 1B), to enhance the mitochondrial copper accumulation and induce intense cuproptosis in cancer cells since mitochondria are the sites where cuproptosis occurs. As shown in Scheme S1, the synthesis of TPP-CuET involved the conjugation of a well-known mitochondrial-targeting cationic lipophilic moiety, triphenylphosphonium,23 to a hydroxyl-derived CuET (HO-CuET). Specifically, HO-CuET was first synthesized by reacting 2-(ethylamino)ethanol with carbon disulfide under alkaline conditions, followed by cupric ion complexation. TPP-CuET was then obtained through an esterification reaction between 3-carboxypropyl triphenylphosphonium bromide and HO-CuET. 1H-NMR spectra (Fig. S1, ESI†) cannot fully characterize the structure of copper coordination compounds owing to the paramagnetic property,24 and other characterization methods were utilized in this work. The FT-IR spectra of CuET and TPP-CuET (Fig. 1C) showed the presence of v(N-CSS) at 1500 cm−1 and vs(CSS) at ∼500 cm−1. In TPP-CuET, the absorption peak at 1728 cm−1 was ascribed to the ester bond vibration, while the absorption peak in the range of 900–650 cm−1 belonged to the out-of-plane bending vibration of C–H on the benzene ring. Mass spectra analysis manifested that HO-CuET and TPP-CuET had m/z values of 390.9695 and 526.6101, respectively, which matched well with the calculated structures (Fig. S2, ESI† and Fig. 1D). Furthermore, CuET, OH-CuET, and TPP-CuET exhibited strong UV-vis absorption at 435 nm (Fig. S3, ESI†), which was ascribed to the ligand-to-metal charge transfer and d–d transition of Cu(II).25 These results demonstrated the successful synthesis of TPP-modified CuET without affecting the coordination structure.
The cytotoxicity of TPP-CuET was assessed by an MTT assay on a panel of cancer cell lines. As shown in Fig. S4 (ESI†), TPP-CuET exhibited a broad-spectrum anticancer activity in B16, CT26, and HepG2 cells, with IC50 values falling below 3 μM. In comparison to CuET, TPP-CuET exhibited a marginal reduction and maintained a considerable cytotoxicity against 4T1 cells (Fig. S5, ESI†). The slightly reduced cytotoxicity of TPP-CuET was probably due to the increased hydrophilicity (Fig. S6, ESI†), as confirmed by a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Po/w value of 1.55 ± 0.02, which is perfectly within the desirable range for drug development (0 to 4),26,27 whereas CuET is beyond the applicable range of logPo/w.28 Notably, TPP-CuET elicited cuproptosis in 4T1 cells with a mitochondrial respiratory dependency similar to CuET, which could be mitigated by UK-5099 (Fig. 1E). It is worth noting that substituting glucose with galactose in the cell culture medium has been observed to shift the cellular energy metabolism from glycolysis to mitochondrial oxidative phosphorylation.29,30 In this context, 4T1 cells relying on mitochondrial respiration were more vulnerable to TPP-CuET compared to those dependent on glycolysis, confirming that TPP-CuET induces cuproptosis with a reliance on mitochondrial metabolism (Fig. 1F and G). Furthermore, TPP-CuET treatment resulted in a decrease in the level of DLAT protein, further confirming the induction of mitochondrial respiratory-dependent cuproptosis in 4T1 cells (Fig. 1H).
Po/w value of 1.55 ± 0.02, which is perfectly within the desirable range for drug development (0 to 4),26,27 whereas CuET is beyond the applicable range of logPo/w.28 Notably, TPP-CuET elicited cuproptosis in 4T1 cells with a mitochondrial respiratory dependency similar to CuET, which could be mitigated by UK-5099 (Fig. 1E). It is worth noting that substituting glucose with galactose in the cell culture medium has been observed to shift the cellular energy metabolism from glycolysis to mitochondrial oxidative phosphorylation.29,30 In this context, 4T1 cells relying on mitochondrial respiration were more vulnerable to TPP-CuET compared to those dependent on glycolysis, confirming that TPP-CuET induces cuproptosis with a reliance on mitochondrial metabolism (Fig. 1F and G). Furthermore, TPP-CuET treatment resulted in a decrease in the level of DLAT protein, further confirming the induction of mitochondrial respiratory-dependent cuproptosis in 4T1 cells (Fig. 1H).
