
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalytic CS2 insertion into epoxides in neat conditions: a straightforward approach for the efficient synthesis of Di- and tri-thiocarbonates†
Marcos López-Aguilar a,
Nicolás Ríos-Lombardíaa,
Miguel Gallegosb,
Daniel Barrena-Espésb,
Joaquín García-Álvarez*a,
Carmen Concellón*a and
Vicente del Amo*a
a,
Nicolás Ríos-Lombardíaa,
Miguel Gallegosb,
Daniel Barrena-Espésb,
Joaquín García-Álvarez*a,
Carmen Concellón*a and
Vicente del Amo*a
aLaboratorio de Química Sintética Sostenible (QuimSinSos), Departamento de Química Orgánica e Inorgánica, (IUQOEM) and ORFEO-CINQA, Facultad de Química, Universidad de Oviedo, E33071 Oviedo, Spain. E-mail: garciajoaquin@uniovi.es
bDepartamento de Química Física y Analítica, Facultad de Química, Universidad de Oviedo, E33071 Oviedo, Spain
First published on 23rd December 2024
Abstract
The straightforward organocatalytic insertion of carbon disulfide (CS2) into epoxides using either choline chloride (ChCl) or tetrabutylammonium chloride (TBACl) is reported, for the first time, under solvent-free (neat) conditions. Fine-tuning of our system allowed us to obtain either dithiocarbonates (DTCs) or trithiocarbonates (TTCs) with high efficiency. Additionally, a mechanistic proposal is presented, supported by experimental evidence, DFT calculations and wavefunction analyses.
C1 heterocumulenes [X
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CX (e.g., CO2, CS2, carbodiimides) or XCY (e.g., isocyanates, thioisocyanates, ketenes)] are versatile building blocks for creating complex organic structures with diverse properties.1 While CO2 chemistry is well-studied,2 its heavier analogue CS2 has been less explored.3 For example, the insertion of CS2 into epoxides to form dithiocarbonates (DTCs) or trithiocarbonates (TTCs) remains limited, despite their interesting applications, partly due to challenges in controlling chemo- and regioselectivity, which often lead to complex product mixtures (Scheme 1).4 Although this highly-challenging reaction can be chemically domesticated, this often requires sophisticated metal-based catalysts.5 Additionally, these CS2 insertion reactions typically use volatile organic compounds (VOCs) as solvents, which are toxic, flammable, and sometimes carcinogenic.6 Moreover, these non-renewable VOCs also contribute significantly to waste generation in this process.7 Considering all these precedents, and aiming for the development of new sustainable methodologies suitable for the fixation and valorisation of heterocumulenes under the Green Chemistry framework,8 herein we report the use of simple, easily-available and cheap organocatalysts for the insertion of CS2 into epoxides. This protocol renders in demand either DTCs or TTCs, in high yields. Going one step further, and bearing in mind the phrase (coined by P. T. Anastas and J. C. Warner, usually considered the fathers of the Green Chemistry concept): “The best solvent is no solvent”,9 we describe a “solventless” protocol (neat conditions) without needing VOC-based reaction media. Finally, and aside from the Green Chemistry point of view, we also make special emphasis on shedding light on the mechanism of our CS2 insertion into epoxides.
CX (e.g., CO2, CS2, carbodiimides) or XCY (e.g., isocyanates, thioisocyanates, ketenes)] are versatile building blocks for creating complex organic structures with diverse properties.1 While CO2 chemistry is well-studied,2 its heavier analogue CS2 has been less explored.3 For example, the insertion of CS2 into epoxides to form dithiocarbonates (DTCs) or trithiocarbonates (TTCs) remains limited, despite their interesting applications, partly due to challenges in controlling chemo- and regioselectivity, which often lead to complex product mixtures (Scheme 1).4 Although this highly-challenging reaction can be chemically domesticated, this often requires sophisticated metal-based catalysts.5 Additionally, these CS2 insertion reactions typically use volatile organic compounds (VOCs) as solvents, which are toxic, flammable, and sometimes carcinogenic.6 Moreover, these non-renewable VOCs also contribute significantly to waste generation in this process.7 Considering all these precedents, and aiming for the development of new sustainable methodologies suitable for the fixation and valorisation of heterocumulenes under the Green Chemistry framework,8 herein we report the use of simple, easily-available and cheap organocatalysts for the insertion of CS2 into epoxides. This protocol renders in demand either DTCs or TTCs, in high yields. Going one step further, and bearing in mind the phrase (coined by P. T. Anastas and J. C. Warner, usually considered the fathers of the Green Chemistry concept): “The best solvent is no solvent”,9 we describe a “solventless” protocol (neat conditions) without needing VOC-based reaction media. Finally, and aside from the Green Chemistry point of view, we also make special emphasis on shedding light on the mechanism of our CS2 insertion into epoxides.
