
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conversion of methanol at copper clusters on TiO2(110) and SiOx: direct dehydrogenation vs. partial oxidation and influence of cluster size and substrate†
Maximilian
Grebien
 and
Katharina
Al-Shamery
*
and
Katharina
Al-Shamery
*
Institute of Chemistry, Carl von Ossietzky University of Oldenburg, Carl-von-Ossietzky Straße 9-11, D-26129 Oldenburg, Germany. E-mail: Katharina.al.shamery@uni-oldenburg.de
First published on 3rd February 2025
Abstract
Copper is known to catalyze the conversion of methanol to formaldehyde in single crystal experiments. Here, we present a systematic study of methanol reactions at different-sized nanoparticulate copper clusters on rutile TiO2(110) as well as a native silicon oxide film on a Si(111) wafer. By temperature-programmed reaction spectroscopy (TPRS), we have identified two different pathways, namely the direct dehydrogenation and in the presence of oxygen the partial oxidation to formaldehyde for large copper clusters. While the silica substrate is nonreactive, for rutile TiO2(110) the competing conversion of methanol to methane dominates the formaldehyde formation, depending on the titania reduction degree. At the same time, the low-temperature formaldehyde formation at the highly reduced TiO2(110) was not observed, suggesting the suppression of the dioxomethylene-like intermediate of this species at the surface. Concurrent with these reactions the high-temperature desorption of CO2 was observed as a side-product on all substrates, which can be correlated with the formation of a formate intermediate decomposing into CO2 and H2 at elevated temperatures.
1. Introduction
Methanol is one of the most used chemicals in the chemical industry. Not only is methanol one of the most promising fuel alternatives to reduce greenhouse gases1 and can be used as an energy carrier for hydrogen storage, it can also be utilized to produce various products in the chemical industry like acetic acid, methyl and vinyl acetates, methylamines, formaldehyde, and methyl tert-butyl ether. Around 65% of the methanol produced worldwide is used for conversion into further products.2 The most important product is formaldehyde which is produced from around 30–35% of methanol worldwide.3–5 Formaldehyde can then be converted into formaldehyde resins and isoprene rubber,4 and also has uses in furniture, cleaning products, paints, plastic materials, and even in medicine and many more fields.6 Methanol is normally synthesized from syngas and CO2 with the help of a copper, zinc oxide, and aluminum oxide catalyst.2,7,8 Selective oxidation of the methanol to formaldehyde is then mostly achieved on a silver9–11 or an iron oxide-molybdenum oxide catalyst.12 The question is whether the formation of formaldehyde directly from syngas and CO2 can be observed. Therefore, a better understanding of the elemental steps is necessary. Oxidation of methanol to formaldehyde has been reported in surface science studies on single crystals, for example TiO2,13–23 Cu,24–33 and silver.34,35 Although these experiments provide a lot of information, transformation to industrial catalysis is not always evident, which is known as the materials and complexity gap.28,36 A possible way to overcome this problem is the preparation of complex metal or metal oxide clusters deposited onto well-defined transition metal oxide surfaces.37,38 Preparation of these materials in UHV can be achieved by evaporation and deposition onto the surface as realized for Cu,39 Fe, Cr,40 Rh, Pd, Co, Pt, V,41 and many others.42–51 These co-catalytic systems can lead to improved catalytic activities in comparison to a single material, as support metal interactions can have an impact on the activity of the catalyst.52–55Reactions of methanol on TiO2(110) are already well described in the literature.13,14,56–58 Even when methanol can react on the surface not everything is converted, and a large amount of methanol is desorbed molecularly. Two different temperatures were described for the molecular desorption of methanol: first the desorption from Ti5C centers between 260 K and 310 K and second the desorption of hydrogen-bonded methanol between 180 K and 210 K from bridging oxygen atoms (Obr). A multilayer of methanol would desorb around 140 K.13,14,56–58 The reactivity and product formation depend mostly on the reduction degree of the TiO2(110) single crystal. In our case, all experiments were performed with a slightly (∼3.6% Ti3+/Ti4+) and a highly reduced (∼5.7% Ti3+/Ti4+) single crystal. Methanol adsorbed on the surface reacts with defects in the crystal forming reactive methoxy species that further react to produce methane.13 For this reason, more methane is formed with increasing defect density at higher temperatures (550–670 K). This deoxygenation reaction is further enhanced by the pre-adsorption of oxygen. The second possible reaction path is the partial oxidation of methanol to formaldehyde in the presence of oxygen. Not only can formaldehyde be formed on the slightly and highly reduced titania at high temperatures (550–700 K), but also at a low temperature of 280 K. Formaldehyde formation is also strongly influenced by the reduction degree; increasing the defect density particularly favors low-temperature formaldehyde formation. This low-temperature formation of formaldehyde is attributed to a dioxomethylene-like species, while the formaldehyde and methane formation at elevated temperatures is related to a methoxy precursor as evidenced by Fourier transform infrared reflection adsorption spectroscopy (FT-IRRAS) investigations.13,23 The conversion of methanol on copper single crystals (Cu(110), Cu(111), Cu(100) and Cu(210)) was also well studied. The most important reaction for methanol is the partial oxidation to formaldehyde in the presence of oxygen. On Cu(110) and Cu(111), methanol dissociates near oxygen islands forming methoxy species on the surface. These species further decompose to formaldehyde and hydrogen between 350 and 450 K.24,32 A reaction via methoxy species intermediates has been well discussed in the literature, evidenced by electron energy loss spectroscopy (EELS) and ultraviolet photoelectron spectroscopy (UPS).27,59–61 At the same time, a small amount of CO2 was reported, possibly due to formate decomposition between 440 K and 490 K.26,62 This formate formation was dependent on the adsorption temperature of methanol where adsorption at higher temperatures enhances this reaction product.63 Formation of formaldehyde without oxygen is also possible, but only on rougher single crystals with a higher Lewis basicity. On Cu(210), enough stable methoxy is formed, which further decomposes to formaldehyde.31 In comparison to this, only a recombination of methoxy with hydrogen ad-atoms was found on clean Cu(110).60
Furthermore, the deposition of tungsten oxide clusters ((WO3)n) influences the thermal and photocatalytic methanol oxidation on titania.56,57 These clusters tend to interact strongly with titania by charge transfer to the titania.64 The deposition of tungsten oxide on titania strongly enhances the thermal partial oxidation to formaldehyde. Recently, we demonstrated that charge transfer also occurs when copper clusters are deposited onto rutile TiO2(110) as well as onto a native silicon oxide thin film on a silicon wafer.65 To elucidate whether rough copper clusters, interacting with TiO2(110), enhance methanol conversion, TPRS experiments are presented and are benchmarked against copper clusters on amorphous silica.
2. Experimental
If not stated elsewhere, all results were obtained in a home-built UHV system with a base pressure below 10−10 mbar. The system consists of several connected UHV chambers to secure in vacuo transfer below 10−9 mbar. Adsorption of high-purity molecules was possible by backfilling through a leak valve or via a pinhole doser in all chambers. The gas systems can be equipped with all common gases (used in this work: oxygen (air liquide, 99.999%) and argon (air liquide, 99.999%)) as well as organic molecules, that have a high vapor pressure in vacuum and are stored in special flasks (used in this work: methanol (Sigma-Aldrich, >99.8%)). Before use, the methanol had to be cleaned by several freeze–pump–thaw cycles until no gas bubbles were visible (at least 4–5 cycles).All temperature-programmed reaction spectroscopy (TPRS) experiments were carried out in a chamber equipped with a commercial low energy electron diffraction (LEED) spectrometer (OCI Vacuum Microengineering, BDL800IR-LMX-ISIJ), an argon ion source (Omnivac) and a quadrupole mass spectrometer equipped with a Feulner-cup (Pfeiffer Vacuum, PrismaPro QMG 250 F2, 200 amu). X-ray photoelectron spectroscopy (XPS) experiments were performed in a second UHV chamber equipped with a low energy electron diffraction spectrometer (Specs ER-LEED 100 optic, ER-LEED 1000 A controller), an argon ion source (OmniVac) and a Specs XPS system consisting of a Focus 500 monochromator, an XR50M X-ray source, a Phoibos 150 electron energy analyzer and a 1D-DLD detector (Surface Concept 1D-DLD64_2-150). The XPS was calibrated to the Pt 4f signal position at 71.1 eV with a cleaned Pt(111) single crystal and the whole measurement range was checked afterward to exclude kinetic energy dependent shifts.