Mitochondrial targeting delivery and damage
We then moved our attention to the mitochondrial accumulation and damage induced by TPP-CuET. Upon treatment with CuET and TPP-CuET, a 4.2-fold and 5.6-fold increase in whole-cell copper levels was observed, as quantified through ICP-MS analysis (Fig. 2A). The whole-cell copper level in TPP-CuET-treated cells was only 32.1% higher than those treated with CuET. However, the mitochondrial copper level in TPP-CuET-treated cells was nearly 197.3% as high as that in CuET-treated cells and a substantial 16.5 times that of untreated cells. This observation suggests that the TPP modification significantly alters the intracellular fate of the copper complex, enabling the preferential delivery of cuproptosis inducers to the mitochondria of cancer cells.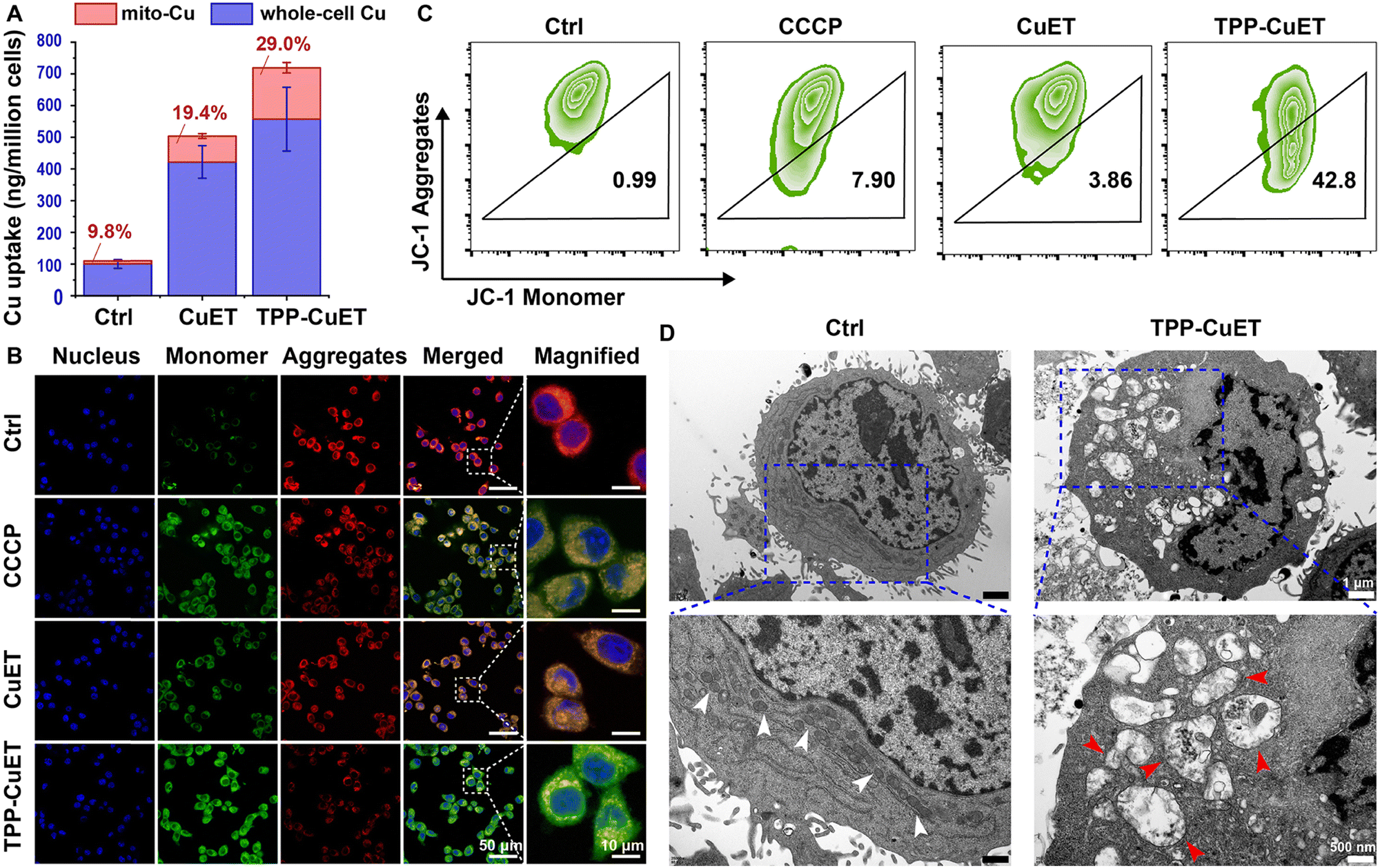 | ||
| Fig. 2 Mitochondrial copper uptake and mitochondrial damage after TPP-CuET treatment. (A) Whole-cell and mitochondrial copper levels of 4T1 cells treated with CuET or TPP-CuET for 12 h (n = 3, mean ± S.D.). (B) Representative CLSM images and (C) flow cytometry quantification of JC-1-labeled 4T1 cells treated with CCCP, CuET, or TPP-CuET. (D) Representative TEM images of 4T1 cells after different treatments for 24 h, wherein normal mitochondria are marked with white arrows and swollen mitochondria with red arrows. | ||
Mitochondrial membrane potential (MMP, ΔΨm) dissipation serves as a crucial indicator of mitochondrial damage. The MMP of CuET-treated 4T1 cells was studied by JC-1 staining using carbonyl cyanide 3-chlorophenylhydrazone (CCCP) as a positive control.31 In normal mitochondria, JC-1 aggregates emit red fluorescence, but when the MMP is dissipated, JC-1 transforms into monomers that emit green fluorescence. Confocal laser scanning microscopy (CLSM) revealed that TPP-CuET accumulation in mitochondria led to severe damage and the emission of green fluorescence, whereas CuET resulted in relatively mild damage to mitochondria (Fig. 2B). Flow cytometry analysis consistently indicated that TPP-CuET treatment severely disrupted MMP homeostasis (Fig. 2C), with 42.8% of cells displaying low MMP, a more substantial effect than CCCP (7.9%). Furthermore, transmission electron microscopy (TEM) provided an intuitive view of the damaged mitochondrial morphology upon TPP-CuET treatment (Fig. 2D), as indicated by the red arrows.32 This disruption included a reduction in the number of mitochondria, rupture and distortion of mitochondrial cristae, and the presence of mitochondria-derived vacuoles. In stark contrast, untreated 4T1 cells exhibited well-structured mitochondrial morphology with dense cristae membranes and a dark matrix, as denoted by the white arrows. In cells treated with CCCP and CuET, some mitochondria displayed a relatively intact morphology, but a considerable number exhibited deformed cristae (Fig. S7, ESI†).
Proinflammatory immunogenic cell death of cancer cells by TPP-CuET
