
Catalytic strategies for detoxifying phosphorus(V) biocides
Larissa Maria S.
de Carvalho†
 ,
Nathália R. D.
de Souza†
and
Eduardo H.
Wanderlind
*
,
Nathália R. D.
de Souza†
and
Eduardo H.
Wanderlind
*
Federal Rural University of Rio de Janeiro (UFRRJ), Institute of Chemistry, Department of Organic Chemistry, BR 465, Km 7, CEP 23.897-000, Seropédica (Rio de Janeiro), Brazil. E-mail: ewanderlind@ufrrj.br
First published on 21st November 2024
Abstract
Organophosphorus substances are employed in several industrial segments, albeit they may feature high toxicity levels depending on their structures. Based on previous extensive investigations of structure–reactivity patterns, researchers have been working on the development of catalysts as a means to detoxify phosphorus(V) organic compounds rapidly and safely through specific reaction pathways. This highlight reviews some recent advances in the utilization of catalytic systems for the decomposition of organophosphorus(V) compounds, in most cases using simulants of nerve agents. The nature of the catalysts is wide, including heterogeneous, colloidal and supramolecular systems, and although not all cases may be practical for the detoxification of phosphorus(V) organic structures, they are certainly useful for future research on this theme.
 Larissa Maria S. de Carvalho | Larissa Maria S. de Carvalho is pursuing her undergraduate degree in Chemistry at the Federal Rural University of Rio de Janeiro (UFRRJ, Brazil). Her research projects are related to the investigation of homogeneous catalytic mechanisms of phosphate transfer reactions employing kinetic and spectroscopic analyses. |
 Nathália R. D. de Souza | Nathália R. D. de Souza is a Chemistry undergraduate student at the Federal Rural University of Rio de Janeiro (UFRRJ, Brazil). She works on the investigation of non-mimetic models of enzyme catalysis using host–guest complexes in solution, with an emphasis on the use of pillar[n]arenes as macrocyclic hosts. |
 Eduardo H. Wanderlind | Prof. Eduardo H. Wanderlind obtained his PhD degree from the Federal University of Santa Catarina (UFSC, Brazil) on Physical Organic Chemistry and started his independent career in 2022 at the Institute of Chemistry of the Federal Rural University of Rio de Janeiro (UFRRJ), where he founded the Catalysis Research Group. In addition to fundamental research on reaction mechanisms, the team also works on the development of novel sustainable catalysts envisioning applications such as the detoxification of organophosphates and energy production. |
1. Introduction
Phosphorus, obtained especially from rocks in the environment, can be transformed into a range of industrially important products, besides being naturally present in a variety of biomolecules.1–3 Considering phosphorus(V) specifically, the range of structures available is quite diverse, with consequent implications in reactivity and applications (Fig. 1), which span from flame retardants and plasticizers4–6 to well-known insecticides and herbicides.1,2 An example of an organophosphorus(V) herbicide is glyphosate, a broad-spectrum herbicide that is most used in the world nowadays, whose mechanism of action is hampering the amino acid synthesis from glucose in plants by inhibiting the 5-enolpyruvyl-shikimate-3-phosphate synthase (EPSPS) enzyme.7,8 Although glyphosate is considered to have low toxicity (LD50 greater than 5000 mg kg−1 for male rats9), possible carcinogenic effects in humans and environmental contamination raise concerns about its widespread use in agriculture.2,10 | ||
| Fig. 1 Examples of organophosphorus compounds, including derivatives of phosphonates, phosphate esters and phosphorothioates. (i) Typical flame retardants and plasticizers. (ii) Examples of pesticides. Parathion is a phosphorothioate that can be found with the two alkyl substituents being both ethyl or both methyl, being referred to as ethyl parathion or methyl parathion, respectively. (iii) Nerve agents of the G-series (tabun and sarin) and the V-series (VX and R-VX). (iv) Simulants of nerve agents: diisopropyl fluorophosphonate (DFP), dimethyl methylphosphonate (DMMP), and p-nitrophenyl diphenylphosphate (PNPDPP). Ethyl paraoxon is the product of the metabolism of ethyl parathion; the same relationship applies to methyl paraoxon with respect to methyl parathion. | ||
The potentialities of using organophosphorus compounds as insecticides were identified in the 1930s by Schrader, with the initial structures evaluated soon found to be very toxic to mammals, with tabun as the first candidate of the P(V) insecticide.1,2,11 This led to the research on more appropriate organophosphorus compounds that could still be used as insecticides but exhibiting lower toxicity to mammals. Unfortunately, the high toxicity of certain representatives was explored in the development of chemical warfare agents, including tabun itself, which turned out to be the first nerve agent produced, and the whole G- and V-series of organophosphorus nerve agents.1,11 The high toxicity of such compounds is due to the inhibition of acetylcholinesterase, responsible for the hydrolysis of acetylcholine. As shown in Scheme 1, when a P(V) organic compound reaches the acetylcholinesterase active site, the nucleophilic reaction of the serine residue with the electrophilic phosphorus leads to the inhibited enzyme, precluding the reaction of the serine group with its target substrate, acetylcholine. If an antidote is not administered at this stage (usually pralidoxime, which is capable of reacting with the phosphorus center), the regeneration of the serine residue will no longer be possible in the aged enzyme. Thus, depending on the degree of intoxication and/or the delay in administering an appropriate antidote, the contamination due to an organophosphorus(V) compound is irreversible and can lead to death.12–14
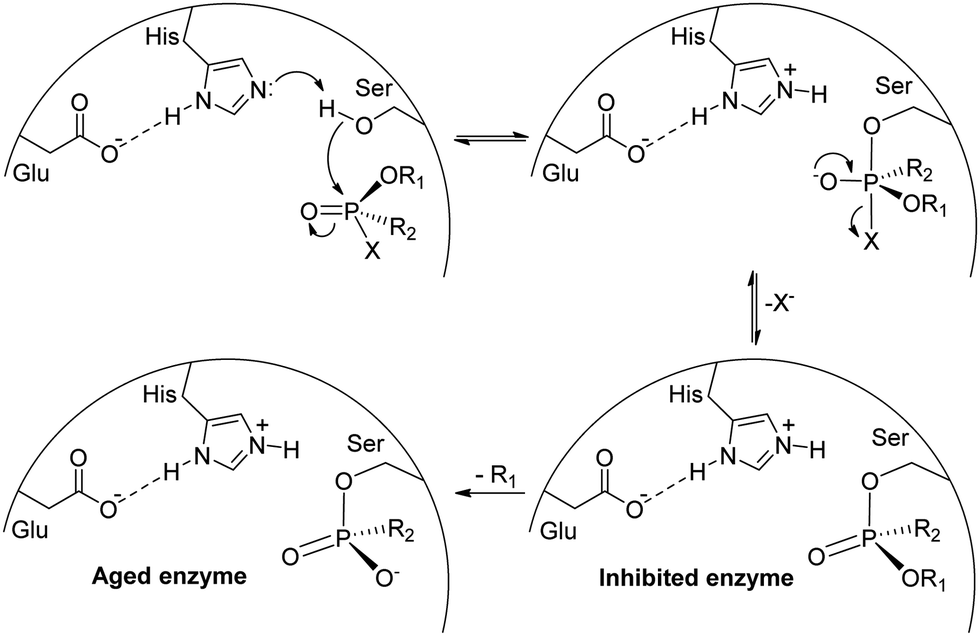 | ||
| Scheme 1 Representation of the inhibition of AChE by a generic organophosphorus(V) compound. The image was adapted from ref. 15. | ||
Thus, although the P(V) chemistry is intrinsically related to life, due to the involvement in fundamental biochemical processes, and even though man-made phosphates are industrially important, there are delicate applications of such compounds that deserve more consciousness when dealing with them, especially regarding pesticides and nerve agents. Even though the industries of insecticides and herbicides led to the commercialization of compounds that are less toxic than tabun, there is still a significant amount of cases of human intoxication by organophosphorus pesticides. Besides, application of P(V) pesticides can lead to environmental contamination, with the presence of these non-natural structures in nature being an important issue.2,10
Regarding chemical warfare agents, the development of structures for this application soon evolved to the scenario of existing stockpiles of organophosphorus nerve agents that later needed to be rapidly and safely detoxified, that is, decomposed into products of lower molecular weight exhibiting lower toxicity than their precursors.1,16 In this sense, the Organisation for the Prohibition of Chemical Weapons (OPCW) turned out to be responsible for the Chemical Weapons Convention (CWC) that entered into force in 1997 as an effort for the worldwide chemical disarmament and security, having 193 States committed to it today.17 The OPCW reports in its website the activities related to the verifiable monitoring of chemical weapons’ destruction progress and of industry verification, with 100% of the chemical weapon stockpiles declared by the State Parties destroyed as of 31 March 2024.17 These non-trivial efforts of OPCW were recognized by the 2013 Nobel Prize for Peace to the institution for “its extensive efforts to eliminate chemical weapons”.18
The risks associated with excessive pesticide utilization and the chemical security of chemical warfare agents have motivated governments, industries and academia in finding appropriate means of detoxifying P(V) compounds. Considering their structural variety, it is quite difficult to propose one method capable of detoxifying several subclasses due to the different patterns of reactivity. For example, alkaline hydrolysis and incineration are technologies employed for the decomposition of P(V) nerve agents, although these strategies suffer from some disadvantages such as the need for the treatment of the afterburner gases in the case of incineration, and the formation of toxic products in the reaction of some representatives with hydroxide, in the case of alkaline hydrolysis.1,16 These situations highlight that investigations on how reaction pathways can be tuned for improving chemical detoxification are required, and one special niche is the use of catalysis for this objective, i.e., using a catalytic species that can lead to a specific set of products through a reaction path that would be otherwise slow in their absence. In this sense, the term “catalytic detoxification” has been used,19–21 not only focusing on P(V) compounds, but also concerning other classes of chemical warfare agents.
Catalysis by itself is a huge subarea within natural sciences and engineering, meaning that the options of catalysts employed for degrading organophosphorus compounds are incredibly large, encompassing several topical areas depending on the nature of the catalysis (homogeneous, heterogeneous, colloidal, supramolecular, etc.). Thus, the purpose of this highlight is to review some recent advances in the catalytic detoxification of P(V) compounds, providing a broad yet concise overview of the diversity of systems that have been explored for achieving this goal, using selected examples of reports by authors from all five continents working on this topic. Because the understanding of the efficiency of catalytic systems is intrinsically related to their comprehension at a molecular level, the discussion will sometimes focus on the description of the reactivity of the species involved considering plausible reaction pathways.
Readers should note that most part of the examples presented in this highlight utilize (either ethyl or methyl) paraoxon as the substrate, which is one of the most common simulants for organophosphorus(V) nerve agents. The strategy of using such surrogates when evaluating the potentialities of new catalytic systems is a recurring precautionary measure that avoids dealing with extremely toxic nerve agents while facilitating permissions for conducting the experiments in research facilities, although, of course, such simulants are still toxic and need to be appropriately handled by trained personnel. We hope that this highlight can be insightful for readers of different areas in the planning of novel catalytic systems that can find applications within the contexts of chemical detoxification and chemical security of P(V) compounds.
2. Catalyzing the decomposition of P(V) compounds
2.1. Reactions with nucleophiles
Reactions of nucleophiles with organophosphorus(V) compounds in solution considering the nucleophilic attack at phosphorus are, by far, the most common reactions envisioned for the decomposition of such substrates. Thus, the cleavage of the bond between phosphorus and a given leaving group with its substitution by a small nucleophile is a direct means of transforming the starting substrate into a “smaller”, and, hopefully, less toxic product. If the detoxification process envisioned is indeed a catalytic one, then the phosphorylated nucleophile is expected to be decomposed into the same products of the uncatalyzed solvolysis, regenerating the free nucleophile in an authentic catalytic cycle that is overall faster than the uncatalyzed reaction. This is represented in Scheme 2 with the general reaction of a nucleophile with a phosphate triester containing an aromatic leaving group (such as methyl or ethyl paraoxon) in an aqueous medium. The hydrolysis of the phosphorylated nucleophile intermediate regenerates the nucleophile and leads to a phosphate diester, which is the phosphorus-containing product of the uncatalyzed hydrolysis of the phosphotriester.3,22,23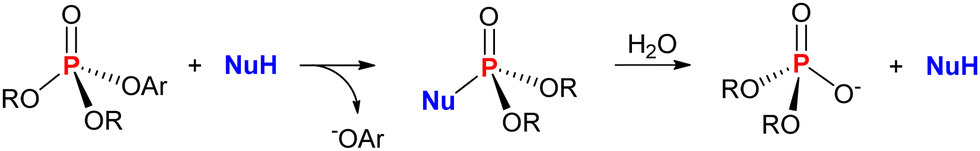 | ||
| Scheme 2 General path of a substitution reaction involving a nucleophile NuH and a phosphate triester with an aromatic leaving group (−OAr) in an aqueous medium. Hydrolysis of the phosphorylated nucleophile leads to the phosphate diester (shown as a monoanion). | ||
Although there is a significant amount of studies that investigated the reactions of phosphate esters with nucleophiles in solution, it is not unusual that authors did not evaluate the reactivity using catalytic quantities of the nucleophilic catalyst or did not investigate the decomposition of the phosphorylated nucleophile in detail. This is mainly because most part of such studies focused on establishing structure–reactivity relationships using classical physical organic investigations, which have provided important paradigms of the reactivities of organophophorus(V) compounds.7,22,24 Thus, we cover such reactions in this first section, even though not in nucleophilic catalysis cycles, because they provide substantial rationales for planning more sophisticated detoxification systems based on nucleophilic reactions at phosphorus.25–28 It is important to highlight that these reactions depend on the solvent properties, so that solvent effects can be rationally explored to optimize the degradation of P(V) compounds in either catalytic or non-catalytic nucleophilic substitution reactions.