The deposition of copper was carried out at room temperature by electron beam evaporation using a FOCUS EFM3 electron beam evaporator equipped with a molybdenum crucible. Pure copper was used (copper pellets, MaTecK, 99.9999%, size 3–5 mm). Further information on the evaporation process and preparation can be found elsewhere.65 The chosen substrates were silicon wafers (Si(111)) with a native oxide layer (Siegert Consulting, 0.5 mm thick) and rutile TiO2(110) single crystals (10 mm × 10 mm, 1 mm thick, surface net GmbH). Before evaporation, the TiO2(110) single crystals were cleaned with several reduction cycles consisting of argon ion bombardment (20 min, 300 K, 1 keV, 6–8 μA, 5.5 × 10−5 mbar argon) and annealing (15 min, 900 K).66 This procedure creates Ti3+ interstitials that even remain after annealing and change the color of the crystal from colorless to blue or black depending on the reduction degree.19 For this work, two different crystals were used: one slightly reduced titania crystal (light blue, around 10–15 reduction cycles, Ti3+/Ti4+ ≈ 3.6%) and one highly reduced crystal (dark blue/black, >100 reduction cycles, Ti3+/Ti4+ ≈ 5.7%). The (110)(1 × 1) surface structure was confirmed via LEED. The silicon wafers were cleaned beforehand with sonication in acetone and isopropanol for at least 15 min to remove a polymer film from the surface. After insertion into the UHV chamber, the wafers were cleaned several times by annealing to 900 K for 20 min. The surface was not sputtered to keep the unreactive native silicon oxide layer intact. The samples were mounted on a home-built sample holder described in earlier publications.13,15,56,64,67 The sample temperature was checked with a K-type thermocouple (CHAL-005, Omega Engineering, 0.75% approx. error) glued inside a small hole at the side of the sample for TiO2 or on top for the wafers with Ceramabond 569 (T-E-Klebetechnik). All TPR spectra were recorded with a temperature ramp of 2 K s−1. If not mentioned elsewhere, one monolayer of methanol was used related to the saturation of Ti5C and Obr centers on the pristine crystal. This corresponds to the deposition for 20 seconds with a pinhole doser and a backing pressure of 10−1 mbar of methanol in the doser compartment for the experiments presented. A possible impact of surface hydroxylation by water adsorption from the chamber background water pressure, prior to methanol or oxygen adsorption, could be excluded, as reported in earlier works.56,57,64 The temperature-programmed X-ray photoelectron spectroscopy measurements were recorded with a ramp of 0.5 K s−1. Further parameters for all XPS measurements can be found in the ESI,† in Tables S1 and S2. All experiments were performed multiple times with freshly prepared copper clusters for reproduction.
3. Results
In the following, we present coverage-dependent TPRS experiments for the conversion of methanol with and without oxygen pre-adsorption at copper clusters on silicon wafers and slightly and highly reduced rutile TiO2(110) single crystals. While methanol reacts at pristine slightly and highly reduced rutile TiO2(110) surfaces, as well as on different copper surfaces,13,14,16,24,31,32,34,56 the amorphous and thin native oxide film on top of Si(111) is not reactive. When methanol is adsorbed onto the clean and oxygen-pre-covered SiO2 surface, only molecular methanol desorption is observed (see Fig. S1 in the ESI†). Copper clusters can be prepared by electron beam evaporation or thermal evaporation exhibiting Volmer–Weber growth.39,68–70 Characterization of these systems prepared by electron beam evaporation has already been performed with X-ray photoelectron spectroscopy, as reported in a recently published report.65 In short, copper clusters prepared on the silicon wafer with a native oxide film, as well as both the slightly and highly reduced titania crystal, exhibited Volmer–Weber growth behavior (cluster/island growth) analyzed by plotting the integrated Cu 2p3/2, Ti 2p3/2 and Si 2p-XPS areas against the nominal thickness of the evaporated copper layer (s–t plot) in accordance with the combined He+ low-energy ion scattering and XPS work of the groups of Diebold, Pan, and Madey.39,40,68 Copper clusters on all three substrates exhibited charge transfer from the copper clusters to the support material depending on their size. In all examples, the ratio of Ti3+/Ti4+ and Si3+/Si4+ rose with increasing coverage, indicating a strong interaction between clusters and the support. For further information on the growth behavior and the structural and electronic properties, we refer readers to a previous publication.653.1 Temperature stability of the as-deposited copper clusters
As the stability of the copper clusters was unknown during TPRS measurements, temperature stability measurements were performed via temperature-programmed XPS. For this purpose, 20.9 Å of copper was deposited onto the silicon wafer with a native oxide film and then heated to 800 K with a ramp of 0.5 K s−1 starting at 120 K. In Fig. 1 the measured Cu 2p3/2 is presented. It is evident from the temperature-resolved plot that the intensity of the Cu 2p3/2 is decreasing at elevated temperatures. Around 500–550 K, the start of the signal intensity decrease is apparent. When the sample was heated to 800 K, the intensity of the Cu 2p3/2 was only two-thirds of the starting intensity. This could also be seen from test TPRS measurements (not shown), where the copper-related products decreased and completely disappeared after some TPRS cycles. XPS measurements performed after the TPRS measurements confirmed the removal of copper, with only residual amounts remaining on the surface. For TiO2, 2.09 Å and 20.9 Å of copper were deposited at room temperature, cooled down to 120 K, and heated to a temperature up to 800 K, while holding the respective temperature for ten minutes (see Fig. S2, ESI†). On TiO2 the same effect is apparent, as the intensity decreases with high temperatures. The total area of both Cu 2p3/2 signals decreases by more than 50% after heating to 800 K. Diebold et al. have shown that annealing copper clusters on non-reduced TiO2(110) already has an effect above 370 K. In their examples, a steady state is reached after some minutes where the area of the Cu 2p is not decreasing anymore.39 This is in contrast to our investigation and could be due to a different reduction degree or differently ordered clusters.65 However, the cause is not fully clear. As the cluster properties influence the TPRS experiments, the temperature limit in these experiments was set to 510 K. In addition, TPRS experiments to 800 K were recorded for the highest coverage (20.9 Å) of copper deposited onto the two different titania single crystals to analyze possible high-temperature desorption species.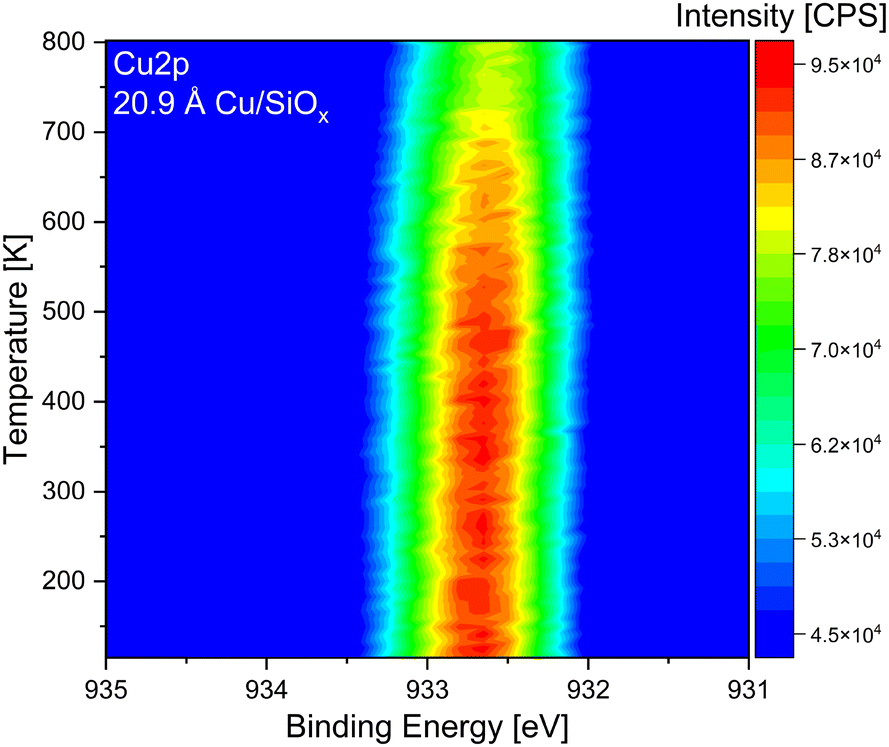 | ||
| Fig. 1 Temperature-programmed X-ray photoelectron Cu 2p3/2 spectra after depositing 20.9 Å of copper onto a silicon wafer with a native oxide film. The sample was first cooled down to 120 K and then slowly heated to 800 K with a heating rate of 0.5 K s−1 while XP-spectra were recorded. All spectra loops were measured with a pass energy of 30, a step size of 0.15 eV and a dwell time of 25 ms. | ||
3.2 Thermal conversion of methanol on Cu/SiOx
First, copper was deposited with different coverages ranging from 0.523 Å to 20.9 Å onto the silicon wafer with a native oxide film. As the surface is not reactive to methanol itself, only the reactivity of the copper clusters themselves is monitored. The conversion of methanol was measured without and with pre-adsorption of 56 L oxygen at T = 115 K. Methanol was adsorbed via pinhole dosing with a pressure of 10−1 mbar methanol in the doser for 20 seconds in the experiments presented. This corresponds to a monolayer of methanol adsorbed on titania and is used for better comparison. For clarity, only two selected coverages are presented, while the other coverages are displayed in the ESI,† Fig. S3 and S4. Fig. 2 shows TPR spectra taken on a wafer with 1.045 Å and one with 20.9 Å of copper deposited for the mass-to-charge ratios m/z = 15 (methanol, methane), m/z = 29 (methanol, formaldehyde), m/z = 30 (methanol, formaldehyde) and m/z = 31 (methanol) (further information on the important fragmentation patterns can be found in the ESI,† Fig. S5). At the lower coverage of 1.045 Å Cu, only molecular desorption of methanol is observed. If compared to the pristine wafer (Fig. S1, ESI†) there are two different desorption peaks for methanol, one at 169 K and one at 190 K. The peak at 190 K is close to the peak observed on the pristine wafer, the peak at 169 K would then correspond to methanol adsorbed on copper or the perimeter of Cu/SiOx. With higher coverages, only one, broader, desorption signal for methanol is apparent, which is shifted to higher desorption temperatures and decreases significantly for the two highest coverages. For 20.9 Å of copper, substantial differences in comparison to 1.045 Å of copper occur. At around T = 189 K and T = 384 K, two new species appear in the spectra, while at T = 201 K and T = 235 K the molecular desorption of methanol is observed. The species at T = 189 K corresponds to carbon monoxide desorbing from the surface of the copper. This species is also observed without methanol, or any other molecules, adsorbed on the surface, as seen in Fig. S6 in the ESI.† The m/z = 28 (main m/z of CO) displays a desorption signal at the same position. m/z = 29 and m/z = 30 would then correspond to isotopes of carbon monoxide like 13C16O for m/z = 29 and 12C18O for m/z = 30, as these are the most common stable isotopes.71 In CO-adsorption experiments, an increase in m/z = 29 and m/z = 30, with the same shape and desorption temperature, is observed simultaneously with the m/z = 28 signal for CO (Fig. S7, ESI†). Apparently, the copper clusters are very sensitive to CO from the background of the chamber, even with pressures at 10−10 mbar. This extra feature is present for all TPR spectra with higher copper coverages and will not be discussed further. The signal at T = 384 K can be correlated with the formation of formaldehyde, as in comparison to the methanol desorption only small increases in m/z = 15 and m/z = 31 are detected. The formation of formaldehyde is observed above a coverage of 10.45 Å of copper. At the same time, signals at m/z = 2, 28, and 44 can be detected (see Fig. 3). While m/z = 28 is apparent in the fragmentation pattern of formaldehyde, m/z = 44 corresponds to carbon dioxide desorption. The signal at m/z = 2 indicates the desorption of hydrogen from the surface. On the clean copper clusters, only the two largest copper coverages (10.45 Å and 20.9 Å) are reactive for the conversion of methanol to formaldehyde, while for smaller coverages only molecular methanol adsorption is apparent. Concurrently with the formaldehyde formation on the large clusters, H2 and CO2 are observed as side products.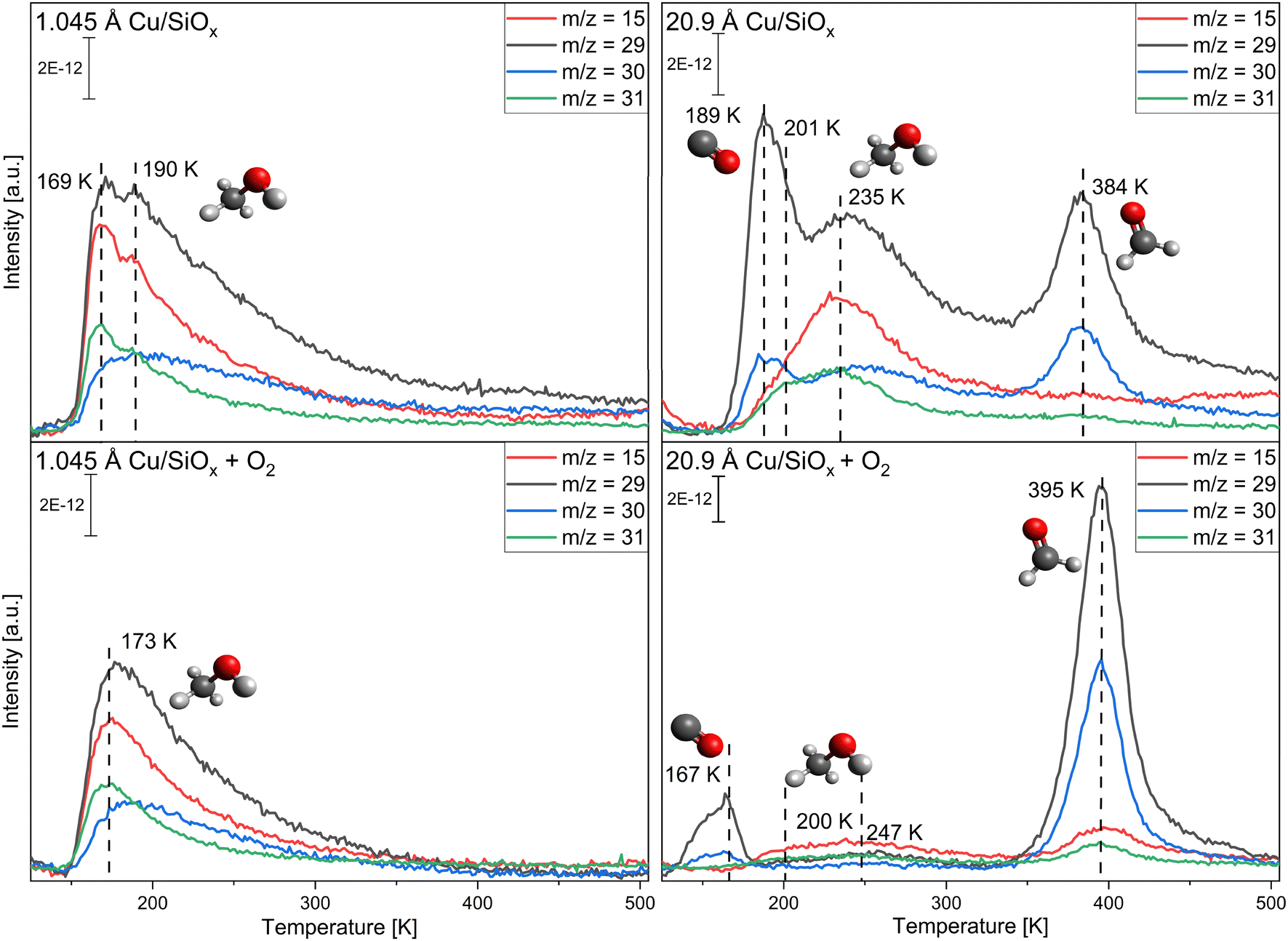 | ||
| Fig. 2 Copper-coverage-dependent TPR spectra of a monolayer methanol (top spectra) adsorbed at T = 115 K and methanol with 56 L of oxygen pre-adsorbed (bottom spectra) on SiOx. Shown are the relevant m/z signals for methanol (m/z = 31), formaldehyde (m/z = 29, 30) and methane (m/z = 15), as well as some contributions from the carbon monoxide isotope desorption (m/z = 29, 30). The desorption spectra are presented for 1.045 Å of copper (left) and 20.9 Å (right) of copper deposited. The heating rate of the sample is 2 K s−1. | ||
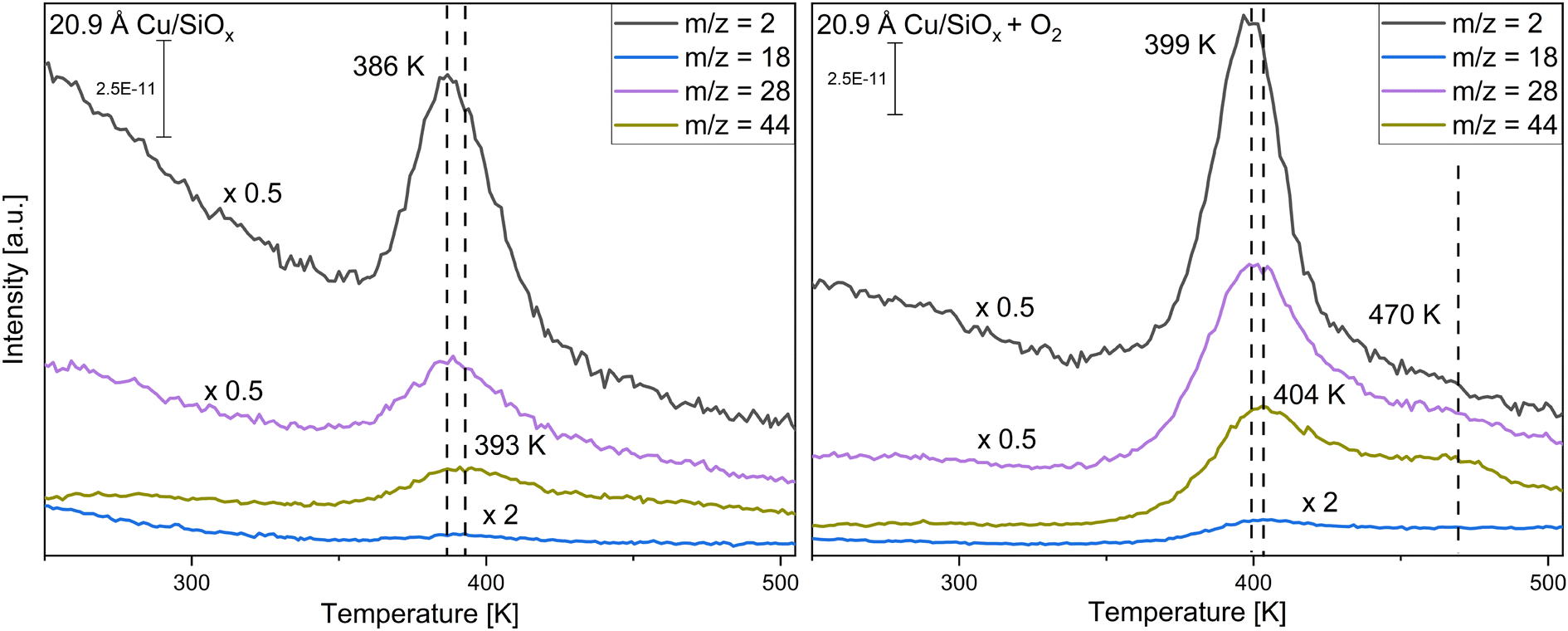 | ||
| Fig. 3 TPR spectra of a monolayer methanol (left spectrum) adsorbed at T = 115 K and methanol with 56 L of oxygen pre-adsorbed (right spectrum) with 20.9 Å of copper being deposited at SiOx. Shown are the responsive m/z for hydrogen (m/z = 2), carbon monoxide (m/z = 28), carbon dioxide (m/z = 44) and water (m/z = 18). For better visibility, the signals of m/z = 2 and m/z = 28 were divided by two and the signal of m/z = 18 was multiplied by two. The heating rate of the sample is 2 K s−1. | ||
In the second step, both surfaces were pre-covered with 56 L of oxygen (Fig. 2). For the small cluster coverage of 1.045 Å copper deposited, only one methanol desorption signal is observed at T = 173 K, even with oxygen. For the larger clusters (10.45 Å and 20.9 Å of copper deposited), the formaldehyde formation is substantially enhanced with oxygen pre-adsorption. At the same time, the desorption signal for methanol nearly disappears at 20.9 Å of copper. The desorption temperature of formaldehyde increases by ∼10 K compared to methanol adsorption on the clean copper clusters. When m/z = 2, 18, 28, and 44 are measured, the intensity of these signals also increases and the signals shift nearly by the same amount as for formaldehyde. In contrast to the reaction without oxygen, water (m/z = 18) is desorbing. A new CO2 species solely at m/z = 44 is observed at 470 K with a desorption peak shifted by ∼70 K to higher desorption temperatures in comparison to the formaldehyde desorption.
3.3 Defect-dependent thermal conversion of methanol on rutile TiO2(110)
In Fig. 4 the TPR spectra for the monolayer adsorption of methanol with and without oxygen pre-adsorption for the slightly reduced (LR) TiO2(110) crystal are presented. As discussed above, TPR spectra were only recorded up to 500 K, so possible methane and formaldehyde species >500 K cannot be observed in these experiments. As before, only the adsorption at 1.045 Å and 20.9 Å of copper are discussed, while the other coverages are presented in the ESI,† Fig. S8 and S9. For the low coverage of 1.045 Å deposited copper, only two signals are observed at T = 220 K and T = 306 K, corresponding to the desorption of molecular methanol. Temperature changes in comparison to the pristine crystal are mostly due to the deposited copper. No extra reaction is observed in the low-temperature regime up to 500 K. This does not change until 20.9 Å of copper is deposited onto the titania. At this copper coverage, mainly four signals are observed: at T = 183 K the CO desorption from residual gas adsorption, at T = 234 K and T = 306 K, the molecular methanol desorption, and at T = 409 K the formaldehyde desorption. The amount of methanol desorbing from the surface decreases with higher copper coverages, and at 20.9 Å of copper, only a fraction of the amount desorbing at the lowest coverage of 0.523 Å of copper is detected. Concomitant with the formaldehyde formation, H2, CO2, and CO desorption are observed (see Fig. S10 in the ESI†). In contrast to Cu/SiOx, formate is apparent at T = 480 K without oxygen pre-adsorption. Only with the highest copper coverage (20.9 Å) the direct dehydrogenation to formaldehyde is observed, and formate formation is evident from the desorbing CO2 decomposition product.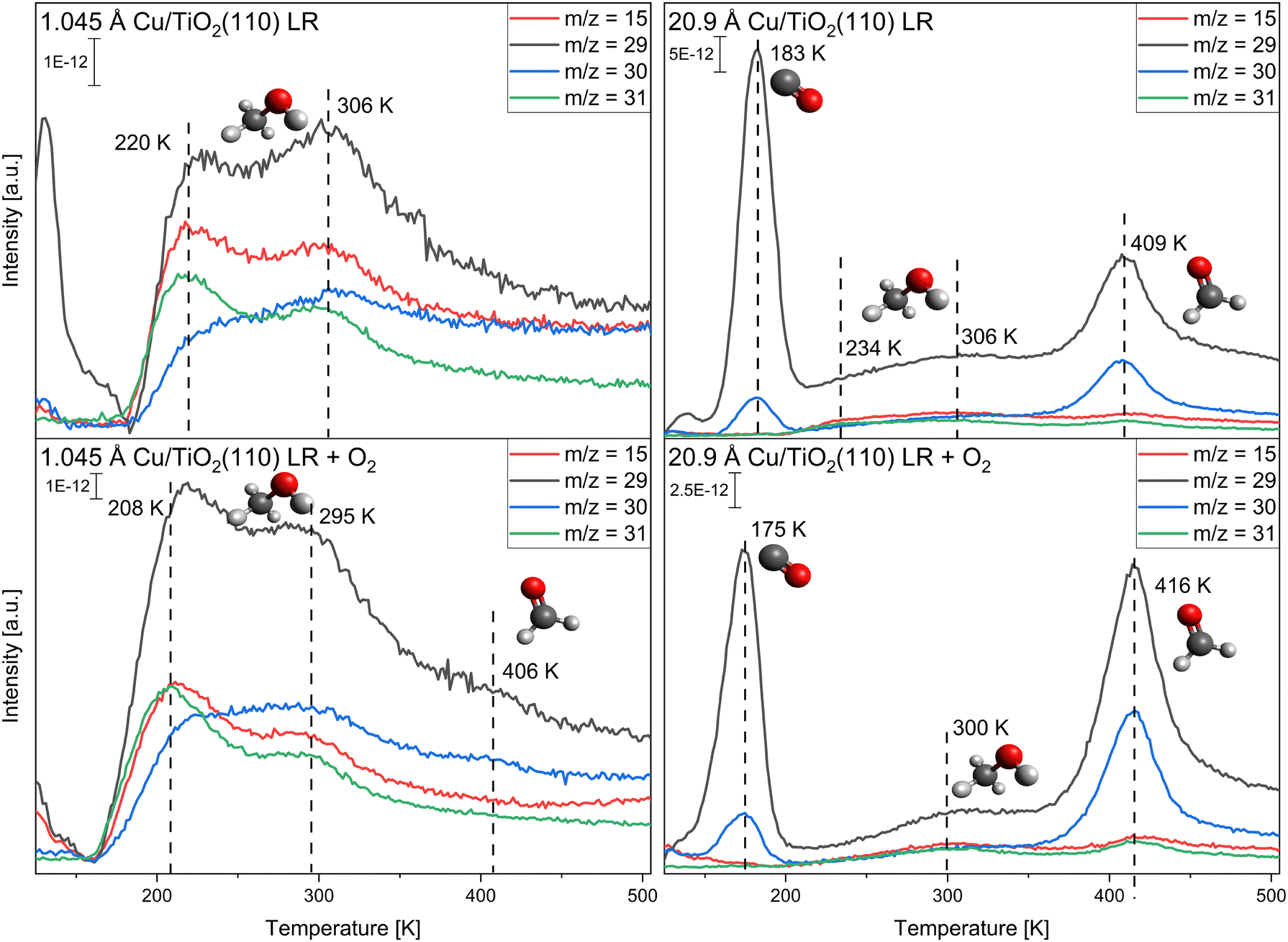 | ||
| Fig. 4 Copper-coverage-dependent TPR spectra of a monolayer methanol (top spectra) adsorbed at T = 115 K and methanol with 56 L of oxygen pre-adsorbed (bottom spectra) at the slightly reduced TiO2(110) crystal. Shown are the relevant m/z signals for methanol (m/z = 31), formaldehyde (m/z = 29, 30), and methane (m/z = 15), as well as some contributions from the carbon monoxide isotope desorption (m/z = 29, 30). The desorption spectra are presented for 1.045 Å of copper (left) and 20.9 Å (right) of copper deposited. The heating rate of the sample is 2 K s−1. | ||
When oxygen is pre-adsorbed on the surface, the amount of methanol desorbing increases for all copper coverages. This could be due to an enhanced sticking probability of methanol with pre-adsorbed oxygen, as the resulting methanol desorption signals should be a combination of both methanol on titania and copper.32 Around T = 406 K weak signals at m/z = 29 and m/z = 30 are apparent for 1.045 Å of copper deposited. This formaldehyde formation is too low to be quantified. With increasing copper coverage, this formaldehyde formation is increasing. At the largest cluster size (20.9 Å of copper deposited) the largest amount of formaldehyde desorbing at T = 416 K is measured, which is substantially enhanced in comparison to the clean copper. Only one, broad and weak methanol desorption signal is observed at 300 K. At the same time H2, CO2, and CO desorption are detected with a small amount of water desorbing (Fig. S10, ESI†). The desorption of CO2 at T = 475 K is concomitant to a possible formate formation, as observed on copper single crystals.26,62
When a monolayer of methanol was adsorbed at copper deposited onto highly reduced TiO2(110), a similar behavior was observed. The spectra are presented in the ESI,† Fig. S11 and S12. Without pre-adsorption of oxygen, only methanol desorption is apparent for clusters with 20.9 Å of copper deposited. At 20.9 Å of copper, a small signal at T = 395 K indicates the desorption of formaldehyde. This amount is insignificant compared to the same coverage on the slightly reduced crystal. Furthermore, no formation of CO2, CO, H2, and formate is observed. In all spectra, a rising m/z = 15 signal is detected, which is cut off at 500 K.
After oxygen is pre-adsorbed onto the surface, formaldehyde is apparent for the two highest copper coverages (10.45 Å and 20.9 Å). This is in contrast to the formation of formaldehyde on the slightly reduced crystal, where formaldehyde was already observed at 1.045 Å of copper deposited. Concurrently CO2, H2, H2O, and CO formation is detected, with CO2 formation at higher temperatures attributed tentatively to formate decomposition (Fig. S13, ESI†). At around T = 460–467 K a second formaldehyde species is observed, which is apparent as a shoulder in the main desorption signal. This shoulder is better observable when only half a monolayer of methanol is adsorbed on the surface (see S14, ESI†). With 10.45 Å of copper, the intensity of the main signal and shoulder are nearly the same, while with 20.9 Å of copper, the main signal increases at 400 K, while the shoulder is stagnant.
For a comparison of Cu/TiO2(110) with the pristine single crystals, the slightly and highly reduced single crystals with 20.9 Å of copper deposited were heated to 800 K in the TPRS measurements. Fig. 5 presents the evolution of the m/z = 15 and m/z = 29 signals with increasing temperature for both the pristine TiO2(110) and Cu/TiO2(110) when a monolayer of methanol with 56 L of oxygen was adsorbed. The spectra, with oxygen pre-adsorbed, were measured directly after the last experiment of heating to 500 K. The corresponding spectra without oxygen pre-adsorption (Fig. S15, ESI†) were measured directly after the first spectra after heating to 800 K. With that, annealing effects could influence these experiments. Since only differences in signal intensities were found in comparison to the unannealed samples, the differences to the pristine single crystal can still be discussed. The complete spectra with the whole m/z range are displayed in the ESI,† in Fig. S16 and S17. For the pristine TiO2(110) single crystals, the desorption of methanol from Ti5C centers is apparent at T = 210 K, while the desorption from Obr is at T = 280 K for the slightly and T = 310 K for the highly reduced TiO2(110). With 20.9 Å of copper deposited onto these substrates, the desorption species of methanol at 210 K disappears, while only small amounts of methanol desorb above 300 K. The drop in methanol desorption is more substantial when the monolayer adsorption of methanol on the clean pristine TiO2(110) is compared with the clean 20.9 Å Cu/TiO2(110) systems, especially at the slightly reduced TiO2(110) (Fig. S15, ESI†). Methane desorption is observed at T = 670 K on the pristine slightly reduced TiO2(110), while the amount desorbing from the highly reduced TiO2(110) is larger with two different species at T = 555 K and T = 600 K. After the deposition of the copper, the amount of methane desorbing decreases, with all desorption signals shifting to higher temperatures. At T = 663 K, formaldehyde is observed on the pristine slightly reduced TiO2(110). On the other hand, low-temperature formaldehyde formation (T = 275 K) is apparent on the pristine highly reduced TiO2(110). With copper deposited onto the titania, the high-temperature formaldehyde signal at T = 663 K is still apparent, but smaller in comparison to the pristine titania. At the same time, a pronounced new formaldehyde desorption signal at T = 421 K is detected. For copper on the highly reduced TiO2(110), a similar trend is apparent. The low-temperature formaldehyde signal at T = 275 K disappears, while a new species at 397 K appears.
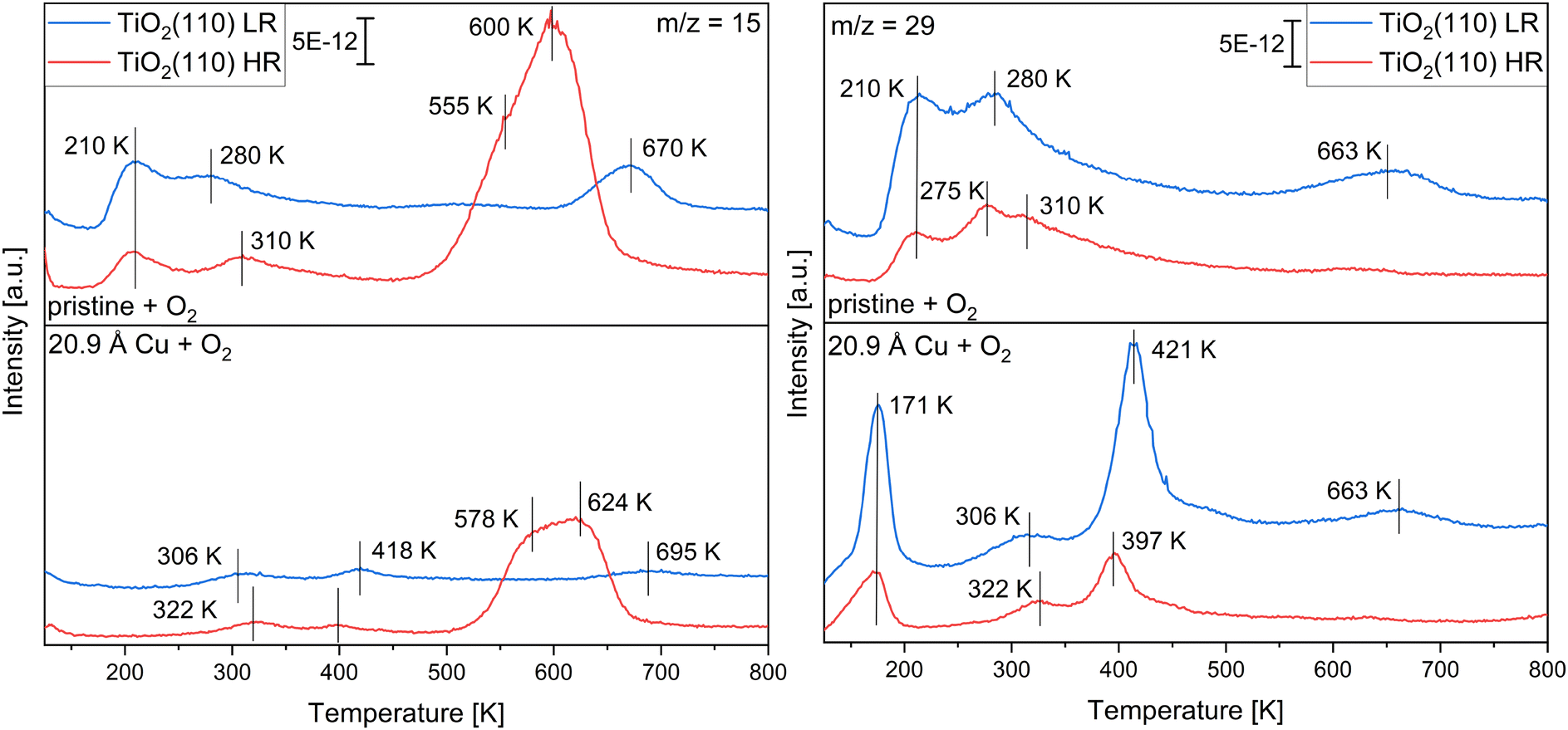 | ||
| Fig. 5 TPR spectra of a monolayer methanol adsorbed at T = 115 K and methanol with 56 L of oxygen pre-adsorbed at the pristine slightly (LR) and highly (HR) reduced TiO2(110) single crystals (top spectra) and 20.9 Å of copper deposited onto the slightly and highly reduced single crystals (bottom spectra). Displayed are the m/z = 15 and m/z = 29 signals for the experiments reaching 800 K at a rate of 2 K s−1. | ||
The desorption temperatures for the formation of formaldehyde on the three substrates with and without oxygen pre-adsorption are presented in Table 1 for a coverage of 20.9 Å of copper. A significant formation of formaldehyde without oxygen is only observed for copper on the silicon wafer and the slightly reduced titania crystal. When oxygen is pre-adsorbed onto the surface, formaldehyde is apparent in considerable amounts at all three samples with the desorption temperature shifted to higher temperatures. The amount of formaldehyde and methanol desorbing from the surface was tracked by the area of the signal at m/z = 29 for formaldehyde and m/z = 31 for methanol (Fig. 6). The formaldehyde desorption is increasing on all samples with higher copper coverages, while oxygen pre-adsorption is further increasing the formation of formaldehyde. The amount of methanol desorbing from the surface decreases with higher coverages and with higher formaldehyde formation.
| Methanol | Methanol + O2 | |
|---|---|---|
| SiOx | 384 K | 395 K |
| TiO2(110) LR | 409 K | 416 K |
| TiO2(110) HR | 395 K | 390 K, 448 K |
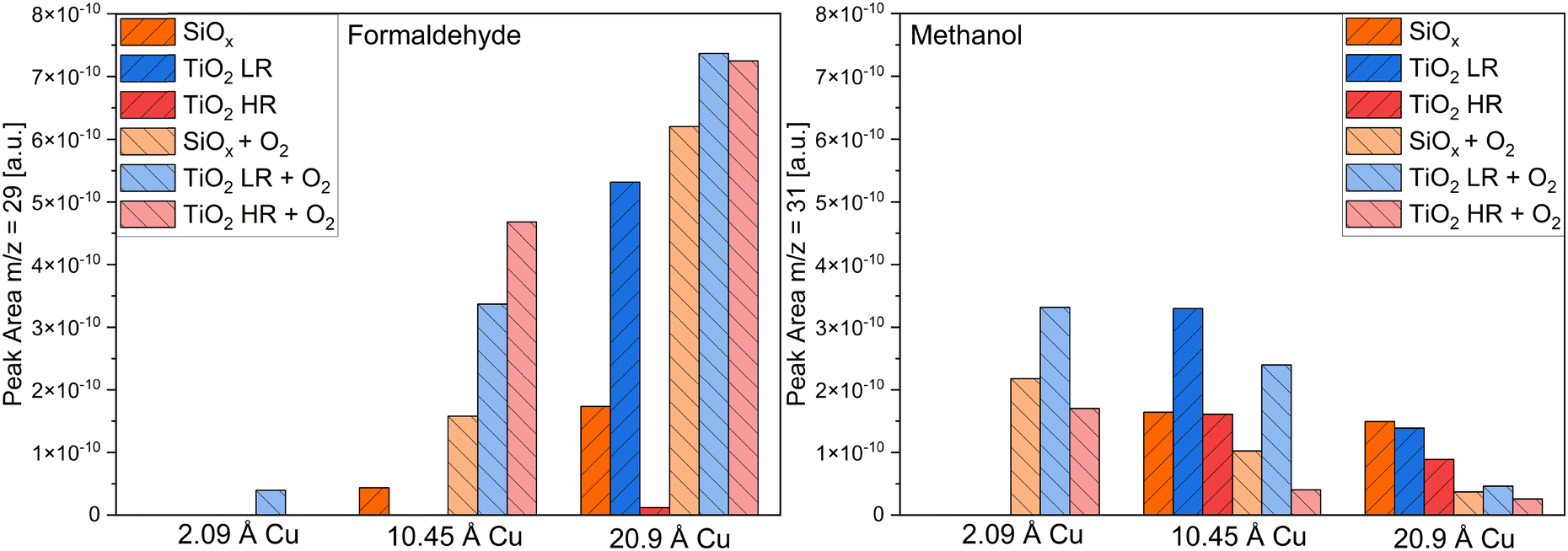 | ||
| Fig. 6 Area of the observed peaks for the formaldehyde formation at m/z = 29 and the methanol formation at m/z = 31 when copper was deposited on the silicon wafer with a native oxide film (orange), on the slightly reduced TiO2(110) (LR) single crystal (blue) and the highly reduced TiO2(110) (HR) single crystal (red). The signal areas were taken from the spectra shown in this work and the ESI.† | ||
4. Discussion
In the following, we want to discuss the trends for the formation of formaldehyde and possible side products on the copper-covered substrates.For copper on a silica surface, the formation of formaldehyde on copper clusters was observed as the sole product. The resulting spectra are in good accordance with TPRS data on a Cu(210) single crystal31 and differ from Cu(111), Cu(110), and Cu(100).26,32 Copper is a weak Lewis base and the interaction of molecules occurs via hydrogen bonding, as described by Chen and Masel.31 Due to this, methoxy is not stable on the clean copper surface and the dehydrogenation to formaldehyde is not favored. A rougher surface would lower the work function and with that the basicity of the surface. Because of this, direct dehydrogenation on rougher copper surfaces is possible. Since supported clusters are much rougher than a clean-cut single crystal, direct dehydrogenation is theoretically possible on the clusters. Even if this is possible in theory, only the two highest coverages (10.45 Å and 20.9 Å) have shown reactivity. It was suggested by Varazo et al.72 that copper clusters, in comparison to copper single crystals, are more reactive owing to a greater amount of step and edge defects and with that more active sites.72–74 The greater activity can then be related to the facilitation of the O–H scission and concomitant stable methoxy formation. In our case, in principle it is possible that smaller clusters do not possess as many defects to stabilize the methoxy formation, and with that, no stable methoxy and formaldehyde are formed. A more likely cause may be related to a possible influence of the different electronic properties of small clusters in comparison to large clusters. As observed in our previous publication,65 small clusters exhibit a more nonmetallic electronic character as apparent from Auger signals, which are reminiscent of copper oxide Auger signals. At higher coverages between 6.27 Å and 10.45 Å, a change to a predominantly bulk-like metallic character was observed. As this change from a more nonmetallic to a metallic electronic structure is observed at a similar copper coverage as the start of formaldehyde formation occurs, we suggest a possible connection. For better insight into the size-dependent reactivity, further studies on the structure, crystallinity, and electronic properties are important. The same effect is evident at oxygen-pre-covered copper clusters. No reaction was observed for clusters below a coverage of 10.45 Å deposited, while formaldehyde formation strongly increased for the two highest copper coverages. The formation of formaldehyde was accompanied by the evolution of hydrogen and water (with oxygen pre-adsorption), as hydrogen gets abstracted from the methoxy and either recombines with adsorbed hydrogen or oxygen on the surface. Simultaneously, the formation of CO2 at T = 470 K indicates the formation and decomposition of formate, as observed on copper single crystals.26,27,32,62 Without oxygen pre-adsorption, this shoulder is either nonexistent or too small to be visible as less formaldehyde is formed to react to formate, which decomposes to CO2. No new reaction products correlated with the reduced silica species were observed. This indicates that either the Si3+ is not reactive or pinned to the copper/silica interface.
The group of Bowker and Madix suggested the following mechanism for the reaction of methanol to formaldehyde:32,60
| CH3OH(g) → CH3OH(ad) | (1) |
| CH3OH(ad) + O(ad) → CH3O(ad) + OH(ad) | (2) |
| CH3O(ad) → H2CO(ad) + H(ad) | (3) |
| H2CO(ad) → H2CO(g) | (4) |
| 2H(ad) → H2(g) | (5) |
| H(ad) + OH(ad) → H2O(ad) | (6) |
For the direct dehydrogenation only H(ad) is produced in (2) as no oxygen is present in the reaction. The other steps are the same as proposed by Chen and Masel.31 For the formation of CO2 through formate, the following mechanism was suggested from the groups of Bowker, Madix and Mariotti:63,75,76
| H2CO(ad) + O(ad) → H2COO(ad) | (7) |
| H2COO(ad) → HCOO(ad) + H(ad) | (8) |
| HCOO(ad) + H(ad) → CO2(g) + H2(g) | (9) |
The mechanism for the formation of formate is still under discussion and does not implement the formation in the absence of adsorbed oxygen. Further studies are needed for a concrete mechanism in this case.
For the conversion of methanol on Cu/TiO2(110), the deoxygenation to methane and other hydrocarbons competes with the formation of formaldehyde through the methoxy intermediate. On the slightly reduced TiO2(110) crystal, only the highest copper coverage (20.9 Å) has shown reactivity for the direct dehydrogenation of methanol to formaldehyde at T = 409 K at the copper surface. One reason for the different behavior in comparison to Cu/SiOx could stem from the different consumption of methoxy on the surface. While methoxy formed on copper should react to form formaldehyde, as seen on Cu/SiOx, the possibility of methoxy diffusing to titania and reacting to form methane is likewise possible. With larger cluster sizes, more active titania centers are covered, while the copper surface is more defect-rich and active, hence, formaldehyde can form, as seen when 20.9 Å of copper is deposited. At the same time, the formation of formate seems to be more preferred on Cu/TiO2(110) than on Cu/SiOx. With oxygen pre-adsorption, the formation of formaldehyde, corresponding to the reaction on copper, was already apparent at T = 406 K when 1.045 Å of copper was deposited. The amount of formaldehyde desorbing from the surface increased with increasing copper coverage. With the O–H scission and concomitant methoxy formation on the surface, enough methoxy is available on the surface, that both the formation of formaldehyde on copper and the formation of methane on titania is enabled. In comparison to Cu/SiOx, more methoxy is likely available, as methoxy can also form on the titania surface. At the same time, the influence of the charge transfer cannot be neglected. It appears that the copper is more reactive on the titania surface with oxygen pre-adsorbed, than on silica.
For the highly reduced TiO2(110), no significant formaldehyde formation was observed in the absence of oxygen, even at the largest coverage (20.9 Å). Only large amounts of methane were observed. This confirms that the formation of methane through the methoxy intermediate is more favorable than the formation of formaldehyde. If the methane evolution with 20.9 Å of copper on the highly reduced TiO2(110) (T = 568 K and T = 637 K) is compared to the pristine single crystal (T = 528 K and T = 581 K), a shift between 40 K and 60 K to higher temperatures is apparent. This shift could be induced by the interaction of the copper clusters with the titania surface, changing the electron density and the binding of the methoxy at the surface. On the oxygen pre-covered surface, formaldehyde was observed for the two largest copper coverages (10.45 Å and 20.9 Å) in similar amounts to the slightly reduced TiO2(110) at T = 390 K. One possibility for the different behavior of copper on the two different defect-rich surfaces, could stem from the varying interaction between copper and the substrate. It appears that formaldehyde formation is favored at smaller copper coverages due to the stronger interaction of the slightly reduced titania with the copper. Another influence should originate from Ti3+ diffusion at elevated temperatures. Within our experiments, it was not possible to discern any interaction of Ti3+ diffusing to the surface and interacting with methoxy or methanol, as a cause for a change in reactivity. As more Ti3+ is available at the highly reduced TiO2(110), this could result in different catalytic behaviors of both systems.77 For this, further methods like FT-IRRAS and scanning tunneling microscopy (STM) would be needed. The methane formation is enhanced with oxygen pre-adsorption. Both the desorption signals at T = 624 K and T = 578 K (20.9 Å Cu/TiO2) are shifted to higher temperatures compared to the pristine highly reduced TiO2(110) (T = 555 K and T = 600 K). At the same time, a second formaldehyde species at T = 467 K, 50–60 K higher than the main formaldehyde signal, was observed, when methanol was adsorbed with oxygen pre-adsorption. This second formaldehyde species could also emanate from adsorption centers at the interfaces of copper and titania, as the signal decreases with larger copper coverages. With larger methanol coverages, the additional signal stagnates in comparison to the main formaldehyde desorption signal at T = 390 K, indicating an earlier saturation of binding sites. As the additional signal was only observed on the highly reduced TiO2(110), intrinsic defects in the titania could influence the formation of this formaldehyde species that is strongly bound to the surface. When the reaction of methanol at the oxygen-pre-covered surface for copper on the highly reduced TiO2(110) is compared with the pristine highly reduced TiO2(110), the low-temperature formaldehyde species at T = 275 K is missing. Complete quenching of the reaction from methanol through the dioxomethylene-like intermediate was already apparent when 0.523 Å of copper was deposited. We suggest that the copper is hindering the formation of the dioxomethylene-like intermediate responsible for this reaction.
Varazo et al. observed small amounts of formaldehyde at lower coverages (2–4 ML) of copper, while at higher coverages (8–12 ML) no formaldehyde was apparent on clean copper clusters deposited onto oxidized TiO2(110).72 The formation of formaldehyde at lower coverages was attributed to the diffusion of oxygen from the titania to copper and was only possible when large fractions of the substrate were uncovered. This is in contrast to our findings, where only at large copper coverages formaldehyde is produced. We suggest that rather than a partial oxidation with oxide from the titania, a direct dehydrogenation reaction is more plausible for our system. The reason for the different behavior is not clear, but XPS experiments have not shown changes in the oxygen signal during the reaction.
Comparing all three substrates, the pristine copper clusters on the silicon wafer with a native oxide film exhibited an earlier direct dehydrogenation of methanol to formaldehyde (10.45 Å Cu), while at 20.9 Å Cu, the slightly reduced Cu/TiO2(110) was the more efficient catalyst, although the desorption temperature was 35 K higher. For the partial oxidation of methanol to formaldehyde, all three copper-covered substrates were comparably efficient for the highest copper coverage (20.9 Å). If the selectivity of the methanol conversion to formaldehyde is compared for all three substrates, the silica substrates are the favored support, as methane was formed on TiO2(110) and especially on the highly reduced TiO2(110), where methane was the favored product and not formaldehyde.
5. Conclusions
In this work, we have shown a systematic TPRS study of the reaction of methanol on different-sized copper clusters deposited onto two oxidic supports (silica and titania) via electron beam evaporation in UHV. The coverage-dependent reactivity of these catalytic systems toward the direct dehydrogenation and partial oxidation was the focus of this research.The direct dehydrogenation toward formaldehyde is possible on copper clusters, in contrast to single crystals, as the surface is more defect-rich and rougher, and with that, most likely exhibits an increased Lewis basicity. At copper clusters deposited onto the unreactive and inert native oxide surface of a silicon wafer, a good conversion to formaldehyde between T = 380 K and T = 400 K for larger copper coverages (10.45 Å and 20.9 Å) was observed, while the reactivity was further enhanced with pre-adsorption of oxygen, owing to the strengthening of the methoxy bonding on the surface. At the same time, smaller clusters are not reactive.
Direct dehydrogenation to formaldehyde was also observed for the largest copper coverage (20.9 Å) on the slightly reduced titania crystal, while no significant dehydrogenation was seen on the highly reduced titania crystal. As a further reaction, deoxygenation to methane is also enabled through a possible methoxy intermediate. Both reactions compete, with methane formation being more favorable with higher defect densities of the support. On the oxygen-pre-covered surface, a reaction to formaldehyde occurs at both the copper-covered slightly and highly reduced TiO2(110). For the slightly reduced TiO2(110), formaldehyde is observed at smaller copper coverages (1.045 Å) compared to the highly reduced TiO2(110) (10.45 Å of copper), possibly influenced by the larger charge transfer from the copper to the slightly reduced TiO2(110).65 A possible low-temperature formaldehyde formation observed for pristine TiO2(110), was in the meantime completely quenched after copper deposition on the highly reduced TiO2(110), indicating the absence of the known dioxomethylene-like intermediate for the methanol adsorption.
This study provides important insights into the oxidation of methanol to formaldehyde at moderate temperatures compared to industrial processes with silver catalysts. We demonstrated that copper clusters deposited on silica are highly reactive in the direct dehydrogenation and especially the partial oxidation of methanol. Compared to this, Cu/TiO2(110) exhibited a worse selectivity, as methane appeared as a side product and charge transfer influenced the reactivity of the copper clusters. The insights gained in this study can be applied to further catalytic research in steady-state flow experiments and the direct conversion of CO2 to formaldehyde.
Author contributions
Maximilian Grebien: investigation, formal analysis, visualization, writing – original draft. Katharina Al-Shamery: conceptualization, project administration, supervision, funding acquisition, writing – review & editing.Data availability
Data for this article, including TPRS and XPS are available at Zenodo at https://zenodo.org/doi/10.5281/zenodo.13767634.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Ralf Nustedt is acknowledged for the technical support and Lars Mohrhusen, Nils Brinkmann, and Jessica Kräuter for the help with installing the XPS system. The DFG is thanked for their funding of this XPS system [INST 184/209-1 FUGG]. Lars Mohrhusen is also thanked for his support and his titania knowledge.Notes and references
- M. Svanberg, J. Ellis, J. Lundgren and I. Landälv, Renewable Sustainable Energy Rev., 2018, 94, 1217–1228 CrossRef CAS.
- F. Dalena, A. Senatore, A. Marino, A. Gordano, M. Basile and A. Basile, Methanol Production and Applications: An Overview, Elsevier B.V., 2018 Search PubMed.
- K. A. Ali, A. Z. Abdullah and A. R. Mohamed, Renewable Sustainable Energy Rev., 2015, 44, 508–518 CrossRef CAS.
- S. N. Khadzhiev, N. V. Kolesnichenko and N. N. Ezhova, Pet. Chem., 2016, 56, 77–95 CrossRef CAS.
- Methanol Institute, https://www.methanol.org/, (accessed 29 February 2024).
- N. T. Subasi, Biochemical Toxicology – Heavy Metals and Nanomaterials, IntechOpen, 2020, vol. i, p. 13 Search PubMed.
- K. Klier, Adv. Catal., 1982, 31, 243–313 CAS.
- J. Schittkowski, H. Ruland, D. Laudenschleger, K. Girod, K. Kähler, S. Kaluza, M. Muhler and R. Schlögl, Chem. Ing. Tech., 2018, 90, 1419–1429 CrossRef CAS.
- G. J. Millar and M. Collins, Ind. Eng. Chem. Res., 2017, 56, 9247–9265 CrossRef CAS.
- G. J. Millar, M. L. Nelson and P. J. R. Uwins, J. Catal., 1997, 169, 143–156 CrossRef CAS.
- M. D. Thomas, J. Am. Chem. Soc., 1920, 42, 867–882 CrossRef CAS.
- D. S. Lafyatis, G. Creten and G. F. Froment, Appl. Catal., A, 1994, 120, 85–103 CrossRef CAS.
- M. Osmić, L. Mohrhusen and K. Al-Shamery, J. Phys. Chem. C, 2019, 123, 7615–7626 CrossRef.
- E. Farfan-Arribas and R. J. Madix, J. Phys. Chem. B, 2002, 106, 10680–10692 CrossRef CAS.
- J. Kräuter, L. Mohrhusen, T. Thiedemann, M. Willms and K. Al-Shamery, Z. Naturforsch., A: Phys. Sci., 2019, 74, 697–707 CrossRef.
- M. A. Henderson, S. Otero-Tapia and M. E. Castro, Surf. Sci., 1998, 412–413, 252–272 CrossRef.
- M. Shen, D. P. Acharya, Z. Dohnálek and M. A. Henderson, J. Phys. Chem. C, 2012, 116, 25465–25469 CrossRef CAS.
- M. Shen and M. A. Henderson, J. Phys. Chem. C, 2012, 116, 18788–18795 CrossRef CAS.
- L. Mohrhusen, J. Kräuter, M. Willms and K. Al-Shamery, J. Phys. Chem. C, 2019, 123, 20434–20442 CrossRef CAS.
- J. Kräuter, L. Mohrhusen, F. Waidhas, O. Brummel, J. Libuda and K. Al-Shamery, J. Phys. Chem. C, 2021, 125, 3355–3367 CrossRef.
- J. Kräuter, E. Franz, F. Waidhas, O. Brummel, J. Libuda and K. Al-Shamery, J. Catal., 2022, 406, 134–144 CrossRef.
- P. M. Clawin, C. M. Friend and K. Al-Shamery, Chem. – Eur. J., 2014, 20, 7665–7669 CrossRef CAS PubMed.
- L. Mohrhusen and K. Al-Shamery, Catal. Lett., 2023, 153, 321–337 CrossRef CAS.
- J. N. Russell, S. M. Gates and J. T. Yates, Surf. Sci., 1985, 163, 516–540 CrossRef CAS.
- J. Greeley and M. Mavrikakis, J. Catal., 2002, 208, 291–300 CrossRef CAS.
- A. F. Carley, A. W. Owens, M. K. Rajumon, M. W. Roberts and S. D. Jackson, Catal. Lett., 1996, 37, 79–87 CrossRef CAS.
- B. A. Sexton, A. E. Hughes and N. R. Avery, Surf. Sci., 1985, 155, 366–386 CrossRef CAS.
- M. Bowker and K. C. Waugh, Surf. Sci., 2016, 650, 93–102 CrossRef CAS.
- R. Ryberg, J. Electron Spectrosc. Relat. Phenom., 1983, 29, 59–60 CrossRef CAS.
- S. Pöllmann, A. Bayer, C. Ammon and H.-P. P. Steinrück, Z. Phys. Chem., 2004, 218, 957–971 CrossRef.
- A. K. Chen and R. Masel, Surf. Sci., 1995, 343, 17–23 CrossRef CAS.
- S. M. Francis, F. M. Leibsle, S. Haq, N. Xiang and M. Bowker, Surf. Sci., 1994, 315, 284–292 CrossRef CAS.
- S. Sakong, C. Sendner and A. Groß, THEOCHEM, 2006, 771, 117–122 CrossRef CAS.
- I. E. Wachs and R. J. Madix, Surf. Sci., 1978, 76, 531–558 CrossRef CAS.
- A. Montoya and B. S. Haynes, J. Phys. Chem. C, 2007, 111, 9867–9876 CrossRef CAS.
- D. Esposito, Nat. Catal., 2018, 1, 807–808 CrossRef.
- H.-J. Freund, M. Bäumer and H. Kuhlenbeck, Adv. Catal., 2000, 45, 333–384 CAS.
- Z. Luo, A. W. Castleman and S. N. Khanna, Chem. Rev., 2016, 116, 14456–14492 CrossRef CAS PubMed.
- U. Diebold, J.-M. Pan and T. E. Madey, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 3868–3876 CrossRef CAS PubMed.
- U. Diebold, J. M. Pan and T. E. Madey, Surf. Sci., 1993, 287–288, 896–900 CrossRef CAS.
- M. Bäumer and H.-J. Freund, Prog. Surf. Sci., 1999, 61, 127–198 CrossRef.
- K. H. Ernst, A. Ludviksson, R. Zhang, J. Yoshihara and C. T. Campbell, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 13782–13796 CrossRef CAS PubMed.
- C. T. Campbell, J. Chem. Soc., Faraday Trans., 1996, 92, 1435–1445 RSC.
- S. C. Parker, A. W. Grant, V. A. Bondzie and C. T. Campbell, Surf. Sci., 1999, 441, 10–20 CrossRef CAS.
- H.-P. Steinrück, F. Pesty, L. Zhang and T. E. Madey, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 2427–2439 CrossRef PubMed.
- F. Pesty, H.-P. Steinrück and T. E. Madey, Surf. Sci., 1995, 339, 83–95 CrossRef CAS.
- P. J. Møller and J. Nerlov, Surf. Sci., 1994, 307–309, 591–596 CrossRef.
- L. V. Koplitz, O. Dulub and U. Diebold, J. Phys. Chem. B, 2003, 107, 10583–10590 CrossRef CAS.
- O. Dulub, M. Batzill and U. Diebold, Top. Catal., 2005, 36, 65–76 CrossRef CAS.
- Q. Fu and T. Wagner, Surf. Sci. Rep., 2007, 62, 431–498 CrossRef CAS.
- A. Winkler, H. Borchert and K. Al-Shamery, Surf. Sci., 2006, 600, 3036–3044 CrossRef CAS.
- S. J. Tauster, S. C. Fung, R. T. K. Baker and J. A. Horsley, Science, 1981, 211, 1121–1125 CrossRef CAS PubMed.
- G. M. Schwab, Adv. Catal., 1979, 27, 1–22 Search PubMed.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- L. Mohrhusen and K. Al-Shamery, Phys. Chem. Chem. Phys., 2021, 23, 12137–12147 RSC.
- L. Mohrhusen, J. Kräuter and K. Al-Shamery, Phys. Chem. Chem. Phys., 2021, 23, 12148–12157 RSC.
- M. A. Henderson, S. Otero-Tapia and M. E. Castro, Faraday Discuss., 1999, 114, 313–329 RSC.
- B. A. Sexton, Surf. Sci., 1979, 88, 299–318 CrossRef CAS.
- M. Bowker and R. J. Madix, Surf. Sci., 1980, 95, 190–206 CrossRef CAS.
- I. E. Wachs and R. J. Madix, J. Catal., 1978, 53, 208–227 CrossRef CAS.
- C. Ammon, A. Bayer, G. Held, B. Richter, T. Schmidt and H. P. Steinrück, Surf. Sci., 2002, 507–510, 845–850 CrossRef CAS.
- P. R. Davies and G. G. Mariotti, J. Phys. Chem., 1996, 100, 19975–19980 CrossRef CAS.
- L. Mohrhusen, M. Grebien and K. Al-Shamery, J. Phys. Chem. C, 2020, 124, 23661–23673 CrossRef CAS.
- M. Grebien and K. Al-Shamery, Surf. Sci., 2024, 749, 122547 CrossRef CAS.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- U. Leist, A. Winkler, J. Büssow and K. Al-Shamery, Rev. Sci. Instrum., 2003, 74, 4772–4778 CrossRef CAS.
- U. Diebold, J. M. Pan and T. E. Madey, Surf. Sci., 1995, 331–333, 845–854 CrossRef CAS.
- S. Peters, S. Peredkov, N. Ferretti, A. Savci and M. Neeb, J. Electron Spectrosc. Relat. Phenom., 2010, 181, 140–144 CrossRef CAS.
- S. Peters, S. Peredkov, M. Neeb, W. Eberhardt and M. Al-Hada, Phys. Chem. Chem. Phys., 2013, 15, 9575–9580 RSC.
- H.-L. Schmidt and E. Schmelz, Chem. Unserer Zeit, 1980, 14, 25–34 CrossRef CAS.
- K. Varazo, F. W. Parsons, S. Ma and D. A. Chen, J. Phys. Chem. B, 2004, 108, 18274–18283 CrossRef CAS.
- S. Schauermann, J. Hoffmann, V. Johánek, J. Hartmann, J. Libuda and H. J. Freund, Catal. Lett., 2002, 84, 209–217 CrossRef CAS.
- I. V. Yudanov, R. Sahnoun, K. M. Neyman, N. Rösch, J. Hoffmann, S. Schauermann, V. Johánek, H. Unterhalt, G. Rupprechter, J. Libuda and H. J. Freund, J. Phys. Chem. B, 2003, 107, 255–264 CrossRef CAS.
- S. Poulston, A. H. Jones, R. A. Bennett and M. Bowker, J. Phys.: Condens. Matter, 1996, 8(49), L765–L771 CrossRef CAS.
- M. Bowker and R. J. Madix, Surf. Sci., 1981, 102, 542–565 CrossRef CAS.
- Z. Zhang, J. Lee, J. T. Yates, R. Bechstein, E. Lira, J. Hansen, S. Wendt and F. Besenbacher, J. Phys. Chem. C, 2010, 114, 3059–3062 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: TPR spectra of methanol on pristine SiOx; Fig. S2: temperature-dependent XPS spectra of copper on TiO2(110); Fig. S3 and S4: additional TPR spectra of methanol on Cu/SiOx with more coverage; Fig. S5: fragmentation patterns of methanol and possible reaction products; Fig. S6: blank TPR spectra of Cu/SiOx; Fig. S7: TPR spectra of CO on Cu/TiO2(110); Fig. S8–S10: additional TPR spectra of methanol on copper on slightly reduced TiO2(110) with more coverage and m/z ratios. Fig. S11–S14: additional TPR spectra of methanol on copper on slightly reduced TiO2(110) with more copper coverage and m/z ratios; Fig. S15: TPR spectra comparison of pristine TiO2(110) with Cu/TiO2(110); Fig. S16: TPR spectra of methanol on copper on slightly reduced TiO2(110) to 800 K; and Fig. S17: TPR spectra of methanol on copper on highly reduced TiO2(110) to 800 K. See DOI: https://doi.org/10.1039/d4cp03904a |
| This journal is © the Owner Societies 2025 |