The proinflammatory demise of cancer cells serves as a pivotal effector mechanism within the realm of innate immunity. The evaluation of cytoplasmic lactate dehydrogenase (LDH) release, as depicted in Fig. 3A, revealed that both TPP-CuET and CuET elicited LDH release in a concentration-dependent manner. Notably, TPP-CuET demonstrated a significantly higher LDH release at a lower concentration; with 2 μM CuET and TPP-CuET yielding 3.5% and 37.5% of LDH release, respectively. Annexin V-FITC and propidium iodide (PI) staining substantiated the lytic death of 4T1 cells induced by both copper compounds (Fig. 3B), with non-viable cells predominantly exhibiting double-positivity.33 Additionally, differential interference contrast imaging illustrated cellular swelling and foaming following CuET treatment (Fig. S8, ESI†), while TPP-CuET resulted in an increased number of burst cells, a distinct departure from the apoptotic morphology induced by hypoxia,34 characterized by intact cell membranes. TEM observations unveiled discontinuous cell boundaries and the release of cellular contents subsequent to TPP-CuET treatment (Fig. 3C). | ||
| Fig. 3 Characterization of inflammatory immunogenic cell death induced by TPP-CuET. (A) LDH release of 4T1 cells with different treatments. (B) Flow cytometry of annexin V-FITC/PI double-stained cells. (C) Representative TEM images of 4T1 cells after different treatments for 24 h. (D) Representative CLSM images for CRT exposure in 4T1 cells. (E) ATP release of 4T1 cells with different treatments (n = 4, mean ± S.D.). Statistical significance was determined by one-way ANOVA. ****P < 0.0001, **P < 0.01. (F) Representative CLSM images and (G) flow cytometry analysis of HMGB1 in 4T1 cells (n = 3, mean ± S.D.). | ||
Next, we assessed whether the lytic cuproptosis by TPP-CuET could induce immunogenic cell death (ICD), typified by the release of damage-associated molecular patterns (DAMPs). In the context of ICD, the endoplasmic reticulum-resident calreticulin (CRT) is translocated to the surface of the cell membrane, thereby conveying an “eat me” signal to macrophages or stimulating immature dendritic cells through receptor binding.35 As exemplified in Fig. 3D, cells treated with TPP-CuET emitted robust green fluorescence upon staining with an Alexa Fluor 488-conjugated CRT antibody, in stark contrast to the comparatively faint fluorescence observed in CuET and the positive control of chemotherapeutic doxorubicin (DOX)-treated cells.36,37 In the culture supernatant of 4T1 cells subjected to TPP-CuET treatment, significantly higher levels of ATP release were detected in comparison to DOX treatment (Fig. 3E). High-mobility group box 1 (HMGB1) represents another crucial marker of ICD, as it is released into the extracellular milieu to activate peripheral immune cells.38 Importantly, TPP-CuET led to a significant reduction in intracellular HMGB1 levels, as confirmed by CLSM and flow cytometry analysis (Fig. 3F and G). Taken together, the pro-inflammatory cuproptosis induced by CuET and TPP-CuET in 4T1 cells exhibits immunogenic characteristics, and the mitochondrial-targeting properties of TPP-CuET amplify its potential in promoting anti-tumor immunity.
In vitro macrophage polarization
DAMPs and other immunostimulatory factors released by cancer cells succumbing to ICD favor the crosstalk between dying cancer cells and innate immune cells, such as macrophages. This interaction serves to initiate an adaptive immune response that holds therapeutic significance.39 A recent investigation by Solier et al. highlighted the central involvement of mitochondrial copper in the activation of inflammatory macrophages.40 In light of this, our research delves into the response of tumor-associated macrophages (TAMs) to the release of immunogenic substances during cancer cell death.As illustrated in Fig. 4A, we collected conditioned medium devoid of drugs, containing DAMPs released by 4T1 cells following various treatments, and employed it to culture RAW 264.7 macrophages. The administration of CuET and TPP-CuET-induced DAMPs led to the reinvigoration of macrophages, resulting in 43.2% and 52.2% populations of CD86+ M1 phenotype cells, respectively (Fig. 4C). Unlike the positive control lipopolysaccharide (LPS), we distinctly observed the peaks of CD86+ macrophages using flow cytometry (Fig. 4B). In a transwell model, the conditioned medium from each group was placed in the lower chamber and RAW 264.7 macrophages were inoculated in the upper chamber to explore the migratory capacity of macrophages after immune stimulation. Micrographic examination revealed that, in comparison to CuET, TPP-CuET treatment attracted more macrophages to migrate through the polycarbonate membrane of the upper chamber towards the lower chamber (Fig. 4D and E). Although LPS induced 59.5% of macrophage M1 polarization, these cells displayed relatively limited antigen responsiveness and exhibited scarce downward migration. Furthermore, we assessed the IL-1β and TNF-α mRNA expression levels in RAW 264.7 cells after various treatments using RT-qPCR measurements. IL-1β holds significant importance in antigen-specific CD8+ T cell-mediated antitumor immune responses.41 The DAMPs derived from TPP-CuET treatment exhibited comparable efficacy to LPS, resulting in a substantial increase in IL-1β mRNA levels by several hundred times (Fig. 4F), potentially attributed to mitochondrial dysfunction.42 Meanwhile, the expression of TNF-α mRNA was significantly up-regulated under the treatment of two copper ICD inducers compared to LPS treatment (Fig. 4G). Overall, the stimulation of TAMs by DAMPs released during the immunogenic death process, particularly induced by mitochondrial-targeting cuproptosis initiator TPP-CuET, was very intense. M1-polarized macrophages actively migrated towards target cell antigens and secreted relevant cytokines, thereby enhancing their participation in anti-tumor immunity.
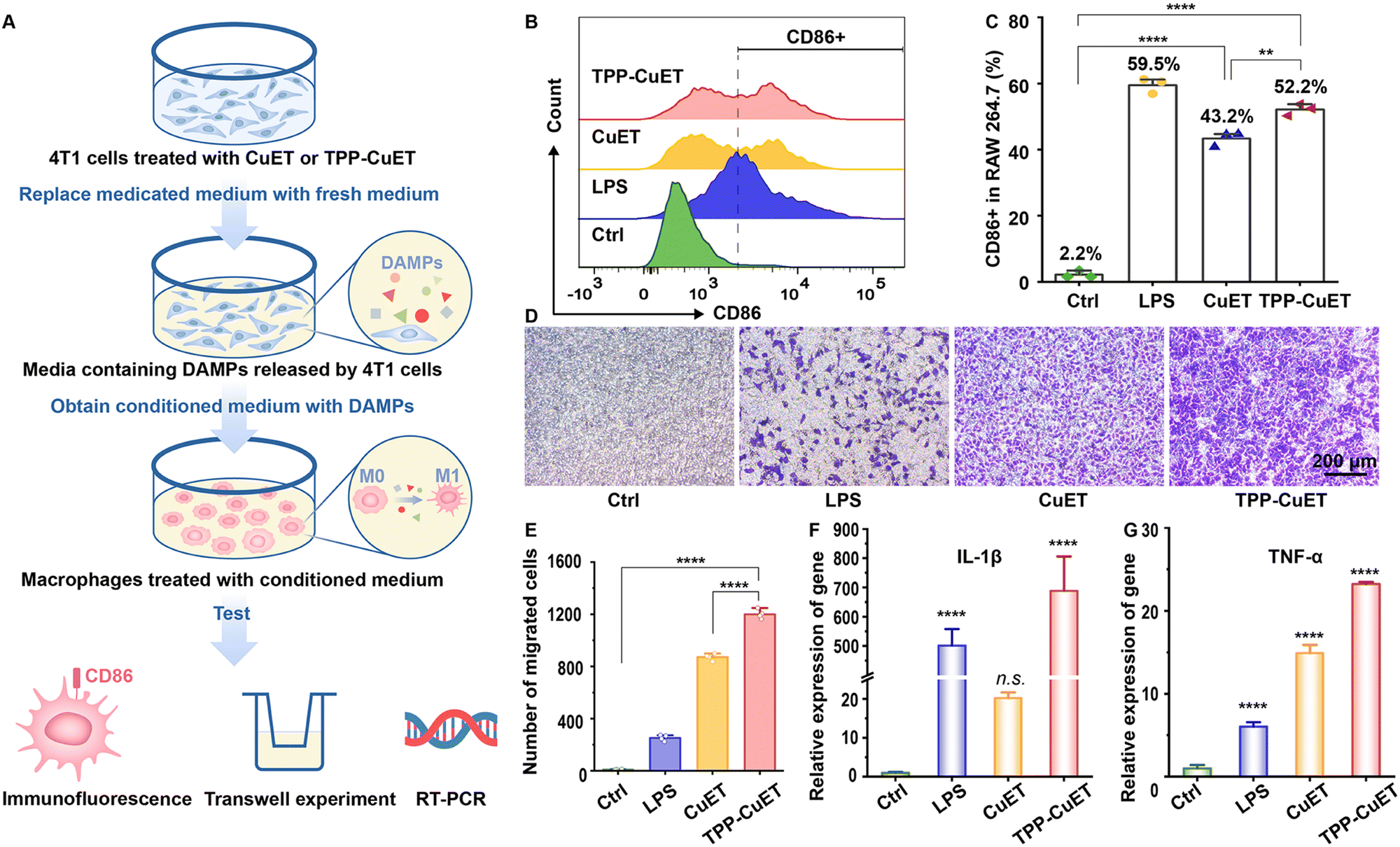 | ||
| Fig. 4 In vitro immune responses of RAW 264.7 cells after various stimulations. (A) Schematic diagram of polarization experiment operation of RAW 264.7 cells in conditioned medium. (B) Representative flow cytometric analysis and (C) relative quantification of M1 macrophages (CD86+). (D) Representative microscopic photos and (E) cell count statistics of macrophages migrating to the Transwell upper chamber after different treatments (n = 4, mean ± S.D.). Relative gene expression of (F) IL-1β and (G) TNF-α in RAW 264.7 cells treated with different conditioned medium or LPS as a positive control (n = 3, mean ± S.D.). Statistical significance was determined by one-way ANOVA. **P < 0.01, ****P < 0.0001. n.s.: not significant. | ||
Transcriptome analysis of antitumor mechanisms
To comprehensively elucidate the anticancer mechanisms of TPP-CuET, we conducted transcriptome analysis to identify differentially expressed genes (DEGs) in 4T1 cells following various treatments. Principal component analysis (PCA) affirmed the reproducibility of samples within the treatment groups (Fig. 5A). Notably, when compared to CuET, TPP-CuET exhibited a longer clustering distance from the control group, signifying a greater degree of differential gene expression. TPP-CuET led to 3391 upregulated or downregulated DEGs in 4T1 cells (Fig. S9A, ESI†), of which 1188 were unique genes. A Venn diagram depicted an overlap of 1941 DEGs due to the structural similarity and 508 unique DEGs between TPP-CuET and CuET (Fig. S9B, ESI†). Among the DEGs influenced by TPP-CuET and CuET treatments (Fig. 5B and Fig. S10A, ESI†), we observed the presence of genes associated with cuproptosis, such as Lipt113 and Rfesd that encodes the Rieske (Fe–S) domain-containing protein. Additionally, numerous innate immune-related genes, including Hspa1a (i.e. HSP70), Hspa1b, Trim15, etc., were significantly upregulated. In contrast to CuET, TPP-CuET notably suppressed a considerable number of genes related to the TCA cycle and respiratory electron transport (Fig. S10B, ESI†), including COX1/2/3, ATP6/8, DN1/2/3, etc., while concurrently upregulating genes associated with both innate and adaptive immunity, such as Prdm1, Mmp25, Siglec15, Blnk, Pik3r3, and Lag3. | ||
| Fig. 5 Transcriptome sequencing analysis of untreated or CuET/TPP-CuET treated 4T1 cells; TPP-CuET was abbreviated as T-Cu in this figure. (A) Principal component analysis of samples from each experimental group. (B) Volcano plots of DEGs in TPP-CuET treated 4T1 cells compared to the control. (C) GO enrichment of the top 30 downregulated genes and corresponding terms of TPP-CuET compared to CuET. (D) KEGG map of the MHC I antigen processing and presentation pathway of the TPP-CuET group. Pink indicates the upregulation of genes, while green indicates both upregulation and downregulation of corresponding genes. (E) Negative enrichment of the TCA cycle in TPP-CuET treated 4T1 cells compared to CuET via GSEA. | ||
To further unravel the functional implications of these DEGs, we employed the hypergeometric distribution algorithm to conduct Gene Ontology (GO), KEGG pathway, and Reactome enrichment analyses. Given the mitochondrial targeting property of TPP-CuET, numerous pathways associated with mitochondrial function and structure were prominently suppressed among the top 30 down-regulated items in GO enrichment analysis (Fig. 5C) and the top 10 down-regulated items in Reactome enrichment analysis (Fig. S11, ESI†), including aspects like mitochondria cristae formation and mitochondrial electron transport. The KEGG pathway analysis revealed that, in comparison to CuET, TPP-CuET upregulated more genes involved in the major histocompatibility complex (MHC) class I pathway, thereby promoting antigen processing and presentation to CD8 T cells and natural killer cells (Fig. 5D and Fig. S12, ESI†). Notably, the substantial downregulation of the Sdhaf1 gene (Fig. 5B), which plays a role in the assembly of mitochondrial respiratory chain complex II, aligns with recent research suggesting that complex II deficiency enhances antigen presentation and T cell-mediated killing.12 In addition, Gene Set Enrichment Analysis (GSEA) demonstrated that TPP-CuET inhibited the cuproptosis target TCA cycle (Fig. 5E), disrupted the mitochondrial inner membrane, and impeded the electron transport chain, thereby substantiating its impact on mitochondrial functions (Fig. S13, ESI†). Both CuET and TPP-CuET significantly upregulated the protein-coding genes involved in antigen processing and presentation (Fig. S14, ESI†). Overall, the transcriptome analysis unveils that cuproptosis induced by mitochondrial-targeting TPP-CuET severely disrupts mitochondrial metabolism while concurrently stimulating both innate and adaptive immune responses in the host.
Conclusions
In summary, we developed a TPP-modified CuET aimed at intensifying copper's interaction with mitochondria and promoting robust immunogenic cuproptosis. Notably, our developed copper complex exhibited a remarkable ability to enhance metal accumulation within mitochondria and induce mitochondrial metabolic blockade in 4T1 breast cancer cells. This heightened mitochondrial interaction led to a strong immunogenic cell death, M1 polarization of macrophages, and stimulation of innate and adaptive immunity, indicating a potent immunostimulatory response. Further research is needed to improve the cell uptake efficiency and tumor targeting in vivo. Overall, our findings uncover the antitumor immune efficacy of cuproptosis, underscoring the potential of cuproptosis as a valuable tool in the fight against cancer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the financial support by the National Natural Science Foundation of China (51903172). We would like to thank the Analytical & Testing Center of Sichuan University for FT-IR (Dr Yani Xie) and ICP-MS (Dr Xi Wu) work. Thanks go to the Center of Growth, Metabolism and Aging, College of Life Sciences, Sichuan University, for flow cytometry analysis (BD FACScalibur and BD LSRfortessa). The kind assistance of Chengdu Lilai Biotechnology Co., Ltd is appreciated.References
- L. Campisi, R. J. Cummings and J. M. Blander, Am. J. Transplant., 2014, 14, 1488–1498 CrossRef CAS PubMed.
- G. Wang, B. Li, H. Tian, L. Xie, J. Yan, W. Sang, J. Li, Z. Zhang, W. Li and Y. Dai, Adv. Funct. Mater., 2023, 33, 2213425 CrossRef CAS.
- R. Tang, J. Xu, B. Zhang, J. Liu, C. Liang, J. Hua, Q. Meng, X. Yu and S. Shi, J. Hematol. Oncol., 2020, 13, 110 CrossRef PubMed.
- H. Li, J. Sun, H. Zhu, H. Wu, H. Zhang, Z. Gu and K. Luo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2021, 13, e1670 CAS.
- H. Li, Y. Feng, Q. Luo, Z. Li, X. Li, H. Gan, Z. Gu, Q. Gong and K. Luo, Theranostics, 2023, 13, 5386–5417 CrossRef CAS PubMed.
- Z. Zhou, H. He, K. Wang, X. Shi, Y. Wang, Y. Su, Y. Wang, D. Li, W. Liu, Y. Zhang, L. Shen, W. Han, L. Shen, J. Ding and F. Shao, Science, 2020, 368, eaaz7548 CrossRef CAS PubMed.
- Q. Wang, Y. Wang, J. Ding, C. Wang, X. Zhou, W. Gao, H. Huang, F. Shao and Z. Liu, Nature, 2020, 579, 421–426 CrossRef CAS PubMed.
- S. E. Weinberg and N. S. Chandel, Nat. Chem. Biol., 2015, 11, 9–15 CrossRef CAS PubMed.
- L. Sainero-Alcolado, J. Liaño-Pons, M. V. Ruiz-Pérez and M. Arsenian-Henriksson, Cell Death Differ., 2022, 29, 1304–1317 CrossRef CAS PubMed.
- Y. Liu, Z. Zhou, J. Hou, W. Xiong, H. Kim, J. Chen, C. Zheng, X. Jiang, J. Yoon and J. Shen, Adv. Mater., 2022, 34, 2206121 CrossRef CAS PubMed.
- J. Yan, W. Li, H. Tian, B. Li, X. Yu, G. Wang, W. Sang and Y. Dai, ACS Nano, 2023, 17, 14667–14677 CrossRef CAS PubMed.
- K. C. Mangalhara, S. K. Varanasi, M. A. Johnson, M. J. Burns, G. R. Rojas, P. B. Esparza Moltó, A. G. Sainz, N. Tadepalle, K. L. Abbott, G. Mendiratta, D. Chen, Y. Farsakoglu, T. Kunchok, F. A. Hoffmann, B. Parisi, M. Rincon, M. G. Vander Heiden, M. Bosenberg, D. C. Hargreaves, S. M. Kaech and G. S. Shadel, Science, 2023, 381, 1316–1323 CrossRef CAS PubMed.
- P. Tsvetkov, S. Coy, B. Petrova, M. Dreishpoon, A. Verma, M. Abdusamad, J. Rossen, L. Joesch-Cohen, R. Humeidi, R. D. Spangler, J. K. Eaton, E. Frenkel, M. Kocak, S. M. Corsello, S. Lutsenko, N. Kanarek, S. Santagata and T. R. Golub, Science, 2022, 375, 1254–1261 CrossRef CAS PubMed.
- Y. Lu, Q. Pan, W. Gao, Y. Pu and B. He, J. Mater. Chem. B, 2022, 6296–6306, 10.1039/D2TB01150F.
- Y. Lu, Q. Pan, W. Gao, Y. Pu, K. Luo, B. He and Z. Gu, Biomaterials, 2022, 281, 121335 CrossRef CAS PubMed.
- W. Chen, W. Yang, P. Chen, Y. Huang and F. Li, ACS Appl. Mater. Interfaces, 2018, 10, 41118–41128 CrossRef CAS PubMed.
- Q. Pan, B. Zhang, X. Peng, S. Wan, K. Luo, W. Gao, Y. Pu and B. He, Chem. Commun., 2019, 55, 13896–13899 RSC.
- P. Zheng, B. Ding, G. Zhu, C. Li and J. Lin, Angew. Chem., Int. Ed., 2022, e202204904, DOI:10.1002/anie.202204904.
- B. Ding, J. Sheng, P. Zheng, C. Li, D. Li, Z. Cheng, P. Ma and J. Lin, Nano Lett., 2021, 21, 8281–8289 CrossRef CAS PubMed.
- Y. Xu, M. Gong, Y. Wang, Y. Yang, S. Liu and Q. Zeng, Sci. Data, 2023, 10, 334 CrossRef PubMed.
- N. Li, Y. Wang, X. Wang, N. Sun and Y. H. Gong, Pharmacol. Res., 2022, 175, 106033 CrossRef CAS PubMed.
- D. Tang, X. Chen and G. Kroemer, Cell Res., 2022, 32, 417–418 CrossRef PubMed.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- N. Pettenuzzo, L. Brustolin, E. Coltri, A. Gambalunga, F. Chiara, A. Trevisan, B. Biondi, C. Nardon and D. Fregona, ChemMedChem, 2019, 14, 1162–1172 CrossRef CAS PubMed.
- Q. Pan, X. Peng, J.-E. Cun, J. Li, Y. Pu and B. He, Chem. Eng. J., 2021, 409, 128222 CrossRef CAS.
- M. Yazdanian, Curr. Protoc. Pharmacol., 2013, 60, 9–19 Search PubMed.
- J. A. Arnott and S. L. Planey, Expert Opin. Drug Discovery, 2012, 7, 863–875 CrossRef CAS PubMed.
- A. Noble, J. Chromatogr., 1993, 642, 3–14 CrossRef CAS.
- R. A. Skolik, J. Solocinski, M. E. Konkle, N. Chakraborty and M. A. Menze, Am. J. Physiol.: Cell Physiol., 2021, 320, C778–C793 CrossRef CAS PubMed.
- D. Grimm, L. Altamirano, S. Paudel, L. Welker, M. E. Konkle, N. Chakraborty and M. A. Menze, Cell Tissue Res., 2017, 369, 641–646 CrossRef CAS PubMed.
- W. Zhou, X. Wang, M. Hu, C. Zhu and Z. Guo, Chem. Sci., 2014, 5, 2761–2770 RSC.