Investigations of phosphoryl transfer to nucleophiles were performed by Pavez and coworkers,29 who investigated the reaction of diethyl 4-nitrophenyl phosphate (ethyl paraoxon) with piperidine in a range of ionic liquids, comparing the results with those of the same reaction in aqueous and organic solvents. The authors showed that the reaction of ethyl paraoxon with piperidine is greatly influenced by the reaction medium, a feature that can be precisely explored to direct the decomposition of the substrate through a specific reaction pathway. When evaluating the reaction in ionic liquids (encompassing five structures of cations with up to four anion counterions) and in three organic solvents (1,4-dioxane, acetonitrile and dimethylsulfoxide), the authors showed that the nucleophilic attack of piperidine can occur in either three nucleophilic centers of the substrate, with the ratio of products varying according to the reaction medium, Scheme 3.29 While the nucleophilic attack of piperidine at phosphorus leads to 4-nitrophenolate displacement and the formation of a phosphoroamidate bond (path A; Scheme 3), the attack of the nucleophile at the aromatic carbon in a SNAr reaction is responsible for the formation of the tertiary amine and diethyl phosphate (path B; Scheme 3). Lastly, piperidine can also attack the alkyl carbon of the ethyl moiety, leading to N-ethyl piperidine and the ethyl 4-nitrophenyl phosphate diester, which is further susceptible to nucleophilic attack by piperidine (in excess in the reaction medium) at phosphorus (path C; Scheme 3).
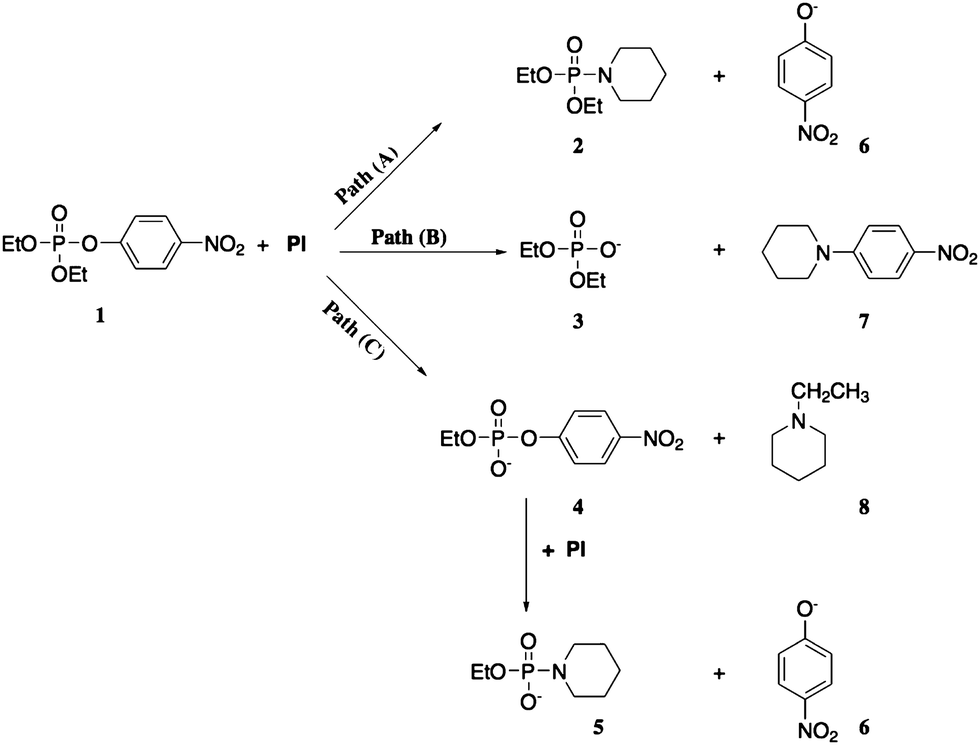 | ||
| Scheme 3 Observed paths in the reactions of ethyl paraoxon with piperidine in ionic liquids and organic solvents at 25 °C. Reprinted with permission from ref. 29. Copyright 2013 American Chemical Society. | ||
While paths B and C in Scheme 3 account for 99% of the reaction of the substrate with piperidine in 1,4-dioxane, 54% of the product distribution comes from path A when using the ionic liquid 1-butyl-3-methylpyridinium dicyanamide ([Bmpy][DCA]) as the reaction medium, this being the greatest contribution of path A in the series of ionic liquids evaluated. In order to understand the trends in reactivity, the authors compared rate constants of the three paths for a series of ionic liquids featuring the same cation, but different anions, and a series of ILs featuring the same anion, but different cations. Specifically, for path A, the authors propose that the ILs may favor the reaction by stabilizing transition states due to hydrogen-bond donating abilities of the cations, as well as their ability for interacting through pi stacking, with the size of the anions influencing the efficiencies of both types of interactions.29
The use of bio-based solvents such as ethyl lactate and gluconic acid has also been evaluated in this reaction by the same group,30 and the product distribution between paths A, B and C (Scheme 3) also varied depending on the nature of the solvent, except for the ethyl lactate, in which case the authors observed the solvent instead of piperidine phosphorylation. In this case, the authors propose a general base catalysis promoted by piperidine in the nucleophilic attack of the hydroxyl group of the solvent, either at the phosphorus, the aromatic carbon, or the aliphatic carbon. Considering the cost of ionic liquids, the authors propose that bio-based solvents might be suitable alternatives for the decomposition of organophosphorus compounds.30
Interesting correlations between the properties of ILs and the reactivity of a substitution reaction at phosphorus were also reported by Butler and Harper (Fig. 2).31–33 The authors chose to evaluate the reaction of diethyl chlorophosphate with ethanol-d6, which has the advantage of forming partially deuterated triethyl phosphate as the sole product, evaluating initially the effect of the ionic liquid 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([bmim][(CF3SO2)2N]) in ethanol or ethanol/acetonitrile.31,34 Then, further studies were performed so that there were kinetic data for the reaction in the presence of five ionic liquids with the same anion, but a different cation,32 as well as the data for the reaction in the presence of five ILs with the same cation, but a different anion.33 For each IL, the mole fraction of the salt was varied in the kinetic investigations, with rate constants generally increasing with the increase in the mole fraction of the IL up to a maximum, followed by a decrease in reactivity. Considering also the correlations between activation parameters for the reactions in the presence of the ILs and the corresponding Kamlet–Taft βN parameters, the results suggest that the molecular origin of the effect of the ILs in the substrate ethanolysis is related to the hydrogen bond accepting ability of the anions, and that additional interactions such as ion-pairing are also probably operative.
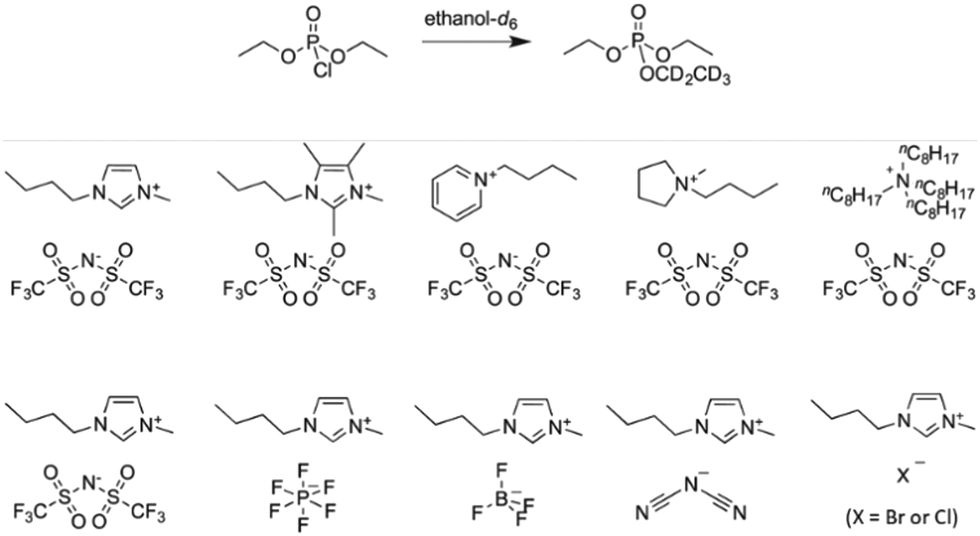 | ||
| Fig. 2 The reaction of diethyl chlorophosphate with ethanol-d6 in the presence of nine ionic liquids, whose structures are shown. Reprinted (adapted) with permission from ref. 32 and 33. Copyright 2016 and 2019 John Wiley & Sons. | ||
2.2. Metal complexes
Metal ions are known to effectively catalyze phosphoryl transfer reactions in a number of enzymatic reactions, so that important investigations are devoted to the elucidation of the roles of the metal ions in such reactions.35,36 In fact, generations of physical inorganic chemistry groups have prepared inorganic complexes as catalysts for the hydrolysis of phosphate esters in insightful biomimetic models of phosphoryl transfer reactions, providing a strong background on the understanding of metal ion catalyzed phosphoryl transfer reactions. In this sense, it is generally accepted that substrate coordination to metal centers may, for example, (i) enhance substrate electrophilicity, (ii) stabilize developing charges along reaction coordinates, and (iii) assist leaving group departure, as well as (iv) favor the reactions of metallo-bound water/hydroxide moieties with the electrophilic center.37–39The investigations of phosphoryl transfer reactions in the presence of metal complexes in homogeneous systems have been mostly devoted to phosphate mono- and diesters as substrates, mainly because these are the most important biological functionalities in enzymatic systems, but still dephosphorylation of phosphate triesters can indeed be accelerated by metal complexation in solution.40–42 A recent example was reported by Luiz and coworkers43 with three mononuclear iron(III) complexes (1, 2 and 3 in Scheme 4) in the hydrolytic decomposition of the phosphate triester DEDNPP. While complex 1 had been reported previously, complexes 2 and 3 were novel and were prepared with the ligand L in complex 1 being further functionalized at the aldehyde functionality to render aminomethylbenzimidazole moieties in the second coordination sphere of the complexes. Such modifications could be advantageous for intramolecular acid/base and/or nucleophilic catalysis, with complex 3 intentionally featuring a longer spacer between the benzimidazole and the phenolic moieties in comparison to complex 2. After evaluating the effect of the pH upon initial velocities, the effect of the substrate concentration was studied at optimum pH values (pH 5.5 for complex 1 and pH 7.0 for complexes 2 and 3) in order to obtain binding and kinetic parameters.43
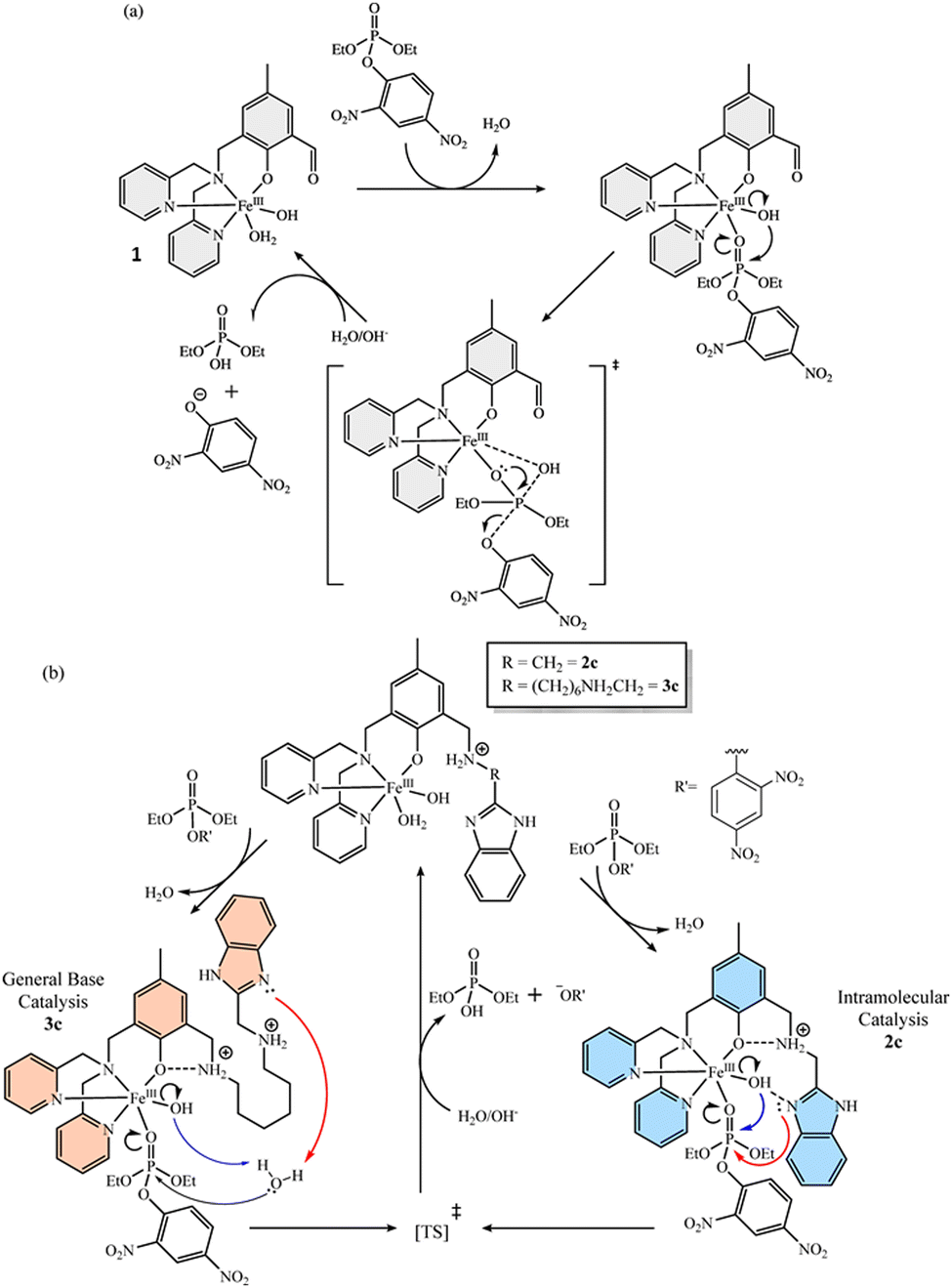 | ||
Scheme 4 Proposed reaction paths for the dephosphorylation of DEDNPP catalyzed by mononuclear iron(III) complexes: (a) 1 and (b) 2 and 3, all in water/acetonitrile 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) considering the complex species present at optimum pH values (pH 5.5 for complex 1 and pH 7.0 for complexes 2 and 3). Reprinted with permission from ref. 43. Copyright 2024 John Wiley & Sons. :1 (v/v) considering the complex species present at optimum pH values (pH 5.5 for complex 1 and pH 7.0 for complexes 2 and 3). Reprinted with permission from ref. 43. Copyright 2024 John Wiley & Sons. | ||
