
Design of functionalized luminescent MOF sensor for the precise monitoring of tuberculosis drug and neonicotinoid pesticide from human body-fluids and food samples to protect health and environment†
Abhijeet
Rana
a,
Nazir Ud Din
Mir
a,
Arpa
Banik
b,
Ananya
Hazra
a and
Shyam
Biswas
 *a
*a
aDepartment of Chemistry, Indian Institute of Technology Guwahati, Guwahati, Assam 781039, India. E-mail: sbiswas@iitg.ac.in
bDepartment of Chemistry, Indian Institute of Science Educational and Research, Berhampur, 760003, Odisha, India
First published on 6th December 2023
Abstract
Selectively detecting pharmaceutical drugs and pesticides from drinking water and environmental water sources with an ultralow detection limit by a simple handling method remains challenging. To circumvent such challenges, we prepared an aluminum metal–organic framework (MOF) using 2-(((2-hydroxy naphthalene-1-yl)methyl)amino)terephthalic acid (H2L) linker. We explored the potential of the guest-free MOF for the fluorescence detection of a broadly utilized neonicotinoid category of pesticide called nitenpyram and a very common antibiotic drug used for curing tuberculosis, namely rifampicin. It is the first MOF-based fluorescent sensor capable of detecting nitenpyram and rifampicin by fluorescent turn-off mode. This probe has lower detection limits (LOD for rifampicin: 11.7 nM and nitenpyram: 13.8 nM) as compared to those of formerly reported probes for nitenpyram and rifampicin sensing. It can detect nitenpyram and rifampicin instantaneously (within 5 s). The ultrafast nature of this probe makes it superior to the previously reported time-consuming probes. This sensor material is recyclable five times for sensing nitenpyram and rifampicin, boosting its cost-effectiveness and sustainability. This chemically robust MOF material was utilized for the fluorometric detection of both analytes in drinking water and environmental water samples. The probe can also precisely quantify rifampicin from human blood serum and urine and nitenpyram from soil and food samples (rice and corn). The mechanistic aspects of the detection process have been unveiled with the help of systematic experimental techniques and molecular simulations. In addition, the repeatability and reproducibility of the probe to sense both analytes were demonstrated. The overall work presents an inexpensive and compatible multiple-usable photoluminescent sensor of nitenpyram and rifampicin to address real-world issues.
Introduction
The detection of pesticides has become increasingly significant in recent years due to their negative influence on non-targeted animals and humans. Nitenpyram ((E)-N-[(6-chloropyridin-3-yl)methyl]-N-ethyl-N′-methyl-2-nitroethene-1,1-diamine) is a neonicotinoid insecticide that is commonly used to protect crops from crop-destroying insects in agricultural fields and veterinary use to treat adult flea infestations.1 The Japanese Takeda Company invented this significant family of pyridine chemicals in 1989. It is very harmful to insects because it primarily inhibits the central nervous system of the target insect. It is highly water-soluble and environmentally stable, with a hydrolytic half-life of 415 days at pH 7. Therefore, it could easily be assimilated into the animal and human body through the food chain.2 Long-term exposure to this insecticide may negatively impact mammals (including humans), causing genotoxicity, neurotoxicity, and endocrine disruption.3 It can also cause environmental pollution.4 Nitenpyram exposure decreases the lifetimes of honey bees, one of the prime members of the natural pollination process. The decrease in the honey bee population directly affects the production rate of agricultural products.5 Therefore, monitoring such pesticides in the environment (soil, natural water, and food samples) is essential.Like pesticides, monitoring pharmaceutical drug waste in different environmental water specimens has also become important to stop environmental pollution. Rifampicin (RIF, IUPAC: 3-[(4-methyl-1-pyrazinyl)imino]methyl), an essential semi-synthetic antibacterial drug belonging to the rifamycin group, is used to cure tuberculosis, leprosy, inactive meningitis, cholestatic pruritus, and HIV.6 It inhibits the production of bacterial DNA-dependent RNA polymerase.7 Rifampicin was discovered in 1965 from the soil bacterium. Long-term use of rifampicin can cause adverse impacts on human health (hepatotoxicity, fever, allergic reactions) and the environment.8 The exposure of rifampicin to a healthy human can lead to the development of antibiotic resistance. The latter creates severe medical issues, as the bacteria will no longer be affected by the previous antibiotics.9 Dogs are also adversely affected by rifampicin, which causes hepatotoxicity.10 Therefore, detecting and monitoring such pharmaceutically active drugs are essential to protect the health of humans and animals.
The development of health science, crop protection, and agricultural research accelerated the production and consumption of drugs, pesticides, and insecticides. The overdoses of drugs and excess consumption of pesticides, herbicides, and insecticides cause their accumulation in water bodies. The excess concentration of such chemicals and drugs directly affects the health of plants, animals, and humans. Therefore, to minimize the toxicity caused by rifampicin and nitenpyram, it is very important to develop some analytical methods for detecting and determining the concentrations of these insecticides and drug compounds in drinking water and environmental water sources. The widely used methods to detect these artificial chemicals are gas/liquid chromatography,11,12 thin layer chromatography,13 high-performance liquid chromatography (HPLC),14 mass spectrometry,15,16 photometry, biological detection techniques, hyperpolarization, and Raman spectroscopy due to their sensitivity and precision.17 However, these methods are time-consuming and expensive. In contrast, with high sensitivity, less cost, and user compatibility, fluorescence sensing has become a more reliable way to detect nitenpyram and rifampicin.
Zhang et al. developed a La(III)/Ta(III) mixed-metal fluorescent MOF for the selective recognition of nitenpyram, but the probe has a higher detection limit (0.63 M).18 Yang et al., on the other hand, developed a functional Zr-MOF (FMOF) to select nitenpyram with a lower detection limit (0.11 μM).19 The presence of nitenpyram at lower concentrations (such as nanomolar) is also hazardous. While both the probes described by Zhang and Yang et al. have good selectivity, their probes are not sensitive enough to detect nanomolar concentrations of nitenpyram. On the other hand, the probe presented in this report can detect nanomolar concentrations of nitenpyram (LOD = 13.8 nM).
Cai et al. synthesized a folic acid-protected copper nanocluster (FA-CuNC) for rifampicin detection in methanol.20 Because of the high LOD (0.07 μM) and sensing in methanol (not a suitable sensing medium), the FA-CuNC's real-field application is limited. Chen et al. also synthesized gold nanoclusters for fluorescence detection of rifampicin in water. Although Chen et al. could overcome the issue of organic solvent and preferentially sense rifampicin in an aqueous medium, their probe has a high detection limit of 0.09 μM.21 To address the issue of organic solvent and sensitivity, we prepared an environmentally friendly functional Al-MOF capable of detecting rifampicin with nanomolar sensitivity in an aqueous medium.
Herein, we report a 2-(((2-hydroxy naphthalene-2-yl)methyl)amino)terephthalic acid (H2L linker) and Al(III) based metal–organic framework for the nanomolar level detection and monitoring of rifampicin and nitenpyram. The probe can detect both analytes (rifampicin and nitenpyram) selectively, even in the presence of their congeners, without significant interferences. The detection time for nitenpyram and rifampicin is only 5 s. The ultrafast nature of the probe towards detecting both analytes made it unique for real field applications as it could detect these analytes instantaneously. The MOF material can detect both analytes at very low concentrations. The LOD values for nitenpyram and rifampicin detection are 13.8 nM and 11.7 nM, respectively. These values are lower than those reported to date for nitenpyram and rifampicin sensors (Tables S11 and S12, ESI†). Our probe's low detection limit makes it advantageous over other reported sensors. This MOF can also detect rifampicin and nitenpyram from environmental water samples (lake, tap, river and distilled water) to safeguard human health and the environment. This luminescent MOF can also precisely quantify rifampicin from human blood serum and urine, and nitenpyram from soil and food samples.
Experimental section
Chemicals and general methods
Related reagents and instruments have been mentioned in the ESI.†Synthesis of [Al(OH)(L)]·0.5H2O (1)
A homogenous suspension of AlCl3·6H2O (56 mg, 0.15 mmol) and H2L linker (50 mg, 0.15 mmol) in water was prepared in a Teflon-lined autoclave by sonicating for 20 min. The uniform combination of metal salt and linker was then exposed to 150 °C for 24 h in a hot air oven. After 24 h, the autoclave was slowly cooled to room temperature, and then the obtained brown residue was washed thoroughly with water, DMF, and acetone and put in an oven for 24 h at 100 °C. The as-synthesized material was termed 1. The as-synthesized form of the material was then solvent-exchanged by stirring in methanol for 24 h and then vacuum dried at 120 °C for 24 h. The solvent-free vacant pore form of the material was termed 1′. The obtained yield was 45.3 g (77.8%). The calculated elemental % was C: 60.16%, N: 3.69, H: 3.72 and the obtained elemental microanalysis was C: 60.01%, N: 3.59, H: 3.64.Results and discussion
Structural elucidation of 1
The PXRD pattern of the as-synthesized MOF matches the PXRD profile of simulated MIL-53 MOF (Fig. S1, ESI†). Again, the Pawley fitting was performed by taking the PXRD pattern of the as-synthesized MOF compared to the crystal structure of previously reported pristine MOF. The obtained Pawley-fit is displayed in Fig. 1a, which suggests good agreement with the parent MOF with very low Rwp (2.6%) and Rp (1.8%) values. The parent MOF was first reported by Férey et al. It comprises an unfunctionalized terephthalic acid linker and Al(III) as the metal center.22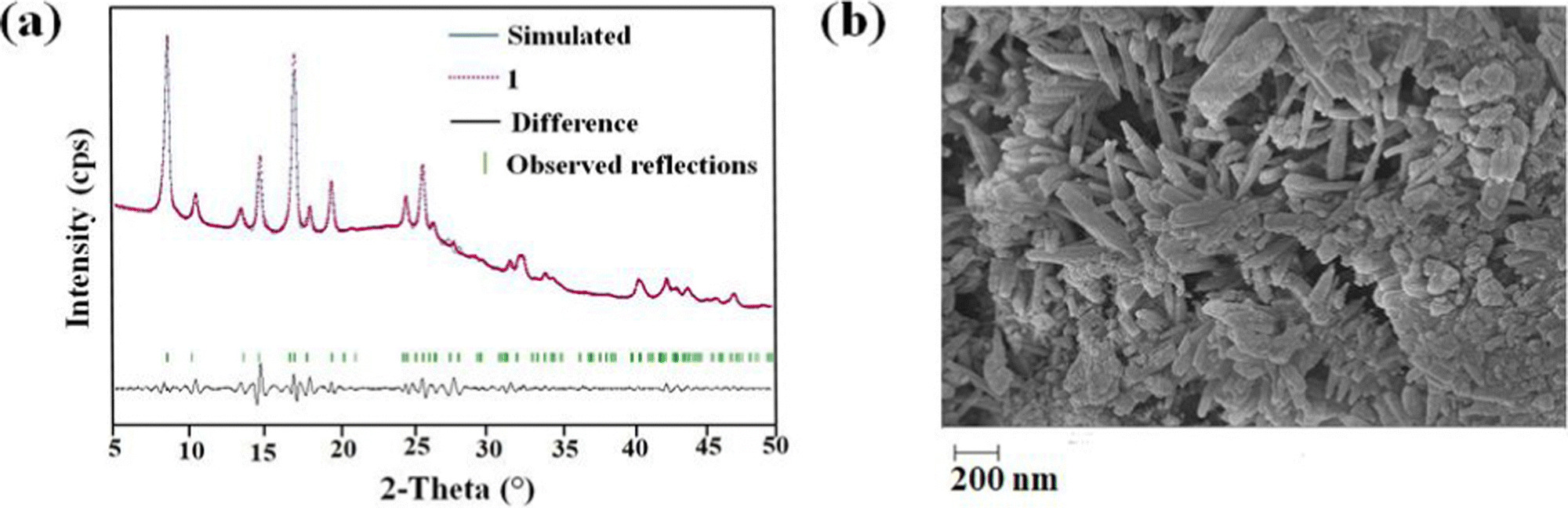 | ||
| Fig. 1 (a) Pawley refinement plot for 1′. (b) FE-SEM image of 1′. | ||
Again, the indexing of the slow scan PXRD profile of the synthesized MOF confirmed that all the PXRD peaks are due to the orthorhombic form of the parent MOF. The obtained indexing data also display the close resemblance of the cell parameters of the synthesized MOF with the parent MOF (Table S1, ESI†). The MOF possesses AlO6 octahedra consisting of four oxygen from the carboxylate of four different terephthalate linkers and two μ2-OH groups, and aluminum is the central metal atom. A chain of inorganic AlO6 octahedra is formed with the help of bridging hydroxy groups (Fig. 2b). The inorganic chain is connected using terephthalate linkers to give a 3D porous framework. The 3D framework possesses rhombic-shaped 1D channels, as shown in Fig. 2a. The porosity of the material was affected due to the bulky naphthalene group occupying the rhombic channels, reducing the material's surface area.
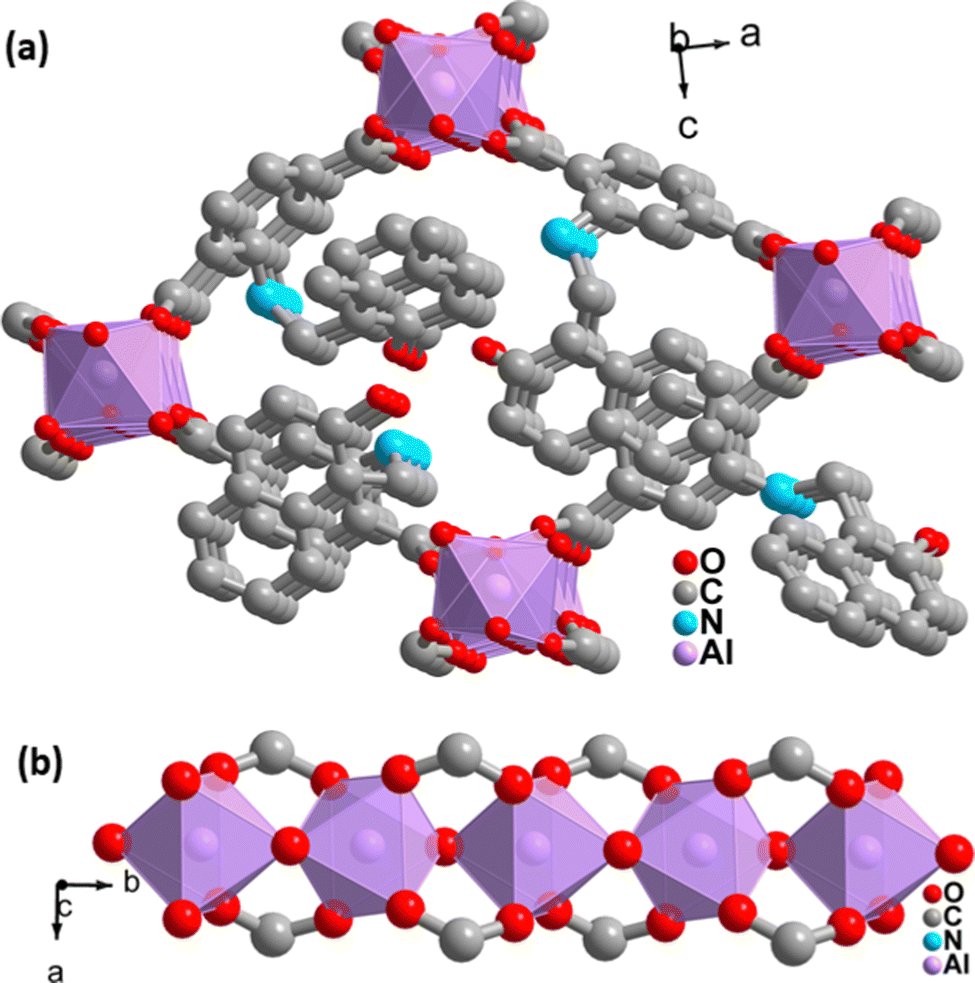 | ||
| Fig. 2 (a) 3D network structure of 1′. (b) 1D inorganic linear chain in the structure of 1′. | ||
Functional group investigation
ATR-IR measurements of 1 and 1′ revealed that the asymmetrical and symmetrical stretching vibrations of the –COO− group (1574 cm−1 and 1439 cm−1) are lower than those of the free carboxylic acid in the linker. The effective attachment of the –COO− group to the Al(III) ion aided in this shift of stretching frequencies. The amide carbonyl group of the adsorbed DMF molecules is responsible for the peak at 1656 cm−1 in the as-synthesized material (1).23 The identical peak, however, is absent in the activated form of material 1′, confirming the proper activation process (Fig. S2, ESI†).To confirm the binding of Al(III) ions with the carboxylate linkers, we performed an X-ray photoelectron spectroscopy (XPS) analysis. The Al 2p XPS spectrum (Fig. S5, ESI†) exhibited two peaks due to the Al–O bond with the carboxylate oxygen of the linker and the Al–O bond of the inorganic sub-units.24 In the O 1s spectrum, the Al-coordinated oxygen peak is at 532 eV. The absence of any peak above 533 eV in the O 1s spectrum confirmed that no free carboxylic acid linker remained in the MOF material.24
Compositional and morphological investigation of 1
The homogenous rod-shaped particles, as displayed in the FE-SEM images, confirmed the purity and homogeneity of 1′ (Fig. 1b). The presence of the necessary elements was confirmed by the EDX spectrum of 1′ (Fig. S3, ESI†) (C: 75.2, O: 15.2, N: 4.9, Al: 4.4%). The EDX elemental mapping validated the homogeneous distribution of desired elements all over the material (Fig. S4, ESI†).Examination of physicochemical stability
The thermal stability of 1 and 1′ in an oxygen environment was examined at a heating rate of 4 °C min−1 (Fig. S6, ESI†). The TG curve of 1 demonstrated a weight loss of 2.2% at 105 °C due to the loss of 0.5 water molecules per formula unit. After 360 °C, the dissociation of linkers from metal centers resulted in a sudden mass loss. Following that, we found an almost flat TG curve for the activated material (1′) up to 360 °C, indicating the absence of any solvent molecule in 1′. According to the findings, the activated and as-synthesized versions of the material are stable in an oxygen atmosphere up to 360 °C.The stability of 1′ in acidic and alkaline pH and organic solvents was demonstrated by stirring it in the respective solutions for 24 h.25 Afterward, it was filtered, and the PXRD pattern was recorded. The PXRD pattern of the recovered sample of 1′ was precisely similar to the activated form of the material, confirming the stability of the material in such chemically harsh conditions (Fig. S7, ESI†).
Investigation of the porosity of 1′
The high surface area and porosity of MOFs are critical properties for their application in various sectors. As a result, we conducted an N2 adsorption analysis of 1′ at −196 °C. The specific BET surface area and pore volume of 1′ were 50 m2 g−1 and 0.31 cm3 g−1 (at p/p0 = 0.5), respectively. The functionalization of MOF by a bulky 2-hydroxy naphthalene group reduces the available pores for the adsorption of N2 gas, which resulted in a decreased surface area of 1′ in comparison to parent MOF (Fig. S8, ESI†).26,27Photoluminescence (PL) detection of rifampicin
Rifampicin is a widely used tuberculosis medication. Excess biomedical and pharmaceutical use and unwanted disposal into the environment aid in the distribution of this drug in the natural environment. The presence of rifampicin in natural water sources may endanger the health of animals, including humans. Because of the above-mentioned issues, we synthesized a luminescent MOF probe in this study. By changing its light absorption and emission properties through functional tuning, we applied it for the selective detection of rifampicin.The MOF suspension was prepared by dispersing 10 mg of MOF in 10 mL of water, sonicating it for 1 h, and then leaving it undisturbed for 24 h to settle the surplus colloidal MOF particles. The stability of the MOF suspension was characterized using a dynamic light scattering (DLS) experiment in an aqueous medium. The obtained results (Fig. S9, ESI†) display that the MOF particles were present in the sensing medium in monodispersed form with an average particle size of 1500 nM.28,29 After obtaining a stable MOF suspension, 2800 μL of mili-Q water and 200 μL of the MOF suspension were used for all sensing studies. An aqueous solution of 1 mM rifampicin was prepared. The aqueous suspension of the probe was excited with fluorescence light at 330 nm, and the PL intensity was measured in a wavelength range of 350 to 600 nm for all sensing experiments (Fig. S10, ESI†).
Rifampicin sensing was accomplished by the gradual addition of 50 μL of 1 mM aqueous solution of rifampicin to a quartz cuvette containing 2800 μL of mili-Q water and 200 μL of MOF suspension. The PL emission intensity was measured after each progressive augmentation. The probe's PL emission intensity was suppressed after each addition step, and saturation in the PL emission intensity was observed (93% quenching) after adding 300 μL of 1 mM rifampicin (Fig. 3a).
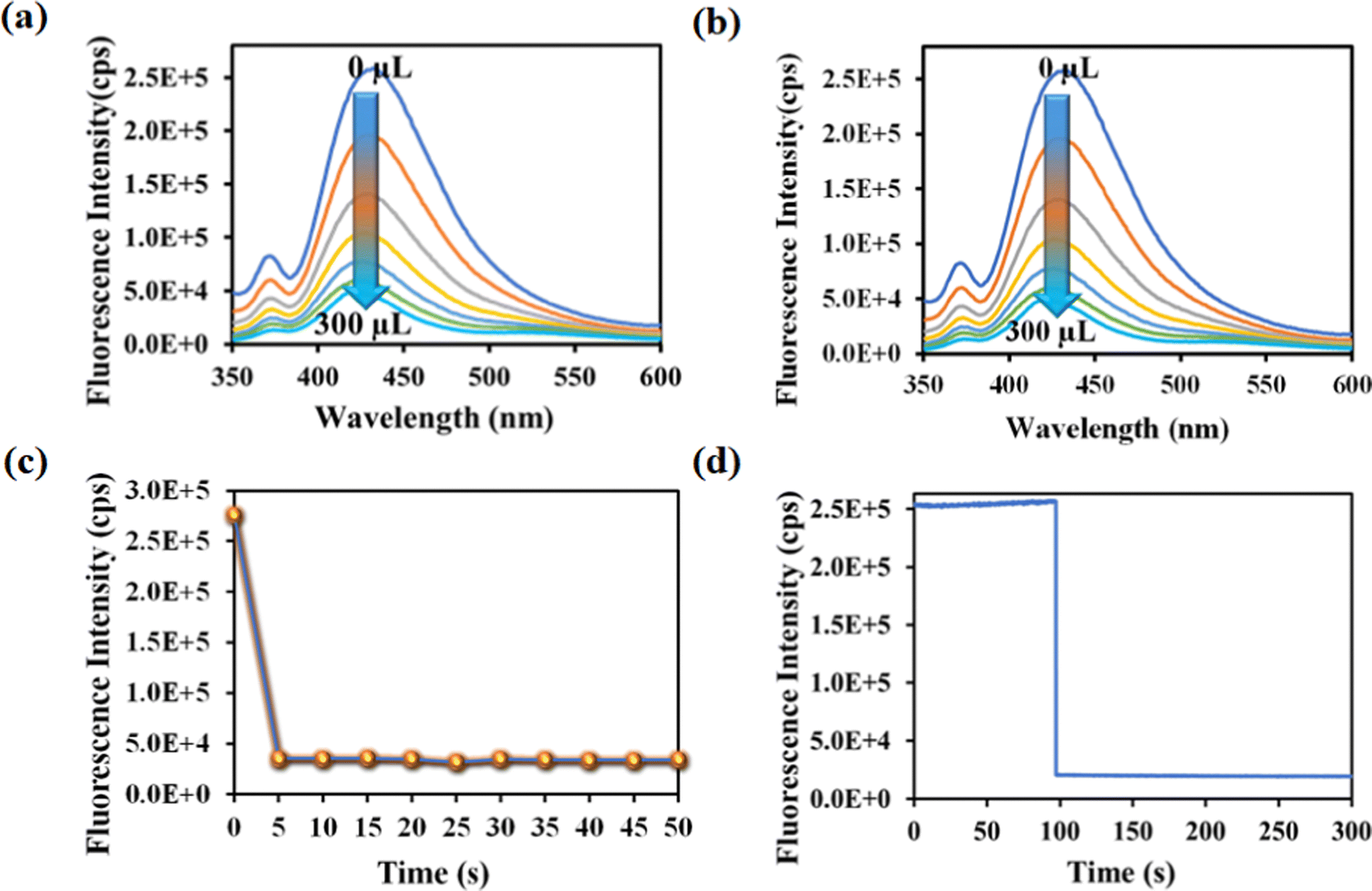 | ||
| Fig. 3 (a) PL emission intensity plot for 1′ with incrementally added rifampicin. (b) Time-dependent PL emission intensity of 1′ in the presence of 300 μL of 1 mM rifampicin. (c) PL emission intensity of the probe with variation in time after adding 300 μL, 1 mM rifampicin. (d) Fluorescence kinetic study of the probe by recording the PL intensity at 430 nm with variation in time after the addition of 300 μL, 1 mM rifampicin. | ||
One of the key features of the probe is its short period of analyte detection. As a result, we conducted a time-dependent fluorescence titration experiment to investigate the detection time of our probe for rifampicin sensing. We introduced 300 μL of 1 mM rifampicin to an aqueous MOF suspension and measured the PL emission intensity over time. After introducing the rifampicin solution, we noted a quick quenching and saturation of the PL emission intensity of our probe within 5 s (Fig. 3b and c). As a result, the detection time for rifampicin was estimated to be 5 s. A fluorescence kinetic experiment confirmed the detection time for rifampicin sensing. The kinetic study was carried out by exciting the probe at 330 nm and collecting the PL emission intensity at the emission maxima, i.e., at 430 nm, before and after the addition of rifampicin. After adding rifampicin, we noticed a rapid drop in PL emission intensity at 430 nm within 5 s. The kinetic fluorescence experiment also confirmed that the probe requires only 5 s to detect rifampicin (Fig. 3d). The rapid sensing ability of the probe boosted its real-world applicability by detecting the drug instantly. Among all the known works on rifampicin sensing, our probe is the fastest.
Numerous sensors are available for detecting drug molecules, but most of them suffer from selectivity over the congeners. As a result, an ideal probe must have selectivity over the congeners of the targeted analyte. To test the selectivity of rifampicin over other analytes, 300 μL of 1 mM aqueous analyte solution was added to a cuvette containing MOF suspension. We have observed that the I/I0 value for competing analytes is close to 1, while the I/I0 value for rifampicin is 0.1 (Fig. 4a). For competitive analytes, the unity value of I/I0 indicates that the PL intensity remained essentially constant both before and after the analyte was added to the MOF suspension. However, the I/I0 became nearer to zero for rifampicin due to a drastic fall in PL intensity of the MOF after the addition of rifampicin.
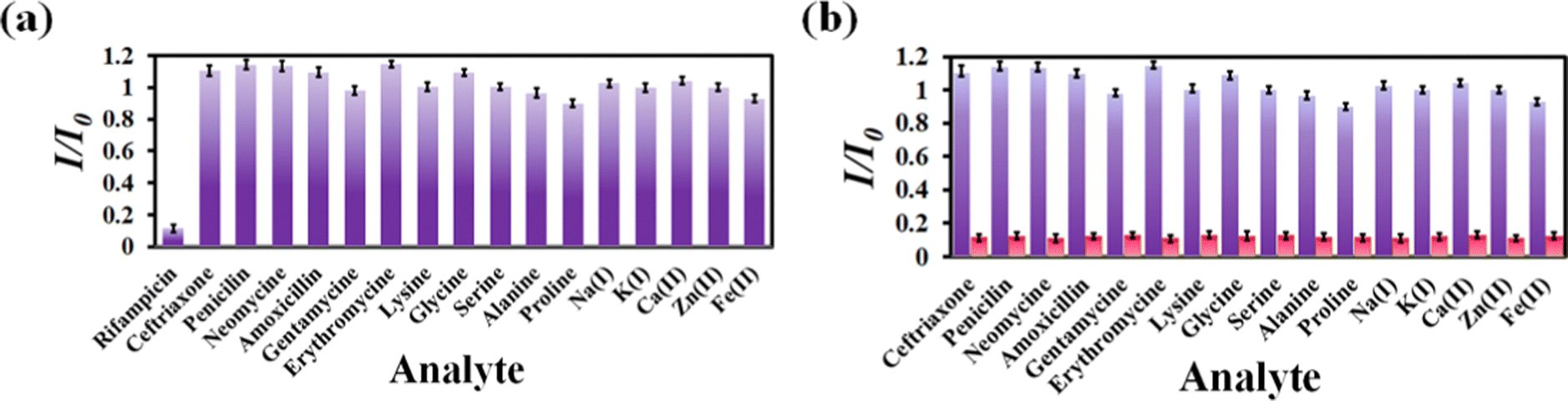 | ||
| Fig. 4 (a) Selectivity plot for rifampicin over its congeners. (b) Selectivity plot for rifampicin in the coexistence of the competitive analytes. | ||
The on-field applicability of a probe requires the probe's selectivity toward sensing a targeted analyte even when competing analytes are present. Therefore, we conducted a three-step selectivity experiment. At first, the PL emission intensity of MOF was measured, and then one of the competitive analytes was introduced to the MOF suspension, and the PL spectrum was recorded. Finally, the targeted analyte, i.e., rifampicin, was introduced to the MOF suspension containing a competitive analyte, and the intensity of the PL emission was measured (Fig. S11–S26, ESI†). We observed that adding a competitive analyte could not alter the PL emission intensity of MOF. With the addition of a competing analyte, the I/I0 value stayed close to one (Fig. 4b). However, it almost reached zero after the addition of rifampicin, which confirmed that our probe could detect rifampicin even in the presence of competitive congeners.
One of the critical factors for an ideal sensor for its real-field usability is the sensitivity of detection. As a result, rifampicin was detected at a lower concentration. The detection limit was derived using the formula 3σ/k, where k is the slope of the linear-fit plot of the concentration of rifampicin versus PL emission intensity of MOF and σ is the standard deviation of ten blank readings of the MOF suspension (Fig. S27, ESI†). Rifampicin's LOD value was found to be 11.7 nM. Our probe has a far lower sensitivity than the previously reported probes for rifampicin sensing (Table S11, ESI†). In contrast to other sensors, our probe's lower detection limit distinguishes this material and makes it more appropriate than others for practical use.
The Stern–Volmer constant (Ksv) was determined using the Stern–Volmer equation (Ksv[Q] + 1 = I0/I). ‘I’ and ‘I0’ are the PL intensities of 1′ in the absence and presence of various analytes, respectively, and ‘Q’ is the analyte concentration (Fig. S28, ESI†). The high Ksv value for rifampicin sensing (1.8 × 106 M−1) substantiated our probe's sensitivity to rifampicin. Fig. 5a depicts the 3D S–V graphs for all of the examined analytes. The 3D S–V graphs demonstrated rifampicin's exceptional selectivity over a wide range of other congeners.
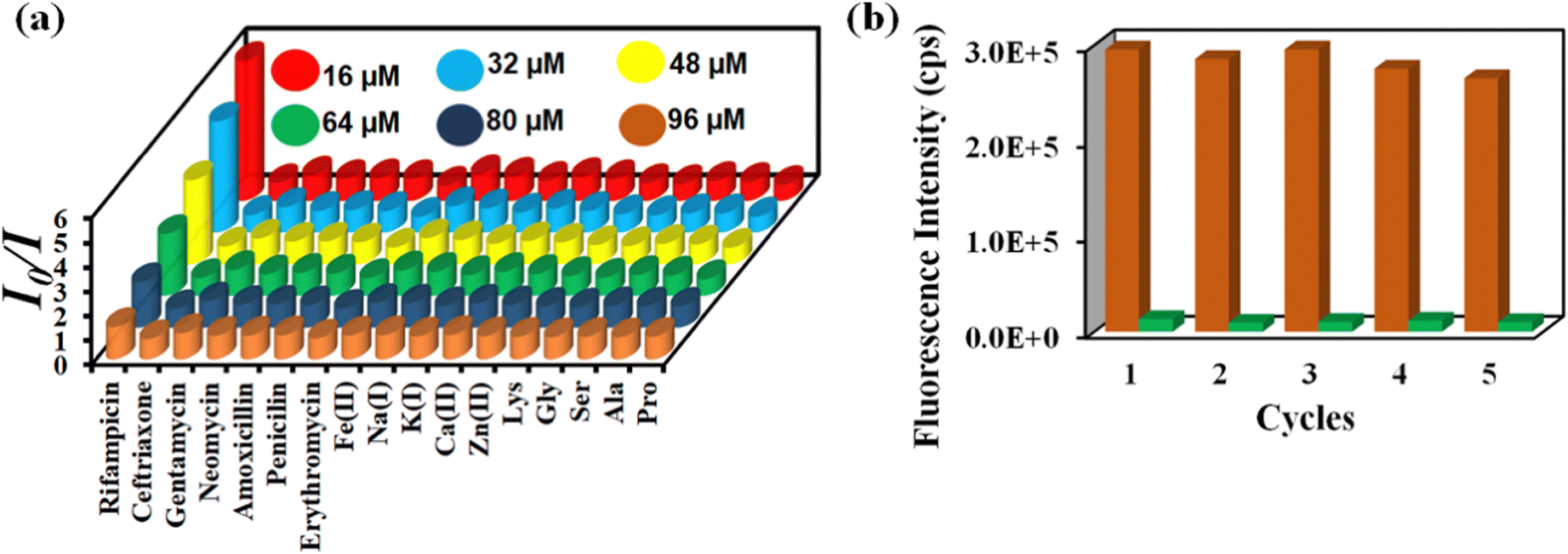 | ||
| Fig. 5 (a) 3D Stern–Volmer plot for the detection of rifampicin by 1′ and (b) recyclability of 1′ towards rifampicin sensing. | ||
The capacity to reuse a sensor due to its recyclability is beneficial over one-time-usable sensors. The sensor's multiple-time reusability reduces expenses by many folds compared to single-use probes. Therefore, we have demonstrated the recyclability of our probe towards rifampicin. After each sensing experiment, the probe was rinsed adequately with fresh water, and identical fluorescence titration studies were repeated to investigate the recyclability behavior. Fig. 5b displays that following the addition of rifampicin, the PL intensity was suppressed significantly and after the washing step, the probe's fluorescence property was recovered after each sensing cycle. The recyclability experiment was conducted five times, and the results confirmed that the quenching efficiency remained nearly the same even after the fifth cycle. The above results demonstrated that the probe could be used for at least five cycles.
Rifampicin sensing from real water samples
Due to the extremely long half-life of the disintegration of medications, their presence in natural water sources creates a variety of health hazards. Therefore, we conducted rifampicin sensing in environmental water samples to protect living things. The PL intensity of the MOF suspension was measured in the presence of varying amounts of rifampicin in environmental water samples (lake water, ocean, river water, and tap water). The MOF's PL emission intensity decreased as rifampicin concentration increased. The ability of the probe to find rifampicin in samples of naturally occurring environmental water demonstrates the applicability of our probe in real-world applications. (Fig. S29, ESI†).Rifampicin sensing from biological fluids
An excess of rifampicin in the body can cause toxicity, and in extreme cases, death.30 Therefore, we conducted PL titration experiments to quantify rifampicin in human blood serum and urine. Rifampicin is stable in pure water but begins to degrade when it comes in contact with blood. Farina et al. discovered that adding ascorbic acid to a rifampicin solution could boost its stability for up to 3 days in ambient conditions.31 As a result, we prepared a mixture of ascorbic acid and rifampicin in human blood serum and urine separately. Rifampicin was quantified in both the mixtures using the PL titration technique. The obtained results revealed that there was no change in the PL intensity of MOF in the presence of only ascorbic acid, but the PL intensity was significantly quenched in the presence of both rifampicin and ascorbic acid (Fig. S30 and S31, ESI†), strongly recommending that the quenching effect is solely due to rifampicin. Tables S2 and S3 (ESI†) present the good recovery percentages of rifampicin from human blood serum and urine, confirming that our probe could detect rifampicin from human blood serum and urine with high accuracy and precision.PL detection of nitenpyram
Nitenpyram is one of the world's most widely used neonicotinoid insecticides, and it is also utilized in veterinary medicine. The presence of nitenpyram in high concentrations is hazardous to non-targeted organisms. One of the most important criteria for ensuring a healthy society is monitoring such harmful compounds in the natural environment. Therefore, we performed the same PL titration studies in an aqueous medium for nitenpyram sensing as we did for rifampicin sensing.The PL emission intensity of the aqueous suspension of 1′ displayed 95% quenching rapidly (5 s) in the presence of 350 μL of 5 mM nitenpyram solution (Fig. 6a–c). Furthermore, a PL kinetic experiment was also performed to verify the actual detection time of nitenpyram by 1′. The PL kinetic experiment confirmed that our probe could detect nitenpyram in less than 5 s (Fig. 6d). The detection of nitenpyram was also performed in the presence of other congeners. We observed that 1′ possesses excellent selectivity towards nitenpyram in comparison to other competitors (Fig. 7a and b and Fig. S32–S36, ESI†). The LOQ and LOD values for nitenpyram detection were determined using the same experimental approach used for rifampicin (Fig. S37, ESI†). The LOQ and LOD values were 45.9 nM and 13.8 nM for nitenpyram, respectively. Our probe's nanomolar LOD and LOQ values confirmed its great sensitivity towards nitenpyram sensing for real-world applications. Moreover, the high value of the S–V constant (Ksv = 3.2 × 106 M−1) for nitenpyram sensing established our probe's great sensitivity and selectivity (Fig. S38, ESI† and Fig. 8a).
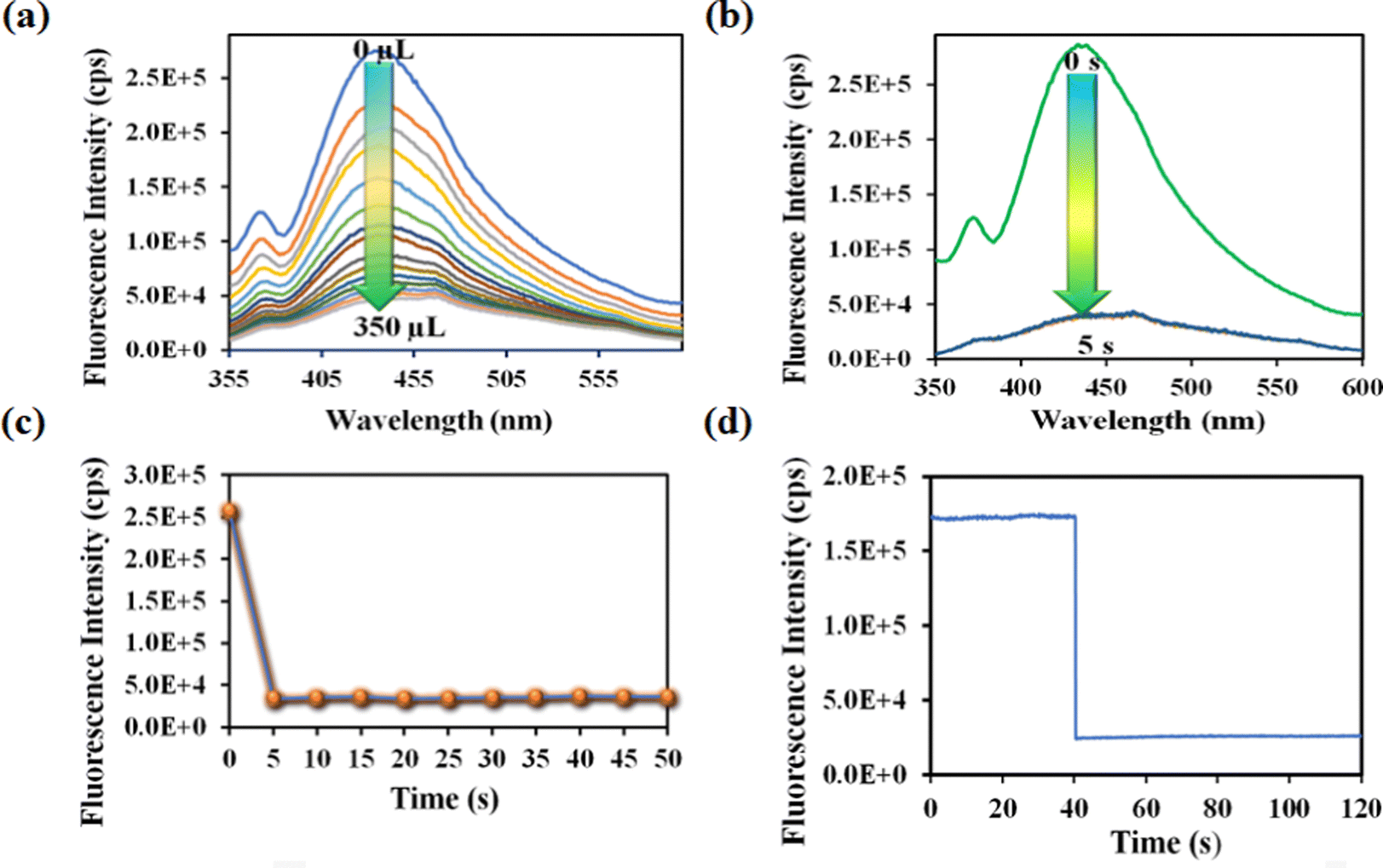 | ||
| Fig. 6 (a) PL emission intensity plot for 1′ with incrementally added nitenpyram. (b) Time-dependent PL emission intensity of 1′ in 350 μL of 1 mM nitenpyram. (c) The PL emission intensity of the probe by changing the time after adding 350 μL, 1 mM nitenpyram. (d) Fluorescence kinetic study of the probe by recording the PL intensity at 430 nm by changing the time after the addition of 350 μL, 1 mM nitenpyram. | ||
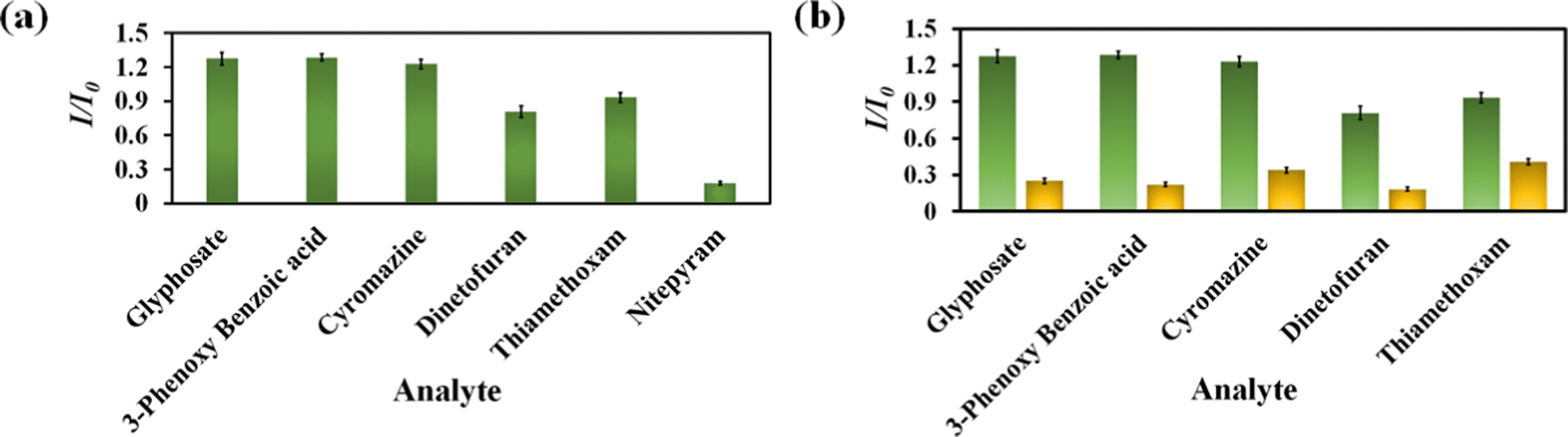 | ||
| Fig. 7 Selectivity plot for nitenpyram over its congeners (a) and selectivity plot for nitenpyram in the coexistence of the competitive analytes (b). | ||
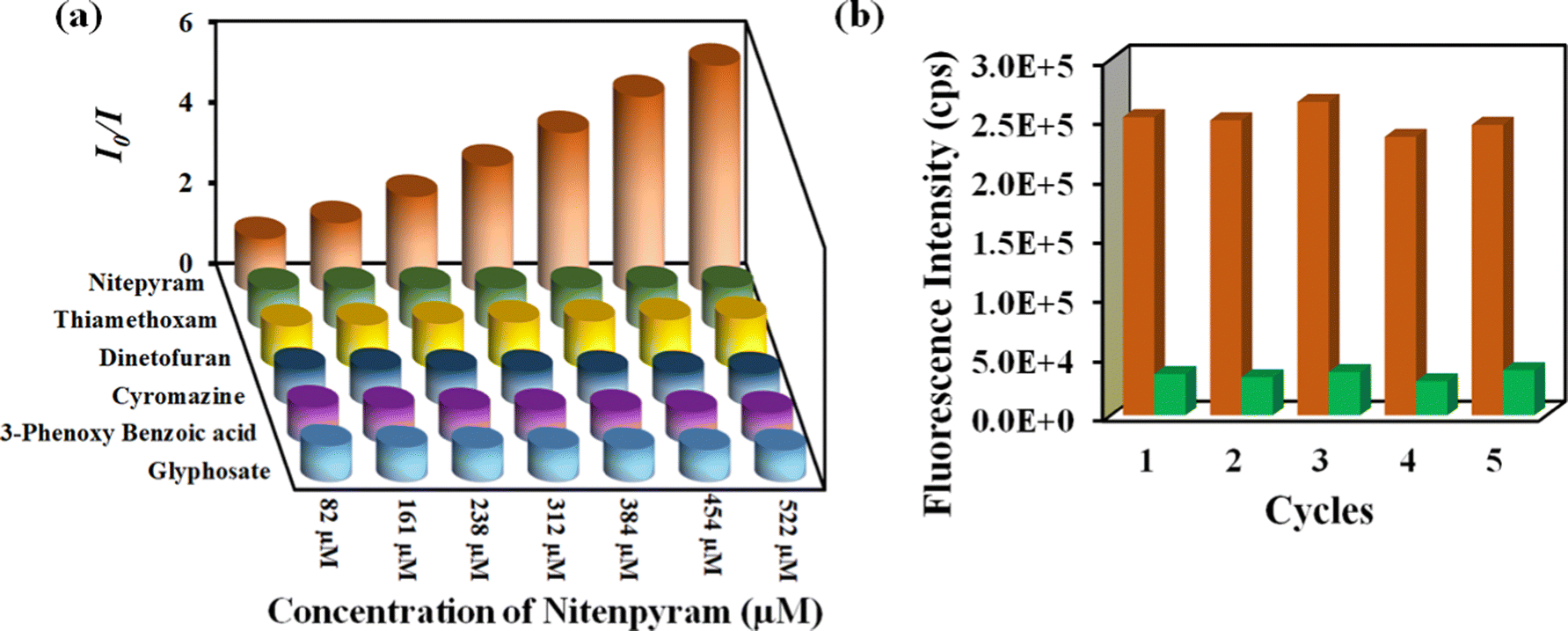 | ||
| Fig. 8 (a) 3D Stern–Volmer plot for detecting nitenpyram by 1′. (b) Recyclability of 1′ towards nitenpyram sensing. | ||
The reusability of 1′ for the sensing of nitenpyram was examined similarly to those described above for rifampicin sensing. The recyclability plot in Fig. 8b confirmed that 1′ could detect nitenpyram at least up to five cycles with excellent efficacy.
Nitenpyram sensing from real water samples
Nitenpyram is a water-soluble pesticide. Its excessive use in crop fields could elevate its concentration in natural water sources, which is a warning towards a safe environment. Therefore, we have carried out a series of PL sensing experiments, taking various concentrations of nitenpyram in natural water specimens (lake, river, tap and distilled water). The obtained results inferred that our probe could detect nitenpyram from natural water sources without interference (Fig. S39, ESI†).Nitenpyram sensing from soil and food samples
The excessive use of pesticides (nitenpyram) could also contaminate soil and crops due to its high water solubility. The presence of such harmful pesticides in soil and food samples will hamper the natural biological cycle of the affected animals. As a result, we performed nitenpyram detection in soil and food samples (rice and corn). The results revealed that 1′ can identify nitenpyram from soil and food samples with high accuracy and precision (Tables S4–S6, ESI†).Reproducibility of PL sensing
We conducted the sensing experiments several times, both on the same day and on other days. The inter-day and intra-day fluorescence detection assays demonstrated that the MOF could repeatedly detect rifampicin and nitenpyram with excellent precision and accuracy (Tables S7 and S8, ESI†).Mechanism of fluorescence quenching by rifampicin
Using a variety of analytical techniques, the mechanism of fluorescence quenching by rifampicin was fully explored. To understand the influence of rifampicin on the crystallinity of the MOF, PXRD investigations were conducted before and after rifampicin sensing. PXRD examination confirmed that the crystallinity of MOF remained unchanged (Fig. S40, ESI†), confirming that the change in PL emission intensity of MOF in the presence of rifampicin is not due to the structural change of MOF. The ATR-IR and EDX analyses were performed before and after treatment with rifampicin. The obtained ATR-IR and EDX spectra of MOF before and after treatment of rifampicin remained the same (Fig. S41 and S42, ESI†). Again, 1′ is recyclable towards rifampicin sensing, as mentioned above in Fig. 4b. All the above-mentioned results confirmed that the fluorescence sensing process could not be due to the formation of a ground state complex or reaction between the MOF and rifampicin.The elimination of reaction-based processes compelled us to consider non-reaction-type fluorescence quenching techniques (IFE, PET, FRET and so on). We started with an excited-state lifetime investigation of 1′ in the presence and absence of rifampicin. The excited state lifetime of 1′ was 11.8 ns in the absence of rifampicin, but it was reduced to 8.0 ns with the addition of rifampicin (Fig. S43 and Table S9, ESI†). The decrease in the probe's lifetime in the presence of rifampicin suggests that the PL quenching is caused by a dynamic process. The IFE mechanism is ruled out by the dynamic process of fluorescence quenching. In addition, to better understand the process of the PL quenching by rifampicin, we plotted the UV-Vis spectra of all analytes, including rifampicin, as well as the PL emission of MOF (Fig. S44, ESI†). The overlap plot confirmed that there is enough overlap between the MOF emission spectrum and the rifampicin absorbance spectrum for energy transfer from the excited state of the probe to the analyte. According to the findings displayed above, the quenching of MOF's PL intensity may be caused due to the FRET process. Again, the UV-Vis spectra of other analytes of rifampicin do not display significant overlap with the emission spectrum of MOF, which is the reason behind the selectivity of rifampicin over other congeners.
In addition, we have performed DFT energy level calculations for rifampicin and H2L linker using Gaussian 6.0 software and B 3 LYP function with 631G++ basic set. We found that the HOMO and LUMO of the linker are located at −5.82 eV and −2.16 eV, and rifampicin at −4.92 eV and −2.82 eV (Fig. 9). The PL quenching will only be possible through the PET-based path if the donor's (probe) LUMO is higher in energy than the acceptor's (analyte: rifampicin) LUMO. The energy of the LUMO of the linker and rifampicin provided above indicates that there is a possibility of photoinduced electron transfer from the linker to rifampicin.32
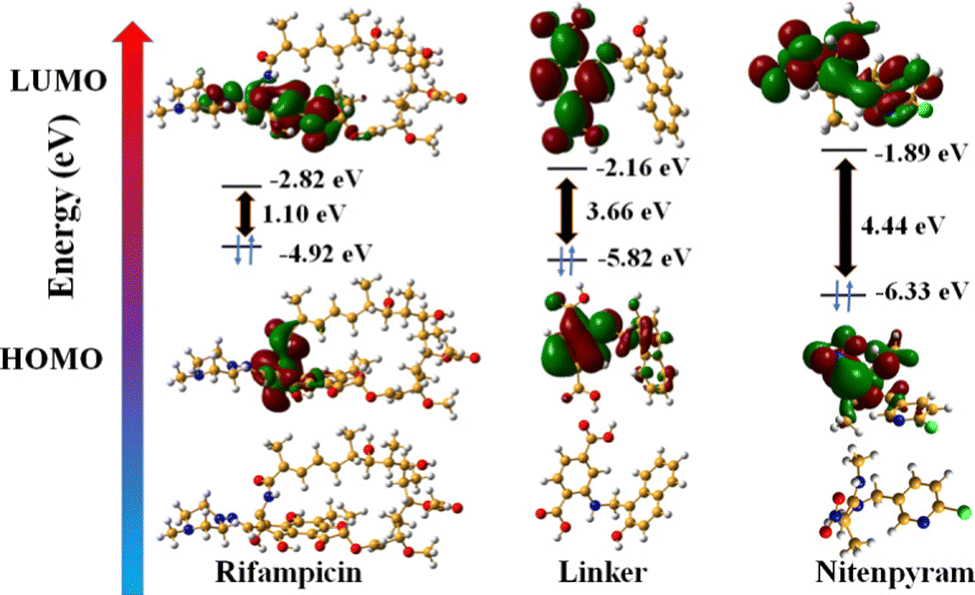 | ||
| Fig. 9 DFT based HOMO and LUMO energy levels of linker, rifampicin, and nitenpyram. | ||
All the above results obtained from the rigorous analytical techniques and theoretical calculations strongly indicate that the quenching of PL intensity of MOF by rifampicin is due to combined FRET and PET mechanisms.
Mechanism of nitenpyram sensing
The PXRD, ATR-IR, and EDX elemental investigation of 1′ before and after nitenpyram sensing were measured and found to be precisely similar (Fig. S40, S41 and S45, ESI†). The similarity in PXRD patterns of 1′ before and after nitenpyram sensing inferred that nitenpyram's quenching of the PL emission intensity of MOF was not because of any structural change of 1′. The resemblance of ATR-IR peaks and retention of all the elements of 1′ before and after nitenpyram sensing indicate that the PL quenching of 1′ by nitenpyram might be due to some non-reaction-based pathways (IFE, FRET, or PET) instead of any reaction or ground state complexation between MOF and nitenpyram. Furthermore, the recyclability of the probe toward nitenpyram sensing also supported the non-reaction type PL quenching.To determine the precise mechanism of the PL quenching by nitenpyram, we measured the excited state PL lifetimes of 1′ in the absence and presence of nitenpyram. In the absence of nitenpyram, the probe's lifetime is 11.8 ns, but it reduces to 6.7 ns (Fig. S46 and Table S10, ESI†) after adding nitenpyram. The decrease in the lifetime of our probe in the presence of nitenpyram revealed that PL quenching occurs via a dynamic mechanism. The dynamic nature of the PL quenching by nitenpyram excluded the possibility of an IFE process. As a result, we calculated the HOMO and LUMO orbital energies of nitenpyram and the linker molecule to understand the possibility of photoinduced electron transfer. The quenching of PL emission intensity of MOF by nitenpyram is only possible when the photoexcited electron from the LUMO of the linker transfers to the LUMO of nitenpyram. But, in the case of nitenpyram, the LUMO is energetically higher than the LUMO of the linker, which is a forbidden condition for the PET process to occur between the probe and nitenpyram.
The results of the aforementioned experiments ruled out the feasibility of PET and IFE. Therefore, we concentrated on the possibility of the FRET process. It is already known that FRET-based quenching occurs when there is enough overlap between the PL emission spectrum of the donor and the UV-Vis absorbance spectrum of the acceptor, which mediates the transfer of energy through a dynamic process.33 Because of that, the PL emission spectrum of MOF was plotted with the UV-Vis absorption spectra of all the analytes, including nitenpyram (Fig. S4, ESI†). The acquired results demonstrated that, among all the analytes, only nitenpyram's UV-Vis absorption spectrum overlaps with the PL emission spectrum of MOF. The dynamic nature of the PL quenching, as well as the overlap of the absorption (nitenpyram) and emission spectrum (MOF), demonstrated that FRET is the sole reason for nitenpyram sensing.
Conclusion
The preparation, complete characterization, and utilization of a 2-hydroxy naphthaldehyde functionalized aluminum MOF for the detection of a popular antibiotic drug for tuberculosis (rifampicin) and a neonicotinoid pesticide (nitenpyram) are presented herein. This is the first luminescent MOF to detect rifampicin through a fluorescence turn-off process. The luminescent, recyclable, and chemically stable MOF can detect rifampicin at an ultra-low concentration (11.7 nM) in an aqueous medium. The probe's detection limit is much lower than the previously reported probes of rifampicin, which establishes its real-world usefulness. It is also the first-ever luminescent MOF-based sensor to detect the nanomolar concentration of nitenpyram via a fluorescence turn-off procedure. This probe can recognize up to 13.8 nM concentration of nitenpyram, which confirms its superior sensitivity over other fluorescent-based nitenpyram sensors. Moreover, it can selectively recognize both these analytes instantaneously (5 s), which is an important property of this probe. Rifampicin and nitenpyram were also detected from various water sources (lake, river, tap, and distilled water) for health and environmental protection. Furthermore, this probe was utilized to precisely quantify rifampicin from human biological fluids (blood serum and urine) and nitenpyram from soil and food samples (rice and corn). The detection of rifampicin and nitenpyram were checked several times to establish the reproducibility and repeatability of the probe to detect both these analytes with high precision and accuracy. The recyclability of the probe up to five cycles was confirmed, which underscores its cost-effectiveness and sustainability. Finally, we have systematically explored the mechanism of the PL quenching of MOF by rifampicin and nitenpyram with the help of modern analytical instruments. The FRET and PET processes are collectively responsible in the case of rifampicin sensing, whereas the FRET process is responsible for nitenpyram sensing. The overall work presents a systematic detection and monitoring of an essential class of tuberculosis drug (rifampicin) and a neonicotinoid pesticide (nitenpyram) to protect the health of animals, including humans, and our environment.Author contributions
AR performed all the experiments and prepared the manuscript, taking scientific ideas and advice from SB.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support for this work was obtained from SERB through grant no. CRG/2021/000080 and EEQ/2021/000013. A. R. is thankful to PMRF for providing financial support.References
- M. Rust, M. Waggoner, N. Hinkle, D. Stansfield and S. Barnett, J. Med. Entomol., 2003, 40, 678–681 CrossRef CAS PubMed
.
- H. Wang, L. Pan, Y. Liu, Y. Ye and S. Yao, J. Electroanal. Chem., 2020, 862, 113955 CrossRef
- W. Liu, Z. Li, X. Cui, F. Luo, C. Zhou, J. Zhang and L. Xing, Toxicol. Appl. Pharmacol., 2022, 446, 116065 CrossRef PubMed
- S. Yan, W. Sun, S. Tian, Z. Meng, J. Diao, Z. Zhou, L. Li and W. Zhu, J. Environ. Sci., 2024, 137, 120–130 CrossRef PubMed
- M. A. I. Ahmed, C. F. A. Vogel and G. Malafaia, Sci. Total Environ., 2022, 804, 150254 CrossRef PubMed
- M. Grobbelaar, G. E. Louw, S. L. Sampson, P. D. van Helden, P. R. Donald and R. M. Warren, Infect., Genet. Evol., 2019, 74, 103937 CrossRef PubMed
- E. A. Campbell, N. Korzheva, A. Mustaev, K. Murakami, S. Nair, A. Goldfarb and S. A. Darst, Cell, 2001, 104, 901–912 CrossRef PubMed
- C. Shen, Q. Meng, G. Zhang and W. Hu, Br. J. Pharmacol., 2008, 153, 784–791 CrossRef CAS PubMed
- Q. Xu, G. Owens and Z. Chen, J. Clean. Prod., 2020, 264, 121617 CrossRef CAS
- J. Bajwa, M. Charach and D. Duclos, Vet. Dermatol., 2013, 24, 570–e136 CrossRef
- A. Srivastava, D. Waterhouse, A. Ardrey and S. A. Ward, J. Pharm. Biomed. Anal., 2012, 70, 523–528 CrossRef CAS PubMed
- B. Milz, I. B. Idros and B. Spangenberg, J. Liq. Chromatogr. Relat. Technol., 2012, 35, 1404–1414 CrossRef CAS
- M. F. Khan, S. A. Rita, M. S. Kayser, M. S. Islam, S. Asad, R. Bin Rashid, M. A. Bari, M. M. Rahman, D. A. Al Aman and N. I. Setu, Front. Chem., 2017, 5, 27 CrossRef PubMed
- S. Ge, Y. Wang, Q. Song, L. Chen, Y. Zhang and D. Hu, Food Addit. Contam., Part A, 2020, 37, 955–962 CrossRef CAS PubMed
- T. Yoshida, H. Murakawa and K. Toda, J. Pestic. Sci., 2013, 38, 27–32 CrossRef CAS
- Ž. Temova Rakuša, R. Roškar, A. Klančar Andrejc, T. Trdan Lušin, N. Faganeli, I. Grabnar, A. Mrhar, A. Kristl and J. Trontelj, Int. J. Anal. Chem., 2019, 2019, 75433 Search PubMed
- Q. Wang, Y. Liu, Y. Bai, S. Yao, Z. Wei, M. Zhang, L. Wang and L. Wang, Anal. Chim. Acta, 2019, 1049, 170–178 CrossRef CAS PubMed
- A. Li, Q. Chu, H. Zhou, Z. Yang, B. Liu and J. Zhang, Inorg. Chem. Front., 2021, 8, 2341–2348 RSC
- J. Liu, W. H. Xiong, L. Y. Ye, W. S. Zhang and H. Yang, J. Agric. Food Chem., 2020, 68, 5572–5578 CrossRef CAS PubMed
- Y. Zhang, Q. Deng, C. Tang, M. Zhang, Z. Huang and Z. Cai, Spectrochim. Acta, Part A, 2023, 286, 121944 CrossRef CAS
- K. Chatterjee, C. W. Kuo, A. Chen and P. Chen, J. Nanobiotechnol., 2015, 13, 1–9 CrossRef CAS
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem. – Eur. J., 2004, 10, 1373–1382 CrossRef CAS
- A. Rana and S. Biswas, Inorg. Chem. Front., 2023, 10, 2742–2753 RSC
- C. M. Moran, J. N. Joshi, R. M. Marti, S. E. Hayes and K. S. Walton, J. Am. Chem. Soc., 2018, 140, 9148–9153 CrossRef CAS
- A. Rana, S. Ghosh and S. Biswas, Inorg. Chem. Front., 2023, 10, 612–620 RSC
- M. Allahbakhshi, N. M. Mahmoodi, M. Mosaferi, H. Kazemian and H. Aslani, Surf. Interfaces, 2022, 35, 102471 CrossRef CAS
- P. Chakraborty, A. Rana, S. Mukherjee and S. Biswas, Inorg. Chem., 2022, 62, 802–809 CrossRef PubMed
- A. Nath, D. V. Gaikwad and S. Mandal, Dalton Trans., 2023, 52, 4303–4308 RSC
- A. Nath, G. M. Thomas, S. Hans, S. R. Vennapusa and S. Mandal, Inorg. Chem., 2022, 61, 2227–2233 CrossRef CAS PubMed
- A. Sridhar, Y. Sandeep, C. Krishnakishore, P. Sriramnaveen, Y. Manjusha and V. Sivakumar, Indian J. Nephrol., 2012, 22, 385 CrossRef PubMed
- A. Santoveña-Estévez, J. Suárez-González, A. R. Cáceres-Pérez, Z. Ruiz-Noda, S. Machado-Rodríguez, M. Echezarreta, M. Soriano and J. B. Fariña, Pharmaceutics, 2020, 12, 195 CrossRef
- S. Mukherjee, S. Ghosh and S. Biswas, Inorg. Chem. Front., 2022, 9, 6288–6298 RSC
- A. Rana, S. Nandi and S. Biswas, New J. Chem., 2022, 46, 10477–10483 RSC
Footnote |
| † Electronic supplementary information (ESI) available: EDX, ATR-IR, TRPL, fluorescence spectra, N2 sorption isotherm, PXRD patterns, TG curves, comparison tables. See DOI: https://doi.org/10.1039/d3tc03712f |
| This journal is © The Royal Society of Chemistry 2024 |