- X. Zhang, Y. Wu, Z. Li, W. Wang, Y. Wu, D. Pan, Z. Gu, R. Sheng, H. Tomás, H. Zhang, J. Rodrigues, Q. Gong and K. Luo, Carbohydr. Polym., 2020, 247, 116749 CrossRef CAS PubMed.
- Y. Wang, W. Gao, X. Shi, J. Ding, W. Liu, H. He, K. Wang and F. Shao, Nature, 2017, 547, 99–103 CrossRef CAS PubMed.
- M. Weinmann, V. Jendrossek, R. Handrick, D. Güner, B. Goecke and C. Belka, Oncogene, 2004, 23, 3757–3769 CrossRef CAS PubMed.
- T. Panaretakis, O. Kepp, U. Brockmeier, A. Tesniere, A.-C. Bjorklund, D. C. Chapman, M. Durchschlag, N. Joza, G. Pierron, P. van Endert, J. Yuan, L. Zitvogel, F. Madeo, D. B. Williams and G. Kroemer, EMBO J., 2009, 28, 578–590 CrossRef CAS PubMed.
- Y. Ou, K. Chen, H. Cai, H. Zhang, Q. Gong, J. Wang, W. Chen and K. Luo, Biomater. Sci., 2018, 6, 1177–1188 RSC.
- W. Zhu, Y. Bai, N. Zhang, J. Yan, J. Chen, Z. He, Q. Sun, Y. Pu, B. He and X. Ye, J. Mater. Chem. B, 2021, 9, 4201–4210 RSC.
- L. Zhang, N. Montesdeoca, J. Karges and H. Xiao, Angew. Chem., Int. Ed., 2023, e202300662, DOI:10.1002/anie.202300662.
- A. D. Garg, L. Galluzzi, L. Apetoh, T. Baert, R. B. Birge, J. M. Bravo-San Pedro, K. Breckpot, D. Brough, R. Chaurio, M. Cirone, A. Coosemans, P. G. Coulie, D. De Ruysscher, L. Dini, P. de Witte, A. M. Dudek-Peric, A. Faggioni, J. Fucikova, U. S. Gaipl, J. Golab, M. L. Gougeon, M. R. Hamblin, A. Hemminki, M. Herrmann, J. W. Hodge, O. Kepp, G. Kroemer, D. V. Krysko, W. G. Land, F. Madeo, A. A. Manfredi, S. R. Mattarollo, C. Maueroder, N. Merendino, G. Multhoff, T. Pabst, J. E. Ricci, C. Riganti, E. Romano, N. Rufo, M. J. Smyth, J. Sonnemann, R. Spisek, J. Stagg, E. Vacchelli, P. Vandenabeele, L. Vandenberk, B. J. Van den Eynde, S. Van Gool, F. Velotti, L. Zitvogel and P. Agostinis, Front. Immunol., 2015, 6, 588 Search PubMed.
- S. Solier, S. Muller, T. Caneque, A. Versini, A. Mansart, F. Sindikubwabo, L. Baron, L. Emam, P. Gestraud, G. D. Pantos, V. Gandon, C. Gaillet, T. D. Wu, F. Dingli, D. Loew, S. Baulande, S. Durand, V. Sencio, C. Robil, F. Trottein, D. Pericat, E. Naser, C. Cougoule, E. Meunier, A. L. Begue, H. Salmon, N. Manel, A. Puisieux, S. Watson, M. A. Dawson, N. Servant, G. Kroemer, D. Annane and R. Rodriguez, Nature, 2023, 617, 386–394 CrossRef CAS PubMed.
- F. Ghiringhelli, L. Apetoh, A. Tesniere, L. Aymeric, Y. Ma, C. Ortiz, K. Vermaelen, T. Panaretakis, G. Mignot, E. Ullrich, J.-L. Perfettini, F. Schlemmer, E. Tasdemir, M. Uhl, P. Génin, A. Civas, B. Ryffel, J. Kanellopoulos, J. Tschopp, F. André, R. Lidereau, N. M. McLaughlin, N. M. Haynes, M. J. Smyth, G. Kroemer and L. Zitvogel, Nat. Med., 2009, 15, 1170–1178 CrossRef CAS PubMed.
- K. Shimada, Timothy R. Crother, J. Karlin, J. Dagvadorj, N. Chiba, S. Chen, V. K. Ramanujan, Andrea J. Wolf, L. Vergnes, David M. Ojcius, A. Rentsendorj, M. Vargas, C. Guerrero, Y. Wang, Katherine A. Fitzgerald, David M. Underhill, T. Town and M. Arditi, Immunity, 2012, 36, 401–414 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb02886k |
| This journal is © The Royal Society of Chemistry 2024 |