
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metallopolymer-based block copolymers for perfluorinated substances (PFAS) and ion removal†
Till
Rittner
 a,
Sebastian
Pusse
a,
Blandine
Boßmann
a,
Kevin
Staudt
b,
Aaron
Haben
c,
Ralf
Kautenburger
c,
Horst P.
Beck
d and
Markus
Gallei
*ae
a,
Sebastian
Pusse
a,
Blandine
Boßmann
a,
Kevin
Staudt
b,
Aaron
Haben
c,
Ralf
Kautenburger
c,
Horst P.
Beck
d and
Markus
Gallei
*ae
aPolymer Chemistry, Saarland University, Campus C4 2, 66123, Saarbrücken, Germany. E-mail: markus.gallei@uni-saarland.de
bPhysical Chemistry, Saarland University, Campus B2 2, 66123 Saarbrücken, Germany
cInorganic Solid State Chemistry, Elemental analysis group, Saarland University, Campus C4 1, 66123, Saarbrücken, Germany
dInorganic and Analytical Chemistry, Saarland University, Campus Dudweiler, Beethovenstrasse Zeile 4, 66125, Saarbrücken, Germany
eSaarene, Saarland Center for Energy Materials and Sustainability, Campus C4 2, 66123, Saarbrücken, Germany
First published on 15th October 2024
Abstract
Similar to the efforts made to combat standard (transition)metal-ion pollution in drinking water, such as chromate or lead, a significant endeavor has been directed towards removing perfluorinated organic substances (PFAS), typically found in very low concentrations. In this study, we have developed asymmetric membranes based on cobaltocenium-containing block copolymers (BCP) that selectively target PFAS but can also be utilized in metal ion removal. These membranes, prepared via the self-assembly and non-solvent-induced phase-separation process (SNIPS), offer a practical and crucial solution to water pollution. Their versatility is demonstrated by their ability to customize the surface and pore size to meet specific filtration requirements and their effectiveness in targeting different pollutants. The metallopolymer membranes we have produced have been tested for applications in a water filtration system and demonstrate a high permeance of 521 ± 49 L h−1 bar−1 m−2. Furthermore, depending on the settings used for the perfluorooctanoic acid (PFOA) retention experiments, a stable permeance of 48 ± 2 up to 171 ± 26 L h−1 bar−1 m−2 as well as a PFOA retention decrease of 99.3% at lower and 96.6% at higher water flux was found. The PFAS and metal ion retention capability is followed by ion chromatography (IC) and liquid chromatography coupled with mass spectroscopy (LC-MS). In addition, the ion-capturing is investigated for sodium chromate and lead nitrate for these porous BCP membranes. Here, remarkable retention for both anionic and cationic metallic pollutants of 46.4% and 99.8%, respectively, is verified by inductively coupled plasma mass spectrometry (ICP-MS). For porous membrane recycling, the selective solubility of the cobaltocenium BCPs is furthermore utilized to form new membranes from recycled material. Finally, by up-cycling the used membranes via calcination under reductive or oxidative conditions, we can prepare porous cobalt-containing ceramics with tailorable ceramic composition and well-defined porous architectures, offering a sustainable solution to material utilization and ceramic production.
Introduction
Complementary to the established elimination of harmful (transition)metal contaminants from drinking water, the removal of per- and polyfluorinated substances (PFAS) is gaining ever-increasing attention as health concerns and governmental regulations rise.1 However, due to the overall low aqueous concentrations of these chemicals of single digit up to several hundreds of nanograms per liter, concentration before a (selective) removal and controlled destruction is necessary.2 For the most prominent compounds, perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS), conventional wastewater treatment with low-cost adsorbers was initially performed.3 Here, materials like granular activated carbon, silica, or others were used, resulting in large amounts of contaminated material leading to an expensive PFAS removal and regeneration process.4,5 To reduce the environmental impact, significant effort towards developing systems based on renewable or natural resources is made.6 On the other hand, polymer-engineered activated carbon was presented as a higher-performing alternative.7 Lately, carbon-based micro architectures like carbon nanotubes (CNT), carbon microspheres, or molecular imprinted polymers are being investigated.8–10 Costly but even more effective are polymer-based adsorbents.11 These, mostly cationic polymers show excellent performance and can be turned into hydrogels or resins for convenient applications.12 For short-chain PFAS, ion exchange resins have proven quite successful in single-use applications but show drastically lower efficiency after regeneration.13 Eliminating these major problems, membrane-based removal strategies present a suitable alternative.2,14,15 In contrast to previously discussed methods, membranes are less susceptible to co-contaminants and fouling due to the selectively tuned membrane parameters like porosity, material, and pore size. Furthermore, a great benefit of membranes is the possibility of using tailored materials for the target application. Several crucial and beneficial influences have been identified for the concentration, capture, and removal of PFAS. The most prominent are ionic interactions.16,17 The negatively charged PFAS are strongly attracted and interact with cationic material via electrostatic interactions, resulting in large cluster formation. Ionic surfaces are additionally utilized for reduced fouling and antimicrobial benefits.18 Furthermore, a hydrophobic and aromatic environment has been shown to be beneficial for PFAS removal by stronger interaction with, for example, the membrane surface.19,20 Here, especially polymeric materials are highlighted due to the advantages in processing, robustness, and their low cost compared to inorganic competitors.2 Moreover, interesting intrinsic polymer properties can be used to improve the membrane formation and its performance further. One of these techniques is the self-assembly and non-solvent-induced phase separation (SNIPS) technique.21–23 Here, the selective solubility of polymer or polymer segments can be exploited to generate asymmetric membranes with (highly) porous surfaces and porous sublayers by self-assembly and introducing a polymer solution to a non-solvent. This intricate process can be influenced by a wide range of factors, opening the possibilities for selective membrane manufacturing.24,25 One additional benefit is demonstrated by the wide range of homopolymers and block copolymers or combinations thereof, which can be utilized. Here, especially, smart polymers that change their properties upon a specific (external) stimulus are of ever-growing interest.26,27 In literature, this upcoming class of functional polymers is known to be affected by a range of external triggers such as temperature, pH value, ion content, and external fields. By including these responsive polymeric materials in membrane formation, a new class of switchable smart membranes has evolved.28–32 Within this field, temperature-responsive polymers have especially been used for flux control and PFAS removal.33,34 In contrast to thermally switchable polymers, redox-active polymers emerged as interesting alternatives. In a recent study, different metallocene-containing polymers were compared in terms of PFOS and PFOA adsorption.35 Surprisingly, not only excellent adsorption but also desorption upon electrochemical switching could be demonstrated. The electrochemical addressability is introduced to the polymer by the metallocene motive, fusing inorganic metal atoms like iron and cobalt with the organic cyclopentadiene ligands.36,37 The formed complexes form a highly stable and redox active complex, which can be switched within polar and non-polar states on demand. For the iron-containing ferrocene polymers, a range of applications from controlling surface wettability over battery and sensor interfaces,38–40 to catalytic, preceramic materials,41,42 and ion-selective chromatography43 are described in the literature. Ferrocene can chemically or electrochemically oxidize to the cationic ferrocenium, which is required for adequate PFAS adsorption and is prone to auto-reduction in an aqueous environment if no permanent current is present.43–45 By using cobalt instead of iron, the isoelectronic cobaltocenium polymers feature a permanent cation compared to the uncharged ferrocene.46 In addition to antimicrobial effects and use in ion exchange resins,47–49 cobaltocenium polymers presented the optimal desorption of PFAS for all metallocene-containing polymers tested.35 On the one hand, cobaltocenium-based applications benefit from the chemical inertness; on the other hand, cobaltocenium-containing material and polymers are challenging to prepare because of the intrinsically lower reactivity compared to the ferrocene motive. Due to the increasing interest in cobaltocenium-containing polymers, new approaches and strategies for introducing metallocene into a polymer backbone or side-chain have been developed.50,51 Furthermore, due to the difficulty of preparing metallocene-containing monomers in high purity required for controlled polymerization methods, new post-modification methods have been developed. By this, the polymer, bearing a convertible functional group, can be adjusted to the required properties before