For complex 1, the main species at pH 5.5 is [FeL(OH2)OH]+ (L is the organic ligand) as determined by potentiometric and spectrophotometric titrations, and in this case the authors propose that the decomposition of DEDNPP arises from the intramolecular nucleophilic attack of the hydroxo group on phosphorus (Scheme 4(a)), with this mechanism being responsible for the 12-fold catalytic effect in comparison to the spontaneous hydrolysis of the substrate. On the other hand, the catalytic effects obtained with complexes 2 and 3 at optimum pH (7.0) are 27 and 38-fold, respectively, i.e., at least twice as that promoted by the complex without the benzimidazole moiety. Provided that aliphatic amines are protonated and benzimidazole rings are neutral at pH 7.0 for complexes 2 and 3 (whose main species, [FeL(OH2)OH]+, are named 2c and 3c in Scheme 4(b), respectively), the authors have outlined the plausible reaction mechanisms shown in Scheme 4(b). Both cases consider the participation of the benzimidazole groups, but with different roles that would explain the different catalytic effects promoted by each complex, as well as the different values of kinetic isotope effects (kH2O/kD2O = 1.43 and 2.11 for the reactions of DEDNPP with complexes 2c and 3c, respectively). While in complex 2 the benzimidazole can be positioned close to the phosphorus electrophilic center for nucleophilic catalysis, the approximation of the benzimidazole ring for a direct nucleophilic attack on phosphorus in 3c should be entropically unfavorable due to the longer carbon spacer, and a general base catalysis pathway was invoked, consistent with the larger kinetic isotope effect. The mechanistic considerations are substantiated by the analyses of the geometries of the complexes formed by the coordination of the substrate trough the phosphoryl oxygen to the iron(III) center of 1, 2c and 3c, obtained by ground state calculations. In any event, the experimental and theoretical evidence does not allow excluding the participation of the hydroxo groups in complexes 2c and 3c, so that the mechanisms outlined by the blue and red arrows are proposed to occur in a parallel fashion in each case.43
Similarly, the exploration of the coordination of phosphate substrates to metal ions and the effect that ligands may have on binding and catalytic activity have been explored by Trinh and coworkers44 using self-assembled peptoid membranes to which cobalt ions were bound. Initially, the authors prepared six peptoids featuring hydrophobic and hydrophilic components, and a ligand portion for later cobalt complexation (Fig. 3). The six initial peptoids featured the same hydrophobic and hydrophilic constitutions, differing in the structure of the ligands in order to evaluate the roles that the sizes and the presence/absence of non-coordinating side chains close to the complexation site could play in catalysis. Then, a further range of peptoids were prepared (up to 15) varying the functional groups and spacers in the ligands, hydrophobic and/or hydrophilic components. Using a range of characterization techniques, including potentiometric titrations and microscopy analyses, the authors proposed that most of the peptoids were able to self-assemble into membranes to which Co2+ can coordinate, the metal center being a binding site for a P(V) compound. Methyl paraoxon was chosen as a model substrate and the overall catalytic cycle proposed by the authors is shown in Fig. 3, with substrate cleavage occurring by the nucleophilic attack of a hydroxo ligand. Peptoid 14, featuring two amino groups in the hydrophilic side chain and aryl groups in the ligand, delivered the best catalytic effect of 219-fold at pH 10. It is noteworthy that the authors showed that the catalytic effect can be modulated by the appropriate choices of the structures of ligands, side chains and spacers, proposing the rationale shown in Fig. 3 for the enzyme mimics.44
 | ||
| Fig. 3 (a) Self-assembly of peptoids and coordination of Co2+. (b) The membranes are used for the catalytic hydrolysis of methyl paraoxon. (c) Structures of some of the peptoids evaluated. Reprinted (adapted) with permission from ref. 44 under a Creative Commons License (American Chemical Society, 2020). | ||
2.3. Metal organic frameworks (MOFs)
Since the first reports of the synthesis and structures of metal organic frameworks (MOFs) in the 1990s, there has been an overwhelmed development in the preparation protocols and applications of these materials within the field of reticular chemistry. A MOF is as a periodic crystalline network composed of organic linkers and metal nodes featuring permanent porosity, typically accompanied by large surface areas, a characteristic that has been particularly attractive for applications in fields spanning from drug delivery to catalysis and energy storage.45–47 Depending on the nature of the organic linkers and the metal nodes and also depending on the synthesis protocol, MOFs’ properties can be tuned to achieve more specific topologies for envisioned applications, and as a result, thousands of MOFs have been known to date. In addition, the number of applications of MOFs is also benefited with the preparation of composite materials with other components such as metal oxides and polymers, as well as through their post-modifications.46,48The use of MOF materials in the catalytic detoxification of chemical warfare agents and some simulants has been explored by a number of groups.49–53 One recurring strategy is the investigation of the effect of MOFs in the hydrolytic decomposition of methyl paraoxon in an aqueous medium as a surrogate of methyl parathion, with the MOFs participating as catalysts for the hydrolysis reaction. The efficiencies of the materials are generally explained in terms of a typical heterogeneous catalytic reaction, initiated by the substrate adsorption onto the MOF surface through coordination to a metal node, which activates the substrate toward the nucleophilic attack by a water molecule or hydroxide. In addition, besides the nature of the metal node, the properties of the pore size and the surface area of the MOF materials are also important for the overall efficiency of the catalytic process.
Practical applications of MOFs in the context of organophosphorus catalytic detoxification include their utilization in composites.49,54–56 One specific example is the work reported by Dwyer and coworkers57 regarding the incorporation of the microporous MOF UiO-66 into a composite fabric containing poly(vinylidene)fluoride (PVDF), Ti(OH)4, and triethanolamine (TEA) as a buffer (Fig. 4). The composite was obtained by a sol–gel synthesis method, followed by an electrospinning step, rendering slightly yellow fibers that were not completely dry due to the presence of TEA. When in distilled water, the fiber promoted the degradation up to ca. 40% of methyl paraoxon within 120 min, and some 10% lower conversion was observed when employing the corresponding fiber prepared in the absence of TEA. This effect was attributed by the authors to a significant catalysis inhibition by product adsorption on the MOF surface in the absence of TEA, which should be responsible for removing adsorbed products from the catalyst surface due to acid/base interactions. The material was further tested with two nerve agents, diisopropyl fluorophosphate and soman, with best results obtained for soman, in which case full consumption of the reactant was achieved with a half-life of 35 min. The same group had previously employed a similar approach for obtaining closely related composites, with polymethylmethacrylate in liu of poly(vinylidene)fluoride and without TEA, which allowed 94% conversion of methyl paraoxon within 120 min.58 These strategies highlight that the chemistry observed in the heterogeneous solid–liquid catalytic systems can be thought to be employed in more realistic scenarios in which detoxification of P(V) biocides might be necessary, such as the use of protective clothing. In addition, it is worth noting that there is some kind of structure–reactivity relationship considering both the catalyst and the substrate, provided that the same material delivers different catalytic effects for different substrates and, correspondingly, the catalytic hydrolysis of a substrate is dependent on the structure of the MOF used, indicating that more studies are required to obtain materials that can be generally used with a broad scope of substrates.57
 | ||
| Fig. 4 Upper image: SEM micrographs of PVDF/Ti(OH)4/UiO-66/TEA (A) and (B) and PVDF/Ti(OH)4/UiO-66 (C) and (D). Lower image: pictures of PVDF/Ti(OH)4/UiO-66 and PVDF/Ti(OH)4/UiO-66/TEA fabrics. Reprinted (adapted) with permission from ref. 57. Copyright 2018 American Chemical Society. | ||