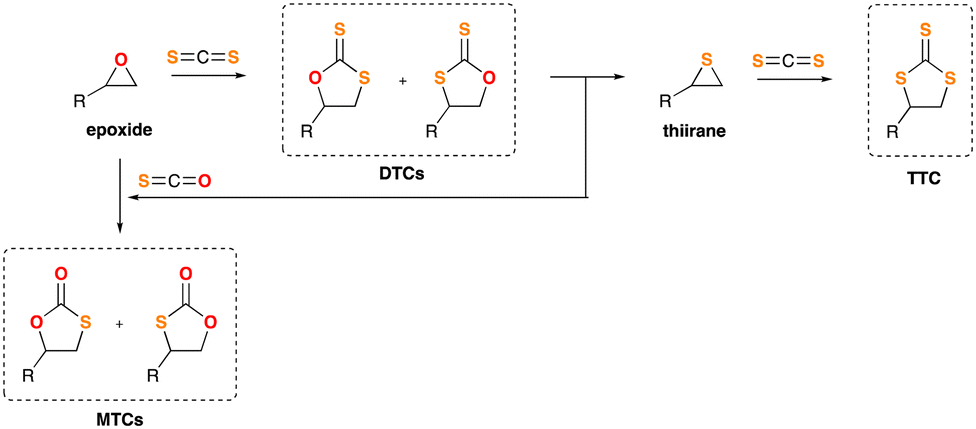 | ||
| Scheme 1 General sequence for the preparation of dithiocarbonates (DTCs) or trithiocarbonates (TTCs). | ||
Based on literature precedents and its analogy to our previous CO2 insertion protocol,8a we chose commercially available styrene oxide (1a) as the model substrate. Thus, 1a was initially dissolved in CS2, treated with a suitable organocatalyst, and subsequently heated up inside a sealed tube under vigorous stirring. After carefully scrutinising all the experimental parameters affecting the course of this transformation (see the ESI† file for details), we were delighted to find an optimised set of conditions. It implies the use of readily available and cheap choline chloride (ChCl, which is a biorenewable salt formerly known as vitamin B4)10,11 or tetrabutylammonium chloride (TBACl) as organocatalysts, and 3 equiv. of CS2 at 100 °C, for 24 hours in the absence of any external VOC-solvent.12 By using this simple and straightforward methodology we were able to afford the corresponding TTC 2a in 93% conversion (in the case of using ChCl as the catalyst; Table 1, entry 1), or 84% (when using TBACl; Table 1, entry 2). Interestingly, we observed experimentally that the presence of water was necessary to achieve optimum results, as the chloride salts are sparingly soluble in CS2. At this point, and in the framework of Green Chemistry, it is important to highlight the environmentally benign character of our protocol as: (i) it occurs under neat conditions (using CS2 as both the reagent and the reaction media); (ii) exhibits a significant atom economy,13 moreover when the transformation 1a → 2a (formation of the TTC) requires two equiv. of CS2; and (iii) employs an organocatalytic method (as assessed by Rothenberg “Catalysis is the key for sustainability”),14 in which simple, non-toxic and cheap ammonium salts (ChCl or TBACl) are employed as readily available off-the-bench catalysts. Going one step further, we decided to critically evaluate the sustainability virtues of our transformation by using Sheldon's E-factor as Green Chemistry Metric.15 In this sense, and taking into account that a remarkably 86% yield of 2a was achieved when water was the only material used for the workup of the reaction and the subsequent purification steps, we calculated an E-factor of 0.945, in between the range of values calculated for previous syntheses of TTCs in neat conditions (0.48 to 6.87; see ESI†).
|
|||
|---|---|---|---|
| Entry | Cat. (mol%) | H2O (equiv.) | Conversionb (%) |
| a General conditions: styrene oxide 1a (0.88 mmol) was dissolved in CS2 and treated with an aqueous solution of the stated catalyst. The mixture was stirred at 100 °C for 24 h, inside a 10 mL sealed tube.b Conversion of styrene oxide into product 2a, as determined by 1H NMR spectroscopy from crude reaction mixtures, using CHBr3 as an internal standard.c 5% of a corresponding monothiocarbonate (5a, a single isomer) was identified.d 7% of a corresponding monothiocarbonate (5a, a single isomer) was identified. | |||
| 1 | ChCl (2.5) | 2 | 93c |
| 2 | TBACl (10) | 1 | 84d |
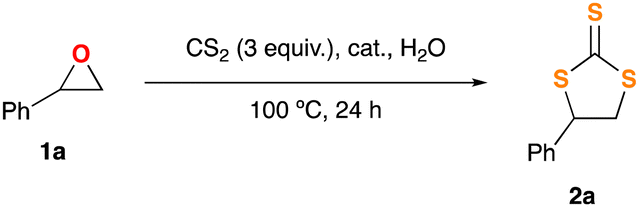
Motivated by our preliminary results, which indicate the possibility of synthesising the desired thiocarbonates under more sustainable reaction conditions, we next decided to explore the scope and limitations of our insertion of CS2 into epoxides. Accordingly, we selected some starting materials 1b–k, decorated with different functional groups and structural patterns, particularly those which proved to be a challenge for other methodologies previously communicated.4,5 Epoxides 1b–k were subjected to the conditions described in Table 1, employing, in turns, either ChCl or TBACl as organocatalysts (Fig. 1). As previously observed for 1a, all the reactions proceeded smoothly. To our surprise, their outcome relies deeply on the nature of the organocatalyst employed. Epoxides 1b and c (which bear a single non-bulky alkyl substituent), afforded the corresponding TTCs 2b and c in good yield when using TBACl as organocatalyst, while no reaction was observed for the case of ChCl. Likely, sterically encumbered epoxides 1d–g rendered TTCs 2d–g in moderate yield upon reaction with TBACl, being ChCl a superior organocatalyst for epoxide 2e. Remarkably, products 2f and g were formed in a diastereo pure form, retaining in both cases the relative spatial configuration of the stereocenter adjacent to the oxirane function. Moreover, and to the best of our knowledge, this is the first occasion in which epoxides 1f and g are transformed into TTCs by either methodology.4,5 The relative stereochemistry of products 2f and 2g was unambiguously disclosed by single crystal X-ray diffraction techniques (Fig. S54 and S55, ESI†). Moreover, and trying to show the importance of trithiocarbonates as building blocks for the synthesis of other organic architectures,1 we designed a two-step synthetic protocol to convert TTC 2e into the corresponding diastereopure bis-thioester 3 (see Scheme 2, and ESI† for details). Compound 3 was fully characterised and its relative spatial configuration unveiled by single crystal X-ray diffraction analyses, which ultimately allowed us to confirm the stereochemistry of TTC 2e (see ESI† for details).
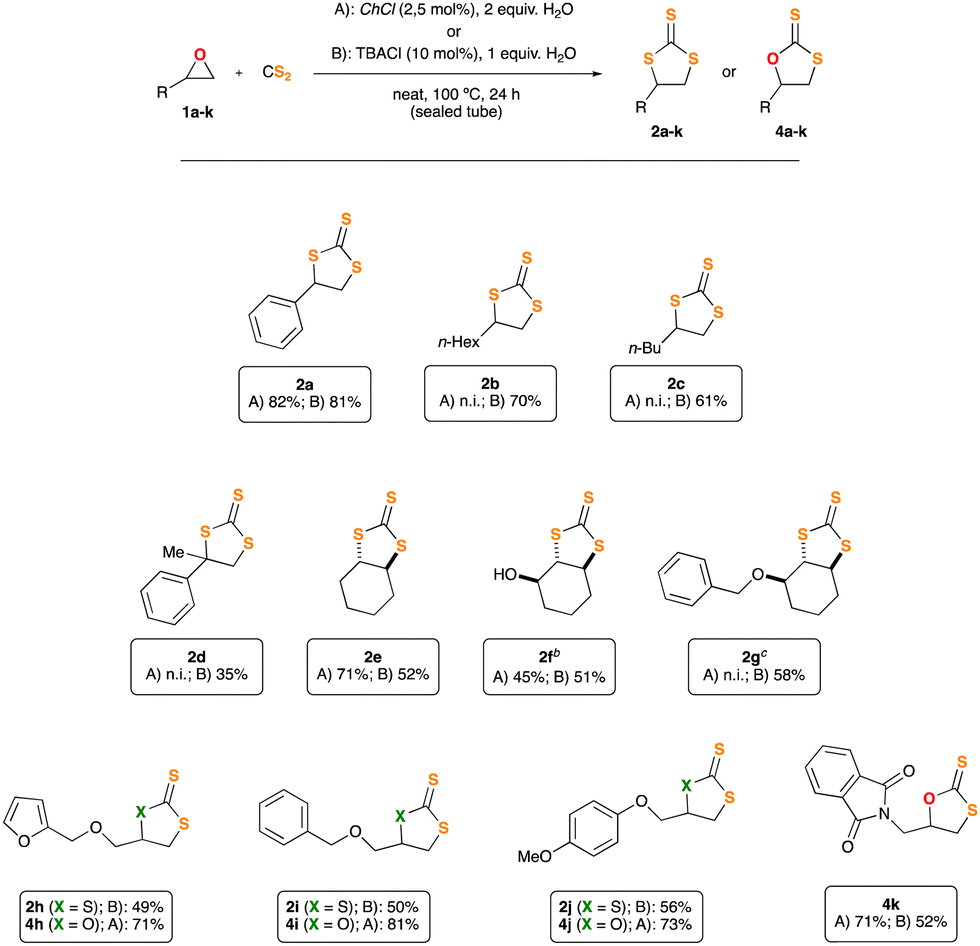 | ||
Fig. 1 Scope of the insertion of CS2 into epoxides. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) General conditions: epoxide 1a–k (0.88 mmol) was dissolved in CS2 (201 mg, 159 μL, 2.64 mmol) and treated with an aqueous solution of the stated catalyst, ChCl or TBACl. Reaction mixtures were stirred at 100 °C for 24 h, inside a 10 mL sealed tube. The yield of analytically pure products, purified by flash chromatography, is given. b(1R*,2R*,6S*)-7-oxabicyclo[4.1.0]heptan-2-ol was employed as the starting material. cO-benzyl-(1S*,2R*,6S*)-7-oxabicyclo[4.1.0]heptan-2-ol was employed as the starting material. General conditions: epoxide 1a–k (0.88 mmol) was dissolved in CS2 (201 mg, 159 μL, 2.64 mmol) and treated with an aqueous solution of the stated catalyst, ChCl or TBACl. Reaction mixtures were stirred at 100 °C for 24 h, inside a 10 mL sealed tube. The yield of analytically pure products, purified by flash chromatography, is given. b(1R*,2R*,6S*)-7-oxabicyclo[4.1.0]heptan-2-ol was employed as the starting material. cO-benzyl-(1S*,2R*,6S*)-7-oxabicyclo[4.1.0]heptan-2-ol was employed as the starting material. | ||
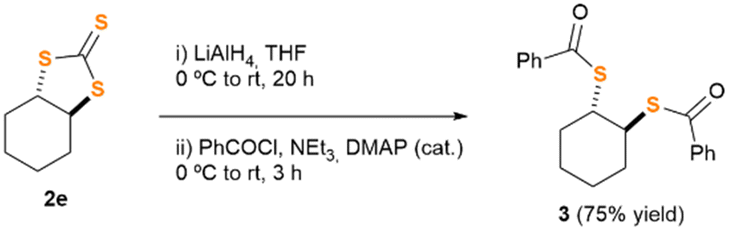 | ||
| Scheme 2 Synthetic scheme for the derivatisation of TTC 2e into bis-thioester 3. | ||
For the epoxides consisting in O-substituted glycidol (1h–j), we observed, as it was expected, the formation of the corresponding TTCs 2h–j when using TBACl as catalyst. Importantly, and showcasing the versatility of our synthetic approach, we were delighted to find that simply replacing TBACl by ChCl (under the same reaction conditions and using the same starting materials), allowed us to obtain the corresponding dithiocarbonates (DTCs 4h–j). These were isolated in good yield and without contamination from their regioisomeric forms, which is relevant for future synthetic application, as the different regioisomers cannot be separated by standard chromatographic techniques. Last, but not least, epoxide 1k, featuring an phthalimide residue, could not be converted into the corresponding TTC, but alternatively its DTC 4k was isolated in good yield, better when ChCl was used.
Based on the aforementioned experimental findings and intrigued by the various reaction pathways observed in the insertion of CS2 into epoxides, which are strongly depending on the nature of the chloride salt used as the catalyst, next we decided to explore the behaviour of an enantiopure substrate, (R)-1i, under the optimised reaction conditions. The resulting crude mixtures were carefully purified by flash chromatography and all the fractions were characterised and analysed by HPLC using a chiral stationary phase. When TBACl was used as the catalyst TTC rac-2i was isolated as the major product, thus jeopardizing the stereochemical integrity of the substrate. Also, the DTCs mixture 4i + 4i′ was isolated and identified. 4i showed an erosion of its stereoinformation, while 4i′ was rendered in racemic form (Scheme 3b). In turn, ChCl gave rise to DTC (S)-4i in enantiopure form (>99% ee) and as a single regioisomer. In this last case the stereogenic carbon of epoxide 1i retains its configuration fully. Moreover, some TTC (R)-2i was also isolated, also in an enatiopure form, implying an inversion of configuration on the pristine stereogenic center (Scheme 3a).
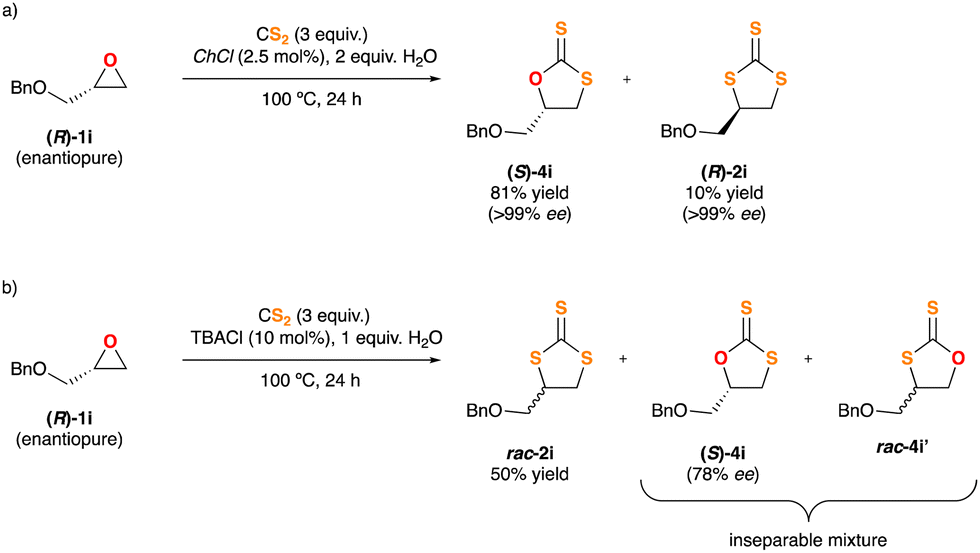 | ||
| Scheme 3 Study of the sterochemical outcome of the insertion reaction of CS2 into chiral epoxide (R)-1i. | ||
With all the above information in hand we propose the following sequence as a tentative mechanism for our insertion of CS2 into epoxides (Schemes 4 and 5), fully supported by DFT calculations (see Appendix for details). Initially, a [CS2·Cl]− adduct, 6, is formed in situ in the reaction medium, as it has been previously postulated.16 It originates from the attack of the halide anion of the catalyst to the electron-cumbered sp carbon of CS2, and would explain the effectiveness of chloride respect to bulkier bromide or iodide ions (see Table S4, ESI†). This short-lived reactive specie, highly nucleophilic, undergoes a SN2-type addition on epoxide 1 from the downward side, opposite to the O atom. This is further supported by wavefunction analysis, which shows ideal electron delocalization values of 0.5 electron pairs at the transition stated, with the corresponding C–S and C–O bonds forming and breaking in a fully concerted and synchronous manner (Appendix Tables S10–S13, and S19–S22, ESI†). Eventually it gives rise to the corresponding DTC 4. The latest, upon vigorous heating, extrudes SCO to afford thiirane 7, which features an inversion of its stereogenic carbons.17 Another attack of adduct 6 on thiirane 7, again by means of a downward-facing SN2-type process (Appendix Tables S28–S31, and S37–S40, ESI†), leads to the formation of TTC 2.18 Overall, this manifold explains the stereochemical outcome observed for the CS2 insertion on cyclohexene oxide derivatives 1e–g (Scheme 4).
 | ||
| Scheme 4 General mechanistic proposal for the insertion of CS2 into epoxides 1 catalysed by chloride anions. | ||
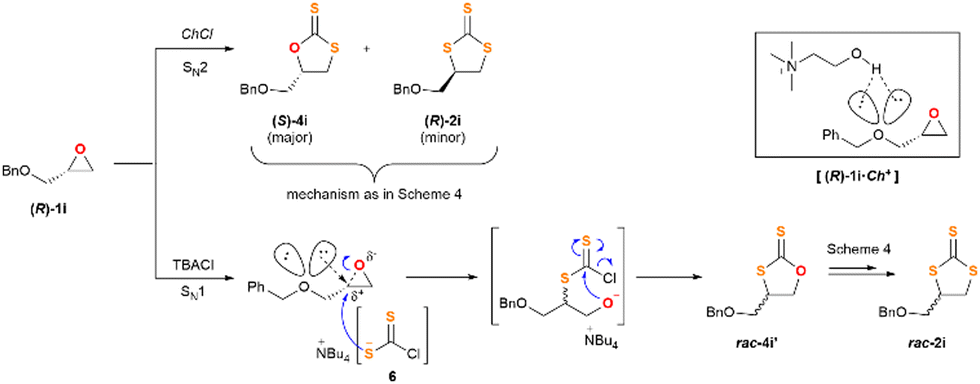 | ||
| Scheme 5 General mechanistic proposal for the insertion of CS2 into epoxide (R)-1i, which explains the unalike outcome of the reaction when TBACl or ChCl are used as catalysts. | ||
The distinct reactivity of TBACl and ChCl is manifested on the results highlighted in Scheme 3, where the CS2 insertion is carried out on the enantiopure substrate (R)-1i. Based on the experimental observations and the computational analysis we believe that ChCl follows the mechanism of Scheme 4, as the chirality of the substrate is transferred entirely to the reaction products (S)-4i (major) and (R)-2i (minor). On the contrary, the use of TBACl as the catalyst leads to a collection of products that lack any chirality, or it has been severely damaged. This experimental fact can be rationalised from the proposal outlined in Scheme 5.
In substrate (R)-1i, the lone pairs of electrons placed on the oxygen atom bearing the benzyl substituent can assist the opening of the epoxide function. Thus, we suggest (R)-1i coexisting with a subsequent flat non-classic carbocationic-type structure, which upon reaction with nucleophile 6 leads to products rac-4i′ and rac-2. It is important to note that this route does only occur when using TBACl as the catalyst, and not ChCl. It therefore implies that the cation accompanying the chloride ion plays a crucial role in the reaction outcome. We suggest that the choline cation can participate in a H-bonding network that comprises the electronic lone pairs of the epoxide and make them unavailable for the aforementioned anchimeric assistance (complex [(R)-1i·Ch]+ in Scheme 5). Notwithstanding with this suggestion, we strongly believe that the extrusion-insertion of SCO17 and CS218 on substrates 4 and 7, respectively, takes place simultaneously and in a quasi-reversible way, as such, it has to be considered if a full picture of the mechanism is pursued.
In conclusion, and edging closer towards the development of a straightforward, more sustainable and solventless-compatible organocatalysed synthesis of di- and tri-thiocarbonates, our experimental findings have uncovered the potential of using simple, readily available and non-toxic quaternary ammonium salts as catalysts to chemically domesticate the highly-challenging insertion of CS2 into epoxides. Fine-tune selection of the catalysts employed (TBACl or ChCl), and the epoxide used as starting material allows the design of “a la carte” synthetic protocols for the formation of di- and tri-thiocarbonates in demand. Also, mechanistic investigations have been disclosed through reactivity studies along with electron population.
All the authors thank: (i) MCIN/AEI/10.13039/501100011033 (project numbers PID2020-113473GB-100, PID2021-122763NB-I00 and PID2023-148663NB-I00) for financial support; and (ii) Universidad de Oviedo SCTIs and Dr D. Elorriaga for technical support. M. L. A. and D. B.-E. acknowledge FICyT for funding (grants PA-22-BP21-088 and PA-23-BP22-168, respectively).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) H. Ulrich, Cumulenes In Click Reactions, Wiley, New York, 2010 Search PubMed; (b) L. Brandsma, Eur. J. Org. Chem., 2001, 4569 CrossRef CAS; (c) J. Louie, Curr. Org. Chem., 2005, 9, 605 CrossRef CAS; (d) S. Schenk, J. Notni, U. Köhn, K. Wermann and E. Anders, Dalton Trans., 2006, 4191 RSC; (e) S. Braverman, M. Cherkinsky and M. L. Birsa, Carbon dioxide, carbonyl sulfide, carbon disulfide, isocyanates, isothiocyanates, carbodiimides, and their selenium, tellurium, and phosphorus analogues. In Science of Synthesis, ed. J. G. Knight, Thieme: Stuttgart, Germany, 2005; 18, 65 Search PubMed.
- (a) C. Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (b) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed; (c) J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434 CrossRef CAS PubMed; (d) W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. W. L. Wang, Y. Liu, B. Louis, J. Scott, A.-C. Roger, R. Amal, H. Heh and S.-E. Park, Chem. Soc. Rev., 2020, 49, 8584 RSC.
- (a) W. A. Schenk and T. Schwietzke, Organometallics, 1983, 2, 190 CrossRef; (b) D. Seyferth, G. B. Womack, M. Cowie and B. W. Hames, Organometallics, 1984, 3, 189 Search PubMed; (c) M. Yokoyama and T. Imamoto, Synthesis, 1984, 797 CrossRef CAS; (d) W. D. Rudorf, J. Sulfur Rep., 1991, 11, 51 CrossRef CAS; (e) W. D. Rudorf, J. Sulfur Chem., 2007, 28, 295 CrossRef CAS; (f) F. J. Iglesias-Sigüenza, Synlett, 2009, 157 Search PubMed.
- (a) H. S. Kim, D. Q. Nguyen, M. Cheong, H. Kim, H. Lee, N. H. Ko and J. S. Lee, Appl. Catal., A, 2008, 337, 168 CrossRef CAS; (b) C. Díez-Poza, L. Álvarez-Miguel, M. E. G. Mosquera and C. J. Whiteoak, Org. Biomol. Chem., 2023, 21, 3733 RSC.
- (a) R. Maggi, C. Malmassari, C. Oro, R. Pela, G. Sartori and L. Soldo, Synthesis, 2008, 53 CrossRef CAS; (b) W. Clegg, R. W. Harrington, M. North and P. Villuendas, J. Org. Chem., 2010, 75, 6201 CrossRef CAS PubMed; (c) C. Beattie and M. North, ChemCatChem, 2014, 6, 1252 CrossRef CAS; (d) J. Diebler, A. Spannenberg and T. Werner, Org. Biomol. Chem., 2016, 14, 7480 RSC; (e) C. Mei, X. Li, L. Liu, C. Cao, G. Pang and Y. Shi, Tetrahedron, 2017, 73, 570 CrossRef; (f) M. Okada, R. Nishiyori, S. Kaneko, K. Igawa and S. Shirakawa, Eur. J. Org. Chem., 2018, 2022 CrossRef CAS; (g) M. Gupta, N. Chatterjee, D. De, R. Saha, P. K. Chattaraj, C. L. Oliver and P. K. Bharadwaj, Inorg. Chem., 2020, 59, 1810 CrossRef CAS PubMed; (h) N. Aoyagi and T. Endo, Synlett, 2020, 92 CAS; (i) X. Du, Z. Liu, Z. Li, X. Yuan, C. Li, M. Zhang, Z. Zhang, X. Hu and K. Guo, RSC Adv., 2024, 14, 10378 RSC.
- (a) R. Heinrich-Ramm, M. Jakubowski, B. Heinzow, J. M. Christensen, E. Olsen and O. Hertel, Pure Appl. Chem., 2000, 72, 385 CrossRef CAS; (b) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2009, 39, 301 RSC.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133 CrossRef CAS.
- For our previous works in the field of CO2 valorisation, see: (a) N. Fanjul-Mosteirín, J. Martín, C. Valdés, C. Concellón and V. Del Amo, Org. Lett., 2020, 22, 6988 CrossRef PubMed; (b) D. Elorriaga, F. de la Cruz-Martínez, M. J. Rodríguez-Álvarez, A. Lara-Sánchez, J. A. Castro-Osma and J. García-Álvarez, ChemSusChem, 2021, 14, 2084 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- Choline chloride (ChCl, an essential micronutrient) is manufactured in the scale of millions of tons with a prize of 1.3 € per Kg J. K. Blusztajn, Science, 1998, 284, 794 CrossRef.
- ChCl has been previously used as organocatalyst for the insertion of CO2 into epoxides to render the corresponding cyclic carbonates. (a) A. Zhu, T. Jiang, B. Han, J. Zhang, Y. Xie and X. Ma, Green Chem., 2007, 9, 169 RSC; (b) K. Wu, T. Su, D. Hao, W. Liao, Y. Zhao, W. Ren, C. Deng and H. Lü, Chem. Commun., 2018, 54, 9579 RSC.
- Notably, we found that reducing the amount of CS2 from 6 to 3 equivalents maintained process activity while improving atom economy and reducing waste (see ESI†).
- (a) B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS PubMed; (b) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS.
- G. Rothenberg, Catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- (a) R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC; (b) R. A. Sheldon, Green Chem., 2017, 19, 18 RSC.
- S. Shirakawa, Chem. Rec., 2023, e202300144 CrossRef CAS.
- The DFT analysis shows that the extrusion of SCO from DTC 4 is, particularly, a reversible process (Appendix: Fig. S1 and S2, ESI,† TS-4C and TS-4D). In this way, the insertion of SCO on thiirane 7 may take place, attacking on either of its strained carbons, by a SN2-type sequence. This fact hinders the outcome of the whole transformations 1 (epoxide) → 4 (DTC) → 7 (thiirane) → 2 (TTC), particularly in those cases in which the intermediate thiirane 7 is unsymmetrically substituted, or possesses a stereogenic centre. Under this scenario, an even-number of downward attacks on the stereogenic carbon of 7 will always preserve the pristine stereochemical configuration. On the contrary, an odd-number of nucleophilic attacks or their combination with SCO/CS2 reinsertions on the same carbon can result in the modification of the starting stereochemistry.
- Similarly, at 100 °C, CS2 could be reversibly extruded from TTC 2, and reincorporated on thiirane 7. The same stereochemical issues arise.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, NMR spectra and computational details. CCDC 2386775–2386777. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc05154h |
| This journal is © The Royal Society of Chemistry 2025 |