introducing the metallocene. Recently, a post-modification method using amine-containing methacrylate block copolymers was developed.52 Here, the cobaltocenium moiety was introduced to form metallocene-containing block copolymers with strong microphase-separation features, ideally to control such block copolymers for applications for convenient membrane formation strategies. Although a series of ion-exchange membranes containing cobaltocenium are known in the literature,53–57 to our knowledge, no block copolymer-based functional membrane formation by the SNIPS process has been attempted so far. Furthermore, PFAS removal using a metallocene-based membrane has not been investigated yet. By using a hydrophobic polystyrene-based polymer with a second ionic cobaltocenium-containing methacrylate block segment, resulting membranes should combine key features required for PFAS removal. In this work, we aim to fabricate novel metallocene BCP-based membranes with ionic and hydrophobic cobaltocenium units at the interface. For this reason, the membrane formation via the SNIPS process of cobaltocenium-containing block copolymers is investigated, and relevant molecular and membrane formation parameters, like the cobaltocenium content, solvent, and non-solvent combinations, are explored. The applicability of the final membranes is tested for PFAS retention, evaluating the optimal configuration that balances retention and permeance. Chromate and lead retention experiments were performed to gain further insight into the membranes’ properties. Lastly, to extend the lifetime of the synthesized material after application, template-based calcination of the used membranes is investigated.Results and discussion
Polymer synthesis
As the introduction briefly addresses, two major factors play a crucial role in addressing the hydrophobic and oleophobic nature of the class of PFAS: (i) hydrophobic, aromatic interactions with the perfluorinated carbon chain and (ii) ionic interactions respecting carbon or sulfonic acid. Cobaltocenium-containing polymers show unique adsorption and desorption properties for PFAS removal. Here, not only is the adsorption promoted compared to other metallocenes, but upon electrochemically switching of the cobaltocenium to cobaltocene, the desorption proceeds readily.35 It is assumed that the combination of the hydrophobicity and ionic character of the metallocene is one major factor playing a crucial role in balancing different types of interactions. The increased hydrophobicity with increasing cobaltocenium content could be demonstrated in our recent study.52 By taking advantage of the ionic character of the cobaltIII as part of the sandwich complex, this unique property can be achieved. An overview of possible interactions of PFOA with the specialized cobaltocenium-containing BCP is given in Fig. 1. For our system, the ionic interactions are prosed to initiate the PFAS adsorption. By Coulomb interactions between the positively charged cobaltocenium complex and the generally negatively charged acid group of the PFAS, a concentration of the pollutant at the membrane's interface is proposed. Furthermore, due to the hydrophobicity of the metallocene and aromatic environment of the polymer backbone, a further increase beyond the charge effect of capacity is anticipated. This influence on different aromatic environments is known from the literature.58–62 After initial adsorption and with higher concentrations, the oleophobic nature of the PFAS becomes more prominent. In this regard, PFAS are known to form selective secondary fluorine–fluorine interactions, leading to second-layer formation. This minimizes other hydrophobic interactions but increases PFAS adsorption.63–65 Due to the high local concentration, further diffusion of PFAS through the membrane is osmotically unfavoured, which should result in a significant decrease in PFAS.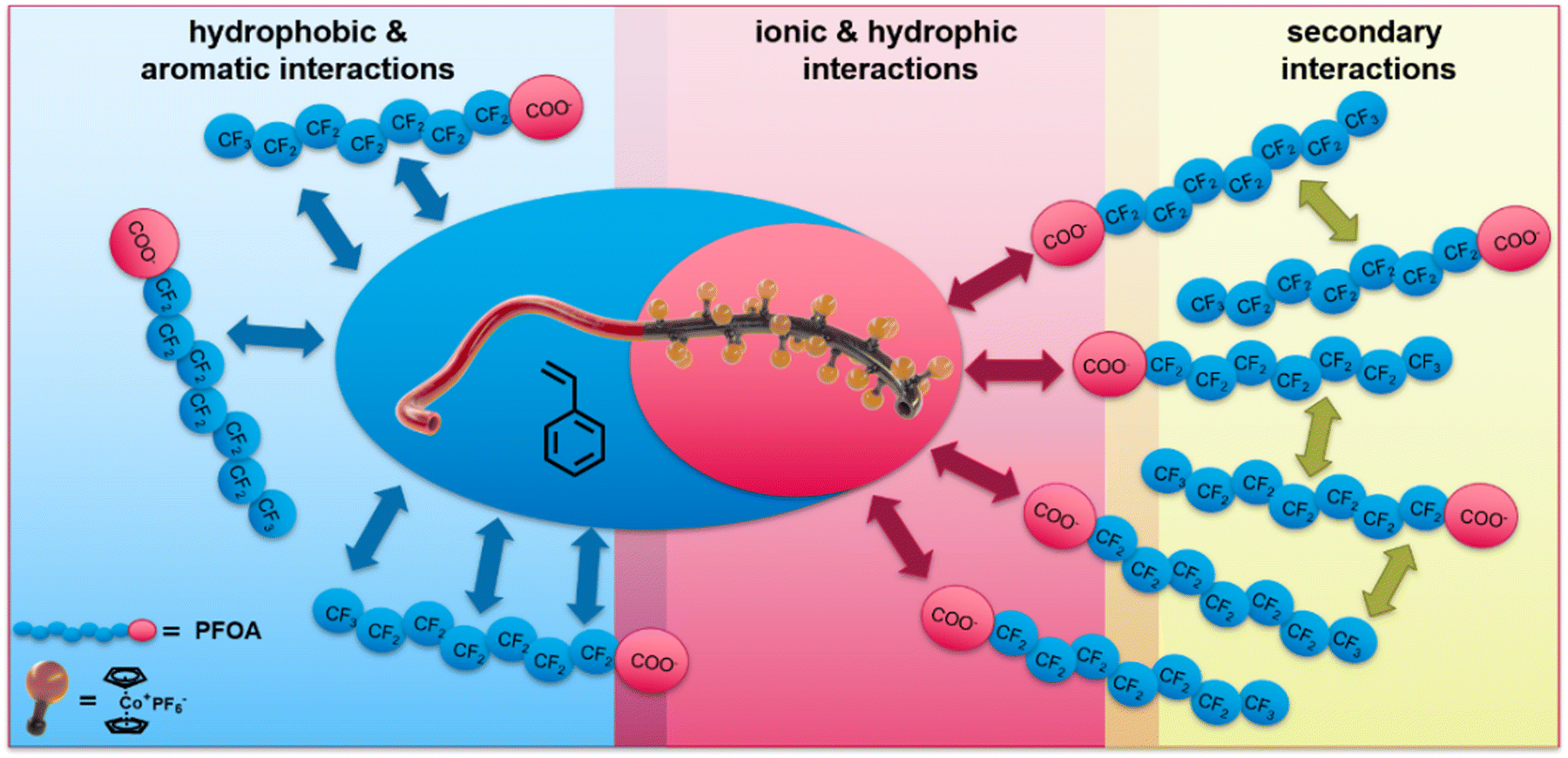 | ||
| Fig. 1 Possible interaction between the synthesized cobaltocenium-containing block copolymer polystyrene-b-(polycobaltoceniumethyl tert-butyl aminoethyl methacrylate-co-polymethyl methacrylate) (PS-b-(PCoEtBAEMA-co-PMMA)) and perfluorooctyl acid (PFOA) and resulting secondary interaction effects. | ||
Within this work, BCPs were synthesized by living anionic polymerization followed by quantitative and block-selective cobaltocenium post-functionalization. In our approach, polystyrene was utilized as the major hydrophobic building block segment to form the membrane matrix. The second minor polymer segment features the ionic cobaltocenium, enabling selective adsorption. At the interface, the cobaltocenium should be utilized for its hydrophobic and ionic interactions.
To evaluate the effect of the cobaltocenium content on membrane formation, BCPs with 5 to 40 wt% and one copolymer with 95 wt% CoEtBAEMA with Mn of 90 to 120 kg mol−1 were synthesized according to the literature. In more detail, after an initial polystyrene block, a second mixed methacrylate block consisting of methyl methacrylate and tert-butylaminoethyl methacrylate was prepared by living anionic polymerization in THF at −78 °C. After workup and analysis, the cobaltocenium was introduced via post-modification. For a more detailed description, the reader is guided to our recent work regarding this topic.52 An overview of the synthesized polymers, the dispersity, and the resulting weight percent (PXX![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) wt%) of cobaltocenium block (PCoEtBAEMA) is presented in Table 1. Additional detailed data on the BCPs used for this study can be found in the ESI† (Fig. S1–S10). As described in previous work, the synthesized polymers excel in high cobaltocenium content, quantitative block functionalization, and resulting self-assembly in the bulk state. They are, therefore, ideal candidates for membrane formation.52
wt%) of cobaltocenium block (PCoEtBAEMA) is presented in Table 1. Additional detailed data on the BCPs used for this study can be found in the ESI† (Fig. S1–S10). As described in previous work, the synthesized polymers excel in high cobaltocenium content, quantitative block functionalization, and resulting self-assembly in the bulk state. They are, therefore, ideal candidates for membrane formation.52
wt% of PtBAEMA and PCoEtBAEMA) of synthesized polymers calculated by SEC measurements and 1H NMR spectroscopy
| Sample | Polymer | M n,NMR | Đ | wt%PCoEtBAEMA(UV-vis)c |
|---|---|---|---|---|
| a PS molar masses were determined by SEC in THF (kg mol−1, PS standards) and used to calculate NMR values for the corresponding block copolymers. b Dispersity of polymers prior to functionalization measured by SEC in DMF with styrene standard. c Weight content of PCoEtBAEMA in % calculated by UV-Vis spectroscopy in THF at 489 nm. d Weight content determined by 1H NMR data of the block copolymers. | ||||
| P6.2 | PS797-b-(PCoEtBAEMA7-co-PMMA8) | 87.8 | 1.08 | 6.2 |
| P20.1 | PS773-b-(PCoEtBAEMA31-co-PMMA12) | 98.7 | 1.08 | 20.1 |
| P27.5 | PS908-b-(PCoEtBAEMA65-co-PMMA28) | 134.4 | 1.10 | 27.5 |
| P31.1 | PS773-b-(PCoEtBAEMA57-co-PMMA27) | 114.4 | 1.11 | 31.1 |
| P33.3 | PS874-b-(PCoEtBAEMA88-co-PMMA48) | 143.3 | 1.10 | 33.3 |
| P40.1 | PS776-b-(PCoEtBAEMA80-co-PMMA30) | 127.0 | 1.10 | 40.1 |
| P92.6 | PCoEtBAEMA387-b-PMMA133 | 225.5 | 1.07 | 92.6d |
Membrane formation
For membrane formation, the self-assembly and non-solvent-induced-phase separation (SNIPS) process was chosen. The general procedure is shown in Fig. 2: to summarize the process, first, the homo- or block copolymers were dissolved in a solvent or solvent mixture. Here, a rather high viscosity was targeted to be ideal for the membrane formation process.25 Next, the solution was cast via a doctor blade with a defined blade gap onto a support. After a defined evaporation time, the as-prepared polymer films were immersed in a non-solvent bath. By this, the final membrane structure is formed via the solvent-nonsolvent exchange, resulting in a typically asymmetric porous membrane structure.66,67 After drying the polymer films, the prepared materials will be investigated with respect to their potential use as membranes. Generally, the membrane formation process is influenced by a range of factors, e.g., solvent, solvent mixtures, the kind of polymer segments, humidity, doctor blade gap, substrate, casting speed, time for solvent evaporation, temperature, and the presence of additives.68 By adjusting these parameters, the membranes’ properties, such as the pore structure, amount of caverns in the sublayer, and resulting water flux, can be strongly influenced.24,69 Due to the novelty of the metallocene-based BCP system, the membrane formation process had to be optimized first. To decrease the overall parameters, we focused on different cobaltocenium contents as well as the solvents used for the polymer solutions. The first membranes were fabricated by using a 31 wt% polymer solution in N-methyl-2-pyrrolidone (NMP) on a nonwoven polyester support. The membrane thickness was adjusted by using a doctor blade with a gap of 200 μm, and the evaporation time was fixed to 15 to 17 seconds before the non-solvent exchange. The polymers were precipitated in salt water with a sodium chloride content of 3.5 wt%. For pore formation, saltwater precipitation and low humidity of below 20% seemed to be essential, as no porous membranes could be obtained without the presence of salt in the bath. For the latter case, without salt additives, only a smooth and dense surface was obtained. Different non-solvents for the BCPs were tested, i.e., ethanol, hexane, and diethyl ether, but no pore formation or integral asymmetric membranes could be obtained. Similar aspects like the ionic solvent requirement and a slow solvent exchange are most likely the reason. For this polymer system, saltwater precipitation was found to form the optimal porous membrane structures. It appeared that the ionic non-solvent was necessary during the pore formation to guide the micelle-to-membrane transformation by ionic stabilization. For other systems, the influence of salt additives on pore formation was explored.25 In our case, due to the ionic cobaltocenium, no further salt was used in the initial casting solution. Interestingly, the casting solutions show strong thixotropic behavior, further improving the formation process. For the SNIPS process, dimethylformamide (DMF), dimethylacetamide (DMAc), and NMP are commonly used.70 For initial testing, NMP was used to evaluate the degree of cobaltocenium required and the influence of the metallocene on the overall pore and membrane formation. Polymers with an amount of 6 to 40 wt% metallocene were evaluated. In contrast to the BCPs, a copolymer with almost quantitative 96 wt% cobaltocenium monomer but without the polystyrene was additionally tested to further elaborate on the advantage of the block copolymer self-assembly process compared to the statistical copolymer. For differentiation, the wt% of cobaltocene-containing monomer in the synthesized polymer is used as indices.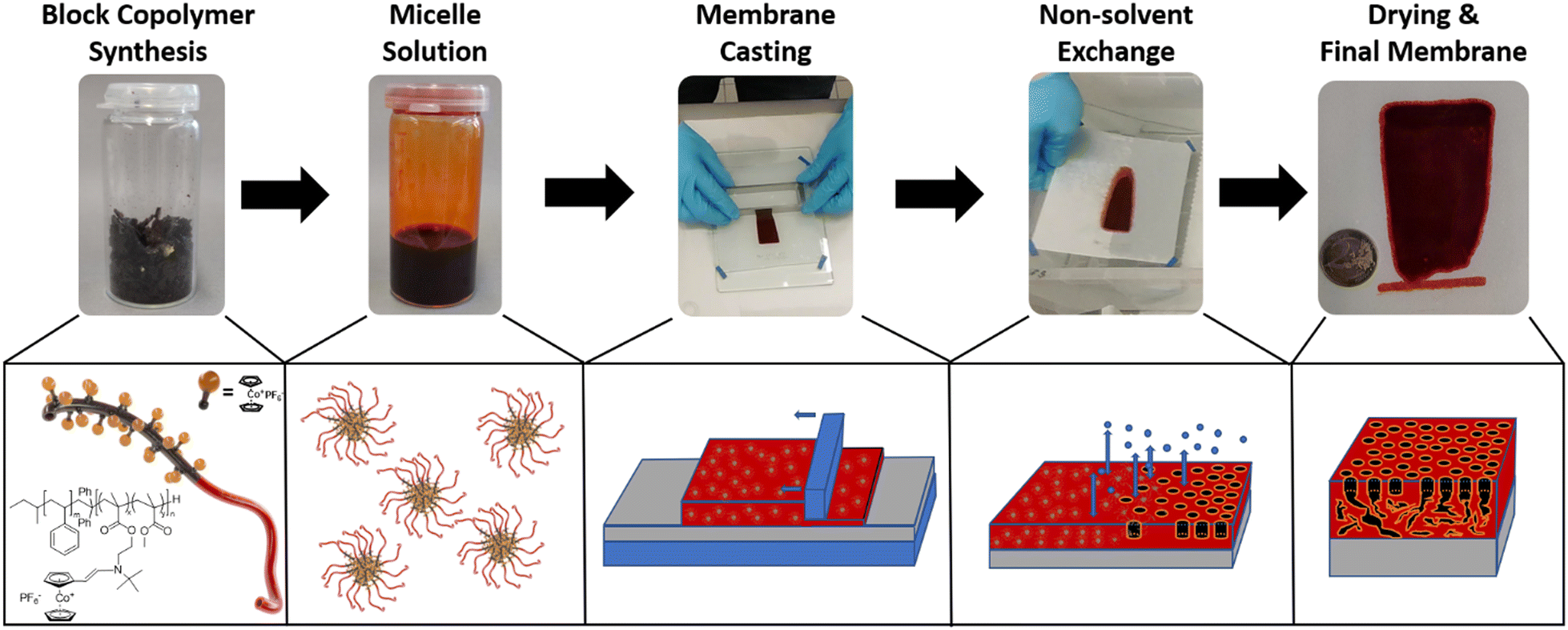 | ||
| Fig. 2 Different steps of the membrane formation using the self-assembly and non-solvent induced phase separation process (SNIPS) starting from block copolymer (BCP) synthesis, over the formation of a micellar solution, membrane casting, non-solvent exchange and drying to form the final porous membrane. | ||
As presented in Fig. S11 (ESI†), a difference in the membrane's macroscopic appearance was found. At low cobaltocenium content of P6.2, the membrane was elastic and homogeneous, whereas, at higher cobaltocenium content, e.g., sample P40, the membrane became brittle. This behavior was expected due to the higher glass transition temperature, Tg, and ionic character of the cobaltocenium block copolymer segment. In comparison to the BCPs, the P92.6 copolymer was very brittle and, therefore, not suitable for membrane formation. A polymer metallocene content between 25 and 35 wt% seemed to be optimal for membrane stability and maximum cobaltocenium incorporation. Next, the membrane surface was investigated in detail by scanning electron microscopy (SEM).
In conclusion, from the SEM investigations of the membranes’ topography given in Fig. S12 (ESI†), a porous surface in all cases was found; therefore, the SNIPS process could be applied to this metallocene-based block copolymer system. Except for P6.2, where porous topography could only be found in certain areas, all other samples showed a continuous porous interface. Furthermore, a disordered sponge-like surface structure was formed in all analyzed samples. The pore size was calculated by analyzing the SEM images with the “imageJ” analytical software (cf. instrumentation section). Here, a random angle model was used for the respective size measurements due to the slanted pore structure. Pores sizes were found to vary only slightly in the range from 17 ± 4 to 23 ± 8 nm with increasing cobaltocenium content. This effect might be caused by the strong ionic interactions of the cobaltocenium moieties, as also previously observed for the microphase-separated morphologies in the bulk state.52 It is assumed that the casting conditions played a crucial role in the formation of the pore structures. In our case, polymers like P31.1 with a cobaltocenium content of 30 wt% revealed the highest pore density and the most homogeneous structure. For the copolymer P92.2, no porous structure was found (Fig. S11, ESI†). Here, a rough surface split by major cracks was found in SEM, rendering these polymers unusable for membrane fabrication and further displaying the benefit of the BCP approach. Due to the novelty of our system and to reduce the number of influencing factors, the optimized casting conditions (200 μm bandgap, evaporation time of 15 seconds, 18% relative air humidity, and precipitation in water with 3.5 wt% NaCl) were kept constant for the following experiments. In summary, the macroscopic and microscopic features of the novel metallocene-based membranes indicate the optimal conditions for obtaining a homogeneous porous layer at a cobaltocenium content of 30 wt%.
To further investigate the nature of the metallocene membrane formation, two BCPs with cobaltocenium contents of 27 (P27.5) and 33 wt% (P33.3) were synthesized in larger quantities by repeating the synthesis. In literature, a dependency of the molecular weight of the BCP on the pore size was proven.25 To test this for the case of metallocene-containing block copolymers and to further tailor the underlying pore structure, a higher molecular weight of approximately 140 kg mol−1 was chosen. Moreover, the solvent dependency on pore formation was further investigated. For this purpose, in addition to the pure NMP, the membrane formation was carried out in DMF and DMAc. Each polymer casting solution was adjusted to roughly match the viscosity of the previous NMP solution. This resulted in a higher polymer content of 42 wt% for DMF and a similar NMP content of 32 wt% for DMAc. All other casting parameters were kept constant and as described above. The respective membrane surfaces and cross-sections analyzed by SEM are given in Fig. 3. Compared to the membranes with 30 wt% cobaltocenium in the block segment in the solvent NMP, a very similar structure with sponge-like slanted pores was found at a higher molecular weight and slightly lower metallocene content of 27.5 wt% for P27.5. With 36 ± 11 nm, the pores, as well as the deviation, were larger compared to the 23 ± 8 nm of P30.1. This further confirms the hypothesis of a strong pore size dependency on the overall polymer length represented rather than the cobaltocenium content. Moreover, by adjusting the polymer size, the pore dimensions can therefore be tailored.
 | ||
| Fig. 3 Scanning electron microscopy (SEM) images of surface and cross sections of membranes of P27.5 (27.5 wt% cobaltocenium block segment) made of casting solutions of N-methyl-pyrrolidone (NMP) (top), dimethyl formamide (DMF) (middle), and dimethyl acetamide (DMAc) (bottom) with respective pore sizes. | ||
Very similar to previous results, a dense porous substructure was found for all investigated samples. A graphical representation of this structure is presented on the left side in Fig. 3. Untypically for SNIPS membranes, the sponge-like surface structure resembled the membrane cross-section very well. For SNIPS membranes, normally, an asymmetric structure is dominant in comparison to the symmetric cross-section architecture found.24 This symmetric structure hints toward a very steady solvent exchange during the formation process. In literature, this dense formation was found for highly concentrated polymer solutions.71 This reason seemed very likely since polymer solutions of 30 to 40 wt% were used. For now, the influence of the respective solvent on the membrane surface was evaluated. By using DMF for the casting solution, a different surface structure was obtained. Here, contrary to the previously found sponge-like structure, a flatter surface with rounder and uniform pores was obtained. Unlike pores formed by applying the NIPS process, where pores are formed via the nonsolvent contact and quick exchange with the solvent, resulting in the breakup of the micellar polymer solution, a different method is suggested here. The structure resembles the arrangements found in the SNIPS process based on the microphase separation of the BCPs.72 To a certain extent, this behavior can also be observed in the substructure architecture. Comparable to the NMP-derived membranes, in DMF, similar pore sizes of 37 ± 11 nm were found. Although the pores looked more uniform, a similar deviation was found compared to the previous membrane.
Last, the influence of DMAc revealed a very similar picture to the previously generated structure using NMP. Here, a slightly smaller pore size of 33 ± 12 nm, but a higher pore density was found. This higher pore density was proposed to be better for an increased water flux and permeability. Contrary to the previously discussed process, the better miscibility of DMAc with water compared to NMP in the SNIPS-type assembly was assumed to be the reason for the denser pore formation. As for the other investigated solvents, a sponge-like continuous substructure was found. Lastly, it shall be noted that only marginal differences between P27.5 and P33.3 were found. To conclude, membranes formed by BCPs containing approximately 30 wt% cobaltocenium monomers were found to possess the best properties for pore development. The formation could further be influenced by the respective solvent used. Here, membrane surfaces from DMF indicated a pore formation process via self-assembly but lower pore density in contrast to membranes cased from NMP or DMAc. For DMAc especially, the number of pores was drastically increased compared to initial NMP membranes.
PFAS retention
To investigate the membranes’ PFAS and ion retention measurements, an 8 × 4 cm membrane was prepared from a 31 wt% DMAc polymer solution of P33.3 by precipitation in saltwater after 15 seconds of evaporation at 21 °C and a humidity of 20%. The resulting membrane after casting, the surface, and the cross-section are presented in the ESI† (Fig. S13). Here, no significant change to the prior fabricated membrane was found.Membrane experiments like the water flux and analyte retention were performed in a dead-end filtration system with 0.4 bar pressure and membrane diameters of 10 mm. The resulting time-resolved permeance and PFOA retention measurements are presented in Fig. 4. First, the neat water permeance was performed as a reference to compare the possible fouling effects caused by the eluent used. The initial water flux measurement is presented as part of Fig. 4a. Here, an initial flux reduction was observed, which is typically found for this kind of membrane.73 Due to the compression of the membrane, a slight decrease in permeance was expected due to the deformation of the polymeric material, resulting in reduced porosity.74 After 110 minutes, the water flux stabilized at a value of 521 ± 67 L h−1 bar−1 m−2. After this membrane stabilization, the flux measurement was stopped, and the ultra-pure water was exchanged for PFOA-spiked water. To simplify the otherwise very complex analysis, we decided to focus only on the retention of PFOA as a representative of this vastly diverse group of pollutants for initial studies.
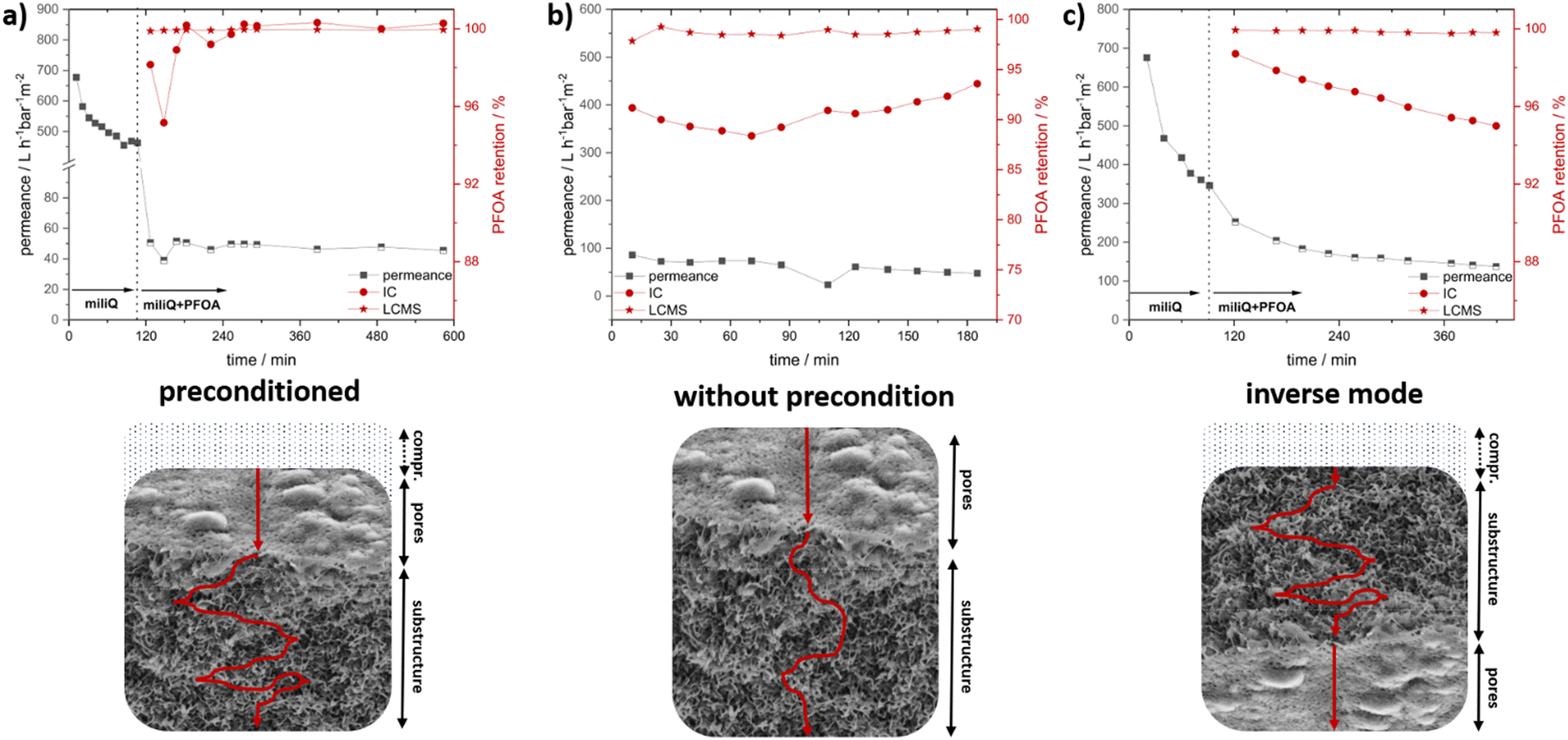 | ||
| Fig. 4 Water flux measurements and PFOA retention experiments of membrane casted from polymer P33.3 in DMAc in different modes: (bottom) of a dead-end setup at pressure of 0.4 bar; (a) initial water flux measurement with pure water, followed by eluent change to PFOA spiked water and resulting water flux and PFOA retention observed by ion chromatography (IC) and liquid chromatography–mass spectroscopy (LC-MS); (b) water flux and PFOA retention measurements with spiked PFOA solution without preconditioning measured by IC and LC-MS; (c) initial water flux measurement with pure water, change to PFOA spiked water and resulting water flux and PFOA retention analyzed with an inverse membrane setup by IC and LC-MS. | ||
It should be noted that more complex perfluorinated compounds, like GenX, were analyzed in a recent study by Medina et al. with very similar polymers.35 For the first insights on the membrane performance and separation capabilities, we focus on PFOA, while future studies will contain complex analyses as well as legacy PFAS.
For our experiments, a concentration of PFOA of 2.556 mg L−1 was chosen to enable several analytical methods to analyze the PFOA reduction after the adsorption and to check the capacity of PFOA adsorption. Liquid chromatography coupled with mass spectroscopy (LC-MS) and ion chromatography (IC) were used for PFOA quantification and for gaining insights into the membranes’ separation performance. The initial PFOA sample was taken from inside the apparatus to verify the correct starting concentration, and no relevant adsorption was found for the apparatus. This initial value was then compared to the results obtained after the filtration steps. Directly after changing to the PFOA-spiked water, a sharp decrease in water flux was noticed. This was rather expected since, due to the ionic nature of the membrane, by introducing a different ionic species, the intrinsic repulsion started to affect the water flux. In addition, for PFAS, which is widely used for its strong surface interactions and combability, a strong fouling effect resulting in a drastically reduced flux is described.73 In our case, it is assumed that due to the strong ionic and hydrophobic interactions, the PFOA adsorbed onto the membrane interfaces, which consisted of cobaltocenium moieties. This increased the local concentration, and the usable pore size decreased, leading to a decrease in water flux. Surprisingly, a very stable mean permeance of 48 ± 2 L h−1 bar−1 m−2 was recorded over 8 hours, which points to the fact that this change of surface polarity occurred in a fast way. Respecting samples at certain periods were analyzed regarding the PFOA content. Here, samples were collected over intervals of 20 minutes, and resulting PFOA retention percentages are presented as an interval average. Over the whole experiment, a mean reduction of 99.3 ± 1.1% compared to the stock solutions was found by IC. Within the first minutes of the experiment, a slightly decreased retention was found. Interestingly, the course of the water flux was represented by the PFOA retention. The reason for this is assumed to be caused either by the change in the eluent or induced by the initial PFAS adsorption accompanied by a layer formation. After one hour, the equilibrium was reached, and over the next six hours, PFOA retention was found to be very close to 100% for all the following samples. Here, for some cases, values slightly above 100% retention appeared, which were caused by calibration and the detection and calibration limit of the instrument. LC-MS was utilized to analyze this specific region better. Here, a very similar trend with an even higher average retention of 99.9 ± 0.1% was found via LC-MS. Interestingly, a small decrease in retention of 0.1% could be found for the initial samples, verifying the observation by IC. The permeance and retention values of all experiments were compiled in Table 2. Next, several experiments were performed to find the optimal configuration combining high water flux and PFOA retention. For this purpose, the first experiment, including extensive preconditioning, was compared to (I) a measurement without preconditioning by directly using a PFOA-spiked solution and (II) an inverse flow, where the membrane is switched upside down. The compiled water flux and retention are presented in Fig. 4b and Fig. 4c, respectively. In these experiments, the mean time of the analyte spent inside the membrane was to be assessed.
| Experiment | Permeancea/L h−1 bar−1 m−2 | PFOA retention ICb/% | PFOA retention LC-MSc/% | Cr retentiond/% | Pb retentiond/% |
|---|---|---|---|---|---|
| a Permeance was determined gravimetrically. b PFOA concentration was analyzed by ion chromatography (IC) (see instrumentation section and main text for more details). c PFOA concentration was determined by liquid chromatography coupled mass spectrometry (LC-MS). d Determined by inductively coupled mass spectrometry. e Under the detection limit. | |||||
| Pure water | 521 ± 49 | — | — | — | — |
| Precondition (PFOA) | 48 ± 2 | 99.3 ± 1.1 | 99.94 ± 0.02 | — | — |
| Without Precondition (PFOA) | 61 ± 13 | 90.6 ± 1.2 | 98.66 ± 0.27 | — | — |
| Inverse membrane (PFOA) | 171 ± 26 | 96.6 ± 1.0 | 99.86 ± 0.06 | — | — |
| Sodium chromate retention | 41 ± 3 | — | — | 46.45 ± 0.05 | — |
| Lead acetate retention | 30 ± 14 | — | — | — | 99.85 ± 0e |
Here, depending on the time, there is a higher possibility of interaction with the membrane interface, and proposed secondary PFOA interactions should lead to increased retention. By first compressing the membrane with pure water during the preconditioning, a higher mean time in the membrane is proposed (Fig. 4b, bottom). Additionally, due to the compression, the number of accessible pores was reduced, forcing the analyte through lesser pores, resulting in a higher local PFOA concentration, which is beneficial for retention. To evaluate this compression effect, the second experiment was performed using PFOA-spiked water directly. This way, a shorter meantime in the membrane (Fig. 4b, bottom) and a resulting worse PFOA retention were expected. To our surprise, a very similar and stable permeance of 61 ± 13 L h−1 bar−1 m−2 was found compared to the measurement right from the start. Here, especially for the initial measurements where the compression of the membrane did not occur, the PFOA retention measured by IC was significantly lower, averaging at 90.6 ± 1.20% over the experiment. This expected trend was further validated by LC-MS, where a slightly lower retention of 98.7 ± 0.3% was found as compared to the original value. Compared to the values found by IC, a larger deviation was expected, but due to the sophisticated PFOA analysis, a direct comparison of both methods turned out to be difficult. Nevertheless, both methods showed similar results, underpinning the important influence of the average time inside the membrane as a key factor for retention and contact time inside the membrane. The permeance is generally an important key parameter for membrane applications. Regarding this, in our next experiments, we aimed to increase the flux without losing the quality of PFOA retention (Fig. 4c, bottom). Since compression affected the retention, a reduction in compression, which would otherwise help the permeance, could not be utilized. For this reason, a different approach was developed. Here, the synthesized membrane is used in an “inverse flow mode” with the supporting substrate and sponge-like substructure directed upwards. The main limiting bottleneck, the porous structures with the smallest pores, was now located at the final filtration point of the integral asymmetric membrane. This way, fouling by concentrating the analyte at the pore surface and the resulting decrease of accessible pores can be drastically reduced since the analyte first had to diffuse through the sublayers. Here, due to the high surface area and narrow network, most of the analyte-cobaltocenium interactions, as well as secondary PFOA interactions, are proposed. Hence, the overall concentration of analyte was assumed to be decreased, reducing the fouling at the porous surface area. By compressing the membrane this way, the force is directed on the substructure rather than on the surface-layer pores, and in combination with the inverse flow direction, leading to less overall pore compression. Nevertheless, compression of the substructure turned out to be advantageous due to the resulting increase in mean dwell time within this layer. Surprisingly, the results derived during the initial stage of the filtration experiment were very similar to the experiment averaging at 441 ± 81 L h−1 bar−1 m−2. After the change to the PFOA-spiked water, a gradual decrease in permeance was found. In contrast to the steep decline for the initial experiment, this was a significant improvement for the filtration experiment. Over the first two hours, the permeance further decreased but stabilized at around 150 L h−1 bar−1 m−2 for the rest of the experiment. By the inverse configuration, a 3-fold increase in permeance compared to the permeance of 48 L h−1 bar−1 m−2 of the normal compressed mode was found. Next, the PFOA retention was analyzed. In contrast to both prior measurements, an average value of 96.6 ± 1.0% was found. This decrease compared to the first measurement might be caused by the increased flow, reducing the mean dwell time in the membrane, and the reduced repelling effects due to the inverse setup. LC-MS found a similar trend. Here, an average retention of 99.8 ± 0.1% was found, which was lower than the initial one but significantly higher than the second experiment. To conclude, we could show the feasibility of metallocene-based membranes for PFOA removal, and we identified key factors like compression and increased dwell time that affect retention. Furthermore, the permeance could be improved 3-fold by a rather simple inverse setup with only minor reductions in retention.
Chromate and lead retention
As the primary hypothesis, namely the adsorbtion of PFAS via the cobaltocenium membrane, has already been validated, the ionic character of the membrane and potential additional applications beyond PFAS adsorption warrant further investigation. To this end, two prominent water contaminants – lead and chromate – were selected for further “proof-of-principle” investigation (Fig. 5). Moreover, the membrane was evaluated for its feasibility in environments with exceedingly high salt concentrations.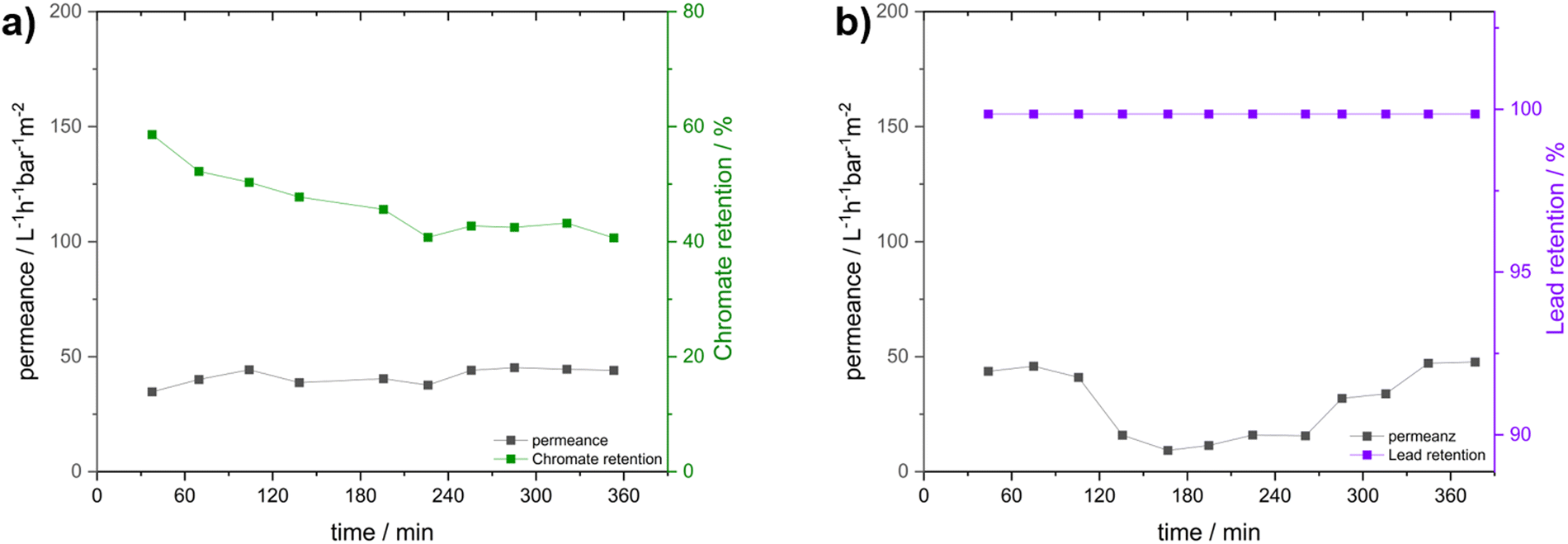 | ||
| Fig. 5 Water flux combined with sodium chromate and lead nitrate retention measurements analysed inductively coupled plasma mass spectroscopy (ICP-MS); (a) compiled water flux and sodium chromate retention; (b) compiled water flux and lead nitrate retention. | ||
In comparison to PFOA, the repelling effects were most likely dominated by ionic interactions here since no further hydrophobic interaction was possible. For this reason, sodium chromate (Na2CrO4) was used as an anionic pollutant. Additionally, the effect of the highly oxidative compound and the effects on the cobaltocenium membrane were investigated. Here, a concentration of two hundred times the regulatory limit (0.05 mg L−1) for drinking water of 10 mg L−1 was chosen, and a very similar concentration of 9.8 mg L−1 was found for the prepared stock solution.75 Similar to the above-described experiments, a dead-end filtration apparatus and a pressure of 0.4 bar were utilized. Inductively coupled plasma mass spectroscopy (ICP-MS) was used for chromate and lead quantification. For Cr, this yielded a rather different result than the hydrophobic PFAS (Fig. 5a). At a permeance of 41 ± 3 L h−1 bar−1 m2, an average retention of 46.4 ± 0.1% was found. Unlike before, where the retention was increased during the experiment, this time, a decrease from 60% to 40% compared to the stock solution was found after four hours. After that time, a stable 40% was found, indicating the equilibration of the system. The mainly cationic membrane interface did not directly repel the negatively charged chromate but rather hindered diffusion through the membrane by creating a high local concentration of the pollutant on the membrane surface and in the pores.76 Additionally, an ion exchange with hexafluorophosphate, the initial cobaltocenium counterion, may increase the local concentration. Nevertheless, we showed that for PFOA retention, not only ionic interactions but hydrophobic and secondary interaction effects played a major role. Additionally, in all cases, the membrane surface was analyzed afterward by SEM (Fig. S14, ESI†), showing no degeneration or loss of the porous membrane structure or significant change, demonstrating the inertness of the synthesized membranes.
Last, to demonstrate the cationic nature of the membrane, the retention of a cationic pollutant was to be tested. Here, we decided to focus on lead since it is one of the prominent examples in literature and causes problems in drinking water purification up to date.77 For this reason, a very similar experiment with the same conditions as for sodium chromate was performed for lead nitrate (Pb(NO3)2). Here, the 100-fold drinking water limit (0.01 mg L−1) of 1.0 mg L−1 was targeted.75 As later revealed by ICP-MS, the concentration of the prepared stock solution was slightly higher at 1.16 mg L−1. Despite this slight deviation, this should not have too much influence on the experiment since the measured stock solution was always used for the ratio determination, and a concentration of the same order of magnitude was used. Similar to prior experiments, the combined water permeance and lead retention are displayed in Fig. 5b.
Again, a similar mean permeance of 30 ± 14 L h−1 bar−1 m−2 was found. Here, initially, there was a reduction of the permeance, but surprisingly, for later samples, an increase was found. Comparable with the other samples, the initial decrease was likely caused by fouling due to the increased lead concentration. For the rise in permeance after the first 180 min, partial destruction of the membrane due to the pressure or a change in formation due to the introduced lead was proposed. However, analysis of the surface after the experiment by SEM did not show any significant change (Fig. S4, ESI†). Here, only the impacts of the initial preconditioning and, therefore, reduced pore content was found, as for every other sample. The increased permeance could also be an effect of the ion exchange from hexafluorophosphate (PF6−) to nitrate, making the membrane more hydrophilic and, therefore, more accessible for water. However, for now, the reason behind the increase has not been identified. More interestingly, in the analyzed residual water after filtration, no lead could be found by ICP-MS, resulting in a quantitative retention of 99.9% for all samples.
Lastly, to view possible limitations with very high salt concentrations, the membrane was tested in a simulated seawater environment (Fig. S15, ESI†). For this, a 3.5 wt% sodium chloride solution was used for the permeance experiment. The results are presented in Fig. S10 (ESI†). To our surprise, even at these high salt concentrations, the permeance stabilized at a value of 18 ± 1 L h−1 bar−1 m−2 after a short compression phase. In this regard, membrane optimization has to be performed as well, but for now, general applicability could be shown.
To conclude, in addition to the excellent PFAS retention, the membrane's ionic nature was demonstrated by significant chromate and quantitative lead nitrate reduction. In addition, the chemical robustness against strong oxidizing agents, sodium chromate, and tolerance of high salt concentrations was shown.
Regeneration, recycling and up-cycling
Lately, recycling and up-cycling of polymer-based materials have gained significant attention, especially due to the high prices for specialized resources like cobalt. To address this aspect, several attempts to prolong the membrane lifetime, recycling, and upcycling the porous cobalt-containing structure are presented. As mentioned earlier, upon electrochemical switching, cobaltocenium polymers can readily release PFAS. In our case and with our setup, electrochemical switching was not possible due to material and apertural restraints. For adequate switchability over the entire membrane, sufficient conductivity was required, which was not intrinsically available. For now, we investigated the redox switchability by surface contact cyclic voltammetry. Here, the initial polymers with 5 to 40 wt% cobaltocenium content were used. In more detail, a platinum working and counter electrode and an Ag/AgCl reference electrode in acetonitrile were used for measurement. Referencing was done separately against ferrocene (Fc/Fc+). As can be concluded from the obtained electrochemical results in Fig. S16 (ESI†), a reversible electron transfer from CoIII to CoII could be found at E1/2 = −1,54 V (vs. Fc/Fc+) and with an ΔEp= 0.07 to 0.2 V. In sample P6.1, next to the primary signal, a smaller signal could be found in the −1.4 V to −1.2 V region, which was most likely caused by the conjugation with the respective amine.78 Overall, all samples showed excellent addressability with increasing cobaltocene content. When less cobaltocenium is present, the overall shape of the respective signal is still heavily influenced by the conjugation to the respecting amine, resulting in broader spectra and, in the case of P6.1, in an additional peak. In contrast, at a higher content of 30 or 40 wt%, only one sharp signal was found. An explanation for this might be a metallocene-to-metallocene charge transfer rather than a transfer to the respective linker. With increasing cobaltocenium content, the local concentration increases, resulting in a more likely charge transfer, and, except for P20, an increase in signal strength was found. It is proposed that this was caused by the very intricate nature of CV measurements. Only slight variations of the measuring environment, like worse contact of the polymer film to the electrode, may yield different results. Overall, the good electrochemical switching capabilities of the synthesized polymers could be demonstrated, paving the way for future electrochemical applications.Next to regeneration and recycling, up-cycling, where used material is repurposed for different applications, presents an alternative. Here, metallocene-containing structures are ideal candidates for template-based calcination.42,78 By calcination in either an oxidative or reductive atmosphere, different ceramics are formed. Possible applications of these materials range from catalytic to energy storage and energy storage solutions.79,80 Due to the uniform and porous structure of the formed membranes, obtained template-based porous ceramics present a high potential. In addition, due to the high temperatures, possibly adsorbed PFAS impurities are either evaporated or mineralized by the calcination process, and purification and up-cycling can thereby be combined, increasing the material's lifetime and reducing costs.
For this reason, calcination of the larger DMAc membrane of P33.3 after initial PFOA, chromate, and lead retention experiments were performed in synthetic air and nitrogen atmosphere. The resulting ceramics synthesized by this template design in a nitrogen atmosphere are displayed in Fig. 6. Here a standard protocol up to 800 °C with 10 K min−1 was used. In all cases, a very similar ceramization profile with a singular degradation step with an onset of 364 °C was found. (Fig. S17, ESI†) In the case of the oxygen atmosphere, a lower ceramic yield of 2% was found in comparison to the 8% in a nitrogen atmosphere. This is rather expected since, in an oxidative environment, the carbon content is reduced by oxidation to carbon dioxide. The ceramics were analyzed via SEM and energy-dispersive X-ray spectroscopy (EDS).
 | ||
| Fig. 6 Scanning electron microscopy (SEM) images of ceramic material formed by template-based calcinating of used membranes in a nitrogen atmosphere to 800 °C. Membrane ceramics after PFOA (MCPFOA), lead nitrate (MCLead) and sodium chromate (MCChromate) retention experiments were analysed. | ||
After treatment in an oxidative environment, a very similar structure was found for all samples (Fig. S18, ESI†). Here, a porous structure featuring a crystalline surface was synthesized, strongly resembling the membrane sublayer. A slightly denser structure was found compared to the precursor membrane, which was expected due to the shrinkage of the overall material. Next, the composition was analyzed by EDS. In literature, the formation of cobalt oxides is mainly found in cobaltocenium polymers.81,82 In our case, a cobalt content of up to 34% could be found. In addition, primarily oxygen (36 wt%), carbon (16 wt%) and phosphorous (14 wt%) contributed to the overall composition. The phosphorous was most likely introduced by the PF6− counter ion; for the ceramic resulting from the lead retention, no residual lead could be detected. In contrast, a significant amount of chrome (3 wt%) was found for the other membrane. Additionally, the phosphorous content was slightly reduced, further facilitating the idea of a counter ion exchange with the respecting PF6−.
In contrast, the bulky but porous structure found for the ceramic prepared in oxygen, ceramics formed in a nitrogen atmosphere, showed a remarkably delicate ordered structure (Fig. 6). This gyroid-like structure could be found under the former pore surface in all ceramics formed in nitrogen. It is, therefore, suggested that the sponge-like sublayer of the membrane is the main driving factor for this formation. In all cases, the resulting ceramic was analyzed via EDS regarding the overall composition. As expected, a lower cobalt content of 10 wt% was found compared to the ceramic formed in oxygen. Additionally, a significant carbon content of 65 wt% alongside oxygen (19 wt%) and phosphorous (6 wt%) was found. A similar composition was found for the ceramic prepared in a nitrogen atmosphere. Like before, no lead but chromate (1 wt%) could be found. Along with the higher ceramic yield, this method showed promise for further template-based investigations. For a more detailed description of the composition (Table S2, ESI†) and EDS mappings (Fig. S19–S24, ESI†), the reader is referred to the ESI.†
Experimental
Materials
All chemicals were purchased from Fisher Scientific (Hampton, NH, USA), TCI (Tokyo, Japan), Sigma-Aldrich (St. Louis, MO, USA), Th. Geyer (Renningen, Germany), Deutero (Kastellaun, Germany), and used as received. For anionic polymerization, THF was dried over 2,2-diphenyl hexyl lithium and directly distilled in the flask for polymerization. Monomers were dried over CaH2 and purified using the standard procedure. Anionic polymerizations were performed at −78 °C inside a glovebox by using a cooling well. The polymerization protocol was reported earlier.52Instruments
Scanning electron microscopy (SEM) was measured on a Zeiss Sigma VP device (GeminiSEM 500) using the software SmartSEM Version 6.07. The samples were mounted on an aluminum stud using adhesive carbon tape and sputter-coated with approximately 6 nm platinum using an Automatic Turbo Coater PLASMATOOL 125 SIN 2020_131 from Ingenieurbüro Peter Liebscher.Cyclic voltammetry (CV) was performed by using a BioLogic SP-150 as the potentiostat in a voltammetry cell with a three-electrode configuration with an Ag/AgCl reference electrode in acetonitrile, a Pt-wire for the counter electrode, and a Pt working electrode with an inner diameter of d = 2 mm. The measurements were conducted with a scan rate of 200 mV s−1 in 0.1 M solution with tetrabutylammonium hexafluorophosphate ([TBA][PF6]). Ferrocene was used as a calibration agent, and the evaluation proceeded with EC-Lab V11.46.
Ion chromatography was performed on a Metrohm Compact IC Flex equipped with an 800 Dosino and 858 Professional Sample Processor. For PFOA analysis, the Metrohm Application 8.000.9053EN for “Trace-level detection of perfluorinated compounds in water by suppressed ion chromatography with inline suppression” was used with slight variations. In more detail, separation was achieved by isocratic elution on a reversed-phase column thermostated at 35 °C using an aqueous mobile phase containing boric acid and acetonitrile. For the column, a NUCLEODUR 100-5 C18 ec, 5 μm, 125 × 4.6 mm from Machery-Nagel was used. For the eluent, 20 mmol L−1 boronic acid and 4 mmol L−1 NaOH (pH-adjusted to 8) in 35% acetonitrile were used. The flow was adjusted to 1 mL min−1 with an injection volume of 200 μL. Boronic acid and sodium hydroxide EMSURE from Merck and pentadecafluorocarprylacid (PFOA) (96%) from Thermo Scientific were used.
For ICP-MS measurements, ultrapure water (0.055 μS cm−1) from a PURELAB® Chorus 1 ultrapure water filtration unit (Elga LabWater) was used to prepare all solutions. From the filtrates, 20 μL were taken for the Cr measurement (dilution factor: 500) and 100 μL for the Pb measurement (dilution factor: 100). A solution with 10 mg L−1 of Sc (1 g L−1 in 5% HNO3, Alfa®), Y (1 g L−1 in 2–3% HNO3, Merck CertiPUR®) and Ho (1 g L−1 in 2–3% HNO3, Merck CertiPUR®) in ultrapure water was prepared as the internal standard stock solution for all ICP-MS measurements. HNO3 (ROTIPURAN® Supra 69%, Carl Roth) was used to acidify the measurement solutions. Argon 5.0 (Ar ≥ 99.999 mol%, ALPHAGAZ™ 1 Argon, Air Liquide) was used as plasma gas for ICP-MS measurements. For quantification purposes, an external calibration was prepared using Cr (1 g L−1 in water, Fluka) and Pb (1 g L−1 in 0.5 mol L−1 HNO3, Merck CertiPUR®) ICP-MS standard solutions. For the ICP-MS measurement, an Agilent ICP-MS system 8900 with triple quadrupole (ICP-QQQ) and SPS4 autosampler was used. 52Cr and 208Pb were measured in He collision gas mode, whereby 208Pb was additionally measured without any cell gas.
Liquid chromatography coupled with mass spectrometry (LC-MS) was performed by using a Shimadzu LC20-AD-XR HPLC system coupled with a Bruker solariX 7T FT-ICR-MS. For MS analysis, 1 mL of each sample measured before by IC was taken and measured in the same individual concentration, avoiding any further influences on the samples. Chromatography was performed as an indicator for qualification due to the retention time. 10 μL of each sample were eluted isocratically with 50% acetonitrile/water (v/v) using a Multospher 120 RP18-AQ-3μ column at 50 °C (CS Chromatographie Service GmbH, Langewehe, Germany). The sample was measured with Electrospray-Ionization (ESI) in negative polarity. All measurements were done as triplicates. The data were processed using Bruker Compass Data Analysis software 6.0. Processing parameters: extracted ion chromatogram (EIC) width: 0.01 Da; smoothing algorithms: Gauss, 1.24s, 10 cycles.
SNIPS process
An appropriate amount of cobaltocenium containing polymer PS-b-(PCoEtBAEMA-co-PMMA) was dissolved in a respective solvent and shaken on a shaking plate for 16 h. A honey-like viscosity was targeted. The casting process was done inside a custom climate chamber made by Florian Frieß (Polymer Chemistry Group, Saarland University). The polymer solution was cast via a doctor blade with a gap width of 200 μm on a nonwoven polyester support and added into a room-temperature water bath after 15 to 17 seconds. Five minutes later, the membranes were removed and dried for 16 h in RT and 4 h at 40 °C in a vacuum. In Table 3, the exact measurements for the respecting polymer solutions are given.| Sample | Polymer/g | NMP/g | DMF/g | DMAC/g | Polymer/% |
|---|---|---|---|---|---|
| P6.2 | 0.065 | 0.141 | — | — | 31.4 |
| P20.1 | 0.094 | 0.205 | — | — | 31.4 |
| P27.5 | 0.100 | 0.219 | — | — | 31.3 |
| P31.1 | 0.099 | 0.217 | — | — | 31.4 |
| P33.3 | 0.100 | 0.219 | — | — | 31.3 |
| P40.1 | 0.098 | 0.214 | — | — | 31.4 |
| P92.6 | 0.100 | 0.219 | — | — | 31.4 |
| P27.5 | 0.100 | — | 0.147 | — | 40.5 |
| P33.3 | 0.100 | — | 0.165 | — | 37.7 |
| P27.5 | 0.073 | — | — | 0.155 | 32.0 |
| P33.3 | 0.072 | — | — | 0.162 | 30.1 |
| P33.3 | 0.200 | — | — | 0.443 | 31.1 |
Water-permeation measurements
The water permeance measurements were carried out in a flow cell with a dead-end configuration, a total volume of 400 mL, and a circular membrane with a diameter of 10 mm. The pressure was generated from nitrogen gas. Pure water or PFOA spiked water, as well as chrome and lead spiked water, was directly added to the flow cell reservoir. The water used was purified by an ELGA CLASSIC UVF water purification system and exhibited a resistance of 16 MΩ cm−1.Conclusions
Membranes capable of removing PFAS are of ever-growing interest due to increasing regulatory and safety standards. In this regard, we reported the first membrane preparation of cobaltocenium-containing block copolymers (BCP) via the self-assembly and non-solvent-induced phase separation (SNIPS) process. Here, we could identify crucial factors like cobaltocenium content, polymer size, and solvent used to influence the membrane and pore formation process. Furthermore, time-dependent permeance and PFOA retention of the optimized membrane were investigated in a dead-end flow cell. By combined analysis using ionic chromatography and liquid chromatography coupled mass spectroscopy, almost quantitative retention of 99.3 ± 1.1% and 99.94 ± 0.04%, respectively, at a permeance of 48 L h−1 bar−1 m2 could be demonstrated. Furthermore, by adjusting the measurement mode to an “inverse flow”, the water flux could be tripled with only a minor loss of retention. To further investigate the stability of chemical oxidizers and the ionic character, sodium chromate and lead nitrate retentions were investigated. Here, partial retention by 46% of sodium chromate and total lead nitrate retention of >99% could be verified by inductively coupled mass spectroscopy. Finally, the first steps towards electrochemical regeneration could be demonstrated by promising electrochemical addressability of the used polymer. In subsequent studies, our objective will be to develop this concept further and create fully electrically switchable membranes for efficient PFAS adsorption and resorption. For upcycling purposes, the membranes used were calcinated using a template-based approach under various conditions, yielding highly ordered gyroid-like cobalt ceramics that are interesting for ceramic catalysis.Author contributions
Till Rittner: conceptualization (lead); data curation (lead); formal analysis (lead); investigation (lead); methodology (lead); validation (lead); visualization (lead); writing – original draft (lead); writing – review and editing (lead). Sebastian Pusse: data curation (supporting); formal analysis(supporting); investigation (supporting); methodology (equal); validation (equal). Blandine Boßmann: data curation (supporting); formal analysis (supporting); methodology (equal); validation (supporting). Kevin Staudt: data curation (supporting); formal analysis (supporting); investigation (supporting); methodology (supporting); validation (supporting); writing review and editing (supporting). Aaron Haben: data curation (supporting); formal analysis(supporting); investigation (supporting); methodology (supporting); validation (equal); visualization (supporting); writing review and editing (supporting). Ralf Kautenburger: supervision (supporting); writing review and editing (supporting). Horst P. Beck: supervision (supporting); writing review and editing (supporting). Markus Gallei: conceptualization (lead); data curation (equal); funding acquisition (lead); investigation (equal); project administration (lead); resources (lead); software (lead); supervision (lead); validation (lead); visualization (equal); writing – original draft (lead); writing – review and editing (lead).Data availability
The data that support the findings of this study are available as ESI† or from the corresponding author upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author thanks Jens Pieschel for his general advice. M. G. and S. P. express their gratitude for the partial financial support provided by the European Union through the European Regional Development Fund (EFRE) and the State of Saarland, Germany, in the SWIMEMSYS project. M. G. is grateful for partial funding of this work in the framework of the cooperation platform iCARE for engineering sciences (Saarland). ICP-MS instrumentation for this work was provided by the Elemental analysis group, with financial support from Saarland University and the German Science Foundation (project number INST 256/553-1).Notes and references
- J. Gluge, M. Scheringer, I. T. Cousins, J. C. DeWitt, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, X. Trier and Z. Wang, Environ. Sci.: Processes Impacts, 2020, 22, 2345–2373 RSC.
- S. Das and A. Ronen, Membranes, 2022, 12, 662 CrossRef CAS PubMed.
- T. D. Appleman, C. P. Higgins, O. Quinones, B. J. Vanderford, C. Kolstad, J. C. Zeigler-Holady and E. R. Dickenson, Water Res., 2014, 51, 246–255 CrossRef CAS PubMed.
- D. P. Siriwardena, R. James, K. Dasu, J. Thorn, R. D. Iery, F. Pala, D. Schumitz, S. Eastwood and N. Burkitt, J. Environ. Manage., 2021, 289, 112439 CrossRef CAS PubMed.
- B. Sonmez Baghirzade, Y. Zhang, J. F. Reuther, N. B. Saleh, A. K. Venkatesan and O. G. Apul, Environ. Sci. Technol., 2021, 55, 5608–5619 CrossRef CAS PubMed.
- I. M. Militao, F. A. Roddick, R. Bergamasco and L. Fan, J. Environ. Chem. Eng., 2021, 9, 105271 CrossRef CAS.
- W. Chen, X. P. Zhang, M. Mamadiev and Z. H. Wang, RSC Adv., 2017, 7, 927–938 RSC.
- C. T. Vu and T. Wu, Crit. Rev. Environ. Sci. Technol., 2020, 52, 90–129 CrossRef.
- H. Guo, Y. Liu, W. Ma, L. Yan, K. Li and S. Lin, J. Hazard. Mater., 2018, 348, 29–38 CrossRef CAS PubMed.
- F. Cao, L. Wang, Y. Yao, F. Wu, H. Sun and S. Lu, Environ. Sci.: Water Res. Technol., 2018, 4, 689–700 RSC.
- R. Amen, A. Ibrahim, W. Shafqat and E. B. Hassan, Sustainability, 2023, 15, 16173 CrossRef CAS.
- M. Ateia, M. Arifuzzaman, S. Pellizzeri, M. F. Attia, N. Tharayil, J. N. Anker and T. Karanfil, Water Res., 2019, 163, 114874 CrossRef CAS PubMed.
- P. Gao, J. Cui and Y. Deng, Chem. Eng. J., 2021, 405, 126698 CrossRef CAS.
- T. Lee, T. F. Speth and M. N. Nadagouda, Chem. Eng. J., 2022, 431, 134023 CrossRef CAS.
- E. W. Tow, M. S. Ersan, S. Kum, T. Lee, T. F. Speth, C. Owen, C. Bellona, M. N. Nadagouda, A. M. Mikelonis, P. Westerhoff, C. Mysore, V. S. Frenkel, V. deSilva, W. S. Walker, A. K. Safulko and D. A. Ladner, AWWA Water Sci., 2021, 3, 1–23 Search PubMed.
- W. Cai, D. A. Navarro, J. Du, G. Ying, B. Yang, M. J. McLaughlin and R. S. Kookana, Sci. Total Environ., 2022, 817, 152975 CrossRef CAS PubMed.
- F. Wang and K. Shih, Water Res., 2011, 45, 2925–2930 CrossRef CAS PubMed.
- X. Zhao, R. Zhang, Y. Liu, M. He, Y. Su, C. Gao and Z. Jiang, J. Membr. Sci., 2018, 551, 145–171 CrossRef CAS.
- T. Jin, M. Peydayesh and R. Mezzenga, Environ. Int., 2021, 157, 106876 CrossRef CAS PubMed.
- J. K. Johnson, C. M. Hoffman, Jr., D. A. Smith and Z. Xia, ACS Omega, 2019, 4, 8001–8006 CrossRef CAS PubMed.
- B. Kunst and S. Sourirajan, J. Appl. Polym. Sci., 1974, 18, 3423–3434 CrossRef CAS.
- M. Müller and V. Abetz, Chem. Rev., 2021, 121, 14189–14231 CrossRef PubMed.
- W.-L. Hung, D.-M. Wang, J.-Y. Lai and S.-C. Chou, J. Membr. Sci., 2016, 505, 70–81 CrossRef CAS.
- K. Foroutani and S. M. Ghasemi, Macromol. Mater. Eng., 2022, 307, 2200084 CrossRef CAS.
- M. Gallei, S. Rangou, V. Filiz, K. Buhr, S. Bolmer, C. Abetz and V. Abetz, Macromol. Chem. Phys., 2013, 214, 1037–1046 CrossRef CAS.
- M. R. Aguilar and J. San Román, Smart polymers and their applications, Woodhead publishing, 2019 Search PubMed.
- A. Kumar, A. Srivastava, I. Y. Galaev and B. Mattiasson, Prog. Polym. Sci., 2007, 32, 1205–1237 CrossRef CAS.
- M. K. Purkait, M. K. Sinha, P. Mondal and R. Singh, Stimuli responsive polymeric membranes: smart polymeric membranes, Academic Press, 2018 Search PubMed.
- B. Saini, M. K. Sinha and A. Dey, Process Saf. Environ. Prot., 2022, 161, 684–702 CrossRef CAS.
- W. Wang, P. F. Li, R. Xie, X. J. Ju, Z. Liu and L. Y. Chu, Adv. Mater., 2022, 34, e2107877 CrossRef PubMed.
- S. Schöttner, R. Hossain, C. Rüttiger and M. Gallei, Polymers, 2017, 9, 491 CrossRef PubMed.
- J. Elbert, F. Krohm, C. Rüttiger, S. Kienle, H. Didzoleit, B. N. Balzer, T. Hugel, B. Stühn, M. Gallei and A. Brunsen, Adv. Funct. Mater., 2014, 24, 1493 CrossRef.
- F. Leniz-Pizarro, R. J. Vogler, P. Sandman, N. Harris, L. E. Ormsbee, C. Liu and D. Bhattacharyya, ACS ES&T Water, 2022, 2, 863–872 Search PubMed.
- M. Ezazi, B. Shrestha and G. Kwon, ACS Appl. Polym. Mater., 2021, 3, 4139–4146 CrossRef CAS.
- P. Baldaguez Medina, V. Ardila Contreras, F. Hartmann, D. Schmitt, A. Klimek, J. Elbert, M. Gallei and X. Su, ACS Appl. Mater. Interfaces, 2023, 15, 22112–22122 CrossRef CAS PubMed.
- M. D. Rausch, Can. J. Chem., 1963, 41, 1289–1314 CrossRef CAS.
- C. U. Pittman, J. Inorg. Organomet. Polym. Mater., 2005, 15, 33–55 CrossRef CAS.
- P. Rohland, E. Schröter, O. Nolte, G. R. Newkome, M. D. Hager and U. S. Schubert, Prog. Polym. Sci., 2022, 125, 101474 CrossRef CAS.
- A. Feuerstein, B. Bossmann, T. Rittner, R. Leiner, O. Janka, M. Gallei and A. Schafer, ACS Macro Lett., 2023, 12, 1019–1024 CrossRef CAS PubMed.
- S. M. Beladi-Mousavi, S. Sadaf, A. K. Hennecke, J. Klein, A. M. Mahmood, C. Ruttiger, M. Gallei, F. Fu, E. Fouquet, J. Ruiz, D. Astruc and L. Walder, Angew. Chem., Int. Ed., 2021, 60, 13554–13558 CrossRef CAS PubMed.
- C. Deraedt, A. Rapakousiou, Y. Wang, L. Salmon, M. Bousquet and D. Astruc, Angew. Chem., Int. Ed., 2014, 53, 8445–8449 CrossRef CAS PubMed.
- D. Schmitt, O. Janka, R. Leiner, G. Kickelbick and M. Gallei, Mater. Adv., 2024, 5, 3037–3050 RSC.
- D. Schmitt and M. Gallei, Desalination, 2024, 583, 117674 CrossRef CAS.
- J. P. Hurvois and C. Moinet, J. Organomet. Chem., 2005, 690, 1829–1839 CrossRef CAS.
- G. Tabbi, C. Cassino, G. Cavigiolio, D. Colangelo, A. Ghiglia, I. Viano and D. Osella, J. Med. Chem., 2002, 45, 5786–5796 CrossRef CAS PubMed.
- H. B. Gray, Y. S. Sohn and N. Hendrickson, J. Am. Chem. Soc., 2002, 93, 3603–3612 CrossRef.
- H. Li, P. Yang, J. Hwang, P. Pageni, A. W. Decho and C. Tang, Biomater. Transl. Med., 2022, 3, 162–171 Search PubMed.
- J. Hwang, Y. Cha, L. Ramos, T. Zhu, L. Buzoglu Kurnaz and C. Tang, Chem. Mater., 2022, 34, 5663–5672 CrossRef CAS.
- S. Gu, J. Wang, R. B. Kaspar, Q. Fang, B. Zhang, E. Bryan Coughlin and Y. Yan, Sci. Rep., 2015, 5, 11668 CrossRef CAS PubMed.
- Y. Wang, A. Rapakousiou, J. Ruiz and D. Astruc, Chemistry, 2014, 20, 11176–11186 CrossRef CAS PubMed.
- Y. Wang, A. Rapakousiou and D. Astruc, Macromolecules, 2014, 47, 3767–3774 CrossRef CAS.
- T. Rittner, K. Ghulam, M. Koch and M. Gallei, Polym. Chem., 2024, 15, 3519–3528 RSC.
- W. Yang, J. Yan, P. Xu, J. Chen, Q. Fang, D. Lin, Y. Yan and Q. Zhang, Macromolecules, 2022, 55, 7763–7774 CrossRef CAS.
- R. Zhang, X. Zhao, W. Li, H. Qian and H. Yang, Chem. Commun., 2023, 59, 5289–5292 RSC.
- H. Lin, L. Ramos, J. Hwang, T. Zhu, M. W. Hossain, Q. Wang, S. Garashchuk and C. Tang, Macromolecules, 2023, 56, 6375–6384 CrossRef CAS.
- T. Zhu, S. Xu, A. Rahman, E. Dogdibegovic, P. Yang, P. Pageni, M. P. Kabir, X. D. Zhou and C. Tang, Angew. Chem., Int. Ed., 2018, 57, 2388–2392 CrossRef CAS PubMed.
- N. Chen, H. Zhu, Y. Chu, R. Li, Y. Liu and F. Wang, Polym. Chem., 2017, 8, 1381–1392 RSC.
- S. Roy, J. Moran, K. Danasekaran, K. O'Brien and S. Dakshanamurthy, Int. J. Mol. Sci., 2024, 25, 8241 CrossRef CAS PubMed.
- R. Chen, J. Feng, J. Jeon, T. Sheehan, C. Rüttiger, M. Gallei, D. Shukla and X. Su, Adv. Funct. Mater., 2021, 31, 2009307 CrossRef CAS.
- X. Su, K.-J. Tan, J. Elbert, C. Rüttiger, M. Gallei, T. F. Jamison and T. A. Hatton, Energy Environ. Sci., 2017, 10, 1272–1283 RSC.
- S. R. Cotty, N. Kim and X. Su, ACS Sustainable Chem. Eng., 2023, 11, 3975–3986 CrossRef CAS.
- M. A. Alkhadra, X. Su, M. E. Suss, H. Tian, E. N. Guyes, A. N. Shocron, K. M. Conforti, J. P. de Souza, N. Kim, M. Tedesco, K. Khoiruddin, I. G. Wenten, J. G. Santiago, T. A. Hatton and M. Z. Bazant, Chem. Rev., 2022, 122, 13547–13635 CrossRef CAS PubMed.
- X. Tan, M. Sawczyk, Y. Chang, Y. Wang, A. Usman, C. Fu, P. Král, H. Peng, C. Zhang and A. K. Whittaker, Macromolecules, 2022, 55, 1077–1087 CrossRef CAS.
- S. Kabiri, C. L. Monaghan, D. Navarro and M. J. McLaughlin, Environ. Sci.: Water Res. Technol., 2024, 10, 420–430 RSC.
- E. Gagliano, M. Sgroi, P. P. Falciglia, F. G. A. Vagliasindi and P. Roccaro, Water Res., 2020, 171, 115381 CrossRef CAS PubMed.
- K. V. Peinemann, V. Abetz and P. F. Simon, Nat. Mater., 2007, 6, 992–996 CrossRef CAS PubMed.
- M. Radjabian and V. Abetz, Prog. Polym. Sci., 2020, 102, 101219 CrossRef CAS.
- L. F. Villalobos, M. Karunakaran and K. V. Peinemann, Nano Lett., 2015, 15, 3166–3171 CrossRef CAS PubMed.
- C.-y Yang, G.-d Zhu, Z. Yi, Y. Zhou and C.-j Gao, Chem. Eng. J., 2021, 424, 128912 CrossRef CAS.
- H. H. Wang, J. T. Jung, J. F. Kim, S. Kim, E. Drioli and Y. M. Lee, J. Membr. Sci., 2019, 574, 44–54 CrossRef CAS.
- V. Abetz, Macromol. Rapid Commun., 2015, 36, 10–22 CrossRef CAS PubMed.
- L. Tsaur and U. B. Wiesner, Polymers, 2023, 15 Search PubMed.
- X. Chen, A. Vanangamudi, J. Wang, J. Jegatheesan, V. Mishra, R. Sharma, S. R. Gray, J. Kujawa, W. Kujawski, F. Wicaksana and L. F. Dumee, Water Res., 2020, 182, 116010 CrossRef CAS PubMed.
- K. M. Persson, V. Gekas and G. Trägårdh, J. Membr. Sci., 1995, 100, 155–162 CrossRef CAS.
- W. H. Organization, Guidelines for drinking-water quality: fourth edition incorporating the first addendum, Geneva, 2017.
- J. Elbert, F. Krohm, C. Rüttiger, S. Kienle, H. Didzoleit, B. N. Balzer, T. Hugel, B. Stühn, M. Gallei and A. Brunsen, Adv. Funct. Mater., 2013, 24, 1591–1601 CrossRef.
- P. Kumkum and S. Kumar, Biomass, 2024, 4, 243–272 CrossRef CAS.
- T. Rittner, J. Kim, A. Haben, R. Kautenburger, O. Janka, J. Kim and M. Gallei, Chem. – Eur. J., 2024, 30, e202402338 CrossRef CAS PubMed.
- L. Xu, L. Liang, C. Chen, Z.-H. Chen, Z.-B. Lv, M.-L. Fu and B. Yuan, J. Environ. Chem. Eng., 2023, 11, 111605 CrossRef CAS.
- I. Luisetto, F. Pepe and E. Bemporad, J. Nanopart. Res., 2008, 10, 59–67 CrossRef CAS.
- J. von Irmer, S. Vowinkel, D. Scheid, S. Schöttner, C. Rüttiger, M. Appold and M. Gallei, Polymer, 2017, 122, 303–311 CrossRef CAS.
- C. Rüttiger, V. Pfeifer, V. Rittscher, D. Stock, D. Scheid, S. Vowinkel, F. Roth, H. Didzoleit, B. Stühn, J. Elbert, E. Ionescu and M. Gallei, Polym. Chem., 2016, 7, 1129–1137 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc03546a |
| This journal is © The Royal Society of Chemistry 2024 |