An understanding of general patterns of reactivity in the decomposition of organophosphorus(V) compounds catalyzed by MOFs can be achieved by kinetic and mechanistic investigations, and this approach has been a focus of some reports, both experimentally and theoretically. For example, first investigations of the catalytic hydrolysis of methyl paraoxon promoted by the UiO-66 material showed that the reaction is pH-dependent, with reaction rates increasing with decreasing pH (Scheme 5).59 Initial rates are ca. 50-fold greater at pH 8.8 compared to the corresponding figure at pH 10.2, with initial rates being linearly dependent upon the hydronium concentration in this pH range. This result was attributed to the fact that zirconium-ligated water molecules on defects are displaced by substrate molecules for catalysis to occur on the material surface, and such aqua ligands are converted into hydroxo groups as the pH increases, rendering substitution-resistant ligands, and thus lowering reaction rates. This proposal is fully consistent with the results of a potentiometric titration of UiO-66 in which an equivalence point at pH 9.34 is observed, associated with the deprotonation of water ligands. Thus, a rationale of the overall catalytic process involves the substrate adsorption on the UiO-66 surface through displacement of water-ligated molecules and coordination to zirconium Lewis acid sites, followed by the nucleophilic substitution of 4-nitrophenoxide by a hydroxide ion, and then desorption of the dimethyl phosphate diester anion should take place.59
 | ||
| Scheme 5 pH-Dependent hydrolysis of methyl paraoxon catalyzed by UiO-66. Reprinted (adapted) with permission from ref. 59. Copyright 2015 American Chemical Society. | ||
A more complete mechanistic investigation of a MOF-catalyzed methyl paraoxon hydrolysis was reported by Chen and coworkers using DFT calculations,60 regarding the hydroxide reaction with the substrate catalyzed by the mesoporous NU-1000 material, and more recently by Liao and coworkers,61 who performed an experimental investigation of the same catalytic system. Scheme 6 summarizes the steps involved in the overall catalytic cycle, while Fig. 5 presents the corresponding calculated energies, considering both regular and distorted structures of NU-1000 nodes. Results show a barrier of ca. 35 kJ mol−1 for node dehydration, allowing for the substrate coordination to a NU-1000 node through the phosphoryl oxygen. Then, the substitution reaction of 4-nitrophenoxide by hydroxide takes place through the nucleophilic attack of hydroxide on the phosphorus atom in a concerted mechanism with a single, early transition state that features a hydrogen bond between the leaving group oxygen and a hydroxo ligand at an adjacent node. The authors found that the transition state is earlier in the catalyzed reaction in comparison to the uncatalyzed one, which also proceeds through a concerted SN2(P) mechanism, a result that is attributed to substrate electrophilicity enhancement and leaving group assistance promoted by the solid material.60
 | ||
| Scheme 6 Alkaline hydrolysis of methyl paraoxon over the NU-1000 surface.60,61 | ||
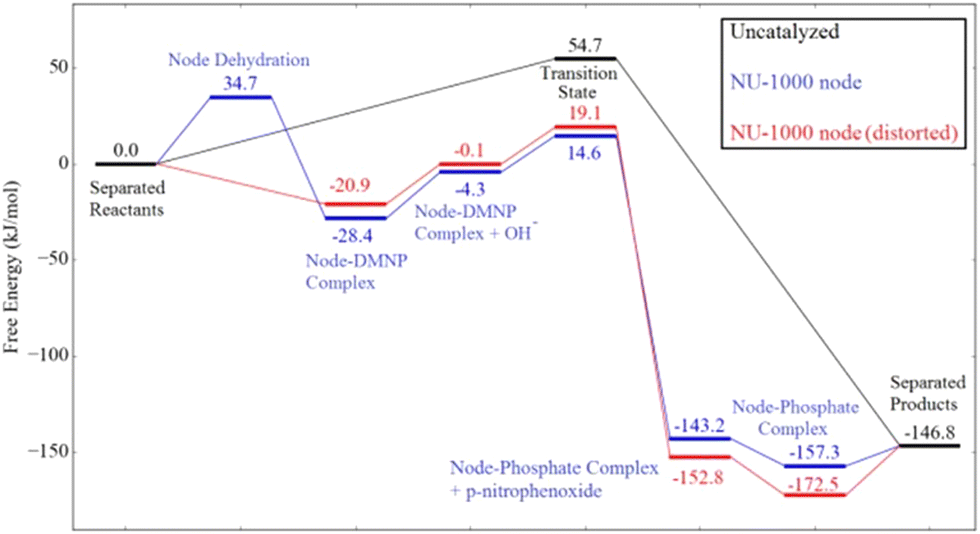 | ||
| Fig. 5 Calculated energies in the methyl paraoxon (named DMNP in the figure) hydrolysis in the absence and on the surface of NU-1000. Reprinted with permission from ref. 60. Copyright 2018 American Chemical Society. | ||
Experimental approaches in monitoring the reaction included the evaluation of the effects of the pH, the amount of the MOF, and the initial concentration of the simulant, and showed that, for NU-1000, the initial rates increase with the pH increase, although there are compensative effects between the fraction of aqua sites and the solution hydroxide concentration. In other words, while the nucleophile concentration increases with pH, the fraction of zirconium sites for coordination is decreased in an equivalent proportion, with the net almost insensitive pH dependence attributed to the equilibrium binding constants of the substrate to the catalyst surface, which increases to 1.8-fold per pH unit.61
2.4. Metal oxides
Besides the use of MOFs, other solid materials featuring metal centers, including simple metal oxides, have shown interesting catalytic activities in reactions involving organophosphorus compounds.62,63 The advantage of using simple metal oxides is that such solids present the known structure and physicochemical properties, so that the choice of solid is facilitated when looking for a good candidate to catalyze a targeted reaction.63,64 For example, the transesterification of methyl paraoxon with 1-propanol over magnesium–aluminum mixed oxides has been investigated by Zimmermann and coworkers65 (Scheme 7), with the solids featuring significant base site densities that facilitate the nucleophilic attack of the alcohol on the phosphorus atom. Thus, the efficiency of the catalysis is interpreted on the basis of substrate adsorption on the catalyst surface through coordination of the phosphoryl oxygen to a metal site, and on the basis of sufficient basicity of the catalyst surface to favor the nucleophilic attack of the alcohol. This system is particularly interesting due to the fact that the transesterified product, a trialkyl phosphate, is structurally analogous to a family of flame retardants, being an insightful result for the planning of future catalytic systems that transform organophosphorus biocides into value-added products.65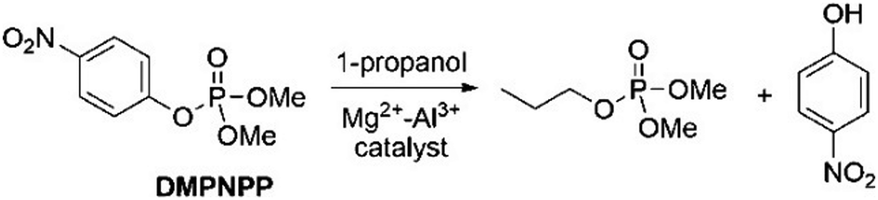 | ||
| Scheme 7 Transesterification of methyl paraoxon (named DMPNPP in the figure) with 1-propanol over Mg2+–Al3+ mixed oxides. Reprinted with permission from ref. 63. Copyright 2020 Royal Society of Chemistry. | ||
Actually, the propanolysis of methyl paraoxon has been shown to be effectively promoted with rate enhancements up to 107-fold by a range of metal oxides featuring different metals, such as magnesium and aluminum in varying proportions (Scheme 7),65 erbium66 and strontium.67 More recently, a closer attention was devoted to the reaction in the presence of the simple MgO,63 with adsorption theoretical calculations aiding the comprehension of the reaction over the catalyst surface. As shown in Fig. 6, by using a 6-atom thick periodic slab model of MgO periclase and allowing the three outermost layers relaxed, the authors showed that methyl paraoxon adsorbs on the catalyst surface through coordination of the phosphoryl oxygen to the Mg2+ sites, with the aromatic ring bending over the catalyst surface so that there is also an interaction between an oxygen atom of the nitro group with a metal site. In its turn, 1-propanol interacts with the solid surface through coordination of its oxygen atom to a metal site, while a hydrogen bond is stablished between the alcohol and an oxygen atom of the surface. Interestingly, a distance map was obtained (Fig. 6), in which case the orange dot represents an adsorption site of the substrate and the blue dots indicate possible positions for 1-propanol, excluding the same coordination site occupied by methyl paraoxon for steric reasons, with the numbers indicating the distance between the nucleophile oxygen and the phosphorus electrophilic center. Thus, the distance map shows that the smallest distance between nucleophilic and electrophilic centers is ca. 2.4 Å, a distance that is favorable for the reaction to occur on the catalyst surface.63
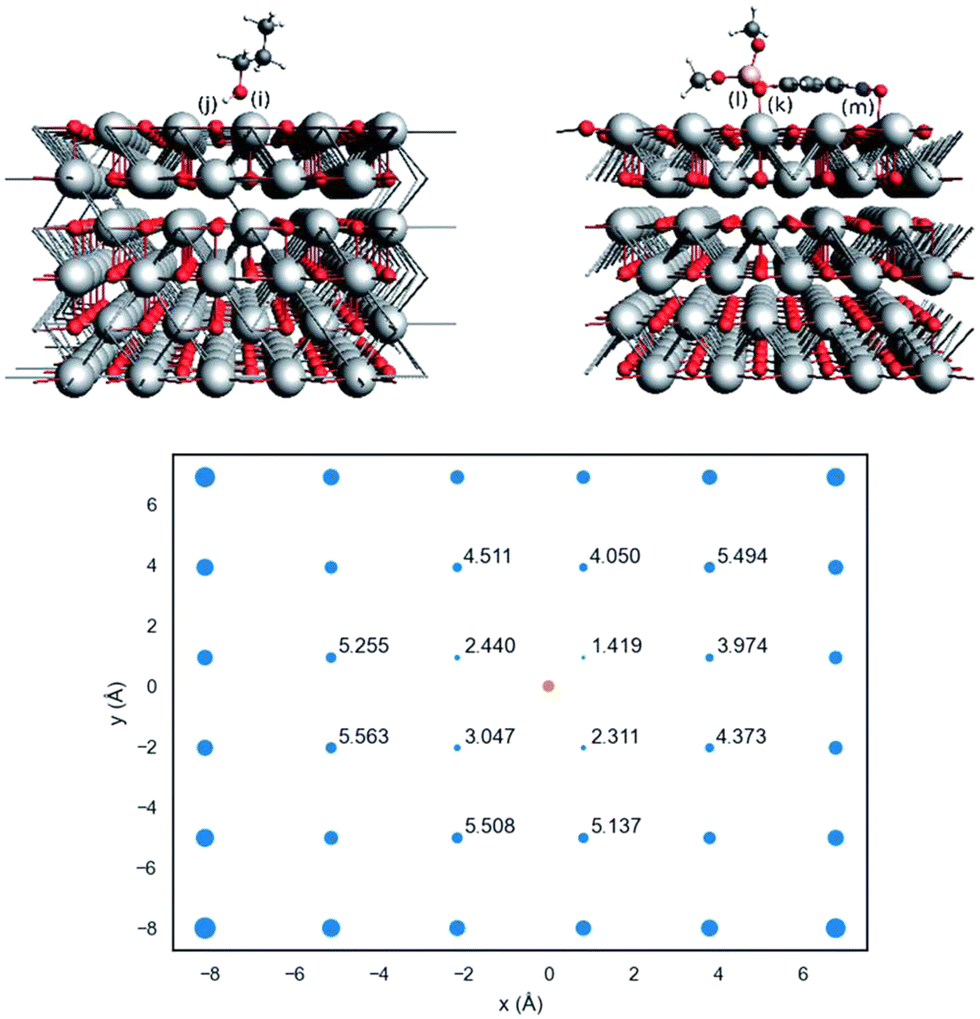 | ||
| Fig. 6 In the upper image, the side views of adsorbed 1-propanol (left) and methyl paraoxon (right) on MgO periclase. In the lower image, the distance map between 1-propanol oxygen (blue dots) and the substrate phosphorus atom (orange dot). Blue dot sizes are proportional to the square of the distances. Reprinted with permission from ref. 63. Copyright 2020 Royal Society of Chemistry. | ||
Transesterification with 1-propanol over metal oxides has also been successful for a series of other phosphate triesters, including triaryl substrates that were converted to tris-n-propyl phosphate in consecutive transesterification reactions.63,68 The disadvantage of the use of metal oxides for the transesterification of aryl organophosphates with alcohols lies on the quite good adsorption capabilities that the products (such as 4-nitrophenol) have towards the surfaces of the catalysts, hampering the use of the materials in catalytic amounts. In this sense, the preparation of metal oxides and similar materials with tuned surface properties seems to be an alternative for envisioning future applications for them.
Similarly to what has been done with MOFs, the propensities of metal oxides in catalyzing the decomposition of simulants of chemical warfare agents have been explored in the preparation of protective and/or decontamination materials. For example, Thomas and coworkers69 have reported recently a peelable gel containing CeO2 nano-octahedra that can exert both the functions of protection from and the decontamination of ethyl paraoxon (Fig. 7). After a careful examination of the possible constituents of the peel-off gel with the aid of a factorial design, the authors found that the best results were obtained with one gram of previously synthesized CeO2, hydroxypropyl cellulose (1150 or 80 kDa), poloxamer 407, Kollidon® SR, water and ethanol. This constitution was found to provide the desired behavior for a peel-off gel, i.e., a gel that dries after a short time after application, keeping its flexibility even after drying, and presenting good skin adhesive behavior. After confirming that CeO2 is able to catalyze the decomposition of ethyl paraoxon in solution (water and ethanol, the solvents of the peelable gel), the authors investigated the efficacy of the gel in skin protection and in the decontamination of the organophosphate after its application. Pig ears were used for this purpose, and the analyses included the evaluation of ethyl paraoxon in the surface, stratum corneum, viable epidermis, dermis, and acceptor medium, after 30 min and 24 h of exposure to the substrate. The results showed that ethyl paraoxon was mostly found in the stratum corneum, the percentage of its recovery in this region being lower when gels with or without CeO2 were used, besides lowering the recovery in the epidermis and dermis. The observations are insightful for the development of future protective materials envisioning a greater range of organophosphorus substrates.69
 | ||
| Fig. 7 In the left, a photograph of the peelable gel containing CeO2 nano-octahedra (NO) employed for the decomposition of ethyl paraoxon. In the right, the comparison of photographs of peelable gels that contain or not the CeO2 NO after exposure to ethyl paraoxon for 30 min. The material containing CeO2 NO turns yellow, indicative of 4-nitrophenol formation. Reprinted (adapted) with permission from ref. 69 under a Creative Commons License (Elsevier, 2024). | ||
Kim and coworkers70 explored the catalytic potential of magnesium oxide coupled to photothermal effects in accelerating the decomposition of methyl paraoxon. After preparing the mesoporous SBA-15 material by the Stöber method, the authors prepared MgO directly on the SBA-15 surface through the precipitation of magnesium from its nitrate salt followed by calcination. Then, the material was reacted with 3-(aminopropyl)triethoxysilane (APTES) in order to provide amine functionalities for further covalent bonding with a previously synthesized graphene oxide (GO), which was performed using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) (Fig. 8). The authors examined the photothermal curves for both MgO@SBA-15 and MgO@SBA15@GO using an infrared irradiation LED (810 nm, 20 mW cm−3) assuring that the heat generated by the LED was eliminated (Fig. 8), and it was observed that the three-component material had its temperature increased to 68 °C in 30 min, maintaining this temperature afterwards. A much lower temperature increase was observed for the material without GO and no significant temperature variation was observed in the control experiment with the three-component material in the absence of the IR-LED. Then, the decomposition of methyl paraoxon was evaluated in the presence of the material in several solvents (pure solvents or binary mixtures), always containing either water or ethanol as the nucleophilic reagent, with the best results being 90% decomposition of methyl paraoxon in ethanol with 1 h of irradiation time. Considering that both metal oxides and carbon materials can be useful towards the catalytic decomposition of organophosphorus(V) compounds, these results provide insights for further developments of novel materials that can combine different modes of catalysis and medium effects in optimized detoxification systems.70
 | ||
| Fig. 8 In the upper image, the preparation protocol of the composite MgO@SBA15@GO. In the lower panel, heating curves of the MgO@SBA15@GO composite and controls (left), and heating curves of the MgO@SBA15@GO composite in three IR irradiation cycles (right). Reprinted (adapted) with permission from ref. 70. Copyright 2023 Elsevier. | ||
2.5. Metal nanoparticles
Features of metal sites are also useful in metal nanoparticles (NPs), either in colloidal solution or supported on solid matrices. One such example is the work reported by Li and coworkers,71 who evaluated the decomposition of some P(V) compounds in the presence of three-dimensional nitrogen-doped graphene (3DNG) in the absence and the presence of cobalt nanoparticles on the surface of the support. After a hydrothermal reduction of graphene oxide (GO) with ammonium hydroxide, cobalt was deposited on the surface of 3DNG by impregnation of Co(Ac)2·4H2O followed by pyrolysis. The materials obtained were named Co/3DNG-1, Co/3DNG-2 and Co/3DNG-3, with the numbers 1–3 denoting increasing amounts of NH4OH used in the preparation of the original 3DNG, while Co/3DG stands for the material not doped with nitrogen. Characterization results showed that the cobalt nanoparticles presented a spherical shape and were uniformly distributed over the 3DNG surface (Fig. 9). Taking methyl parathion as a representative P(V) compound, the authors show that its hydrolysis is greater for the materials containing the Co NPs as compared to the support alone, with the catalyst being active for further cycles, both at 20 and at 80 °C. Although the cobalt content in 3DNG-3 is slightly greater than that in 3DNG-2, the mean size of the nanoparticles in the latter is smaller, an argument that authors used to interpret the greater activity provided by the catalyst 3DNG-2. In addition, because the amount of Co–N bonds in 3DNG-2 is also slightly greater as compared to that in 3DNG-3, authors investigated the Lewis acidity of the catalysts in order to determine if the presence of the Co–N bonds facilitates the hydrolysis reaction. Using temperature-programmed desorption of ammonia (NH3-TPD), authors concluded that 3DNG-2 presents the highest Lewis acidity and suggested that catalysis is also influenced by the support due to its interaction with the metal NP. It is interesting to note that further organophosphorus compounds, such as methyl paraoxon and chlorpyrifos, could also be cleaved by the 3DNG-2 material through hydrolysis at both 20 and at 80 °C with conversions depending on the substrate structure.71 | ||
| Fig. 9 TEM images of (a) Co/3DG, (b) Co/3DNG-1, (c) Co/3DNG-2, and (d) Co/3DNG-3. Reprinted (adapted) with permission from ref. 71. Copyright 2021 American Chemical Society. | ||
Chitin was reported as a support for the preparation of heterometallic composites which were employed as catalysts for the hydrolysis of prothiofos (O-2,4-dichlorophenyl O-ethyl S-propyl phosphorodithioate).72 After suspending chitin in water and adding sodium hydroxide to alkalinize the medium, a solution of a metal salt (silver, gold or palladium) was added dropwise rendering the homo-metallic@chitin composites, and the salt solution of the second metal was added dropwise after five minutes for obtaining the hetero-metallic@chitin composites (Fig. 10). Color changes were indicative of the nucleation of the reduced metals, and a total of six materials were obtained, including one composite featuring the three metals intercalated as Pd@Au@Ag@chitin. When evaluating the hydrolysis of prothiofos in the presence of the materials, the authors highlighted that the catalytic efficiency of the hetero-metallic@chitin composites followed the order Au@Ag@chitin < Pd@Ag@chitin < Pd@Au@Ag@chitin, and that these materials led to greater first-order rate constants and conversions when compared to the corresponding composites featuring only one of the metals. Although the characterization of the reaction products was not performed in depth, HPLC analyses suggested that the initial products of the hydrolysis are 2,4-dichlorophenol and O-ethyl S-propyl phosphorodithioate, consistent with P–O bond cleavage.72
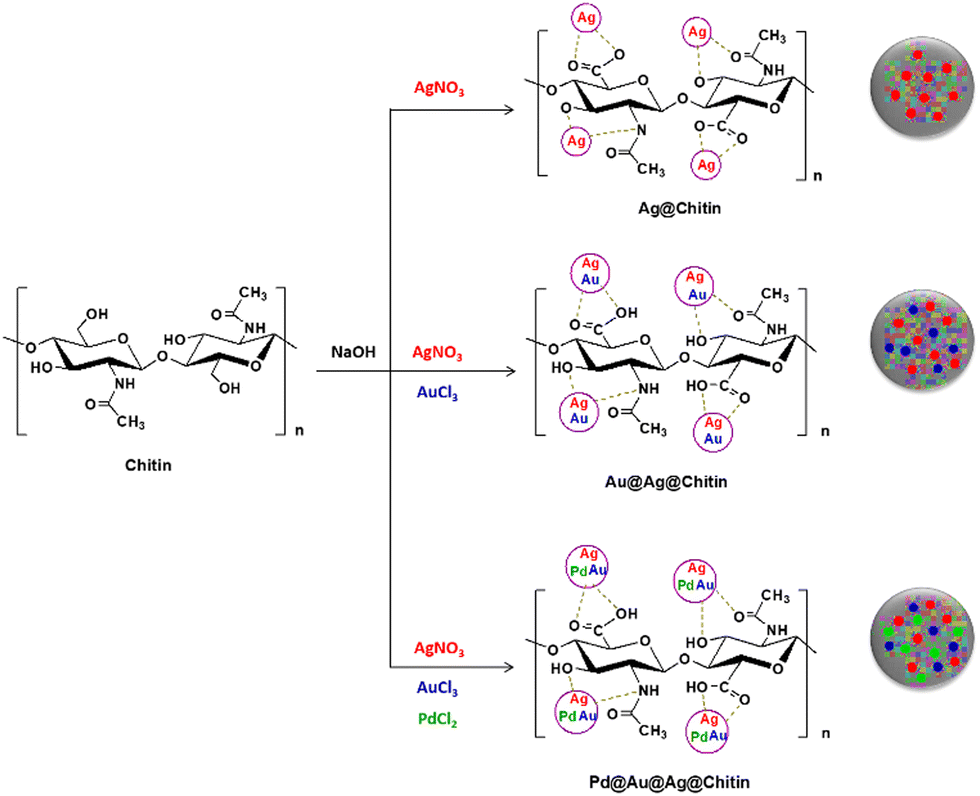 | ||
| Fig. 10 Representation of the formation of silver and heterometallic nanoparticles in the presence of chitin. Reprinted with permission from ref. 72. Copyright 2021 Elsevier. | ||
2.6. Functionalized polymers
Polymeric materials have also been explored for the catalytic detoxification of organophosphorus compounds as a platform for anchoring specific functional groups available for the reaction at desired positions and amounts, with the precise manipulation of the synthesis protocols offering possibilities for the optimization of a given targeted application.73–75 Besides purely synthetic polymers, bioinspired structures and natural polymers are also of interest, e.g., cellulose. Besides the feasibility of obtaining cellulose from renewable sources, this rather general polymer is attractive as a range of materials can be prepared depending on its specific raw source and the preparation protocols, making it able to tune the materials’ properties.76–78 Of course, the simplest and most straightforward way of turning cellulose materials useful for catalytic detoxification purposes is the anchoring of functional groups able to react with targeted substrates.In a recent example, commercial regenerated cellulose beads were used for the anchoring of amidoxime groups and the material was employed in the decomposition of methyl parathion, soman and VX.79 The material was prepared through a two-step procedure, the reaction of the cellulose hydroxyl groups with acrylonitrile being the first to render cyano functional groups available for the reaction with hydroxylamine in the second step (Scheme 8). The final heterogeneous material showed to be reactive towards the decomposition of the substrates, with the effect of the amidoxime functionalized cellulose being superior to that of the non-treated cellulosed beads. Although the authors considered only one mechanistic possibility for the dephosphorylation reaction, i.e., general base catalysis promoted by amidoxime in the hydrolysis of methyl parathion and VX, the results are insightful for future developments in this theme.79
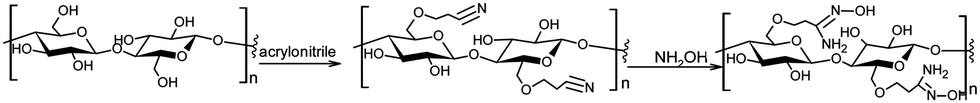 | ||
| Scheme 8 Functionalization of regenerated cellulose with amidoxime in a two-step protocol. Reprinted with permission from ref. 79. Copyright 2021 Royal Society of Chemistry. | ||
Cellulose-based materials have also been prepared using industrial residues as the raw source of cellulose. For example, rice husk, an agriculture residue that accounts for some 22 wt% of the rice seed, has been utilized for anchoring reactive functional groups, such as imidazole,80,81 hydroxamic acid82 and amidoxime.83 In a two-step protocol (Fig. 11), rice husk was reacted with monochloroacetic acid in order to provide carboxylate functional groups in the cellulose structure, and then amidation with 1-(3-aminopropyl)imidazole (API) using NHS and EDC was performed for the functionalization with N-alkyl imidazole. Using potentiometric titration, the authors showed that a significant part of the carboxylate groups was indeed functionalized, with the carboxylic acid and imidazolium groups showing mean pKa values of 6.14 and 7.44, respectively, for their acid dissociation equilibria. The material was evaluated in the decomposition of both DEDNPP and ethyl paraoxon in an aqueous medium at 25 °C, and the kinetic results indicated that the neutral imidazole is able to catalyze the decomposition of both substrates, with rate enhancements over than 107-fold for paraoxon in comparison with the spontaneous hydrolysis of the substrate.80
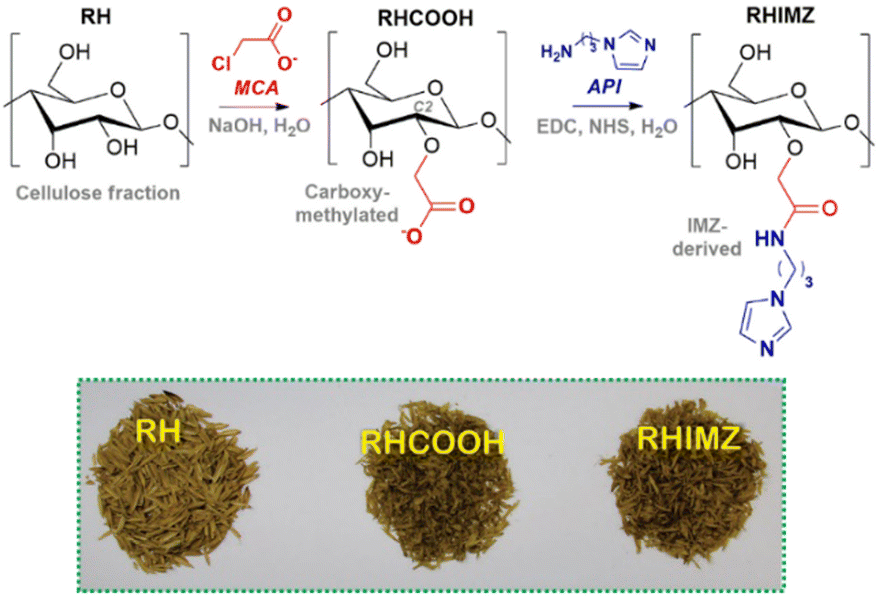 | ||
| Fig. 11 In the upper panel, the two-step protocol for the functionalization of the rice husk cellulose with N-alkyl imidazole. In the lower panel, a photograph with the three materials, untreated rice husk (RH), carboxy-methylated rice husk (RHCOOH) and imidazole-functionalized rice husk (RHIMZ). In the final material, imidazole corresponds to ca. 11% of the functional groups. Reprinted (adapted) with permission from ref. 80 under a Creative Commons License (Brazilian Chemical Society, 2017). | ||
The catalytic hydrolysis of phosphotriesters promoted by imidazole has also been accessed using a synthetic polymer. Through the anchoring of API in commercial poly(ethylene-alt-maleic anhydride), the authors were able to synthesize the PEIIm polymer on a gram scale in a two-step procedure (Scheme 9).84 The polymer was initially evaluated as a heterogeneous catalyst in the hydrolysis of DEDNPP, promoting rate enhancements up to ca. 110-fold at 25 °C (considering 1.0 g L−1 of the catalyst), with the catalyst being recovered with centrifugation. By determining the solvent kinetic isotope effect (SKIE), the authors proposed that the imidazole ring acts as a nucleophile (Scheme 9), leading to a phosphorylated imidazole as an intermediate moiety, which is sequentially hydrolyzed and regenerates the catalytic units. The polymer was shown to maintain the catalytic effect up to 9 cycles using excess of DEDNPP. The polymer could also be used for catalyzing ethyl paraoxon hydrolysis, lowering some 30-fold its half-life at pH 8.0 and 25 °C.84
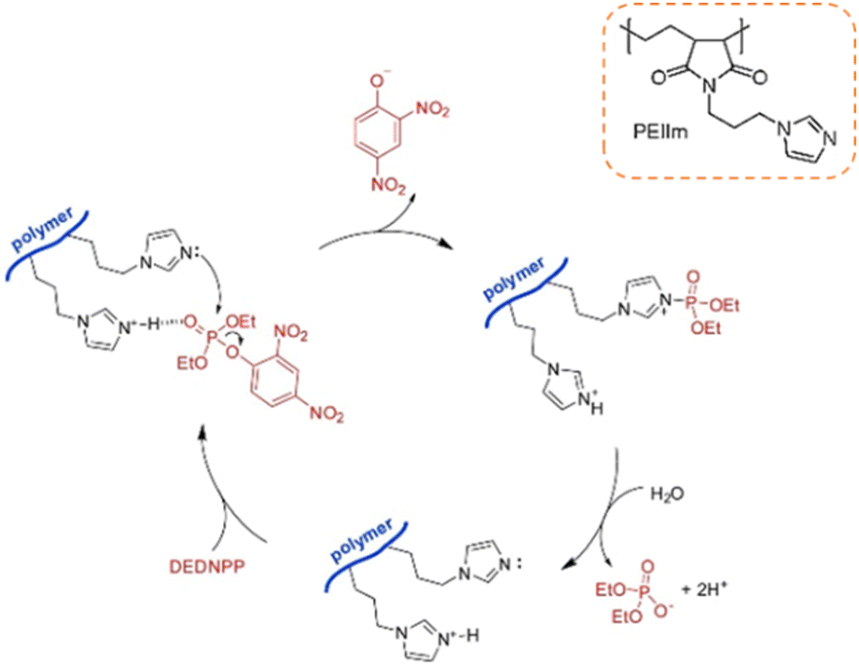 | ||
| Scheme 9 Catalytic cycle of the hydrolysis of DEDNPP promoted by the PEIIm polymer, whose imidazole-containing repeating unit is shown in the inset. Reprinted (adapted) with permission from ref. 84. Copyright 2021 Elsevier. | ||
2.7. Supramolecular colloidal systems
Seminal studies of reactivity of simple micellar and polymeric systems in the last century have shown that such systems exploit some of the unique characteristics responsible for the great efficiency of enzyme catalysis, in particular electrostatic and hydrophobic effects.73,75,85 Because phosphoryl transfer reactions from mono- and diesters occur in various important biological processes, a range of colloidal systems, with or without metal ions, were designed as nanoreactors to accelerate targeted reactions using model phosphate ester substrates, with a range of scenarios explored depending on the nature and charge of polymers, amphiphiles and reactants involved.86,87 Such colloidal systems are somewhat an evolution of what was termed as “micellar catalysis” in the last century, although such a term is recognized as a misnomer in some instances, when the observed increases in rate constants are rather a result of compartmentalization effects and not from actual catalytic pathways.88–90 In any event, both approaches are useful and can deliver reasonable observed rate enhancements, a result that is intrinsically related to the binding of the substrates and reactants to the supramolecular aggregates, arising from effects such as electrostatics, solvophobics, etc, and favoring the appropriate contact between reactive functional groups. In addition, the properties of the local environments to which the substrates bind may also be important to explain enhancements in reactivity.The design of systems composed specifically of amphiphilic and polymeric components, without metal ions, has been creatively explored to deliver supramolecular colloidal systems with potential for the decomposition of organophosphorus compounds. Provided that the substrates of choice may be neutral, the electrostatic features can be rather explored for the proper assembly of the nanoreactor components, with the binding of the substrate to the supramolecular aggregate being the consequence of non-covalent weak forces. One recent example is the complexation of cationic cetyltrimethylammonium (CTA+) with the polymer PAIM (Fig. 12) that features 50% of its functional groups in its backbone as carboxylate anions at pH values greater than ca. 5.5.91 Provided that the imidazolium ring also dissociates as the pH increases, the formation of the supramolecular complex should not be hampered by electrostatic repulsion interactions. It is noteworthy that the aggregation between the two components, performed in the presence of bromide as CTA+ counterion, occurs at CTAB concentrations lower than that of the critical micelle concentration (CMC) of the free surfactant in an aqueous medium, highlighting that the formation of the nanoreactors occurs with low amounts of the amphiphile.91
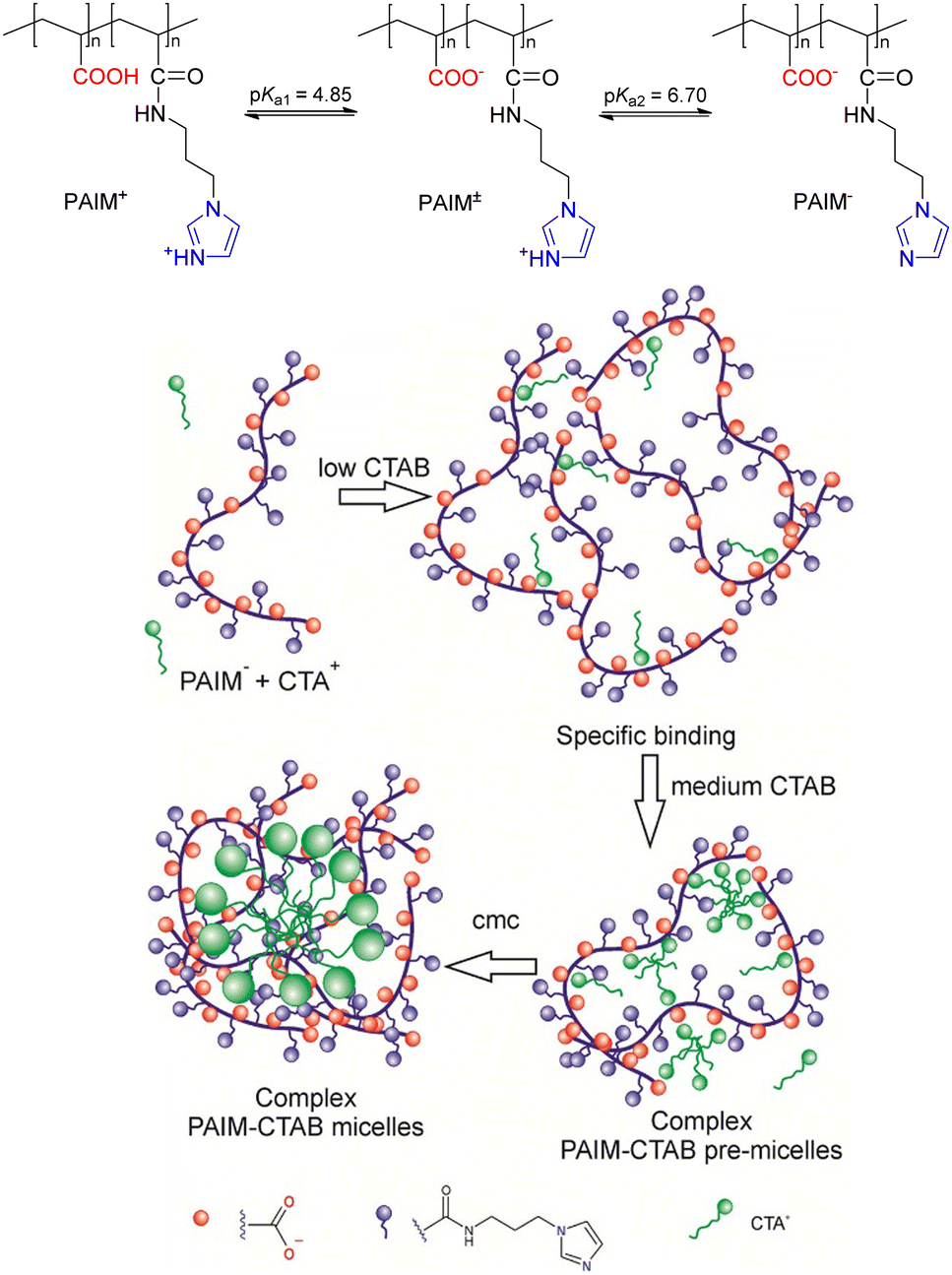 | ||
| Fig. 12 In the upper panel, the structure of the PAIM polymer, which features a 1:1 ratio between carboxylic acids and N-alkylimidazole. In the lower panel, a representation of the proposed aggregation steps between the anionic species of PAIM and CTA+ as a function of the CTA+ concentration, in an aqueous medium at 25 °C. Micelles of CTAB are also present at concentrations greater than its CMC. Reprinted (adapted) with permission from ref. 91. Copyright 2017 American Chemical Society. | ||
The supramolecular aggregate described in Fig. 12 was employed as a catalytic nanoreactor for the dephosphorylation of the phosphate triester DEDNPP, provided that the substrate binding to the hydrophobic pockets of the aggregates promotes the approximation of the substrate to the carboxylate and imidazole nucleophilic functionalities. In fact, considering the aggregates with the CTAB concentration below the CMC, rate enhancements in second order rate constants are up to 6.0 × 104 and 6.0 × 105-fold for the decomposition of DEDNPP through the reactions with carboxylate and imidazole, respectively, in comparison to the spontaneous hydrolysis of the substrate, with these effects being up to ca. 50-fold greater considering the second order rate constants for the reactions of DEDNPP with the polymer alone.91
Further investigations have shown that it is also straightforward to associate CTAB and pure poly(acrylic acid) polymers (PAA) featuring a range of molecular weights. In these cases, significant reactivity in the dephosphorylation of DEDNPP is promoted by two populations of carboxylates, formed by the dissociation of the corresponding acids with apparent pKa values of 3.80 and 5.78 for the PAA of 2 kDa.92 Interesting effects were also reported by the proper assembly of CTAB, PAA and N-alkyl imidazole monomers,93 as well as CTAB and N-alkyl imidazoles alone.94
In addition to aggregates prepared using two or more components, the use of self-assembled comb-like polyelectrolytes is also an alternative for the preparation of nanoreactors. One recent example employs the quaternary ammonium-containing polymers shown in Fig. 13 that also feature hydrophobic carbon tails,95 so that the compounds can aggregate in the aqueous medium providing both hydrophobic pockets for substrate compartmentalization and positive charges for the approximation of anionic reactants such as hydroxide. After evaluating the critical aggregation concentrations for polyelectrolytes, for both the intra- and intermolecular aggregation steps, the authors evaluated the reactivity of the alkaline hydrolysis of three phosphonates in the presence of the aggregates. By invoking a pseudophase model approach to interpret the kinetic results, the authors propose that the aggregates are able to compartmentalize both the moderately hydrophobic substrates and the anionic nucleophile, promoting rate enhancements up to two orders of magnitude depending on the polymer and the substrate.95
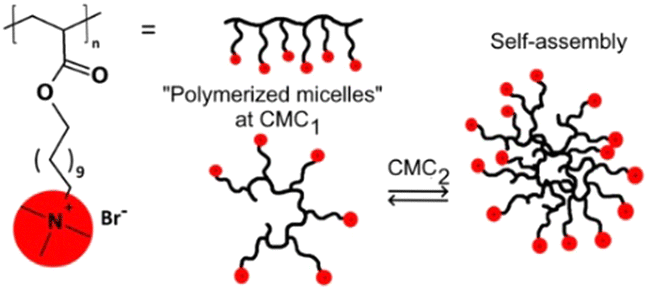 | ||
| Fig. 13 General structure of comb-like polyelectrolytes and the representation of their aggregation. Reprinted with permission from ref. 95. Copyright 2020 John Wiley & Sons. | ||
3. Conclusions and outlook
While some organophosphorus(V) compounds have important practical applications, other toxic structures had or still have been used in risky applications, and to deal with this scenario diverse catalytic systems for their detoxification have been studied to date. Testing such catalysts with simulants of agrochemicals and chemical warfare agents is the most common approach, and even with simulants in lieu of real targets the investigations must be conducted by trained personnel for security reasons. Creativity in the proposal of new systems has been observed, accomplishing a variety of reactants and reaction media, including, but not limited to, nucleophiles, metal ions, solid materials, the use of phosphotriesterases, and so forth.Some major conclusions can be summarized based on the examples discussed herein: (i) polarity and specific interactions with charged species are of paramount importance in the reaction medium for nucleophilic reactions, either catalytic or not, implicating a further opportunity in exploring solvent effects to optimize the degradation of P(V) compounds; (ii) metal centers are quite valuable in manipulating the catalytic performances either in complexes in solution, or as Lewis acid sites on solids, as well as in the form of metal nanoparticles, with several mechanistic explanations being possible; (iii) materials with practical applications can be obtained, such as peelable gels, by the incorporation of catalysts into appropriate matrices; (iv) polymers can be used as catalysts, either in solution, in supramolecular aggregates, or as heterogeneous catalysts; and (v) supramolecular colloidal aggregates offer platforms for enhancing reactivity by approximation of functional groups and/or changes in local medium properties.
Direct comparisons between figures of merit for different systems are sometimes difficult to establish. This is because some investigations are devoted to understanding the catalytic mechanisms, so that excess catalysts may be employed. In other instances, parallel reaction pathways occur with one or more of them being non-catalytic reactions. Also, it is important to note that complete investigations of the effects of the reaction variables are sometimes not performed, so that comparing the rate constants does not give information about the intrinsic reactivity of the system, but rather reflects observed rate constants under specific experimental conditions. Another important issue is that heterogeneous catalysts might have significant part of their reactive sites not available at their surfaces for the reaction, a situation usually not important in the homogenous counterparts. However, due to the binding of substrates at surfaces and interfaces, catalytic performances under heterogeneous conditions can frequently surpass those of the corresponding homogeneous systems. Not infrequently, investigation of reactivity in homogeneous media facilitates the development of heterogeneous catalysts with same functionalities, which can lead to better performances, combined with advantages such as catalyst recovery and reuse.
At any event, we believe that it is important for readers to present some figures of merit of the articles reviewed herein, such as values of half-life, the ratio between rate constants for the catalyzed and uncatalyzed reactions, second order rate constants, turnover numbers and frequencies, etc. This allows one to differentiate between a set of works under investigation and define the most suitable catalyst/reaction medium for specific substrates. Table 1 presents some of the figures of merit accompanied by detailed information such as the substrate and catalyst concentrations, pH and temperature. Readers should note that some systems are interesting regarding the catalytic performance but are somewhat limited to being employed in a considerably basic medium, while others are able to operate at pH values close to neutrality. For readers less familiar with kinetic measurements and interpretation of rate constants, it will be interesting to note the values of half-life, which indicate indirectly the enormous reduction in the time required for the decomposition of P(V) organic compounds.
| Catalyst/reactant | Substrate | Experimental conditions | Figures of merit | Ref. |
|---|---|---|---|---|
| a Figures of merit reported for the first use of the catalyst. b Catalyst regeneration was not evaluated. c First order rate constant for the spontaneous hydrolysis of the substrate (k0 = 7.92 × 10−10 s−1 at 25 °C) was taken from the literature.96 d k cat determined using Michaelis–Menten or Lineweaver–Burk equations; authors used the k0 for DEDNPP hydrolysis in pure water (8.0 × 10−6 s−1 at 25 °C).97 e k cat/kuncat is the ratio between the rate constants for the catalyzed and uncatalyzed hydrolysis of the substrate under the specified experimental conditions. f TOF is the turnover frequency. g First order rate constant for the solvolysis of methyl paraoxon with 1-propanol is estimated to be <1.50 × 10−9 s−1.65 | ||||
| Piperidineb | Ethyl paraoxon | 0.4 M of piperidine in ionic liquids and organic solvents, at 25 °C | For nucleophilic substitution on P(V) specifically, rate enhancementsc are between 80 and 6.5 × 104-fold, compared to the substrate hydrolysis | 29 |
| Piperidineb | Ethyl paraoxon | 2.8 M of piperidine in bio-based solvents, at 25 °C | Values of half-life in the range of <1–27 min, while the value for the spontaneous hydrolysis is ∼28 yearsc | 30 |
| Ethanol-d6 | Diethyl chlorophosphite | Acetonitrile and ionic liquids, at 25 °C | Second order rate constants increase up to 68-fold depending on the ionic liquid and its molar fraction | 31–33 |
| Mononuclear iron(III) complexes | DEDNPP | CH3CN:buffer (1:1, v/v) at 25 °C; buffer is either MES or HEPES at 50 mM |
Values of kcat/k0 in the range of 12–38 and in the pH range of 5.5–7.0d | 43 |
| Metal-containing peptoid membranes | Methyl paraoxon | NEM buffer at pH 10.0 and 25 °C | k cat/kuncat up to 219e | 44 |
| Composite of UiO-66, PVDF, Ti(OH)4, and TEA | Methyl paraoxon, diisopropyl fluorophosphate (DFP) and soman | Distilled water, 25 °C | Degradation of up to ca. 40% of methyl paraoxon in 2 h, and full consumption of DFP and soman with half-life values of 12.4 h and 35 min, respectively | 57 |
| UiO-66 | Methyl paraoxon | 0.45 M of N-ethyl morpholine buffer, at 25 °C | Initial rates increase with decreasing pH (from 10.2 to 8.8). At pH 8.8, the TOF is 0.12 s−1f (considering the available sites at the surface, the TOF is estimated to be 16 s−1) |
59 |
| NU-1000 | Methyl paraoxon | Aqueous medium, 25 °C | TOF values increase sharply with pH, exceeding 30000 s−1 at pH 10.5 |
61 |
| MgO | Methyl paraoxon | 1-Propanol, at 80 °C | Rate enhancements can be of the order of 3 × 106-fold, compared to the substrate spontaneous propanolysisg | 63 |
| CeO2 nano-octahedra incorporated in peelable gel | Ethyl paraoxon | Water or ethanol, at room temperature | Values of half-life are 48 and 34 min in water and ethanol, respectively | 69 |
| MgO@SBA15@GO composite | Methyl paraoxon | Irradiation with an infrared LED (810 nm, 20 mW cm−3) in ethanol | Conversion of 93.1% after 1 h, and 99% after 2 h | 70 |
| CoNPs supported on 3D-nitrogen-doped graphene | Methyl parathion, methyl paraoxon and others | Aqueous medium, at pH 11.3 (50 mM of NH4OH), 80 °C | After 1 h, conversions are 97.5% and 82.3% for methyl parathion and methyl paraoxon, respectively | 71 |
| Hetero-metallic composites with chitin | Prothiofos | Aqueous medium, at room temperature | Conversion of 99% after 2.5 h with the best catalyst (Pd@Au@Ag@chitin) | 72 |
| Amidoxime anchored on regenerated cellulose beads | Methyl parathion, soman and VX | Aqueous buffer at 50 mM for methyl parathion, and unbuffered media for soman and VX | A half-life of 27.7 h at pH 7.22 for methyl parathion. Values for soman and VX were estimated to be 1–10 min | 79 |
| Imidazole-functionalized rice husk | DEDNPP and ethyl paraoxon | 10 mM phosphate buffer (pH 7.5–8.0), 21–30 °C | First order rate constants (per gram of catalyst) are 0.43 and 6.36 × 10−3 min−1 g−1 for DEDNPP and ethyl paraoxon, respectively | 80 |
| Imidazole-rich heterogeneous polymer (PEIIm) | DEDNPP and ethyl paraoxon | pH 8.0, buffered with 50 mM tris buffer containing 1 M KCl, 25 °C | First order rate constants (per gram of catalyst per liter) are 9.1 × 10−4 and 6.18 × 10−8 L−1 g−1 s−1 for DEDNPP and ethyl paraoxon, respectively | 84 |
| Aggregates of CTAB and imidazole-functionalized PAA | DEDNPP | Aqueous medium. Reactivity increases with pH; plateau at pH > 8.5 | Second order rate constants are in the range 0.37–9.23 M−1 s−1, depending on the pH and the CTAB concentration | 91 |
| Quaternary ammonium polyelectrolytes | O-alkyl O-4-nitrophenyl chloromethyl phosphonate (alkyl = C2H5, C4H9, C6H13) | 1 mM NaOH, 25 °C. | Catalytic effects in the range of 5 and 107-fold, depending on the polyelectrolyte | 95 |
In general, some catalytic approaches have been more explored than others, so that there are still horizons for the elaboration of new optimized systems in the near future. For example, to the best of our knowledge, some kinds of materials that have not been explored for organophosphorus detoxification, such as covalent organic frameworks (COFs), and the use of complementary strategies such as microwaves and photothermal effects are also envisioned. Further developments in the understanding of phosphotriesterase enzymes (not covered in this highlight) are expected, and this can also be useful for the planning of more robust and less sensitive artificial enzymes that can mimic features of natural enzymes and deliver comparable rate enhancements. Also, it is worth noting that the development of technologies for the decomposition of P(V) compounds in flow reactors can be an interesting niche of investigation in the future. In addition, it is expected that all the accumulated knowledge in organophosphorus detoxification should evolve towards devices or materials for specific applications. One such niche is the development of technologies of protective clothing using fabrics that contain catalytic species capable of degrading a variety of P(V) compounds rapidly, bearing in mind partnerships between universities and chemical security institutions that possess appropriate installations for developing such projects. Our group is one of those currently exploring some of the above-mentioned options and others, and we look forward to seeing more advances in these growing niches of catalytic detoxification of P(V) compounds that have also been a focus of other groups worldwide.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. M. S. C. and N. R. D. S. thank scholarships from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and the Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), respectively. The authors are also thankful to Central Analítica Multiusuário (CAM-UFRRJ), which is financed by Financiadora de Estudos e Projetos (FINEP), CNPq and FAPERJ.Notes and references
- Y. J. Jang, K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2015, 115, PR1–PR76 CrossRef CAS PubMed.
- S. P.-M. Ung and C.-J. Li, RSC Sustainability, 2023, 1, 11–37 RSC.
- S. C. L. Kamerlin, P. K. Sharma, R. B. Prasad and A. Warshel, Q. Rev. Biophys., 2013, 46, 1–132 CrossRef CAS PubMed.
- A. Blum, M. Behl, L. S. Birnbaum, M. L. Diamond, A. Phillips, V. Singla, N. S. Sipes, H. M. Stapleton and M. Venier, Environ. Sci. Technol. Lett., 2019, 6, 638–649 CrossRef CAS.
- I. Pantelaki and D. Voutsa, Sci. Total Environ., 2019, 649, 247–263 CrossRef CAS PubMed.
- G. Poma, C. Sales, B. Bruyland, C. Christia, S. Goscinny, J. Van Loco and A. Covaci, Environ. Sci. Technol., 2018, 52, 2331–2338 CrossRef CAS PubMed.
- G. P. Horsman and D. L. Zechel, Chem. Rev., 2017, 117, 5704–5783 CrossRef CAS PubMed.
- J. A. Sikorski and K. J. Gruys, Acc. Chem. Res., 1997, 30, 2–8 CrossRef CAS.
- C. MacBean and British Crop Protection Council, The pesticide manual: a world compendium, British Crop Protection Council, Alton, Hampshire, 16th edn, 2012 Search PubMed.
- R. Kanissery, B. Gairhe, D. Kadyampakeni, O. Batuman and F. Alferez, Plants, 2019, 8, 499 CrossRef CAS.
- C. P. Holstege, M. Kirk and F. R. Sidell, Crit. Care Clin., 1997, 13, 923–942 CrossRef CAS PubMed.
- A. A. D. Castro, I. G. Prandi, K. Kuca and T. C. Ramalho, Ciênc. Agrotec., 2017, 41, 471–482 CrossRef.
- T. Čadež, D. Kolić, G. Šinko and Z. Kovarik, Sci. Rep., 2021, 11, 21486 CrossRef PubMed.
- J. J. Sejvar, Neurol. Clin., 2020, 38, 881–896 CrossRef.
- E. H. Wanderlind, M. Medeiros, B. S. Souza, H. D. Fiedler and F. Nome, Rev. Virtual Quim., 2014, 6, 632–652 Search PubMed.
- J. B. Domingos, E. Longhinotti, V. G. Machado and F. Nome, Quim. Nova, 2003, 26, 745–753 CrossRef CAS.
- Organisation for the Prohibition of Chemical Weapons, https://www.opcw.org/node/2632, (accessed June 25, 2024).
- The Nobel Peace Prize 2013, https://www.nobelprize.org/prizes/peace/2013/summary/, (accessed June 25, 2024).
- A. Ebrahimi, E. Nassireslami, R. Zibaseresht and M. Mohammadsalehi, Mol. Catal., 2020, 490, 110965 CrossRef CAS.
- L. Zhang, H. Murata, G. Amitai, P. N. Smith, K. Matyjaszewski and A. J. Russell, Biomacromolecules, 2020, 21, 3867–3877 CrossRef CAS PubMed.
- F. M. Raushel, Nature, 2011, 469, 310–311 CrossRef CAS.
- A. J. Kirby and F. Nome, Acc. Chem. Res., 2015, 48, 1806–1814 CrossRef CAS.
- J. K. Lassila, J. G. Zalatan and D. Herschlag, Annu. Rev. Biochem., 2011, 80, 669–702 CrossRef CAS.
- A. M. Manfredi, W. Demos, E. H. Wanderlind, B. V. Silva, A. C. Pinto, B. S. Souza and F. Nome, J. Phys. Org. Chem., 2016, 29, 600–603 CrossRef CAS.
- N. Dey, J. Kulhánek, F. Bureš and S. Bhattacharya, J. Org. Chem., 2021, 86, 14663–14671 CrossRef CAS PubMed.
- P. T. Wong, S. Bhattacharjee, J. Cannon, S. Tang, K. Yang, S. Bowden, V. Varnau, J. J. O’Konek and S. K. Choi, Org. Biomol. Chem., 2019, 17, 3951–3963 RSC.
- P. T. Wong, S. Tang, J. Cannon, K. Yang, R. Harrison, M. Ruge, J. J. O’Konek and S. K. Choi, ACS Appl. Mater. Interfaces, 2020, 12, 33500–33515 CrossRef CAS.
- S. Zboray, K. Efimenko, J. L. Jones and J. Genzer, Ind. Eng. Chem. Res., 2021, 60, 8799–8811 CrossRef CAS.
- P. Pavez, D. Millán, J. I. Morales, E. A. Castro, A. C. López and J. G. Santos, J. Org. Chem., 2013, 78, 9670–9676 CrossRef CAS.
- P. Pavez, G. Oliva and D. Millán, ACS Sustainable Chem. Eng., 2016, 4, 7023–7031 CrossRef CAS.
- B. J. Butler and J. B. Harper, New J. Chem., 2014, 39, 213–219 RSC.
- B. J. Butler and J. B. Harper, J. Phys. Org. Chem., 2016, 29, 700–708 CrossRef CAS.
- B. J. Butler and J. B. Harper, J. Phys. Org. Chem., 2019, 32, e3819 CrossRef.
- B. J. Butler and J. B. Harper, New J. Chem., 2015, 39, 1525 RSC.
- Y. Lyu and P. Scrimin, ACS Catal., 2021, 11, 11501–11509 CrossRef CAS.
- L. A. Wilson, M. M. Pedroso, R. A. Peralta, L. R. Gahan and G. Schenk, J. Inorg. Biochem., 2023, 238, 112061 CrossRef CAS.
- L. Jiang, Y. Sun, Y. Chen and P. Nan, ChemistrySelect, 2020, 5, 9492–9516 CrossRef CAS.
- E. H. Wanderlind, C. R. Bittencourt, A. M. Manfredi, A. P. Gerola, B. S. Souza, H. D. Fiedler and F. Nome, J. Phys. Org. Chem., 2019, e3837 CrossRef.
- L. R. Gahan, S. J. Smith, A. Neves and G. Schenk, Eur. J. Inorg. Chem., 2009, 2745–2758 CrossRef CAS.
- B. Meza-González and F. Cortés-Guzmán, Phys. Chem. Chem. Phys., 2023, 25, 18652–18658 RSC.
- M. A. R. Raycroft, C. T. Liu and R. S. Brown, Inorg. Chem., 2012, 51, 3846–3854 CrossRef CAS PubMed.
- J. C. Lugo-González, P. Gómez-Tagle, M. Flores-Alamo and A. K. Yatsimirsky, Dalton Trans., 2020, 49, 2452–2467 RSC.
- E. Luiz, G. Farias, M. Alzamora, D. R. Sánchez, F. R. Xavier and R. A. Peralta, Eur. J. Inorg. Chem., 2024, e202300650 CrossRef CAS.
- T. K. H. Trinh, T. Jian, B. Jin, D.-T. Nguyen, R. N. Zuckermann and C.-L. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 51191–51203 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- V. F. Yusuf, N. I. Malek and S. K. Kailasa, ACS Omega, 2022, 7, 44507–44531 CrossRef CAS.
- J. Liu, T. A. Goetjen, Q. Wang, J. G. Knapp, M. C. Wasson, Y. Yang, Z. H. Syed, M. Delferro, J. M. Notestein, O. K. Farha and J. T. Hupp, Chem. Soc. Rev., 2022, 51, 1045–1097 RSC.
- X. Li, R. Zhou, Z. Wang, M. Zhang and T. He, J. Mater. Chem. A, 2022, 10, 1642–1681 RSC.
- K. Ma, Y. H. Cheung, K. O. Kirlikovali, X. Wang, T. Islamoglu, J. H. Xin and O. K. Farha, Acc. Mater. Res., 2023, 4, 168–179 CrossRef CAS.
- D. F. S. Gallis, J. A. Harvey, C. J. Pearce, M. G. Hall, J. B. DeCoste, M. K. Kinnan and J. A. Greathouse, J. Mater. Chem. A, 2018, 6, 3038–3045 RSC.
- D. Ma and Z. Cao, J. Phys. Chem. C, 2022, 126, 19159–19168 CrossRef CAS.
- C. Vieira Soares, G. Maurin and A. A. Leitão, J. Phys. Chem. C, 2019, 123, 19077–19086 CrossRef CAS.
- J. Yang, M. Gao, M. Zhang, Y. Zhang, M. Gao, Z. Wang, L. Xu, X. Wang and B. Shen, Coord. Chem. Rev., 2023, 493, 215289 CrossRef CAS.
- C. Zhou, S. Zhang, H. Pan, G. Yang, L. Wang, C. Tao and H. Li, RSC Adv., 2021, 11, 22125–22130 RSC.
- K. O. Kirlikovali, Z. Chen, T. Islamoglu, J. T. Hupp and O. K. Farha, ACS Appl. Mater. Interfaces, 2020, 12, 14702–14720 CrossRef CAS.
- P. Tang and G. Sun, Mater. Sci. Eng., R, 2021, 145, 100626 CrossRef.
- D. B. Dwyer, N. Dugan, N. Hoffman, D. J. Cooke, M. G. Hall, T. M. Tovar, W. E. Bernier, J. DeCoste, N. L. Pomerantz and W. E. Jones Jr., ACS Appl. Mater. Interfaces, 2018, 10, 34585–34591 CrossRef CAS PubMed.
- D. L. McCarthy, J. Liu, D. B. Dwyer, J. L. Troiano, S. M. Boyer, J. B. DeCoste, W. E. Bernier, J. Wayne and E. Jones, New J. Chem., 2017, 41, 8748–8753 RSC.
- M. J. Katz, R. C. Klet, S.-Y. Moon, J. E. Mondloch, J. T. Hupp and O. K. Farha, ACS Catal., 2015, 5, 4637–4642 CrossRef CAS.
- H. Chen, P. Liao, M. L. Mendonca and R. Q. Snurr, J. Phys. Chem. C, 2018, 122, 12362–12368 CrossRef CAS.
- Y. Liao, T. R. Sheridan, J. Liu, Z. Lu, K. Ma, H. Yang, O. K. Farha and J. T. Hupp, ACS Catal., 2024, 14, 437–448 CrossRef CAS.
- S. B. B. B. Bahia, C. A. Amaya Vargas, Â. M. L. Denadai, M. H. Araujo, A. H. Moraes and T. A. S. Brandão, J. Phys. Chem. C, 2020, 124, 17111–17120 CrossRef CAS.
- G. I. Almerindo, S. C. Buratto, E. H. Wanderlind, L. M. Nicolazi, P. Sangaletti, M. Medeiros, F. S. S. Schneider, G. F. Caramori, R. L. T. Parreira, G. A. Micke, H. D. Fiedler and F. Nome, J. Mater. Chem. A, 2020, 8, 19011–19021 RSC.
- J. C. Védrine, Chin. J. Catal., 2019, 40, 1627–1636 CrossRef.
- L. M. Zimmermann, G. I. Almerindo, J. R. Mora, I. H. Bechtold, H. D. Fiedler and F. Nome, J. Phys. Chem. C, 2013, 117, 26097–26105 CrossRef CAS.
- G. I. Almerindo, P. Bueno, L. M. Nicolazi, E. H. Wanderlind, P. Sangaletti, S. M. Landi, L. A. Sena, B. S. Archanjo, C. A. Achete, H. D. Fiedler and F. Nome, J. Phys. Chem. C, 2016, 120, 22323–22329 CrossRef CAS.
- J. V. Hemmer, E. H. Wanderlind, H. A. G. Bazani, C. E. M. Campos, T. M. Wagner, A. K. Emmerich, J. R. U. Adão and G. I. Almerindo, Braz. J. Chem. Eng., 2020, 37, 533–541 CrossRef CAS.
- L. M. Zimmermann, G. I. Almerindo, M. Medeiros, R. F. Affeldt, E. H. Wanderlind, A. P. Gerola, R. A. Nome, M. A. Tumelero, R. Faccio, A. A. Pasa, H. D. Fiedler and F. Nome, J. Phys. Chem. C, 2018, 122, 25530–25538 CrossRef CAS.
- E. Thomas, C. Bordes, F. Chaput, D. Arquier, S. Briançon and M.-A. Bolzinger, Colloids Surf., A, 2024, 687, 133520 CrossRef CAS.
- Y. J. Kim, S. Y. Heo, W. J. Jang and S. J. Lee, Microporous Mesoporous Mater., 2023, 356, 112588 CrossRef CAS.
- Y. Li, J. Yan, D. Yu, P. Lei, W. Shen, M. Zhong, J. Zhang and S. Guo, Inorg. Chem., 2021, 60, 17635–17640 CrossRef CAS.
- H. E. Emam, H. B. Ahmed and R. M. Abdelhameed, Carbohydr. Polym., 2021, 266, 118163 CrossRef CAS PubMed.
- T. Chen, Y. Lu, X. Xiong, M. Qiu, Y. Peng and Z. Xu, Adv. Colloid Interface Sci., 2024, 323, 103072 CrossRef CAS PubMed.
- D. Jung, P. Das, A. Atilgan, P. Li, J. T. Hupp, T. Islamoglu, J. A. Kalow and O. K. Farha, Chem. Mater., 2020, 32, 9299–9306 CrossRef CAS.
- M. D. Nothling, Z. Xiao, A. Bhaskaran, M. T. Blyth, C. W. Bennett, M. L. Coote and L. A. Connal, ACS Catal., 2019, 9, 168–187 CrossRef CAS.
- S. Magalhães, C. Fernandes, J. F. S. Pedrosa, L. Alves, B. Medronho, P. J. T. Ferreira and M. da G. Rasteiro, Polymers, 2023, 15, 3138 CrossRef PubMed.
- T. Aziz, A. Farid, F. Haq, M. Kiran, A. Ullah, K. Zhang, C. Li, S. Ghazanfar, H. Sun, R. Ullah, A. Ali, M. Muzammal, M. Shah, N. Akhtar, S. Selim, N. Hagagy, M. Samy and S. K. Al Jaouni, Polymers, 2022, 14, 3206 CrossRef CAS.
- V. Thakur, A. Guleria, S. Kumar, S. Sharma and K. Singh, Mater. Adv., 2021, 2, 1872–1895 RSC.
- P. Janoš, O. Tokar, M. Došek, K. Mazanec, P. Ryšánek, M. Kormunda, J. Henych and P. Janoš, RSC Adv., 2021, 11, 17976–17984 RSC.
- J. G. L. Ferreira and E. S. Orth, J. Braz. Chem. Soc., 2017, 28, 1760–1767 CAS.
- L. Hostert, R. B. Campos, J. E. S. Fonsaca, V. B. Silva, S. F. Blaskievicz, J. G. L. Ferreira, W. Takarada, N. Naidek, Y. H. Santos, L. L. Q. Nascimento, A. J. G. Zarbin and E. S. Orth, Pure Appl. Chem., 2018, 90, 1593–1603 CrossRef CAS.
- J. G. L. Ferreira, W. H. Takarada and E. S. Orth, J. Hazard. Mater., 2022, 427, 127885 CrossRef CAS PubMed.
- J. G. L. Ferreira and E. S. Orth, Environ. Pollut., 2023, 330, 121802 CrossRef CAS PubMed.
- E. C. Leopoldino, G. Pinheiro, R. J. Alves, A. Gerola and B. S. Souza, Mater. Today Commun., 2021, 26, 101904 CrossRef CAS.
- J. Pan, S. Liu, H. Jia, J. Yang, M. Qin, T. Zhou, Z. Chen, X. Jia and T. Guo, J. Catal., 2019, 380, 83–90 CrossRef CAS.
- G. A. Monti, R. D. Falcone, F. Moyano and N. M. Correa, RSC Adv., 2023, 13, 1194–1202 RSC.
- F. Mancin, P. Scrimin, P. Tecilla and U. Tonellato, Coord. Chem. Rev., 2009, 253, 2150–2165 CrossRef CAS.
- C. A. Bunton, F. Nome, F. H. Quina and L. S. Romsted, Acc. Chem. Res., 1991, 24, 357–364 CrossRef CAS.
- L. S. Romsted, C. A. Bunton and J. Yao, Curr. Opin. Colloid Interface Sci., 1997, 2, 622–628 CrossRef CAS.
- C. A. Bunton, Adv. Colloid Interface Sci., 2006, 123–126, 333–343 CrossRef CAS PubMed.
- A. P. Gerola, E. H. Wanderlind, Y. S. Gomes, L. A. Giusti, L. Garcia-Rio, R. A. Nome, A. J. Kirby, H. D. Fiedler and F. Nome, ACS Catal., 2017, 7, 2230–2239 CrossRef CAS.
- C. R. Bittencourt, W. Demos, E. H. Wanderlind, F. Nome and A. P. Gerola, ACS Appl. Polym. Mater., 2022, 4, 9313–9322 CrossRef CAS.
- W. Demos, C. R. Bittencourt, L. Nardino, F. Nome and A. P. Gerola, ACS Appl. Nano Mater., 2021, 4, 644–651 CrossRef CAS.
- C. R. Bittencourt, M. H. de Souza, M. Medeiros, F. H. Quina, B. S. Souza and A. P. Gerola, J. Mol. Liq., 2022, 360, 119348 CrossRef CAS.
- T. N. Pashirova, P. A. Fetin, A. A. Lezov, M. V. Kadnikov, F. G. Valeeva, E. A. Burilova, A. Yu. Bilibin and I. M. Zorin, ChemPlusChem, 2020, 85, 1939–1948 CrossRef CAS PubMed.
- A. J. Kirby, M. Medeiros, P. S. M. Oliveira, E. S. Orth, T. A. S. Brandão, E. H. Wanderlind, A. Amer, N. H. Williams and F. Nome, Chem. – Eur. J., 2011, 17, 14996–15004 CrossRef CAS PubMed.
- E. H. Wanderlind, E. S. Orth, M. Medeiros, D. M. P. O. Santos, E. Westphal, H. Gallardo, H. D. Fiedler and F. Nome, J. Braz. Chem. Soc., 2014, 25, 2385–2391 CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |