
Facets of click-mediated triazoles in decorating amino acids and peptides
Subhendu Sekhar
Bag
 *ab,
Aniket
Banerjee
b,
Sayantan
Sinha
b and
Subhashis
Jana
a
*ab,
Aniket
Banerjee
b,
Sayantan
Sinha
b and
Subhashis
Jana
a
aChemical Biology/Genomics Laboratory, Department of Chemistry, Indian Institute of Technology Guwahati, 781039, India. E-mail: ssbag75@iitg.ac.in
bCentre for the Environment, Indian Institute of Technology Guwahati, 781039, India
First published on 31st October 2024
Abstract
Decorating biomolecular building blocks, such as amino acids, to afford desired and tuneable photophysical/biophysical properties would allow chemical biologists to use them for several biotechnological and biosensing applications. While many synthetic methodologies have been explored in this direction, advantages provided by click-derived triazole moieties are second to none. However, since their discovery, click-mediated triazoles have been majorly utilised as linkers for conjugating biomolecules, creating materials with novel properties, such as polymers or drug conjugates. Despite exploring their profound role as linkers, click-mediated triazoles as an integral part of biomolecular building blocks have not been addressed. 1,2,3-Triazole, a transamide mimic, exhibits high aromatic stacking propensity, high associability with biomolecules through H-bonding, and high stability against enzymatic hydrolysis. Furthermore, triazoles can be considered donors useable for installation/modulation of the photophysics of a fluorophore. Therefore, triazole with a chromophoric unit may rightly be utilised as an integral part of biomolecular building blocks to install microenvironment-sensitive solvofluorochromic properties suitable for biological sensing, studying inter-biomolecular interactions and introducing novel physicochemical properties in a biomolecule. This review mainly focuses on the facets of click-derived triazole in designing novel fluorescent amino acids and peptides with a particular emphasis on those wherein triazole acts as an integral part of amino acids, i.e. the side chain, generating a new class of fluorescent unnatural triazolyl amino acids. Thus, fluorescent triazolyl unnatural amino acids, peptidomimetics with such amino acids and aliphatic/aromatic triazolyl amino acids as scaffolds for peptidomimetics are the central part. However, to start with, a brief history, followed by a discussion on various other relevant facets of triazoles as linkers in various fields ranging from therapeutics, materials science, diagnostics, and bioconjugation to peptidomimetics, is cited. Additionally, the possible roles of CuAAC-mediated triazoles in shaping the future of bioorganic chemistry, medicinal chemistry, diagnostics, nucleoside chemistry and protein engineering are briefly discussed.
1. Introduction
Synthetic organic chemistry has contributed significantly to the advancements in science, technology, and medicine.1,2 In the modern day, it focuses on creating unique molecules with specific features, such as heterocyclic compounds.3 Furthermore, a lot of interest has been observed in developing novel techniques for functionalizing and conjugating several molecules, including biomolecules.Among various heterocycles, the creation of a five-membered triazole ring system is one of the most important fundamental developments in organic chemistry, which is frequently employed to create molecules/molecular libraries. Such libraries have essential relevance in research and developments in both academia and industry because triazoles are found to be suitable candidates in various applications across several domains of research, including as agents with potent biological activity. The term triazole was initially coined by Bladin in the late 19th century to denote a five-membered heterocyclic aromatic-ring-system containing three nitrogen atom residues with the molecular formula C2H3N3.4 The five-membered ring is capable of forming two major isomers, 1,2,4-triazole (s-triazole) and 1,2,3-triazole (v-triazole), depending on the two possible spatial arrangements of nitrogen atoms.5 Based on the hydrogen atoms linked to the ring nitrogen, each of them can display primarily two tautomers, which are depicted in Fig. 1(A).
 | ||
| Fig. 1 Structures of (A) the tautomers of triazoles and (B) a few triazole-based drugs. | ||
Out of these two isomers, the 1,2,3-triazole moiety has found its purpose as a crucial functional group in modern-day chemistry just after the introduction of the “click” reaction in 2001 by Sharples and Meldal. Several beneficial characteristics of 1,2,3-triazoles include large dipole moment, aromatic π-stacking ability, high chemical as well as biological stability, and high associability via H-bonding interaction. Because of these remarkable features, the substituted 1,2,3-triazole motif mimics an E- or Z- amide bond. Thus, the 1,2,3-triazoles serve as good foundations in the field of drug discovery research. Numerous well-known drugs with 1,2,3-triazole moiety are now commercially available, such as the β-lactam antibiotic tazobactam, the anticonvulsant medicine rufinamide, and the anticancer medication carboxyamidotriazole, to name a few [Fig. 1(B)].6
2. Methods of formation of 1,2,3-triazoles
Initially, the azide–alkyne cycloaddition reaction was rapidly found to be the most effective method for the creation of 1,2,3-triazoles used for conjugating two or more molecules, including bioconjugation. However, this reaction lacked generality and broad applicability because steric and electronic factors heavily influenced its regioselectivity. While Huisgen and co-researchers conducted extensive research on the 1,3-dipolar cycloaddition reaction between a terminal alkyne and an azide over half a century ago;7 the reaction necessitated heating and lacked regioselectivity as both 1,5- and 1,4-disubstituted-1,2,3-triazoles were produced. Subsequently, Sharpless and Meldal established a significant breakthrough by finding extraordinary regioselectivity in copper-catalyzed alkyne–azide cycloaddition (CuAAC) and presented the notion of “click chemistry”.8,9 The schematic of the CuAAC reaction in comparison to the Huisgen cycloaddition reaction is depicted in Fig. 2(A).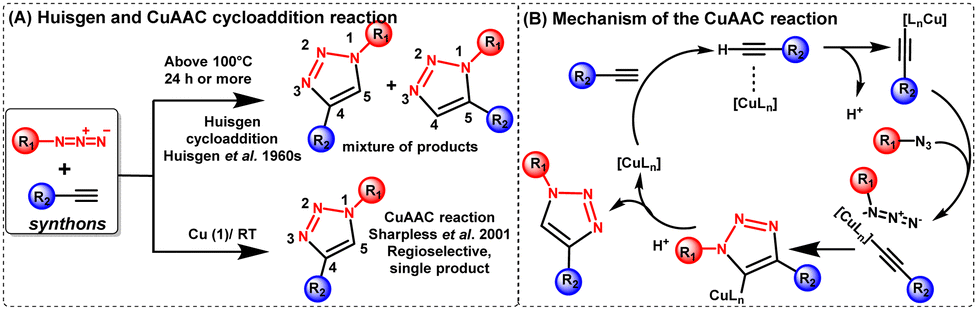 | ||
| Fig. 2 (A) A schematic of the CuAAC reaction in comparison to the Huisgen cycloaddition reaction and (B) mechanism of the CuAAC reaction. | ||
Afterwards, Sharpless demonstrated the selective formation of the 1,4-disubstituted triazole at room temperatures in the presence of Cu(I) as a catalyst with up to 107 times rate enhancements. The reaction is exothermic, and a six-membered Cu(III)-metallacycle is proposed to form when the Cu(I)-acetylide intermediate is attached to the azide moiety [Fig. 2(B)].10
Over the years, these triazole moieties have been synthesized using various forms of click chemistry. The triazoles thus formed have been primarily utilised for their applications as mere linkers in the fields of novel drug discovery, material sciences, biosensor development and so on. However, despite the extensive use of triazole subunits as linkers, their intrinsic properties have not been explored to their fullest potential. Therefore, in this article, we will first briefly introduce the various forms of click reactions and applications of click-derived triazoles as mere linkers in various fields, especially in peptide/protein chemistry. Following this, most of the emphasis has been given to showcasing the unexplored crucial role as an integral part of biomolecular building blocks, especially of amino acids and peptidomimetics.
3. Non-triazole and triazole forming click reactions
As discussed previously, click chemistry refers to a class of high-yielding, selective, and efficient chemical strategies that are widely employed in several fields, such as organic synthesis, drug discovery, material chemistry, chemical biology and decorating biomolecular building blocks. Such reaction processes are distinguished by their capacity to produce desired stable molecular ensembles under mild experimental conditions, making them highly beneficial in several biological and industrial applications. Although our focus lies on the CuAAC-derived triazole moiety, we will initially introduce the various other types of click reactions. This includes (a) non-triazole forming click reactions such as (i) thiol–ene reaction,11 (ii) Diels–Alder reaction,12 (iii) native chemical ligation (NCL),13 (iv) Staudinger ligation,14 (v) thiol-Michael addition,15 and (b) triazole-forming click reactions such as strain-promoted azide–alkyne cycloaddition (SPAAC)16 and copper-catalyzed alkyne–azide cycloaddition (CuAAC).173.1. Non-triazole forming click reactions
Along with the CuAAC reaction, the thiol–ene reaction is also an important form of click chemistry. Triggered by ultraviolet (UV) light, a thiyl radical from thiol adds to an alkene, known as a thiol–ene reaction, which is commonly utilised in bioconjugation reactions (Table 1). However, the process can be hampered by side reactions such as alkene polymerisation and thiol oxidation reactions, which need precise regulation of pH and the wavelength of UV-irradiation.18–20 Thermal [4+2] cycloaddition of a diene and a dienophile, popularly known as the Diels–Alder reaction, is another example of a click reaction,21 which is widely recognised for its excellent selectivity and catalyst-free nature for the production of complex molecular frameworks. However, since the reaction speed is inadequate at room temperature, other forms of click reactions are often preferred (Table 1).21,22| Type of click reaction | Reaction scheme/key features |
|---|---|
| Thiol–ene reaction |

|
| • No metal catalyst • Side reactions: thiol oxidation/alkene polymerization • Application: bioconjugation. | |
| Diels–Alder reaction |
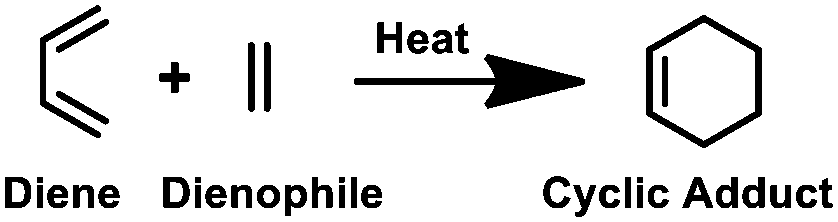
|
| • Catalyst-free • Highly selective • Temperature-dependent. | |
| Native chemical ligation (NCL) |
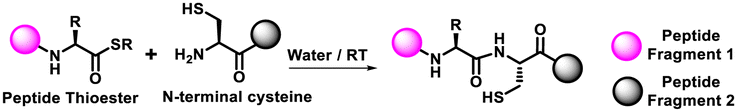
|
| • Physiological pH • Application: total protein synthesis. | |
| Staudinger ligation |
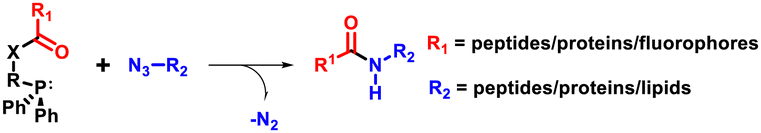
|
| • Bioorthogonal • Traceless and non-traceless variants • Application: bioconjugation and protein labelling. | |
| Thiol-Michael addition |
|
| • Solventless • Fast | |
| Strain-promoted azide–alkyne cycloaddition (SPAAC) |
|
| • No metal catalyst • Biocompatible • High selectivity • Low background labelling • Application: labeling biomolecules in live cells. | |
| Copper-catalyzed azide–alkyne cycloaddition (CuAAC) |
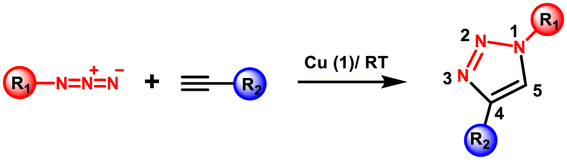
|
| • Highly regioselective • Application: amino acids, peptidomimetic, biosensors, and therapeutics. | |
| Photocatalytic (CuII@TiO2) click reaction |
|
The third category of non-triazole forming click reaction includes native chemical ligation (NCL) that combines a C-terminal peptide thioester with an N-cysteinyl peptide to form a native peptide.23–25 This strategy is commonly used to produce full-length proteins from smaller peptide fragments under aqueous and physiological conditions. Among the various forms, one of the most popular forms of click reaction is the bioorthogonal Staudinger ligation,26,27 which produces an amide linkage between an azide and a phosphine. The extreme selectivity and traceless nature make it highly suitable for bioconjugation reactions and protein assembly using smaller peptide fragments (Table 1).
The thiol-Michael addition reaction is another form of click reaction that falls within the intersection of the thiol-Click and Michael addition reactions. The reaction between thiol and an alkene containing an electron-withdrawing group generates a thiol–ester bond under mild conditions28,29 widely utilised in organic chemistry and materials science (Table 1).
3.2. Triazole forming click-reaction
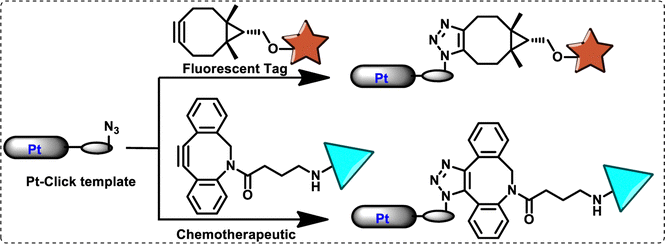
The click-derived 1,2,3-triazoles have drawn much attention over the past two decades mainly due to their biological activities and roles as amide bond isosteres. The concept was initially explored by Huisgen in the 1960s producing a mixture of 1,4- and 1,5-regioisomers.30 Later, advancements in the field by Sharpless and Meldal paved the way for copper-catalyzed azide–alkyne cycloaddition (CuAAC), facilitating the regioselective synthesis of 1,4-disubstituted 1,2,3-triazoles.31 Among all, the strain-promoted azide–alkyne cycloaddition (SPAAC) and CuAAC- reaction for the formation of click-derived triazoles are the most extensively documented and effective protocols widely utilised in the fields of therapeutics, diagnostics, chemical biology and material sciences, which are summarised as follows (Table 1).However, the use of the CuAAC reaction tends to be limited in biological systems owing to the toxicity of copper. This was resolved after the discovery of SPAAC, which utilises strained cyclooctynes such as dibenzocyclooctyne (DIBO), difluoromethylidene cyclooctane (DIFO), azadibenzylcyclooctyne (DIBAC) and 1-butyl-1-cyclooctene (BCN),33 in the absence of a catalyst.34 Moreover, the cyclooctynes guarantee sufficient hydrophilicity and exhibit remarkable reactivity with azides that allows easy and efficient labelling of azide-functionalized molecules/biomolecules with much better specificity and efficiency in comparison to CuAAC processes.35,36 The most prominent instance of the use of this technique has been reported by Bertozzi et al., who recognised the promise of the SPAAC methodology37 in live cells in a manner similar to the CuAAC reaction while avoiding potentially hazardous consequences of copper catalysts (Table 1).38
The reaction is categorized as a kind of Huisgen 1,3-dipolar cycloaddition42 that proceeds smoothly at room temperature. As a result, a wide range of copper catalysts and copper-free, base-free and other methodologies focusing on fulfilling ultimate research demands surrounding click reaction have been developed.43–48
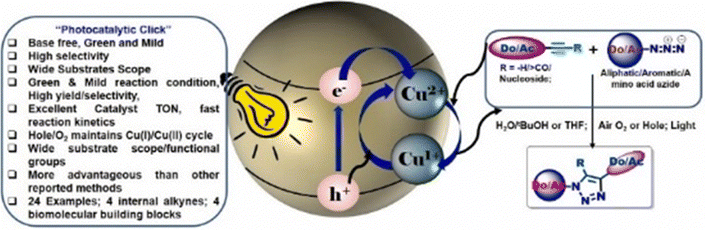
Towards this end, our group has also reported the formation of a new smart photocatalytic system capable of catalysing a base-free click reaction with excellent selectivity, efficacy and rapid reaction kinetics.49 We have created a new and novel photocatalyst for the CuAAC reaction in a green solvent under mild reaction conditions. The catalyst Cu@TiO2 converts Cu(II) to Cu(I), upon light exposure to create electrons from TiO2 semiconductor. Conversely, the produced holes or the oxygen in the air oxidises Cu(I) into Cu(II), which is vital for sustaining a steady supply of feed for the photocatalytic process. A comparative study of the schemes of the various click reactions and their distinct features have been highlighted in Table 1.
Therefore, despite the existence of various types of click reactions, the CuAAC reaction has been primarily used by scientists in multiple fields in recent times. Overall, the triazole moiety generated from copper-catalyzed azide–alkyne cycloaddition (CuAAC) has been found to have applications in various fields due to its high efficacy, associability, and stability. The various facets of click-mediated triazoles are discussed in the following section.
4. Various facets of click-mediated triazoles
As discussed earlier, the highly beneficial properties of triazoles have made them ideal candidates for a wide range of applications. Some of the significant fields where triazoles are used are- (i) drug discovery and development, (ii) material sciences, (iii) bioconjugation chemistry, (iv) supramolecular chemistry and (v) chemical biology. For most of these cases, the click-derived triazole unit serves the role of a linker for tagging/conjugating various units as needed by research. However, the potential of the triazole moiety cannot be limited to their roles solely as linkers. Therefore, in this particular section, emphasis has been given to those facets of triazoles that are less explored or rarely thought of. Thus, the examples of triazoles as linkers in various research fields other than amino acid and peptides will be briefly introduced first. Next, highlights of the examples of peptides containing triazoles as linkers will be discussed. Finally, this section will grossly emphasise the examples where the triazole group has been utilised not as a mere linker but appear as a unique entity or an integral part of a molecule with its unique properties, especially in the field of amino acids and peptidomimetics. In this regard, efforts from our group have been highlighted, showcasing triazole units as integral parts of biomolecular building blocks. Since long we have been involved in utilising the triazole unit not as a mere linker but as an integral part of molecules and exploring its inherent physicochemical properties to (a) create unnatural biomolecular building blocks, such as unnatural amino acids and nucleosides with novel physicochemical and photophysical properties, (b) use as scaffold for inducing protein's secondary structure in short peptidomimetics, (c) exploit its electron donation ability for the installation/modulation of the photophysics of chromophoric molecules. Finally, the atropoisomerism in the designed aromatic triazolo amino acid scaffold has been exploited as a sensor for discriminating methanol from ethanol.Furthermore, triazole units have also been exploited as the backbone of oligonucleotides by Brown et al.50,51 Other facets of triazoles such as (i) in constructing triazole conjugated oligomeric/polymeric materials for organic optoelectronics applications, (ii) in designing cyclic anion receptors and linear foldamers for cation/anion-recognition,52 are beyond the scope of this feature article. A graphical representation of the various facets of click-derived triazoles is depicted in Fig. 3 and described in the following sections.
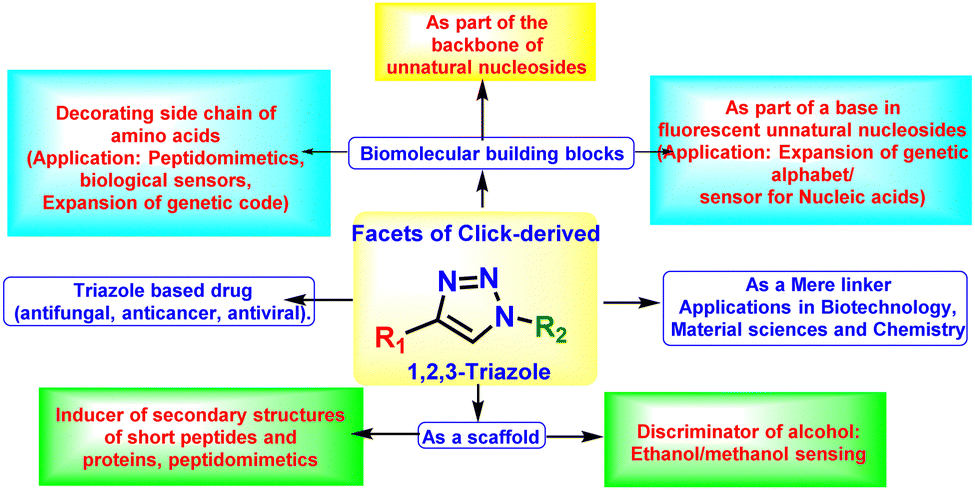 | ||
| Fig. 3 A schematic depicting the various facets of the click-derived triazole moiety. | ||
5. Facets of click-derived triazoles as mere linkers
The triazoles as linkers, as reported by Sharpless and Meldal, won them the Nobel Prize in Chemistry in 2022. Briefly, in 2001, Meldal and his colleagues invented the Cu(I)-catalysed 1,3-dipolar- cycloaddition reaction between azides and terminal alkynes to generate 1,4-disubstituted 1,2,3-triazoles at high yields (∼80–95%) under mild reaction conditions.40,53Sharpless and colleagues simultaneously demonstrated the same using in situ generated Cu(I) as a catalyst, improving the purity and cost-effectiveness of the reaction.41,54 The two processes outlined by Meldal and Sharpless are depicted in Fig. 4(A) and (B).
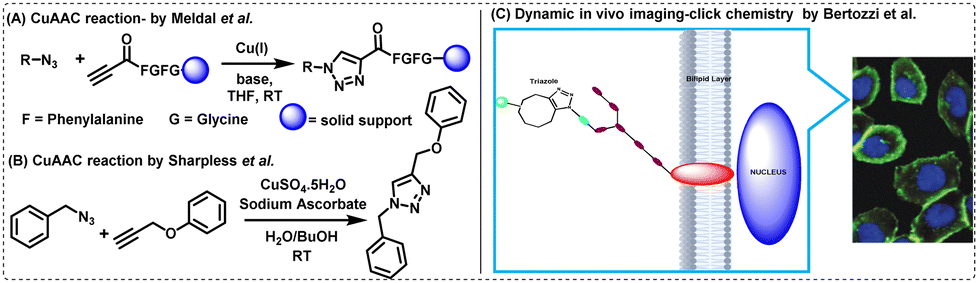 | ||
| Fig. 4 CuAAC reaction reported by (A) Meldal et al.40 and (B) Sharpless et al.41 (C) Dynamic in vivo imaging using click chemistry. The microscopic image is reprinted with permission from Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797.55 | ||
Although CuAAC reaction continued to be widely used since its inception, the cytotoxicity associated with copper often limits the applications of the reaction in biological systems. This led to the development of the copper-free SPAAC by Bertozzi, who also shared the Nobel Prize in Chemistry 2022 along with Sharpless and Meldal. Through her research,38,56,57 Bertozzi showed that azide-labelled glycoproteins on human Jurkat cells were capable of conjugating with biotinylated cyclooctynes, thereby enabling the imaging of dynamic biological processes in vivo [Fig. 4(C)].55
The triazole unit, thus formed as the end product of click chemistry, finds its versatile applications in various domains of chemistry, biology, material science and medical science owing to its high stability, stacking propensity and easy associability with other molecules/biomolecules via H-bonding interaction.
5.1. Triazole as a linker: in drug discovery and development
The CuAAC reaction has become a foundation of biorthogonal chemistry owing to its advantages, such as broad functional group compatibility, high reaction efficiency and regioselectivity under moderate conditions.58 In medicinal chemistry, CuAAC-derived triazoles serve as important linkers with applications in the domain of drug development, serving as either a scaffold or a pharmacophore. The triazole moiety is beneficial in the formation of hydrogen bonds, promotes hydrophobic interactions and is capable of linking probes or other conjugated molecules. In general, there are three types of click chemistry-based drug discovery: (1) high-throughput screening, (2) fragment-based drug discovery, and (3) dynamic template-assisted techniques in fragment-based drug development.59–61The high throughput screening (HTS)62 in pharmaceutical industries became very fast after the discovery of CuAAC click chemistry.63 For example, the CuAAC-generated small molecule triazole libraries could easily be screened for identifying inhibitor profiles for medically-important enzymes. Thus, the CuAAC approach was used for developing an inhibitor against Mycobacterium tuberculosis protein tyrosine phosphatase B (mPTPB), which exhibited a 25-fold specificity for mPTPB in comparison to the other inhibitors.64,65 In a similar work, Wang and colleagues reported the CuAAC-mediated synthesis of tyrosine-aryl C-glycoside hybrids acting as strong inhibitors of protein tyrosine phosphatase 1B (PTP1B).66 The CuAAC method has also been instrumental in generating inhibitors against, HIV-1 protease,67,68 cysteine proteases,69–71 aspartic proteases72 and plasmepsin in malarial parasites.73
Generally, cysteine proteases are crucial drug targets, such as treating Chagas disease caused by Trypanosoma cruzi. Thus, Baskin-Bey and associates synthesized an irreversible 1,2,3-triazole-based tetrafluorophenoxymethyl ketone inhibitor capable of eliminating T. cruzi parasites in cell cultures with excellent IC50 values.74 Furthermore, Yao's group synthesized small molecule inhibitor-peptide conjugates for targeting cancer cells’ lysosomes, which showed potent inhibitory effects on cysteine cathepsins.75 Alongside the production of protease inhibitors, CuAAC has also been used for developing inhibitors against oxidoreductases, such as Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase (CpIMPDH)76 and human lactate dehydrogenase-5 (hLDH-5).77
Barring protease and oxidoreductase inhibitor synthesis, CuAAC has also been employed to create transferase inhibitors.78,79 As an example, Wong and colleagues reported the synthesis of an α-1,3-fucosyltransferase inhibitor [Fig. 5(A)] that exhibited high potency at nanomolar levels, in addition to high specificity for human α-1,3-fucosyltransferase VI (FUT6).
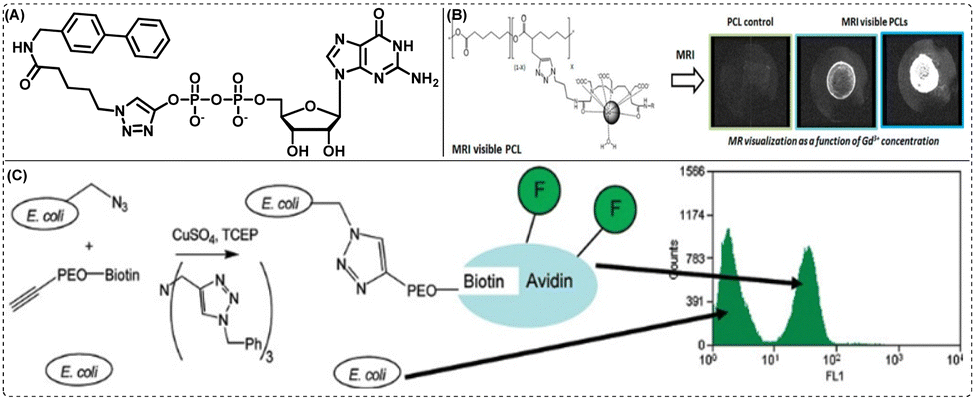 | ||
| Fig. 5 (A) CuAAC click-derived α-1,3-fucosyltransferase inhibitor, (B) MRI visible PCL for enhanced visualization in a temporary imaging application. Reprinted with permission from Biomacromolecules, 2013, 14(10), 3626–363480 and (C) labelling the cell surface of E. coli cells with a CuAAC click-derived methionine analogue, azidohomoalanine (AHA). Reprinted with permission from J. Am. Chem. Soc., 2003, 125(37), 11164–11165.81 | ||
Apart from inhibitor design, the Cu(I)-catalysed click chemistry has also aided in advancing fragment-based drug discovery (FBDD)82 because of its high efficiency and compatibility in aqueous environments, facilitating the direct testing of products without purification.82–85 Although the FBDD method results in fragments with weak binding affinities, the cost-effective way to find diverse hits makes the process useful. In contrast, the dynamic template-assisted strategy complements FBDD by utilising target proteins as templates for the optimization of fragment assembly in a CuAAC-dependent method.86
5.2. Triazole as a linker: labelling with fluorophores for bioimaging
Besides drug discovery and development, click chemistry has also been widely utilised to generate novel and non-toxic bioimaging agents. Thus, in addition to being useful in creating contrast agents for magnetic resonance imaging (MRI), click reactions have also been known to be useful for creating radiopharmaceutical agents for nuclear imaging utilising single-photon emission computed tomography (SPECT) and positron emission tomography (PET).87 Towards this end, the preliminary assessment and click production of 18F-labeled prostate-specific membrane antigen (PSMA)-Tracers [18F]PSMA-MIC01 for prostate cancer imaging have been described by Boehmer and colleagues.88 The target compound was synthesized at a yield of 81% using CuAAC of alkyne functionalized amine-Glu-urea-Lys with fluorinated azide. The fabricated compound was recorded to have imaging potential comparable to the clinically used [68Ga]PSMA-11 radiotracer. Additionally, the PSMA scaffold functionalized with an alkyne showed a significant affinity for binding PSMA. In a different study, Habnouni and colleagues reported the synthesis of a poly(ε-caprolactone) (PCL) carrier functionalized with a propargyl group and an azide-comprising Gd-DTPA contrast molecule for the creation of MRI-PCLs.80 The bioimaging agent, as shown in Fig. 5(B), was produced by a CuAAC reaction between the alkyne groups of PCLs containing a propargyl group and the azide groups of diazido functionalized diethylenetriamine penta-acetic acid (diN 3-DTPA) produced MRI-PCLs in a quantitative yield. Promising outcomes from in vitro cytocompatibility experiments further supported the prospect of new MRI-visible PCLs as cutting-edge click-biomaterials for applications in the field of biomedicine.5.3. Triazole as a linker: in bioconjugation
CuAAC has been frequently exploited for bioconjugation reactions. The idea of bioconjugation lies at the intersection of biology and chemistry and is in the process of developing new drug conjugates. Thus, the bioconjugation on the outer surface of Cowpea Mosaic Virus (CPMV) was the first instance of effective bioorthogonal CuAAC demonstrated by Wang and his co-workers.89 In this approach, azides or alkynes were used for decorating the reactive cysteine or lysine residues. Fluorescein derivatives with complementary reactive groups were subsequently ligated with the residues. In an effort to the expansion of genetic code, Schultz et al. could show site-specific incorporation of alkynyl-/azido-amino acids that enable the labelling of the translated protein via “click” chemistry.90 Hanson et al., showed that TBTA-assisted CuAAC enhanced the identification and screening of sialylated N-linked glycoproteins in Ac4ManNAl-incubated PC3 cells.91 Interestingly, Tirrell and his colleagues demonstrated the successful utilisation of TBTA as a ligand to mark the surface of E. coli cells with azidohomoalanine (AHA) and to label freshly expressed protein inside both E. coli and mammalian cells [Fig. 5(C)].81,92–945.4. Triazole as a linker: in material sciences
CuAAC click chemistry has also been employed by various research groups to produce new-age materials, such as triazole-decorated nanoparticles (NPs) and polymers. The method is frequently used for functionalizing the surfaces of gold and silver nanoparticles with triazoles, which poses significant challenges in surface modification. The nanoparticles with triazole-modified surfaces are shown in Fig. 6(A).95 The same strategy has also been adapted to silicon-based, magnetic and semiconductor NPs. For instance, researchers also developed triazole-linked Au-nanoparticles as nanosensors for detecting proteins, which facilitated the estimation of the total protein concentration.96 Apart from decorating the surface of NPs with triazoles, CuAAC click chemistry is also instrumental in creating a wide variety of tuneable materials such as gels, block polymers and dendrimers.97,98 | ||
| Fig. 6 (A) Triazole-linked functional silver nanoparticles and (B) triazole-derived cyclic anion receptor. | ||
Additionally, surface modification using CuAAC click chemistry has also been extensively researched. For tuning the biological activities for antimicrobial applications, Nottelet and colleagues reported the modification of polylactide (PLA) surfaces with triazoles upon click reaction with azide-terminated biomolecules.99 Interestingly, DNA could be anchored onto the solid support via a triazole ring as a linker for the fabrication of DNA microarrays.100
The functional materials produced from triazoles can also bind anions, cations, or even both. Thus, in the field of host–guest chemistry, Rowan and colleagues reported the synthesis of a triazole-based cyclic compound with improved halide binding properties and demonstrated strong affinities for Br− and Cl− anions in comparison to I− and F− anions [Fig. 6(B)].52 Therefore, it is clear that apart from surface modification, click-derived triazoles have been regularly used for the creation of novel sensors, self-assembling materials and lithography. All these examples highlight the immense potential of CuAAC-derived triazoles in the field of material science.
6. Facets of click-derived triazoles as integral part
In the previous section, we have discussed the various applications of click-derived triazoles as mere linkers. However, the utility of CuAAC-derived triazole moieties cannot be limited to their functions as linkers. To utilise the fullest potentiality, triazole might be considered an integral part of biomolecular building block, such as amino acids and/or other classes of molecules to confer additional novel properties. Having a triazole moiety as an integral part of the designer molecule would expectedly increase the stability of the molecule, enhance the resistance to degradation by enzymes, improve the interaction with biomolecules, and may also add novel photophysical properties to the molecules. A few instances of the triazoles functioning as integral parts of molecules are described in the upcoming sections.6.1. Triazoles as integral part: as donors-installation/modulation of photophysics
The synthesis of small organic fluorescent molecules can be facilitated by “click chemistry” due to its superior regioselectivity, simplicity, and flexibility of reaction conditions. Triazole, being the unique scaffold with electron donation property, has attracted our attention to be utilised for the generation of small molecules with improved or modulated fluorescence photophysics. Such triazolyl molecules would expectedly show solvatochromic photophysics and would allow for sensing biomolecular microenvironment. This idea would considerably improve the efficacy of biological research to generate signals that help comprehend natural biological processes.101–104 Therefore, our attempt was to utilise click reaction towards the installation of fluorescence properties onto non-fluorogenic precursors or modulation of the emission property of weak fluorescent compounds.105Briefly, in our design, a triazole moiety acts as a donor attached to an aromatic scaffold with donation/accepting property, enabling a charge transfer mechanism that installs a fluorescence emission onto a non-fluorescent precursor or modulates the photophysics of a fluorophoric aromatic, such as pyrene as represented in Fig. 7(A). Towards this end, a series of triazolyl donor–acceptor molecules with the general structure, D–T–A/A–T–D/D–T–D/A–T–A, [D = donor; A = acceptor; T = triazole] were synthesized, and their photophysical properties were evaluated. The chemical structures of the synthesized compounds are shown in Fig. 7(B).
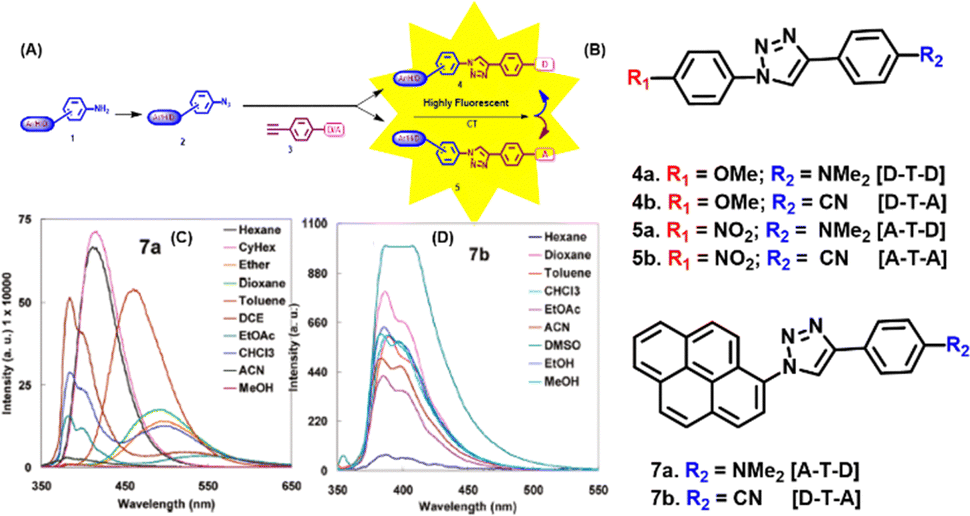 | ||
| Fig. 7 (A) Schematic representation of the fluorescence installation/modulation via the click reaction. (B) Chemical structures of the synthesized click fluorophores. Fluorescence spectra of 7a (C) and 7b (D) in different solvents. Reprinted with permission from J. Org. Chem., 2011, 76(9), 3348–3356.105 | ||
It was observed that whereas solvent polarity has little impact on the absorption property of nearly all fluorophores, it has a significant impact on their emission characteristics. By comparing the triazolyl molecules to their parent amine or the alkyne precursors, we found that each product exhibited modulated and interesting fluorescence properties. Further research has revealed that the dielectric constant of a solvent has a significant impact on the responses of some of the products, resulting in a red-shifted emission.
A solvatochromicity test validated the inferred intramolecular charge transfer (ICT) property of the lowest-lying excited state among the D–T–A/DTD/A–T–D molecules.54,106–110 In an effort to modulate the photophysics of widely used fluorophore pyrene, we came up with a fascinating probe, triazolyl pyrene. We thought that an electronically linked pyrene's emission response may be efficiently modulated by the electron-donating characteristic triazolyl ring. Thus, fluorophore 7a, an A–T–D system, which is a triazolyl-modified pyrene, shows an intriguing fluorescence photophysical feature with dual emission (LE and CT states) properties. In summary, compounds 4a, 4b, and 6b exhibited a red-shifted pattern, broad band shape, and high quantum yield, all of which point to a highly emissive, pure ICT state when the solvent polarity is raised. The nonpolar π–π* state was suggested to be the emitting state by the solvent polarity-independent LE emission of 7b. The dual emission of 7a indicates that mixed LE and CT states are present. Additionally, the solvent polarity and structure of the molecule determine the transition between the two states. The fluorescence spectra of 7a and 7b are depicted in Fig. 7(C) and (D) respectively.
As a result, our research unequivocally demonstrated that the “click” reaction between an azide and an alkyne effectively modifies the emission response of a pyrene fluorophore and adds a fluorescence emission feature to a non-fluorescent precursor. The direct trend between the fluorescence intensities and solvent dielectric constants in these fluorophores is noteworthy. A close look at the fluorophores’ band shape and fluorescence quantum yields showcased the intricate relationship between the structure and emissive states, which make these compounds suitable for a wide range of applications. Molecular recognition systems may be designed using the dual emissive fluorophore since the ICT to LE intensity ratio could be used as a sensing index.
Additionally, in a separate work, the photophysical response of the same probe in a micellar microenvironment was also investigated.111 The probe displayed a dual emission (LE and ICT) at low concentrations of the surfactant in an ionic surfactant solution. Further, the probe displayed dual emission at low concentrations of the non-ionic surfactant TX-100, with ICT emission predominating over LE emission. The emission spectra of the probe in the presence of various surfactants are shown in Fig. 8(a)–(c). Further, it was demonstrated that the order of the binding constant of the probe with non-ionic, anionic and cationic micelles was TX-100 > SDS > CTAB. These results suggested that our probe may find application in biological systems for membrane probing, which would naturally provide a unique fluorescence signal for such contact events and provide insight into the surrounding cell environment.
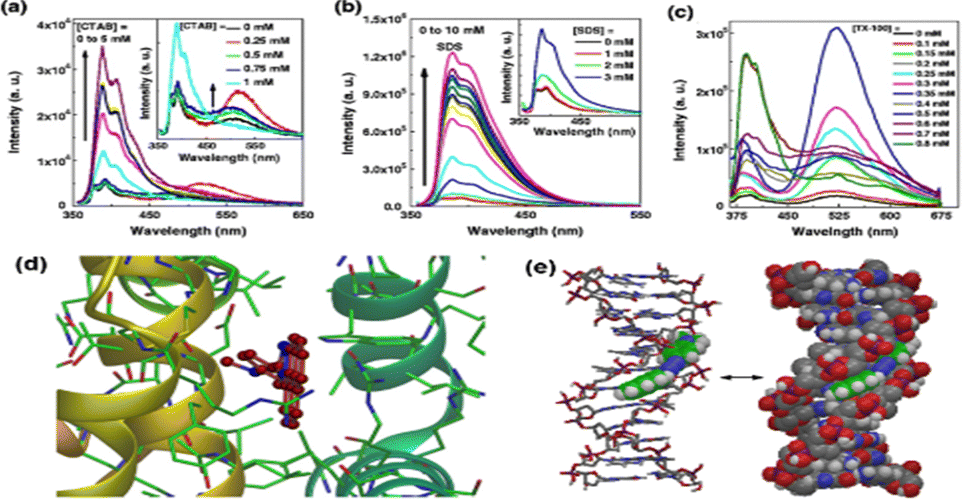 | ||
Fig. 8 Fluorescence spectra (λex![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) = 343 nm) of TNDMBPy in an aqueous solution of varying surfactant concentrations of (a) CTAB, (b) SDS and (c) TX-100. (d) Docking pose of TNDMBPy in the presence of BSA and (e) AMBER* energy minimized geometry of TNDMBPy with ctDNA, showing the minor groove binding of the probe TNDMBPy. Reprinted with permission from J. Fluoresc., 2013, 23, 929–938,111Tetrahedron Lett., 2012, 53, 5875–5879 and Tetrahedron Lett., 2013, 53, 2627–2632. = 343 nm) of TNDMBPy in an aqueous solution of varying surfactant concentrations of (a) CTAB, (b) SDS and (c) TX-100. (d) Docking pose of TNDMBPy in the presence of BSA and (e) AMBER* energy minimized geometry of TNDMBPy with ctDNA, showing the minor groove binding of the probe TNDMBPy. Reprinted with permission from J. Fluoresc., 2013, 23, 929–938,111Tetrahedron Lett., 2012, 53, 5875–5879 and Tetrahedron Lett., 2013, 53, 2627–2632. | ||
Our group also used the TNDMBPy molecule as a fluorescent light-up probe for the effective fluorescence switch-on sensing of copper(II) ions.112 Additionally, we demonstrated that the probe could selectively modify its fluorescence pattern to detect the presence of Cu2+ ions inside the nanocore of a micelle in response to changes in the system's pH. The same probe was also utilised for the sensing of BSA and Calf thymus DNA (CT-DNA). The molecular interactions with BSA and CT-DNA are depicted in Fig. 8(d) and (e), respectively.113
6.2. Triazoles as integral part: as nucleobases-CT-mediated DNA duplex stabilisation
While we have worked extensively towards the generation of novel triazolyl amino acids and scaffold amino acids using click chemistry for use in the fields of peptidomimetics, genetic code expansion and sensors, we have also utilised click chemistry for several other purposes. One of the main aspects of the usage of click chemistry by our group involves the creation of novel triazolyl nucleosides wherein the triazole acts as a nucleobase, with tuned photophysical properties exhibiting a variety of applications.114 Some of these applications are (i) for the detection and stabilization of abasic sites, (ii) dual door entry to exciplex formation in chimeric DNA, (iii) as a fluorescent sensor to detect G-quadruplex (iv) to establish the interaction profile with target proteins, and (v) genetic alphabet expansion.One of our most prominent works in this field involves the design and development of novel triazolyl nucleo-base pairs, wherein, we reported for the first time, the creation of a triazolyl donor/acceptor unnatural base pair (UNBP), namely TphenBDo (triazolylphenanthrene) – TNBBAc (triazolylnitrobenzene) base pair [chemical structures shown in Fig. 9(A)].115
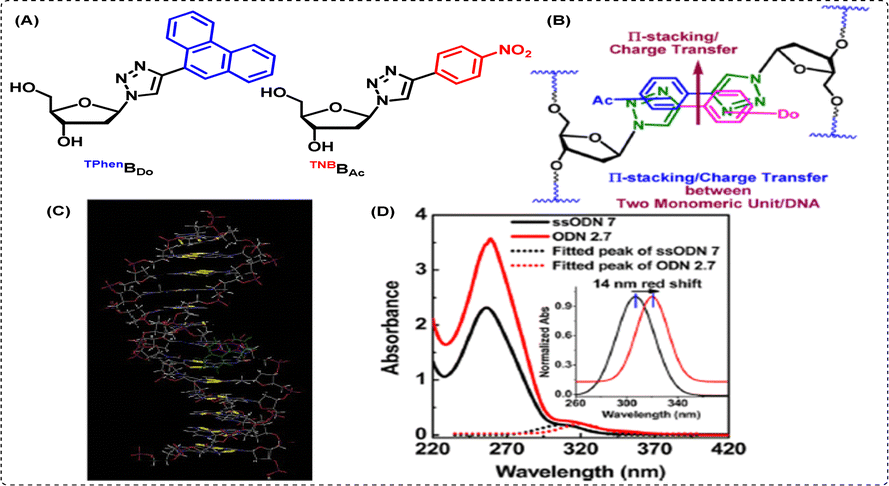 | ||
| Fig. 9 (A) Unnatural triazolyl donor/acceptor nucleobase pairs TPhenBDo and TNBBAc, (B) the π-stacking/charge transfer (CT) mechanism between the two bases in the duplex, (C) AMBER* energy-minimized conformations in the water of the heteropair (TPhenBDo/TNBBAc) duplex, and (D) UV-vis and spectra of probe DNA and heteroduplex DNA at room temperature confirming the formation of ground state CT complexation. Reprinted with permission from J. Org. Chem., 2013, 78(2), 278–291.115 | ||
This work also highlighted our pioneering efforts towards the stabilization of two unnatural heteroduplexes via π-stacking and charge transfer (CT) interactions, as depicted in Fig. 9(B). To summarize, we observed that our unnatural bases are more suitable for hetero-pairing, potentially by interaction with two donor or acceptor pairing partners inside a duplex through π-stacking and/or charge transfer. Our observations were also supported by AMBER*-optimized geometry of the self-pairs heteroduplexes, as depicted in Fig. 9(C).
Herein, we found that in the most stable heteropair TNBBAc/TPhenBDo, the third ring of the phenanthrene unit is involved in major groove binding, both triazoles are involved in intrastrand stacking, and nitrobenzene is involved in intercalative stacking between phenanthrene and its other natural flanking base pair. The nitrobenzene unit also participates in charge transfer interactions with the phenanthrene unit via stacking of the polar –NO2 and polarizable phenanthrene unit. A static quenching of the intrinsic fluorescence of TPhenBDo in the heteropair comprising these two UNBPs, together with the greatest stabilization of the heteropair, also supported our theory of duplex stabilization through the probable involvement of the ground state charge transfer interactions and/or π-stacking. Finally, the thermal stability offered by the TPhenBDo base was found to be comparable to that of a natural A/T base pair. Furthermore, a red shift in the absorption and emission spectra upon heteroduplex formation confirmed the ground state complexation, as revealed from the data shown in Fig. 9(D). Additionally, fluorescent triazolyl nucleosides and their several other classes such as (i) fluorescent unnatural fused-triazolyl-nucleosides116 and (ii) fluorescent unnatural triazolyl C-nucleosides117 have also been used for the detection of target proteins.118
7. Facets of triazoles in decorating amino acids and peptides
In addition to the role as a mere linker in various fields or as an integral part, as shown in nucleoside chemistry and discussed earlier, the click-derived triazoles have also established a significant impact in the design and development of novel amino acids and peptides/peptidomimetics. The triazoles play vital roles towards tuning the structural and functional properties of amino acids and peptides/peptidomimetic designs. Such triazole-embedded peptidomimetics are beneficial in the fields of drug design, biomaterials and structural biology because of the advantageous properties of triazoles, such as high thermal stability, high resistivity toward enzymatic degradation, strong associability and ability to induce preferred conformational and photophysical properties.7.1. Triazoles in decorating amino acids and peptides
One of the major fields in which click-derived triazoles may find their use is the decoration of novel amino acids and peptides. Such designer amino acids and peptides may find applications in chemical biology and showcase several important biophysical functions, a few of which have been cited here. Towards this end, Kee and colleagues have developed a triazolyl analogue of phosphorylated histidine (pHis) utilising CuAAC reaction for facilitating the study of histone-histidine phosphorylation.37,119 The modified amino acid was used to generate antibodies for specifically identifying pHis residues in phosphoproteins and for assisting in protein semi-synthesis.Triazolyl amino acids synthesised via CuAAC-reaction have been reported by Gajewski and colleagues for studying the binding interactions with the SN1 transporter, a crucial protein in L-glutamine transport.120 They have demonstrated that these synthetic amino acids took part in hydrogen bonding with the transporter, and hence opened a gateway for studying proteins involved in glutamine transport pathways. Stanley et al., synthesized a CuAAC-derived triazolyl analogue of glutamic acid to function as a ligand for the AMPA receptor (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor), an ionotropic transmembrane receptor for glutamate (iGluR). Through this study, they demonstrated that the triazole moiety is crucial for binding with essential residues located in the binding pocket of the receptor, and their claims were further validated by molecular docking studies.121
Recently, Hall et al., reported the synthesis of a 1-4-disubstituted 1,2,3-triazole analogue of L-tryptophan using CuAAC.122 The molecule was tested for the activity of L-type amino acid transporter 1 (LAT1), whereby it was found that the analogue displayed an enhanced rate of absorption in comparison to the natural tryptophan amino acid. In a different finding, Niu et al., utilised a one-pot Ugi multicomponent reaction combined with the CuAAC reaction to create unnatural triazolyl amino acids and peptidomimetics [Fig. 10(A)], thereby supplementing this research area.123 Rutjes and colleagues also utilised CuAAC to develop glycoamino acid with triazole linkages capable of acting as acyl donors in chemoenzymatic peptide formation processes.124
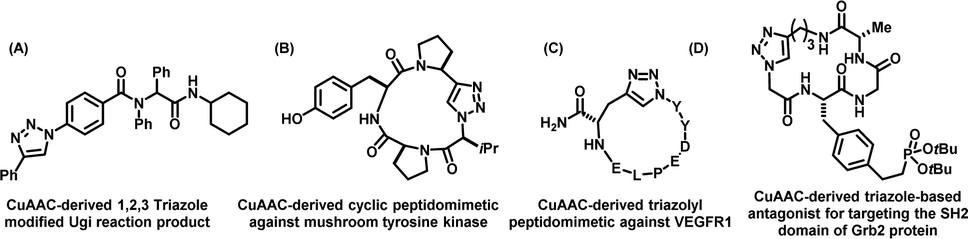 | ||
| Fig. 10 Chemical structures of (A) a CuAAC-derived 1,2,3-triazole-modified Ugi reaction product. (B) A CuAAC-derived cyclic peptidomimetic against mushroom tyrosine kinase. (C) A CuAAC-derived triazolyl peptidomimetic against VEGFR1 and (D) CuAAC-derived triazole-based antagonist for targeting the SH2 domain of the Grb2 protein. | ||
In addition to playing a crucial and purposeful role in decorating amino acids, the CuAAC-mediated 1,2,3-triazoles are also found to have a potential impact in designing and developing novel peptidomimetics, including the cyclic peptides that display enhanced biological activity, resistance to enzyme degradation and high stability.125 Thus, the strategies have been adopted to prepare cyclic peptides via the formation of 1,2,3-triazole through inter-/intramolecular click-chemistry among the clickable azide/alkyne counterparts. Thus, the creation of cyclic triazolo-peptidomimetics, such as a strained cyclic tetrapeptide analogue, that is known to inhibit mushroom tyrosinase, has been reported.126 The beauty and efficiency of triazole formation to bring the cyclic analogue lie in the fact that the traditional lactamization procedures failed to bring the cyclic network, as shown Fig. 10(B). Furthermore, the therapeutic potential of this cyclic troazolo inhibitor was recorded to be three times higher than that of its parent natural product, demonstrating the impact of CuAAC-derived triazoles in boosting biological activity.
The trans-amide bond mimicry of triazole has been exploited in designing peptidomimetics. Thus, Ghadiri et al. reported the synthesis of triazolo-cyclic peptidomimetics, including apicidin analogues as potent histone deacetylase (HDAC) inhibitors.127 Mimicking the trans-amide bonds triazole moiety helped maintain the backbone structure of the cyclic peptide and increase the biological efficacy. They have also synthesised sixteen stereoisomeric triazolyl peptidomimetics as ligands for somatostatin receptors. Their study revealed the variable selectivities for various receptor subtypes of somatostatin and, therefore, provided valuable insights into receptor–ligand interactions.127
Interestingly, triazoles have also been thought to mimic an E-configured tri-substituted double bond that has structural parameters similar to those of a trans-amide bond. Therefore, replacing trans-alkenes with triazole would most likely provide lipophilicity to account for the third substituent. Thus, Waldman and colleagues rightly showcased the practicality of the concept with their designed CuAAC-derived triazole-jasplakinolide analogues wherein the triazole group replaced a double bond of the natural antitumor macrocycle with full retainment of biological activity.128
Goncalves et al., developed triazolo-cyclic-peptidomimetics for targeting vascular endothelial growth factor receptor 1 (VEGFR1), thereby providing valuable structural insights into VEGFR1 binding [Fig. 10(C)].129
In 2008, Liu and colleagues synthesized click-derived triazole-based cyclic peptidomimetics with an RGD motif capable of targeting integrins and membrane proteins involved in tumour-induced apoptosis.130 These triazolo-peptidomimetics exhibited biological activities comparable to the familiar integrin ligand, cRGDFK. Further, Choi and associates designed and developed triazole-based antagonists for targeting the SH2 domain of Grb2 protein involved in signal transduction [Fig. 10(D)]. They showed that the triazole-stabilized β-turn structures could efficiently inhibit the SH2 domain of the Grb2 protein.131 Guan et al., also synthesized CuAAC-derived triazolyl β-turn mimics by conjugating peptide strands and successfully emulated the size and atomic structures of natural β-turns [Fig. 11(A)].127
 | ||
| Fig. 11 Chemical structures of (A) CuAAC-derived triazolyl β-turn mimics synthesized by Guan et al., (B) CuAAC click-derived triazolyl peptidomimetic analogue of parathyroid hormone (PTHrP), and (C) (I) and (II) peptidomimetics showing antimicrobial properties synthesized by Guell and colleagues. | ||
Because of structural similarities, disubstituted triazole rings formed by CuAAC reaction have been explored as biomimetic peptide surrogates in various secondary structural forms of peptides132–138 and prosthetic proteins.139 Inspired by this concept, scientists are successful in applying this chemistry to induce the formation of high-order structures in synthetic peptide- polymers. Thus, the triazole forming CuAAC-mediated click reaction has been utilised by Zhibin Guan et al., for the polymerization of short-chain peptides with clickable azide and alkyne at the two termini. Such peptide polymers with triazole moiety were shown to adopt antiparallel β-strands mimicking β-turns, ultimately forming β-sheet nanofibrils.140 The 1,2,3-triazole ring was considered to induce β-turn conformation,138 which in turn induces folding of the peptide polymer into antiparallel β strands. Essentially, the induced extensive β sheets self-assemble, forming high-order nanofibrils.
In a different study, Cantel and colleagues utilised a CuAAC-derived triazole ring to bridge peptides via their side chains, which introduced conformational hindrances to stabilize α-helical structures. The strategy was applied to a peptidomimetic analogue of parathyroid hormone (PTHrP), towards a triazole-cyclization mediated stabilization of its α-helix [Fig. 11(B)].127,141 CuAAC-derived triazoles have also been used to replace amide linkages in peptidomimetics known as Smac mimics, enhancing their cancer cell inhibition properties. Cyclized Smac mimics containing triazole moieties were reported to be at least five to eight times more efficient in suppressing cancer cell proliferation in comparison to their linear counterparts.142
Triazole peptidomimetics derived via the CuAAC reaction have also found immense significance towards the development of antimicrobial and antifungal agents. In this regard, Meldal and colleagues reported the synthesis of a library of triazolyl peptidomimetics as potent inhibitors of the protease enzymes, thereby targeting the parasite Leishmania mexicana.71 In a different work, Guell et al., utilised CuAAC for substituting peptide bonds with 1,4-substituted triazole moieties for creating peptidomimetics against bacteria (Erwinia amylovora, Pseudomonas syringae and Xanthomonas axonopodis) and fungus (Fusarium oxysporum) that were resistant to proteolytic degradation.143 The chemical structures of two such peptidomimetics are shown in Fig. 11(C).
Apart from peptides, the Cu(I)-catalysed click-derived triazoles are found to have a significant impact in modulating the structures and functions of the peptoid class of designs. Towards this, Althuon and colleagues reported the synthesis of novel linear 1,4- cell-penetrating peptidomimetics labelled with rhodamine B for tracking.144 The peptoids showed high potency as molecular transporters in zebrafish embryos. Furthermore, Jiang et al. developed a CuAAC-derived cyclic peptoid, which demonstrated improved stability and thermostability compared to the parent molecules.145
In summary, the CuAAC-derived triazolyl amino acids and triazolyl peptidomimetics shed light on synthesizing molecules with high stability against enzymatic degradation and enhanced biological activities. The triazole forming Cu(I)-catalysed click reaction has thus opened new avenues for receptor–ligand interaction studies, biosensing and antimicrobial, antifungal and anticancer therapies across various research domains. Although, all the examples mentioned earlier indicated the crucial applications and roles of CuAAC-derived triazoles in developing new amino acids and peptidomimetics, the role of triazole rings as an integral part of biomolecular building blocks is relatively unexplored. Therefore, the usage of triazoles as unique building blocks functioning as an integral part of amino acids and peptides is discussed in the upcoming sections.
7.2. Triazoles as linker in genetically expanded protein
The genetic code expansion (GCE) techniques allow the site-specific incorporation of unnatural amino acids, such as alkynyl- or azido-amino acids and offer the opportunity to label the expressed protein via Cu(I)-catalysed click chemistry for further study of properties of the engineered protein. For example, Chen and colleagues utilised an orthogonal tRNA synthetase (aaRS) derived from Methanococcus barkeri (M. barkeri) and incorporated azide-functionalized lysine into the epidermal growth factor receptor (EGFR). Afterwards, the modified protein was labelled with Cy5 dye via CuAAC reaction involving Cy5-alkynes for further study.146 In a similar work, Vreja et al., made use of a propargyl lysine incorporated in target proteins in eukaryotic and prokaryotic cells using M. mazei aaRS. Click-mediated triazole formation upon reaction with an azide-containing fluorescent probe enables labelling of the protein and, thereby, visualization under a high-resolution microscope.147,148Recently, Meineke and colleagues incorporated picolyl azido lysine (PazK) into HEK293T cells, accomplishing better reactivity with alkyne via a CuAAC reaction.149 In another work, Hao et al., reported the incorporation of an azide-containing pyrrolysine analogue (ACPK) into proteins expressed in both bacterial and mammalian systems that facilitate labelling with a probe via triazole forming CuAAC reaction.150 Further, Lin et al., incorporated the same ACPK at a specific location of T3S effector protein OspF in Shigella and labelled it with a biotin tag via TBTA-assisted Cu(I)-catalysed-triazole forming click reaction.42 This eventually allowed them to study the pathogenesis and protein functions under stress conditions (Fig. 12).
 | ||
| Fig. 12 Site-specific incorporation of photo cross-linker and unnatural amino acids into Shigella. Reprinted with permission from J. Am. Chem. Soc., 2011, 133(50), 20581–20587.42 | ||
7.3. Triazole – a unique building block as an integral part of amino acids and peptides
From the examples discussed above, it is clear that the triazole unit derived via the CuAAC technique has been mostly used as a linker for conjugation and labelling purposes in various fields, including peptide chemistry and protein engineering. While some of the reported works are directed towards the development of CuAAC-derived triazolyl amino acids and peptidomimetics, the triazole moieties in all these cases functioned as mere linkers or as ligands as a whole. The role of triazole rings as an integral part of biomolecular building blocks is limited or not explored critically, considering the potent intrinsic properties of triazole. However, we envisaged that the applications of the click-derived triazole moiety could not be limited to a mere linker since the far-reaching potential of the triazole moiety as an integral part of building blocks has not been greatly studied across the globe. Towards this end, our group is engaged in exploring potential intrinsic properties of the Cu(I)-catalysed click-derived triazole as an integral part of amino acids, peptides and nucleosides. Therefore, the following sections demonstrate our efforts to showcase the unexplored role and facets of triazoles in amino acids and peptides and one relevant and essential example in nucleoside chemistry.Towards this end, we have created triazolyl donor–acceptor (Do–Ac) unnatural amino acids with novel photophysical characteristics. Our observations on the installation/modulation of the emission response and the need for solvatofluorochromic biomolecular building blocks, such as amino acids and nucleosides, have led us to design such triazolyl donor–acceptor (Do–Ac) unnatural amino acids and nucleosides using click chemistry wherein triazoles act as a side chain of amino acids151 and as nucleobases in nucleosides.152 In one of our findings, it was revealed that the triazolyl–donor–acceptor chromophore-decorated unnatural amino acids exhibited highly solvatochromic photophysical properties by virtue of the triazole's donation properties.153
We also applied such amino acids in the design of short peptidomimetics. We hypothesised that the creation of a β-turn conformation may be facilitated by backbone H-bonding interactions when two fluorescent uAAs were incorporated into the two termini of a tripeptide separated by a natural alanine residue. We also anticipated that the two terminal triazolyl aromatics would be able to participate in dipolar photophysical interactions, such as the Förster resonance energy transfer (FRET) mechanism, owing to the π–π-stacking, hydrophobic, and van der Waals interactions in the side chain stabilizing the conformation. We established that the fluorescent solvatochromic Do–Ac amino acid tripeptide adopted a β-turn conformation and exhibited the FRET process between the TPhen unit of TPhenAlaDo [the donor; structure shown in Fig. 13(a)] and the TCNB moiety of TCNBAlaAc [the acceptor; structure shown in Fig. 13(b)]. The schematic presentation of click chemistry-derived unnatural amino acids and fluorescent unnatural peptides is depicted in Fig. 13(c). To the best of our knowledge, this is the first instance of a fluorescent unnatural peptide demonstrating the FRET mechanism to a β-turn conformation. The chemical structure of the peptide is depicted in Fig. 13(d).
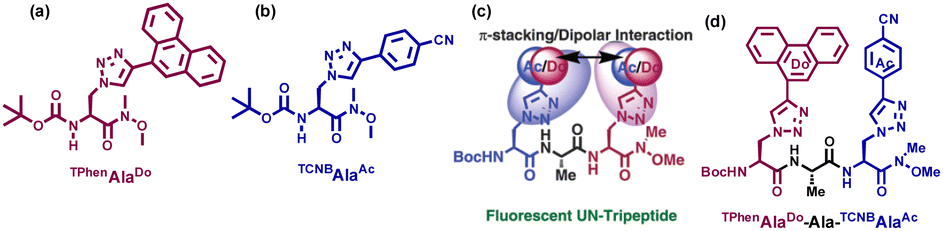 | ||
| Fig. 13 (a) and (b) Chemical structures of click chemistry-derived unnatural amino acids TPhenAlaDo and TCNBAlaAc. (c) and (d) Chemical structure of unnatural amino acids containing peptides showing through space stacking/charge transfer/photophysical interactions. | ||
We believed that adding 1,2,3-triazole residues to an amino acid would significantly enhance its interaction property with other amino acid residues in a protein through stacking or H-bonding interactions, potentially improving activity across a broad spectrum of chemical and biological processes. Therefore, in a separate study, we reported the synthesis, photophysical properties and application of a few more triazolyl unnatural amino acids151 all of which showed solvatochromic photophysical properties.
Amongst all, the photophysical properties of triazolyl perylene amino acid and its application are also highlighted herein. Thus, when compared to perylene, the triazolyl perylene amino acid (TPerAlaDo) [Fig. 14(a)] showed tuneable and solvatochromic absorption and fluorescence properties, suggesting a donor–acceptor nature. The electron charge density of triazole moiety was most likely redistributed within the electron-deficient perylene, resulting in highly solvatochromic ICT absorption and emission.
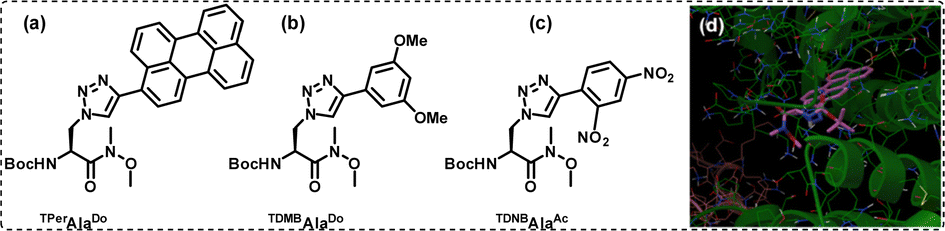 | ||
| Fig. 14 (a)–(c) Chemical structures of the unnatural triazolyl amino acids synthesized using click chemistry. (d) Docking pose of TPerAlaDo in the hydrophobic pocket of BSA. Reprinted with permission from Bioorg. Med. Chem., 2016, 24, 3579–3595. | ||
Subsequently, from our findings, we could establish that the triazolyl perylene produced an intensified fluorescence signal in response to the interaction with BSA.154,155 Molecular docking experiments revealed that the triazolyl perylene unit (TPer) of TPerAlaDo bound inside the hydrophobic pocket of subdomain IB of site I of BSA with high binding efficiency [Fig. 14(d)].
We further exploited two of our donor–acceptor triazolyl aromatic amino acids and incorporated them in a short pentapeptide sequence that was an analogue of a Leu-enkephalin peptide. Opioid peptides, including Leu-enkephalin (sequence: Tyr-Gly-Gly-Phe-Leu), are known to be involved in a wide range of biological activities and responsible for several processes in living beings.156,157 As a result, these endogenous opioid peptides are frequently significant targets with enormous potential towards the creation of novel therapeutics. Unfortunately, due to low metabolic stability in its original form, many analogues must be created in order to get beyond pharmacokinetic and pharmacological obstacles in therapeutic settings. Therefore, with this research gap in mind, we set out to replace the two aromatic amino acids Tyr and Phe with our TDMBAlaDo [the donor amino acid; structure shown in Fig. 14(b)] and TDNBAlaAc [the acceptor amino acid; structure shown in Fig. 14(c)], respectively. All the experiments suggested that the pentapeptide adopted a type II β-turn conformation similar to the less rigid Leu-enkephalin pentapeptide.138,158 Therefore, being an integral part of the amino acids, click-derived triazoles served an important role in installing various properties onto the triazolyl amino acids. Such amino acids found applications in various fields, such as in sensing BSA, in peptidomimetic designing and in the generation of novel FRET peptides useable for studying interbiomolecular interactions, such as protein–peptide/DNA–peptide interactions.
 | ||
| Fig. 15 (A) Chemical structures of aliphatic and aromatic scaffolds amino acids AlTAA and ArTAA, the CD/UV spectra and helical arrangement of ArTAA in the crystal. (B) Chemical structures of designed peptides. Reprinted with permission from Chem. Commun., 2015, 51, 5242.159 | ||
In our pioneering design, the triazole moiety acted as a trans-amide mimetic, enabling the molecular scaffold to nucleate β-turn conformation when present in the backbone of a short peptide sequence. Thus, a leu-enkephalin analogue [sequence: Boc-Tyr-AlTAA-Phe-Leu-CO2Me] [structure depicted in Fig. 15(B)] containing the aliphatic scaffold amino acid, AlTAA in the middle revealed that the synthetic peptide exhibited 100% β-turn structures, as depicted by the CD spectra, wherein Gly–Gly dipeptide of the Leu-enkephalin was replaced by the scaffold amino acid. AlTAA acts as the mimic of this dipeptide. The type-II β-turn conformation was further supported by 2D NMR and molecular dynamics simulation.
Along with the aliphatic triazolo amino acid, AlTAA, its aromatic counterpart, ArTAA, was also utilised as a novel constrained scaffold amino acid for use in peptidomimetic studies.159 The hairpin-shaped scaffold showed atropoisomerism with an aminophenyl unit out of the plane. In line with our previous observation, this molecule, depicted in Fig. 15(A), containing constrained backbone angles ω(i) and Φ(i + 1), was also envisaged to induce the formation of folded conformations in linear peptide chains. The molecular arrangement, model shape of linearly arranged “S” units (two molecules of the unit cell) and UV-visible and CD spectra of ArTAA are depicted in Fig. 15(A). Towards this end, we studied the conformation of our designed tripeptide sequence [sequence: Boc-Tyr-ArTAA-Phe-Leu-CO2Me] [structure depicted in Fig. 15(B)] containing our scaffold revealing its ability to induce a β-sheet-like conformation with a 20% turn. The tripeptide analogue containing the aromatic scaffold amino acid was less likely to adopt a complete turn structure due to its short chain length and the strong rigidity of the scaffold, which could function as a β-turn mimetic β-sheet folding nucleator in turn conformation.
However, there has not been much focus on designing a fluorescent peptide that, in a specific conformation like the β-sheet, may come up with new and novel photophysical phenomena. Such basic photophysics might have broad implications in designing novel fluorescent probes for investigating many biological processes.
With this aim and utilising β-turn mimetic aliphatic triazolo amino acid scaffold (AlTAA), we created a trichromophoric β-sheet pentapeptide and successfully demonstrated the phenomenon of dual door entry to exciplex emission as a new and novel photophysical phenomenon. In short, we created an aliphatic triazolo amino acid scaffolded pentapeptide159 [sequence: Boc-TPyAlaDo-Leu-AlTAA-Leu-TPhenAlaDo-CO2Me] [Fig. 15(B)]. Consequently, we established the theory of the dual mechanism of exciplex emission, which relies on either direct excitation of the FRET acceptor or the donor. We hypothesised that careful design and appropriate placement of a donor/acceptor pair in relation to a third chromophore in a probe where a pair of chromophores engage in π–π-stacking contact would enable the process of excimer/exciplex production via FRET. Thus, to the best of our understanding, this foundational phenomenon is novel and will inspire researchers to develop probes and sensors for biochemical applications.
We have also utilised an aromatic triazolyl amino acid scaffold for the development of a trichromophoric β-sheet pentapeptide to generalise the concept of dual door entry to exciplex/excimer emission.160 Thus, the fluorescent Leu-enkephalin analogue peptide [sequence: Boc-TPyAlaDo-Leu-ArTAA-Leu-TPyAlaDo-CONMe(OMe)] was synthesized, which exhibited β-sheet conformation. In the adopted conformation, the excimer emission was established via two mechanisms: either via FRET or through direct excitation of a FRET acceptor.
A solvent-induced emission study of our pentapeptide also revealed that the increase in monomer emission was because the TPy residues accommodated themselves in the hydrophobic pocket of the BSA protein, thereby putting away the peptide chain on the surface. The structures of the amino acid scaffold and the peptides are shown in Fig. 16(a).
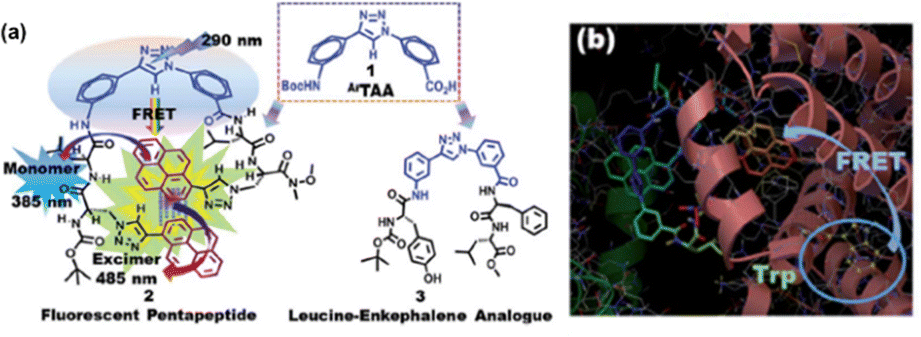 | ||
| Fig. 16 (a) Chemical structures of (1) the amino acid scaffold ArTAA, (2) the trichromophoric fluorescent pentapeptide showing the photophysical aspects and (3) leucine–enkephalin analogue peptide. (b) Molecular docking pose of the pentapeptide in the presence of BSA. Reprinted from S. S. Bag, S. Jana, M. K. Pradhan and S. Pal, RSC Adv., 2016, 6, 72654, DOI: https://doi.org/10.1039/C6RA14084J.160 | ||
Subsequently, we exploited the excimer emissive pentapeptide for sensing BSA protein. Our study revealed that the triazolylpyrene unit of TPyAlaDo resides near the tryptophan residues (Trp) inside the hydrophobic pocket of BSA. Fascinatingly, we also visualized a FRET process from Trp to TPy when excited at the BSA absorption maximum (280 nm), along with a simultaneous drop in the intensity of donor Trp emission at 345 nm and a two- to three-fold elevation of the TPy monomer emission at 385 nm, which was acting as an acceptor. There was a modest rise in the excimer intensity at 470 nm as well. A molecular docking calculation using the Autodock programme supported the close proximity of our probe's TPy moiety to tryptophan (Trp residue number 134), supporting the FRET process. The probe's C-terminal TPy moiety was found in the vicinity of tryptophan and continued to be surrounded by various hydrophobic amino acids of the hydrophobic region of subdomain IB of site I of BSA. The molecular interactions revealed from molecular docking studies are depicted in Fig. 16(b).
Thus, all these findings unequivocally demonstrated that excimer emission occurs in the triazolo aromatic amino acid scaffolded trichromophoric β-sheet fluorescent pentapeptide via both FRET and FRET acceptor excitation. Additionally, it was explored that the probe was effective for sensing BSA protein by increasing TPy fluorescence by FRET and FRET-mediated TPy–TPy excimer emission. Also, to the best of our knowledge, this is a pioneering work to present a dual-route approach to excimer emission using a new generation of peptide-probe.
Towards this end, we found that our achiral aromatic triazolyl unnatural amino acid scaffold (ArTAA) exhibited optical chirality in a hairpin configuration. With the amino-phenyl moiety, at 25.1°, the out-of-plane and the in-plane methyl benzoate unit in relation to the triazole unit, the scaffold showed axial chirality in the solid state. The UV-visible spectra in different organic solvents showed two different absorption bands at 234 and 297 nm, which also supported our findings. Our theory was inspired by information from CD spectra and certain rotational values, which suggested that our axially chiral scaffold in the shape of a bent hairpin may interact towards differentiating and discriminating between homologous alcohols with slight polarity differences by producing a unique fluorescence signal. Photophysical studies of the scaffold amino acid revealed a hyperchromic effect in addition to a red-shift (∼8 nm) for the longer wavelength absorption band at 294 nm in ethanol in comparison to methanol. Furthermore, our findings demonstrated that the scaffold's in-plane conjugated triazolyl benzoic acid unit and the out-of-plane aminophenyl unit interacted differently in the ground state with ethanol/methanol. At 280 nm, the ArTAA probe exhibited a quantum yield boost of 1650% and an approximately 15-fold increase in emission intensity in ethanol relative to methanol. Thus, differential interactions and the ability to distinguish methanol from ethanol may be caused by the limited rotation of the out-of-plane aminophenyl unit in both the excited and ground states, respectively.
Two other sensors involving our scaffold amino acid were also synthesized. This includes PyAm-ArTAA (sensor 2) and Py2Am-ArTAA (sensor 3). Structures of the axially chiral fluorescent ethanol sensors: scaffold ArTaa and its mono- and bis-pyrenyl amides (sensors 2 and 3) are depicted in Fig. 17(I). In the case of sensor 2, it was possible to distinguish between methanol and ethanol by using emissions from both the scaffold and the pyrene moiety. This was achieved by the significant enhancement of the fluorescence intensity, quantum yield, and wavelength shift. Comparable outcomes were obtained from the third sensor, which demonstrated a significant excimer emission at 465 nm and little to no emission from the scaffold or the pyrene monomer in methanol. On the other hand, all of the emission bands showed a substantial enhancement in intensity in ethanol. A Gaussian-optimized geometry further supported our theory for excimer emission in ethanol. The presence of pyrenyl moieties in both sensors 2–3 also did not cause the scaffold unit's chirality to change, resulting in nearly identical interactions with methanol and ethanol and a less significant shift in the pyrene emission region. Data from photophysical studies and titration experiments suggested that the axial chirality of the hairpin-shaped scaffold amino acid or scaffold moiety in both sensors played a role in differential solvation and H-bonding interactions with methanol/ethanol. While the axial chirality in ethanol was maintained, resulting in uneven solvation of the two halves and H-bonding interactions in ethanol; in methanol, both halves of the molecules may have become proximal to the planarity of the triazole ring, and as a result, solvation occurred throughout the entire molecular system. Fig. 17(II) depicts the steady-state emissions (λex = 280 nm) and photograph obtained under UV-irradiation of ArTAA (a) and (d), sensors 2 (PyAm-ArTAA) (b) and (e) and 3 (Py2Am-ArTAA) (c) and (f) in methanol with increasing v/v% ethanol.
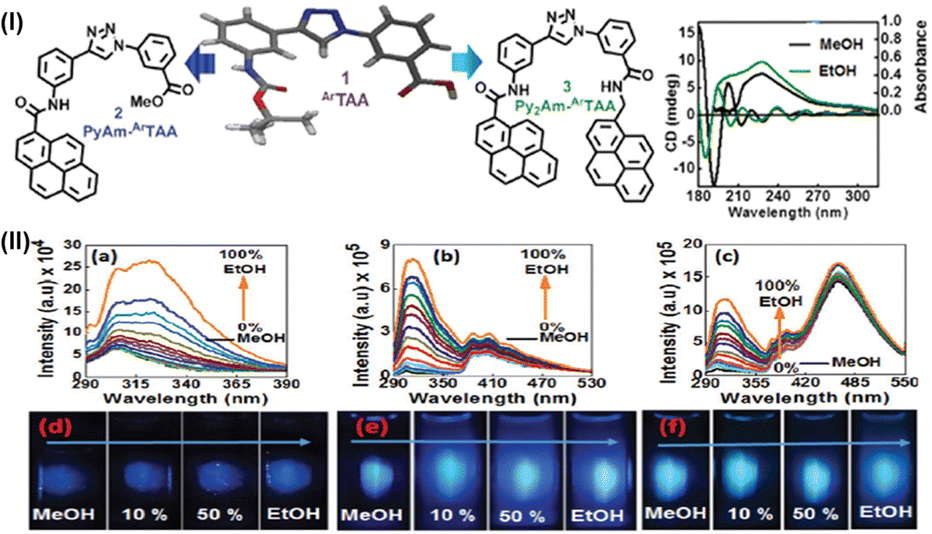 | ||
| Fig. 17 (I) Structures of the axially chiral fluorescent ethanol sensors: scaffold ArTaa and its mono- and bis-pyrenyl amides (sensors 2 and 3) and CD spectra; (II) steady state emissions (λex = 280 nm) and photograph obtained under UV-irradiation of ArTAA (a) and (d), sensors 2 (PyAm-ArTAA) (b) and (e) and 3 (Py2Am-ArTAA) (c) and (f) in methanol with increasing v/v% ethanol. | ||
Next, the sensors’ capacity to detect ethanol vapour was examined. In order to potentially sense ethanol vapour using a fluorescence spectroscopic methodology, our group created solid films of the aforementioned sensors using a spin coating process. In a mixture of methanol and ethanol, it was found that as the ethanol vapour pressure grew, so did the fluorescence intensity of the ArTAA-coated solid film. When it came to detecting ethanol vapour from a methanol–ethanol combination, the two additional pyrenyl derivatives exhibited identical behaviour. The dispersion force between the ethanol/methanol vapour and the amine/acid functionality of the scaffolds, H-bonding, and dipole–dipole interaction may have contributed to molecular adsorption on the sensor surface. Hansen polarity values also indicated that ethanol has a larger molecular affinity for the solid scaffold surface than methanol, which is likely the cause of the enhancement of fluorescence intensity with the gradual increase in ethanol vapour pressure.
To summarize, by just observing fluorescence, it was possible to conclude that the axially-chiral scaffold amino acid and the other two sensors had high sensitivity in discriminating ethanol from methanol (0.5–2.1 v/v%). Furthermore, the sensing of ethanol in the vapour phase was made possible by integrating the ethanol sensors onto a solid sheet.
The scaffold ArTAA or the scaffold moieties in the sensors were important in the fluorometric separation of methanol from ethanol through the differential solvation-mediated hydrogen bonding interaction. In terms of practicality, we think it would be appealing to create a system like this fundamental system, that could instantly determine the amount of ethanol present.
The design concept is depicted in Fig. 18(A). The absorption and emission spectra of the FRET pairs are shown in Fig. 18(B). Towards this end, the pentapeptide was installed with a triazolylperylene amino acid (TPerAlaDo) at the N-terminus and triazolylpyrene amino acid (TPyAlaDo) at the C-terminus. The secondary structural analyses established a hairpin turn conformation of the scaffold and the peptide demonstrated turn induced β-sheet-like structures.
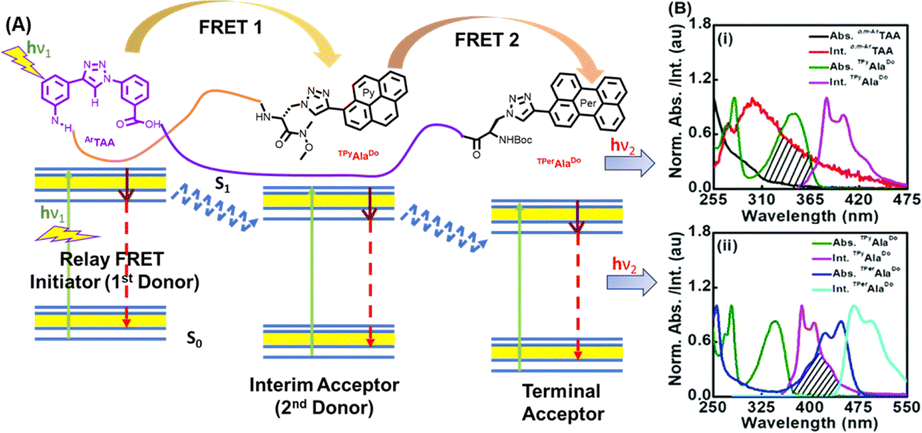 | ||
| Fig. 18 (A) The concept of relay FRET in a peptide containing three different chromophores. (B) (i) and (ii) The absorption and emission spectra of shown FRET pairs, respectively. Reprinted with permission from Chem. Commun., 2018, 54, 9765–9768.162 | ||
We designed a trichromophoric fluorescent pentapeptide [sequence: {BocNH-TPerAlaDo-Leu-o,m-ArTAA-Leu-TPyAlaDo-CONMe(OMe)}] to study the photophysics. Thus, we were able to establish a relay FRET process wherein the scaffold (o,m-ArTAA) serves as the FRET source, the triazolylpyrene (TPy) of TPyAlaDo acts as the interim chromophore, and the triazolyl perylene (TPer) of TPerAlaDo serves as the final emitting FRET terminal. The generation of FRET events was validated by time-resolved fluorescence studies and steady-state fluorescence studies. These results further motivated us to explore whether the FRET events exhibited by our tailored pentapeptide were preserved in a protein microenvironment. An overlap between the emission spectra of the BSA protein and the absorption spectra of our pentapeptide was suggestive of a potential energy transfer event from the tryptophan residues of the BSA protein to the pyrenyl moiety of the pentapeptide. Our observations were further validated by photophysical studies. Finally, the molecular docking studies also revealed the interaction of the pyrene residue of our pentapeptide with the Trp134 residue, located in the hydrophobic pocket of the BSA protein. In principle, a relay FRET of this type in a short, tailored peptide sequence is effective in providing insights into more advanced probe design for the investigation of intricate inter-biomolecular interactions that extend beyond standard FRET dimensions within a living cell.
8. Future prospects
1,2,3-Triazole-a unique building blocks, derived via Cu(I)-catalysed click chemistry, are well-known to exhibit several important physicochemical properties such as trans amide mimic, enhanced stability, greater biorthogonality, better resistance to enzymatic degradation, H-bonding/stacking ability and high bio-associability. Such properties make them ideal candidates for applications in biomedical science, biotechnology, material and environmental science. Several triazoles are found to have the potential to tackle various emerging threats of drug resistance to human health in clinical science. It may therefore be expected that the click-derived triazoles have an immense role to play in the advancement of therapeutics, diagnostics and biosensing applications in current times. In the field of therapeutics, the CuAAC-derived triazole molecules can function as protease-resistant peptidomimetics with more fabulous biological activities in vivo, such as in the reduction of frequency of dose administration and improving the patient's compliance. This feature is very essential in treating chronic diseases whereby the drug's efficacy is needed for longer time intervals. Furthermore, the development of prodrugs ideal for selected physiological environments is also possible by tuning the parent molecule with click chemistry-derived triazole-based moieties. Additionally, the triazole-containing molecules have immense potential for reducing the toxicities associated with conventional chemotherapy, thereby allowing the development of more specific and safer anti-cancer therapies.Furthermore, the promising ability of the CuAAC-derived triazoles towards the formation of biorthogonal bonds under physiological parameters makes them suitable for use in the field of biosensing. These molecules can be utilised to detect cancer biomarkers and facilitate the early diagnosis and treatment of the patient. Additionally, triazole-containing biosensors can also be used for detecting the presence of environmental pathogens and toxins, offering significant benefits in the public healthcare sector. Triazoles can also be used for developing real-time imaging agents163,164 for the precise visualization of cancer cells or inflammation sites using techniques such as PET (positron emission tomography) or MRI (magnetic resonance imaging), thus enhancing diagnostic precision and monitoring of the treatment process.
Along with therapeutics and biosensors, genetic code expansion (GCE) and synthetic biology are the two major fields where CuAAC-derived triazoles hold enormous potential. This is because, in GCE,165 the synthesis and incorporation of novel triazolyl unnatural amino acids with tuned photophysical/physicochemical properties enable researchers to create proteins with desired properties. Such proteins may exhibit enhanced stability, catalytic activities, or added photophysical properties, making them highly beneficial for both therapeutic and diagnostic purposes. Further, this has the potential to study the protein–protein interactions in greater detail, which could ultimately help gather a deeper understanding of various cellular processes and in the discovery of novel therapeutic targets. Currently, our research group is actively involved in the site-specific incorporation of fluorescent triazolyl unnatural amino acids into desired protein sequences for adding new functionalities/properties to the proteins of interest.165 Similarly, in the domain of synthetic biology, the triazolyl molecules can be utilised to create proteins for serving specific functions such as targeted drug delivery and in situ biosensing.
The CuAAC-derived triazolyl molecules can also find their use in replacing the phosphodiester linkages or nucleobases towards becoming an integral part of the DNA or a gene.166 The triazolo-nucleosides can be utilised for expanding the genetic alphabet, and thus modified DNA or gene can be recognized by corresponding DNA and RNA polymerases, opening up a gateway of combining genetic alphabet expansion (GAE) with the GCE strategy, therefore potentially allowing the development of semi-synthetic organisms.114 The triazoles can also be used for replacing nucleobases in nucleic acids for the development of novel nucleic acid drugs against cancer and pathogenic viruses. The triazole groups may also find their use towards the creation of C-nucleosides117 or N-nucleosides167,168 with tuned photophysical properties for further use as nucleoside drugs or in the field of GAE. Another field where click-derived triazoles can be used is towards the formation of dipeptide mimics, wherein the triazolyl scaffold amino acids, being an integral part of the peptide chain, can be used towards inducing secondary structures in proteins in an effort towards protein engineering and drug development.
More research efforts are to be initiated to broaden the scope of utilisation of triazole as an integral part of the biomolecular building blocks. Such triazolo-building blocks expectedly would enable the development of therapeutics, diagnostics, and biosensors to their full potential and flourish in the fields of genetic code expansion, genetic alphabet expansion and synthetic biology. The applications of triazolo-building blocks in those fields are expected to continue and even increase to find molecular-level solutions to the existing biomedical hurdles. Finally, triazoles are also expected to shape the future of targeted drug delivery, bioimaging, diagnosis, treatment regimen, understanding the molecular pathways of complex diseases and thereby personalized medicines.
9. Conclusions
To conclude, this feature article summarizes the various types of click chemistry in brief, it emphasised a few of the novel applications of CuAAC-mediated triazoles in several domains and elaborates on the applications in the field of amino acids and peptides. Mostly, the researchers have used and continue to use CuAAC-mediated triazoles as linkers mainly for conjugating two molecular fragments, in order to develop a molecule with tailored properties. Towards this end, several proteins and other biomolecular conjugates have been labelled with different fluorophores. The profound role of click-triazoles as mere linkers has been widely explored. However, the intrinsic properties of the triazole moiety as an integral part of amino acids and peptides have not been looked upon or utilised to their full potential. A few of our efforts in this endeavour, thus, have been recorded through this article to showcase the facets of click-triazoles other than their role as a mere linker.The unique potentiality of click-derived triazole as a sole part of novel biomolecular building blocks has been demonstrated. Triazoles have also been showcased as a scaffold for inducing protein's secondary structure. Based on the innumerable possibilities and the flexibilities of adding photophysical properties using CuAAC click chemistry, our research group has created a novel class of biomolecular building blocks such as triazolyl unnatural amino acids and used them for various applications. These amino acids and scaffolds have been used as peptidomimetics, as sensors, as modulators of photophysical activities and also in the field of genetic code expansion, towards site-specifically incorporating them in desired protein sequences in vivo. Additionally, our group has also utilised click chemistry for the creation of novel triazolyl unnatural nucleobase pairs with the ultimate goal of genetic alphabet expansion aimed towards the creation of a semi-synthetic organism. Overall, the applications of the triazole subunit derived from click chemistry are uncountable, and we believe that the answer to a lot of biomedical problems (such as therapeutics/drugs and biosensors) lies in the correct utilisation of this triazole moiety.
Author contributions
Subhendu Sekhar Bag: supervision, conceptualization, visualization, writing, editing, drawing, and review. Aniket Banerjee: writing – draft, drawing, and review. Sayantan Sinha: validation. Subhashis Jana: validation.Data availability
No primary research work has been included, and no new data have been generated or analysed as part of this Feature Article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank former and current members of the Bag's group for their contributions to the research highlighted in this Account. SSB is thankful to his family members for their constant inspiration and support.References
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS PubMed.
- K. C. Nicolaou, Chem, 2016, 1, 331–334 CAS.
- T. Eicher, S. Hauptmann and A. Speicher, The chemistry of heterocycles: structures, reactions, synthesis, and applications, John Wiley & Sons, 2013 Search PubMed.
- J. A. Bladin, Ber. Dtsch. Chem. Ges., 1885, 18, 1544–1551 CrossRef.
- M. M. Matin, P. Matin, M. R. Rahman, T. Ben Hadda, F. A. Almalki, S. Mahmud, M. M. Ghoneim, M. Alruwaily and S. Alshehri, Front. Mol. Biosci., 2022, 9, 864286 CrossRef CAS PubMed.
- A. A. Ali, 1,2,3-Triazoles: Synthesis and Biological Application, IntechOpen, 2020 Search PubMed.
- R. Huisgen, Angew. Chem., 1963, 75, 604–637 CrossRef CAS.
- M. V. Gil, M. J. Arévalo and O. Lopez, Synthesis, 2007, 1589–1620 CrossRef CAS.
- N. K. Devaraj and M. G. Finn, Chem. Rev., 2021, 121, 6697–6698 CrossRef CAS.
- V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS PubMed.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J. C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- M. Köhn and R. Breinbauer, Angew. Chem., Int. Ed., 2004, 43, 3106–3116 CrossRef.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- N. E. Mbua, J. Guo, M. A. Wolfert, R. Steet and G. J. Boons, ChemBioChem, 2011, 12, 1912–1921 CrossRef CAS.
- H. M. Pineda-Castañeda, Z. J. Rivera-Monroy and M. Maldonado, ACS Omega, 2022, 8, 3650–3666 CrossRef PubMed.
- N. B. Cramer, J. P. Scott and C. N. Bowman, Macromolecules, 2002, 35, 5361–5365 CrossRef CAS.
- W. Song, Y. Wang, J. Qu and Q. Lin, J. Am. Chem. Soc., 2008, 130, 9654–9655 CrossRef CAS.
- N. Floyd, B. Vijayakrishnan, J. R. Koeppe and B. G. Davis, Angew. Chem., Int. Ed., 2009, 48, 7798–7802 CrossRef CAS PubMed.
- F. Fringuelli and A. Taticchi, The Diels–Alder reaction: selected practical methods, John Wiley & Sons, 2002 Search PubMed.
- A. Oluwasanmi and C. Hoskins, Int. J. Pharm., 2021, 604, 120727 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- E. C. B. Johnson and S. B. H. Kent, J. Am. Chem. Soc., 2006, 128, 6640–6646 CrossRef CAS.
- W. Tang and M. L. Becker, Chem. Soc. Rev., 2014, 43, 7013–7039 RSC.
- C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS.
- C. Bednarek, I. Wehl, N. Jung, U. Schepers and S. Bräse, Chem. Rev., 2020, 120, 4301–4354 CrossRef CAS PubMed.
- J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Am. Chem. Soc., 2009, 131, 5751–5753 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- V. Nair and T. D. Suja, Tetrahedron, 2007, 63, 12247–12275 CrossRef CAS.
- J. Totobenazara and A. J. Burke, Tetrahedron Lett., 2015, 56, 2853–2859 CrossRef CAS.
- A. Michael and U. D. E. V. D. Auf, J. Prakt. Chem., 1893, 48, 94–95 CrossRef.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- D. H. Ess, G. O. Jones and K. N. Houk, Org. Lett., 2008, 10, 1633–1636 CrossRef CAS PubMed.
- M. F. Debets, S. S. Van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. Van Hest and F. L. Van Delft, Chem. Commun., 2010, 46, 97–99 RSC.
- A. Kuzmin, A. Poloukhtine, M. A. Wolfert and V. V. Popik, Bioconjugate Chem., 2010, 21, 2076–2085 CrossRef CAS PubMed.
- P. Prasher and M. Sharma, MedChemComm, 2019, 10, 1302–1328 RSC.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS.
- R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1967, 100, 2494–2507 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- S. Lin, Z. Zhang, H. Xu, L. Li, S. Chen, J. Li, Z. Hao and P. R. Chen, J. Am. Chem. Soc., 2011, 133, 20581–20587 CrossRef CAS PubMed.
- N. Nebra and J. García-Álvarez, Molecules, 2020, 25, 2015 CrossRef CAS.
- Y. Chen, T. Cheng, A. Qin and B. Z. Tang, Macromol. Chem. Phys., 2019, 220, 1900064 CrossRef.
- P. Espeel and F. E. Du Prez, Macromolecules, 2015, 48, 2–14 CrossRef CAS.
- A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388–1405 RSC.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- V. Rigolot, C. Biot and C. Lion, Angew. Chem., 2021, 133, 23268–23289 CrossRef.
- S. S. Bag, S. Sinha, S. Singh and A. K. Golder, Tetrahedron, 2023, 149, 133703 CrossRef CAS.
- S. Epple, A. Modi, Y. R. Baker, E. Wȩgrzyn, D. Traoré, P. Wanat, A. E. S. Tyburn, A. Shivalingam, L. Taemaitree and A. H. El-Sagheer, J. Am. Chem. Soc., 2021, 143, 16293–16301 CrossRef CAS PubMed.
- Y. R. Baker, D. Traoré, P. Wanat, A. Tyburn, A. H. El-Sagheer and T. Brown, Tetrahedron, 2020, 76, 130914 CrossRef CAS.
- M. Juríček, P. H. J. Kouwer and A. E. Rowan, Chem. Commun., 2011, 47, 8740–8749 RSC.
- C. W. Tornøe and M. Meldal, Peptides: The Wave of the Future: Proceedings of the Second International and the Seventeenth American Peptide Symposium, June 9–14, 2001, Springer, San Diego, California, USA, 2001, pp. 263–264 Search PubMed.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS PubMed.
- L. K. Mahal, K. J. Yarema and C. R. Bertozzi, Science, 1997, 276, 1125–1128 CrossRef CAS.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- X. Jiang, X. Hao, L. Jing, G. Wu, D. Kang, X. Liu and P. Zhan, Expert Opin. Drug Discovery, 2019, 14, 779–789 CrossRef CAS.
- P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS.
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef CAS PubMed.
- N. Borshell, T. Papp and M. Congreve, Nat. Rev. Drug Discovery, 2011, 10, 167–168 Search PubMed.
- R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. S. Green, R. P. Hertzberg, W. P. Janzen and J. W. Paslay, Nat. Rev. Drug Discovery, 2011, 10, 188–195 CrossRef CAS PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- R. He, Z. Yu, Y. He, L. Zeng, J. Xu, L. Wu, A. M. Gunawan, L. Wang, Z. Jiang and Z. Zhang, ChemMedChem, 2010, 5, 2051–2056 CrossRef CAS PubMed.
- L. Lu and M. Zhu, Anti-Cancer Agents Med. Chem., 2011, 11, 164–171 CrossRef CAS PubMed.
- J. Wang, X. He, L. Gao, L. Sheng, X. Shi, J. Li and G. Chen, Chin. J. Chem., 2011, 29, 1227–1232 CrossRef CAS.
- A. Brik, J. Alexandratos, Y.-C. Lin, J. H. Elder, A. J. Olson, A. Wlodawer, D. S. Goodsell and C.-H. Wong, ChemBioChem, 2005, 6, 1167–1168 CrossRef CAS PubMed.
- A. Brik, J. Muldoon, Y. C. Lin, J. H. Elder, D. S. Goodsell, A. J. Olson, V. V. Fokin, K. B. Sharpless and C. H. Wong, ChemBioChem, 2003, 4, 1246–1248 CrossRef CAS PubMed.
- R. A. Smith, L. J. Copp, P. J. Coles, H. W. Pauls, V. J. Robinson, R. W. Spencer, S. B. Heard and A. Krantz, J. Am. Chem. Soc., 1988, 110, 4429–4431 CrossRef CAS.
- K. Brak, I. D. Kerr, K. T. Barrett, N. Fuchi, M. Debnath, K. Ang, J. C. Engel, J. H. McKerrow, P. S. Doyle and L. S. Brinen, J. Med. Chem., 2010, 53, 1763–1773 CrossRef CAS PubMed.
- C. W. Tornøe, S. J. Sanderson, J. C. Mottram, G. H. Coombs and M. Meldal, J. Comb. Chem., 2004, 6, 312–324 CrossRef PubMed.
- B. Schmidt, ChemBioChem, 2003, 4, 366–378 CrossRef CAS PubMed.
- K. Liu, H. Shi, H. Xiao, A. G. L. Chong, X. Bi, Y. Chang, K. S. W. Tan, R. Y. Yada and S. Q. Yao, Angew. Chem., 2009, 121, 8443–8447 CrossRef.
- E. S. Baskin-Bey, K. Washburn, S. Feng, T. Oltersdorf, D. Shapiro, M. Huyghe, L. Burgart, M. Garrity-Park, F. G. I. Van Vilsteren and L. K. Oliver, Am. J. Transplant., 2007, 7, 218–225 CrossRef CAS PubMed.
- S. L. Ng, P.-Y. Yang, K. Y.-T. Chen, R. Srinivasan and S. Q. Yao, Org. Biomol. Chem., 2008, 6, 844–847 RSC.
- S. K. Maurya, D. R. Gollapalli, S. Kirubakaran, M. Zhang, C. R. Johnson, N. N. Benjamin, L. Hedstrom and G. D. Cuny, J. Med. Chem., 2009, 52, 4623–4630 CrossRef CAS PubMed.
- A. D. Moorhouse, C. Spiteri, P. Sharma, M. Zloh and J. E. Moses, Chem. Commun., 2010, 47, 230–232 RSC.
- L. V. Lee, M. L. Mitchell, S.-J. Huang, V. V. Fokin, K. B. Sharpless and C.-H. Wong, J. Am. Chem. Soc., 2003, 125, 9588–9589 CrossRef CAS.
- R. Roychoudhury and N. L. B. Pohl, Curr. Opin. Chem. Biol., 2010, 14, 168–173 CrossRef CAS PubMed.
- S. El Habnouni, B. Nottelet, V. Darcos, B. Porsio, L. Lemaire, F. Franconi, X. Garric and J. Coudane, Biomacromolecules, 2013, 14, 3626–3634 CrossRef CAS PubMed.
- A. J. Link and D. A. Tirrell, J. Am. Chem. Soc., 2003, 125, 11164–11165 CrossRef CAS PubMed.
- M. Bissaro, M. Sturlese and S. Moro, Drug Discovery Today, 2020, 25, 1693–1701 CrossRef CAS.
- B. C. Doak, R. S. Norton and M. J. Scanlon, Pharmacol. Ther., 2016, 167, 28–37 CrossRef CAS PubMed.
- G. M. Keseru, D. A. Erlanson, G. G. Ferenczy, M. M. Hann, C. W. Murray and S. D. Pickett, J. Med. Chem., 2016, 59, 8189–8206 CrossRef CAS PubMed.
- C. W. Murray and D. C. Rees, Nat. Chem., 2009, 1, 187–192 CrossRef CAS PubMed.
- M. F. Schmidt and J. Rademann, Trends Biotechnol., 2009, 27, 512–521 CrossRef CAS PubMed.
- J. Kaur, M. Saxena and N. Rishi, Bioconjugate Chem., 2021, 32, 1455–1471 CrossRef CAS PubMed.
- V. I. Böhmer, W. Szymanski, K. van den Berg, C. Mulder, P. Kobauri, H. Helbert, D. van Der Born, F. Reeβing, A. Huizing and M. Klopstra, Chem. – Eur. J., 2020, 26, 10871–10881 CrossRef PubMed.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192–3193 CrossRef CAS PubMed.
- A. Deiters, T. A. Cropp, M. Mukherji, J. W. Chin, J. C. Anderson and P. G. Schultz, J. Am. Chem. Soc., 2003, 125, 11782–11783 CrossRef CAS PubMed.
- S. R. Hanson, T.-L. Hsu, E. Weerapana, K. Kishikawa, G. M. Simon, B. F. Cravatt and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 7266–7267 CrossRef CAS PubMed.
- A. J. Link, M. K. S. Vink and D. A. Tirrell, J. Am. Chem. Soc., 2004, 126, 10598–10602 CrossRef CAS PubMed.
- K. E. Beatty, J. C. Liu, F. Xie, D. C. Dieterich, E. M. Schuman, Q. Wang and D. A. Tirrell, Angew. Chem., 2006, 118, 7524–7527 CrossRef.
- K. E. Beatty, F. Xie, Q. Wang and D. A. Tirrell, J. Am. Chem. Soc., 2005, 127, 14150–14151 CrossRef CAS.
- N. Li and W. H. Binder, J. Mater. Chem., 2011, 21, 16717–16734 RSC.
- Y. Chen, Y. Xianyu, J. Wu, B. Yin and X. Jiang, Theranostics, 2016, 6, 969 CrossRef CAS PubMed.
- G. Chen, L. Tao, G. Mantovani, V. Ladmiral, D. P. Burt, J. V. Macpherson and D. M. Haddleton, Soft Matter, 2007, 3, 732–739 RSC.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS PubMed.
- S. El Habnouni, V. Darcos, X. Garric, J. Lavigne, B. Nottelet and J. Coudane, Adv. Funct. Mater., 2011, 21, 3321–3330 CrossRef CAS.
- B. Uszczyńska, T. Ratajczak, E. Frydrych, H. Maciejewski, M. Figlerowicz, W. T. Markiewicz and M. K. Chmielewski, Lab Chip, 2012, 12, 1151–1156 RSC.
- F. Amblard, J. H. Cho and R. F. Schinazi, Chem. Rev., 2009, 109, 4207–4220 CrossRef CAS PubMed.
- A. Okamoto, Y. Saito and I. Saito, J. Photochem. Photobiol., C, 2005, 6, 108–122 CrossRef CAS.
- V. Ljosa and A. E. Carpenter, Trends Biotechnol., 2008, 26, 527–530 CrossRef CAS PubMed.
- M. Sawa, T.-L. Hsu, T. Itoh, M. Sugiyama, S. R. Hanson, P. K. Vogt and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12371–12376 CrossRef CAS.
- S. S. Bag and R. Kundu, J. Org. Chem., 2011, 76, 3348–3356 CrossRef CAS PubMed.
- T. H. N. Pham and R. J. Clarke, J. Phys. Chem. B, 2008, 112, 6513–6520 CrossRef CAS PubMed.
- T. Mitsui, A. Kitamura, M. Kimoto, T. To, A. Sato, I. Hirao and S. Yokoyama, J. Am. Chem. Soc., 2003, 125, 5298–5307 CrossRef CAS PubMed.
- E. Perez-Inestrosa, J.-M. Montenegro, D. Collado and R. Suau, Chem. Commun., 2008, 1085–1087 RSC.
- C. H. Chau, P. S. Steeg and W. D. Figg, Lancet, 2019, 394, 793–804 CrossRef CAS PubMed.
- X. Chen, Y. Zhao and Z. Cao, J. Chem. Phys., 2009, 130(14), 144307 CrossRef PubMed.
- S. S. Bag and R. Kundu, J. Fluoresc., 2013, 23, 929–938 CrossRef CAS PubMed.
- S. S. Bag, R. Kundu and S. Talukdar, Tetrahedron Lett., 2012, 53, 5875–5879 CrossRef CAS.
- S. S. Bag, R. Kundu and S. Jana, Tetrahedron Lett., 2013, 54, 2627–2632 CrossRef CAS.
- S. S. Bag, A. Banerjee and S. Sinha, Synlett, 2024, 35(11), 1195–1227 CAS.
- S. S. Bag, S. Talukdar, K. Matsumoto and R. Kundu, J. Org. Chem., 2013, 78, 278–291 CrossRef CAS PubMed.
- S. S. Bag, S. K. Das and H. Gogoi, Tetrahedron, 2018, 74, 2218–2229 CrossRef CAS.
- S. S. Bag and S. K. Das, Tetrahedron, 2019, 75, 3024–3037 CrossRef CAS.
- S. S. Bag, S. Talukdar, S. K. Das, M. K. Pradhan and S. Mukherjee, Org. Biomol. Chem., 2016, 14, 5088–5108 RSC.
- J.-M. Kee, B. Villani, L. R. Carpenter and T. W. Muir, J. Am. Chem. Soc., 2010, 132, 14327–14329 CrossRef CAS PubMed.
- M. Gajewski, B. Seaver and C. S. Esslinger, Bioorg. Med. Chem. Lett., 2007, 17, 4163–4166 CrossRef CAS PubMed.
- N. J. Stanley, D. S. Pedersen, B. Nielsen, T. Kvist, J. M. Mathiesen, H. Bräuner-Osborne, D. K. Taylor and A. D. Abell, Bioorg. Med. Chem. Lett., 2010, 20, 7512–7515 CrossRef CAS PubMed.
- C. Hall, H. Wolfe, A. Wells, H.-C. Chien, C. Colas, A. Schlessinger, K. M. Giacomini and A. A. Thomas, Bioorg. Med. Chem. Lett., 2019, 29, 2254–2258 CrossRef CAS PubMed.
- T. Niu, C. Cai and L. Yi, Helv. Chim. Acta, 2012, 95, 87–99 CrossRef CAS.
- B. H. M. Kuijpers, S. Groothuys, C. Hawner, J. ten Dam, P. J. L. M. Quaedflieg, H. E. Schoemaker, F. L. van Delft and F. P. J. T. Rutjes, Org. Process Res. Dev., 2008, 12, 503–511 CrossRef CAS.
- N. Agouram, E. M. El Hadrami and A. Bentama, Molecules, 2021, 26, 2937 CrossRef CAS PubMed.
- V. D. Bock, D. Speijer, H. Hiemstra and J. H. van Maarseveen, Org. Biomol. Chem., 2007, 5, 971–975 RSC.
- D. S. Pedersen and A. Abell, Eur. J. Org. Chem., 2011, 2399–2411 CrossRef CAS.
- T.-S. Hu, R. Tannert, H.-D. Arndt and H. Waldmann, Chem. Commun., 2007, 3942–3944 RSC.
- V. Goncalves, B. Gautier, A. Regazzetti, P. Coric, S. Bouaziz, C. Garbay, M. Vidal and N. Inguimbert, Bioorg. Med. Chem. Lett., 2007, 17, 5590–5594 CrossRef CAS PubMed.
- Y. Liu, L. Zhang, J. Wan, Y. Li, Y. Xu and Y. Pan, Tetrahedron, 2008, 64, 10728–10734 CrossRef CAS.
- W. J. Choi, Z.-D. Shi, K. M. Worthy, L. Bindu, R. G. Karki, M. C. Nicklaus, R. J. Fisher and T. R. Burke Jr, Bioorg. Med. Chem. Lett., 2006, 16, 5265–5269 CrossRef CAS PubMed.
- Y. Angell, D. Chen, F. Brahimi, H. U. Saragovi and K. Burgess, J. Am. Chem. Soc., 2008, 130, 556–565 CrossRef CAS PubMed.
- Y. Angell and K. Burgess, J. Org. Chem., 2005, 70, 9595–9598 CrossRef CAS PubMed.
- N. G. Angelo and P. S. Arora, J. Am. Chem. Soc., 2005, 127, 17134–17135 CrossRef CAS PubMed.
- W. S. Horne, M. K. Yadav, C. D. Stout and M. R. Ghadiri, J. Am. Chem. Soc., 2004, 126, 15366–15367 CrossRef CAS PubMed.
- J.-F. Lutz and Z. Zarafshani, Adv. Drug Delivery Rev., 2008, 60, 958–970 CrossRef CAS PubMed.
- Y. L. Angell and K. Burgess, Chem. Soc. Rev., 2007, 36, 1674–1689 RSC.
- K. Oh and Z. Guan, Chem. Commun., 2006, 3069–3071 RSC.
- A. Tam, U. Arnold, M. B. Soellner and R. T. Raines, J. Am. Chem. Soc., 2007, 129, 12670–12671 CrossRef CAS PubMed.
- T. Yu, J. Z. Bai and Z. Guan, Angew. Chem., 2009, 121, 1117–1121 CrossRef.
- S. Cantel, A. Le Chevalier Isaad, M. Scrima, J. J. Levy, R. D. DiMarchi, P. Rovero, J. A. Halperin, A. M. D’Ursi, A. M. Papini and M. Chorev, J. Org. Chem., 2008, 73, 5663–5674 CrossRef CAS PubMed.
- H. Sun, L. Liu, J. Lu, S. Qiu, C.-Y. Yang, H. Yi and S. Wang, Bioorg. Med. Chem. Lett., 2010, 20, 3043–3046 CrossRef CAS PubMed.
- I. Güell, L. Micaló, L. Cano, E. Badosa, R. Ferre, E. Montesinos, E. Bardají, L. Feliu and M. Planas, Peptides, 2012, 33, 9–17 CrossRef PubMed.
- D. Althuon, F. Rönicke, D. Fürniss, J. Quan, I. Wellhöfer, N. Jung, U. Schepers and S. Bräse, Org. Biomol. Chem., 2015, 13, 4226–4230 RSC.
- L. Jiang and K. Kirshenbaum, Org. Biomol. Chem., 2020, 18, 2312–2320 RSC.
- Y. Ge, X. Fan and P. R. Chen, Chem. Sci., 2016, 7, 7055–7060 RSC.
- I. C. Vreja, I. Nikic, F. Gottfert, M. Bates, K. Krohnert, T. F. Outeiro, S. W. Hell, E. A. Lemke and S. O. Rizzoli, ACS Nano, 2015, 9, 11034–11041 CrossRef CAS PubMed.
- I. C. Vreja, S. Kabatas, S. K. Saka, K. Kröhnert, C. Höschen, F. Opazo, U. Diederichsen and S. O. Rizzoli, Angew. Chem., Int. Ed., 2015, 54, 5784–5788 CrossRef CAS PubMed.
- B. Meineke, J. Heimgärtner, A. J. Craig, M. Landreh, L. W. K. Moodie and S. J. Elsässer, Front. Chem., 2021, 9, 768535 CrossRef CAS PubMed.
- Z. Hao, Y. Song, S. Lin, M. Yang, Y. Liang, J. Wang and P. R. Chen, Chem. Commun., 2011, 47, 4502–4504 RSC.
- S. S. Bag, S. Jana and M. K. Pradhan, Bioorg. Med. Chem., 2016, 24, 3579–3595 CrossRef CAS PubMed.
- S. S. Bag, S. Talukdar and S. K. Das, Curr. Protoc. Nucleic Acid Chem., 2014, 58, 1–32 Search PubMed.
- S. S. Bag, S. Jana, A. Yashmeen, K. Senthilkumar and R. Bag, Chem. Commun., 2014, 50, 433–435 RSC.
- J. Shi, Y.-Y. Zhu, J. Wang, J. Chen and Y.-J. Shen, Spectrochim. Acta, Part A, 2013, 103, 287–294 CrossRef CAS PubMed.
- M. Nag, K. Bera, S. Chakraborty and S. Basak, J. Photochem. Photobiol., B, 2013, 127, 202–211 CrossRef CAS PubMed.
- E. Vass, M. Hollósi, Š. Horvat and M. Roščić, Croat. Chem. Acta, 2008, 81, 647–656 CAS.
- J. Hughes, Brain Res., 1975, 88, 295–308 CrossRef CAS PubMed.
- H. Yang, S. Yang, J. Kong, A. Dong and S. Yu, Nat. Protoc., 2015, 10, 382–396 CrossRef CAS PubMed.
- S. S. Bag, S. Jana, A. Yashmeen and S. De, Chem. Commun., 2015, 51, 5242–5245 RSC.
- S. S. Bag, S. Jana, M. K. Pradhan and S. Pal, RSC Adv., 2016, 6, 72654–72658 RSC.
- S. S. Bag and S. Jana, New J. Chem., 2017, 41, 13391–13398 RSC.
- S. S. Bag and A. Yashmeen, Chem. Commun., 2018, 54, 9765–9768 RSC.
- T. Puthiyedath and D. Bahulayan, Sens. Actuators, B, 2018, 272, 110–117 CrossRef CAS.
- Q. Liu, M. Zhao, C. Song, J. Sun, J. Tao, B. Sun and J. Jiang, Molecules, 2023, 28, 2758 CrossRef CAS PubMed.
- S. S. Bag, I. Saraogi and J. Guo, Front. Chem., 2022, 10, 958433 CrossRef PubMed.
- A. H. El-Sagheer and T. Brown, Q. Rev. Biophys., 2015, 48, 429–436 CrossRef CAS PubMed.
- N. A. Al-Masoudi and Y. A. Al-Soud, Nucleosides, Nucleotides Nucleic Acids, 2002, 21, 361–375 CrossRef CAS PubMed.
- Z. Wen, P. R. Tuttle, A. H. Howlader, A. Vasilyeva, L. Gonzalez, A. Tangar, R. Lei, E. E. Laverde, Y. Liu and J. Miksovska, J. Org. Chem., 2019, 84, 3624–3631 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |