
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The influence of fluorine spin-diffusion on 13C solid-state NMR line shapes of CF3 groups†
Ettore Bartalucci ab,
Calogero Quarantac,
Fabio Manzonid,
Igor d'Anciães Almeida Silva‡
a,
Mirijam Zobelde,
Carsten Bolmc,
Matthias Ernst*f and
Thomas Wiegand*ab
ab,
Calogero Quarantac,
Fabio Manzonid,
Igor d'Anciães Almeida Silva‡
a,
Mirijam Zobelde,
Carsten Bolmc,
Matthias Ernst*f and
Thomas Wiegand*ab
aMax Planck Institute for Chemical Energy Conversion, Stiftstr. 34–36, 45470 Mülheim/Ruhr, Germany. E-mail: thomas.wiegand@cec.mpg.de
bInstitute of Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
cInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany
dInstitute of Crystallography, RWTH Aachen University, Jägerstrasse 17–19, 52066 Aachen, Germany
eJCNS-3: Neutron Analytics for Energy Research, Forschungszentrum Jülich GmbH, Wilhelm-Johnen-Straße, 52428 Jülich, Germany
fPhysical Chemistry, ETH Zürich, Vladimir-Prelog-Weg 2, 8093 Zürich, Switzerland. E-mail: maer@ethz.ch
First published on 27th February 2025
Abstract
Indirect spin–spin couplings (“J-couplings”) lead to well-known multiplet patterns in nuclear magnetic resonance (NMR) spectra that are also observable in non-decoupled solid-state NMR spectra, if the J-coupling constant exceeds the linewidth. Such J-multiplet line shapes in the solid state might however be affected by spin diffusion (SD) on the passive nuclei. When the SD rate constant is fast compared to the J-coupling constant, the multiplet resolution can be lost due to a so-called “self-decoupling” mechanism as has been already reported in the context of decoupling and for proton SD in solid adamantane. We herein report on the influence of 19F SD on 13C-detected solid-state NMR spectra of a small organic molecule bearing a trifluoromethyl group. The target compound is the chiral α-(trifluoromethyl)lactic acid (TFLA). Enantiopure phases ((R) or (S), respectively) of TFLA are composed of homochiral dimers whereas the racemic phase consists of heterochiral dimers in the solid state. Despite their structural similarity, the 13C line shapes of the CF3 group in cross-polarization spectra recorded at slow to medium magic-angle spinning (MAS) frequencies – in the range between 14.0 kHz and 60.0 kHz – differ substantially. By combining experimental observations, analytical calculations based on the Bloch–McConnell equations, and numerical spin-dynamics simulations, we demonstrate that differences in the 19F SD rate constant between racemic and enantiopure TFLA-phases significantly affect the respective solid-state 13C NMR spectral line shapes. Slowing down SD by increasing the MAS frequency restores the quartet line shape for both phases of TFLA.
Introduction
The presence of fluorine has significant effects on the physicochemical properties of molecules (for some selective reviews see ref. 1–9). From a chemical point-of-view, the high electronegativity of the fluorine atoms leads to compounds with polarized carbon-fluorine bonds and, therefore, partially negatively charged fluorine atoms,10 which might cause electrostatic repulsion. In medicinal chemistry, the trifluoromethyl (–CF3) group is often used as a bioisostere for a methyl (–CH3) group.2,5,11–13 As a consequence, the molecule becomes more lipophilic, which often improves the desired bioactivity,14 the latter being, however, still a matter of debate.15 The presence of a CF3 group also changes other physicochemical properties, such as volatility, boiling and melting point, as well as solubility.16,17 Furthermore, CF3 groups have been reported to be engaged in amphiphilic noncovalent bonding, acting as both electrophiles and nucleophiles.18 In addition, the role of CF3 groups as hydrogen-bond acceptors has recently been discussed (for a recent example see ref. 19).Fluorine also plays an important role in sublimations of chiral compounds, where self-disproportionations of enantiomers have been observed.16,20 In such processes, the sublimation rates of enantiopure solid entities and their racemic counterparts differ.21 Soloshonok and co-workers have observed such phenomena for various compounds bearing CF3 groups.16 For α-(triflouromethyl)lactic acid (TFLA) discussed here, this behaviour leads to an enantiomeric enrichment over time for the remaining solid.20 Because the crystal structure of the enantiopure compound contains homochiral dimers, whereas heterochiral dimers are found in the racemic form,20 it was suggested that the solid-state molecular packing might explain the differences in the sublimation behaviour, for instance caused by electrostatic repulsions of CF3 groups.16,20,22,23
We have recently employed NMR spectroscopy in solution using an in operando setup24 and in the solid state to investigate the influence of mechanochemistry, e.g., ball milling and resonant-acoustic mixing, on the self-disproportionation process and on molecular-recognition events involving TFLA in general.24,25 In the course of such studies, we noticed that the 13C magic-angle spinning (MAS) NMR line shapes for the CF3 groups differ significantly between the enantiopure and racemic phases of TFLA.25 We herein report 13C-detected solid-state NMR MAS experiments to probe how differences in the 19F spin-diffusion (SD) rate constants for enantiopure and racemic TFLA affect the 13C NMR resonances of the corresponding CF3 groups. Spin diffusion, which describes the energy-conserving transfer of polarization among dipolar-coupled nuclear spins,26 might lead to the so-called “self-decoupling” mechanism27–31 affecting the 19F–13C J-multiplet line shape. Similar effects on 13C line shapes have for instance already been reported for fluorinated single-walled carbon nanotubes32 or flurbiprofen.28
Theory
As reported previously, the effect of SD on J-coupled multiplet lines in solid-state NMR spectra can conveniently be described as an exchange process between the multiplet components using the Bloch–McConnell equations.27,33 For this purpose, a symmetry-adapted basis set for the three magnetically equivalent fluorine atoms in the CF3 group needs to be constructed (which is obviously identical to the ones of protons in a methyl group). In CF3, the three fluorine nuclei with C3v symmetry generate eight spin states, which can be grouped by their symmetry into the three irreducible representations A, Ea and Eb, for which the symmetry-adapted basis sets are listed below.34–36The A manifold (symmetric to spin exchange), with four states:
 | (1) |
 | (2) |
 | (3) |
The four A states have a total group spin 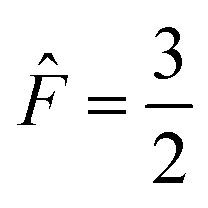 , while the four Ea and Eb states have a total group spin of
, while the four Ea and Eb states have a total group spin of 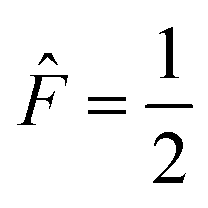 . The only observable transitions are single-quantum transitions within states of the same irreducible representation, and transitions between different manifolds are forbidden. This leads to the expected quartet line shape in the 13C spectra of molecules bearing CF3 groups with a 1
. The only observable transitions are single-quantum transitions within states of the same irreducible representation, and transitions between different manifolds are forbidden. This leads to the expected quartet line shape in the 13C spectra of molecules bearing CF3 groups with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:3:1 integral ratio (1:1:1:1 quartet caused by the A states and two 1:1 doublets caused by the two E-states). A schematic representation of the spin states for a CF3 group is shown in Scheme 1 and the transition probabilities between these states under a perturbation of strength ωi (dipolar coupling) is given by:27
:3:3:1 integral ratio (1:1:1:1 quartet caused by the A states and two 1:1 doublets caused by the two E-states). A schematic representation of the spin states for a CF3 group is shown in Scheme 1 and the transition probabilities between these states under a perturbation of strength ωi (dipolar coupling) is given by:27
 | (4) |
 | ||
| Scheme 1 Spin-states ground state energy level scheme for an isolated CF3 group (without including the coupling to the 13C spin, adapted from ref. 38). The energy difference between the A and Ea, Eb manifolds is the tunneling splitting Λ0. | ||
Therefore, the Bloch–McConnell equations to describe SD for the  group spin are given by:
group spin are given by:
 | (5) |
 group spin by:
group spin by:
 | (6) |
Results and discussion
The line shapes of the CF3 group in 13C-detected spectra of enantiopure and racemic TFLA differ
Fig. 1 shows the chemical structure of TFLA together with the experimental 1H–13C cross-polarization (CP) solid-state NMR spectra of the racemic and enantiopure (S)-TFLA crystalline phases recorded at 17.5 kHz MAS frequency and at the same probe temperature. Note that the spectra were recorded under 1H SPINAL-64 decoupling,39 but no 19F decoupling was applied. Fig. 1a clearly shows that the 13C line shape for the CF3 resonances at ∼124 ppm differs significantly between the two samples. While the expected 1:3:3:1 quartet caused by the three one bond 19F–13C scalar spin–spin couplings for the three magnetically equivalent fluorine nuclei is observed for the CF3 group of rac-TFLA, the enantiopure sample shows strong line broadening with a “quartet-like” line shape buried underneath a broad feature. Fig. S1 (ESI†) shows the comparison of the spectra of (S)-TFLA and (R)-TFLA, illustrating that this line shape is observed for both enantiomeric compounds whose spectral properties are, as expected, identical.
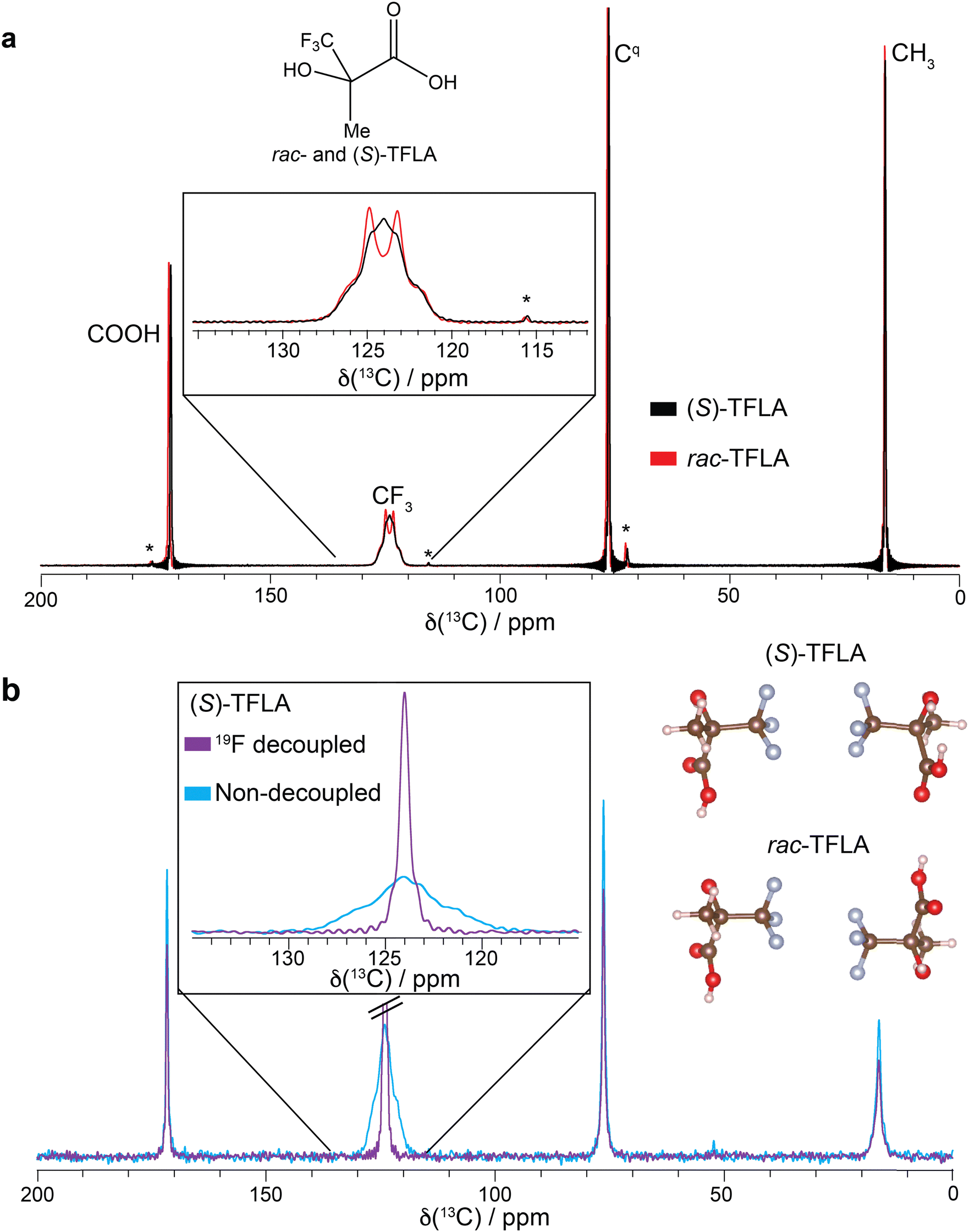 | ||
| Fig. 1 (a) 1H–13C CP spectra of (S)- and rac-TFLA recorded at 17.5 kHz and 16.4 T static magnetic-field strength. The chemical structure of TFLA is shown. (b) 19F–13C CP spectra of (S)-TFLA recorded at 15.0 kHz and 11.7 T with and without 19F high-power decoupling during data acquisition. The homochiral and heterochiral dimeric units taken from the crystal structures of (S)- and rac-TFLA (CSD numbers: (S)-TFLA refined from 666327 – for more details see Experimental section – and rac-TFLA 666328), respectively, are additionally shown.20 * denotes MAS spinning sidebands. Signal truncation in a results from too short acquisition times. | ||
To determine whether this broadening originates from carbon–fluorine indirect spin–spin interactions (J-couplings), 13C-detected 19F–13C CP spectra of (S)-TFLA with and without 19F high-power decoupling during data acquisition have been recorded and are shown in Fig. 1b (note that in these experiments no 1H decoupling was applied). Indeed, a similar line shape as in Fig. 1a can be observed in case of non-19F decoupled spectra, while the expected significantly sharper singlet is detected in the 19F-decoupled spectra, pointing to 19F–13C spin–spin couplings contributing to the broad 13C resonance. This motivated us to further investigate the reasons behind the observed line shape differences between the enantiopure and racemic compounds.
The effect of 19F SD on the 13C NMR multiplet line shape of CF3 groups
To address this observation, we simulated how the 19F SD affects the CF3 multiplet line. The role of 1H SD on 13C NMR line shapes has been reported in detail for adamantane, for which the influence of “self-decoupling” of the 1H–13C J-coupling by proton SD has been discussed.27 Also in context of 19F–13C spin pairs this effect has been reported for the monofluorinated molecule flurbiprofen.28Fig. 2 shows simulated 13C spectra for the CF3 multiplet line as a function of the SD rate constant kex (fixing the transverse 13C relaxation time, T2, to a constant value) using Bloch–McConnell equations as introduced above.27 Depending on the ratio of kex and the magnitude of the 19F–13C J-coupling constant, different line shapes are observed, consistent with three exchange-like regimes. For the case of kex ≪ 2πJCF (similar to the slow exchange regime), the result is a perfectly resolved quartet multiplet. In case of coalescence kex ≈ 2πJCF, only a single line is observed whose linewidth decreases for kex ≫ 2πJCF (fast exchange regime). In the fast exchange regime, the sharp resonance is denoted as “self-decoupled”.
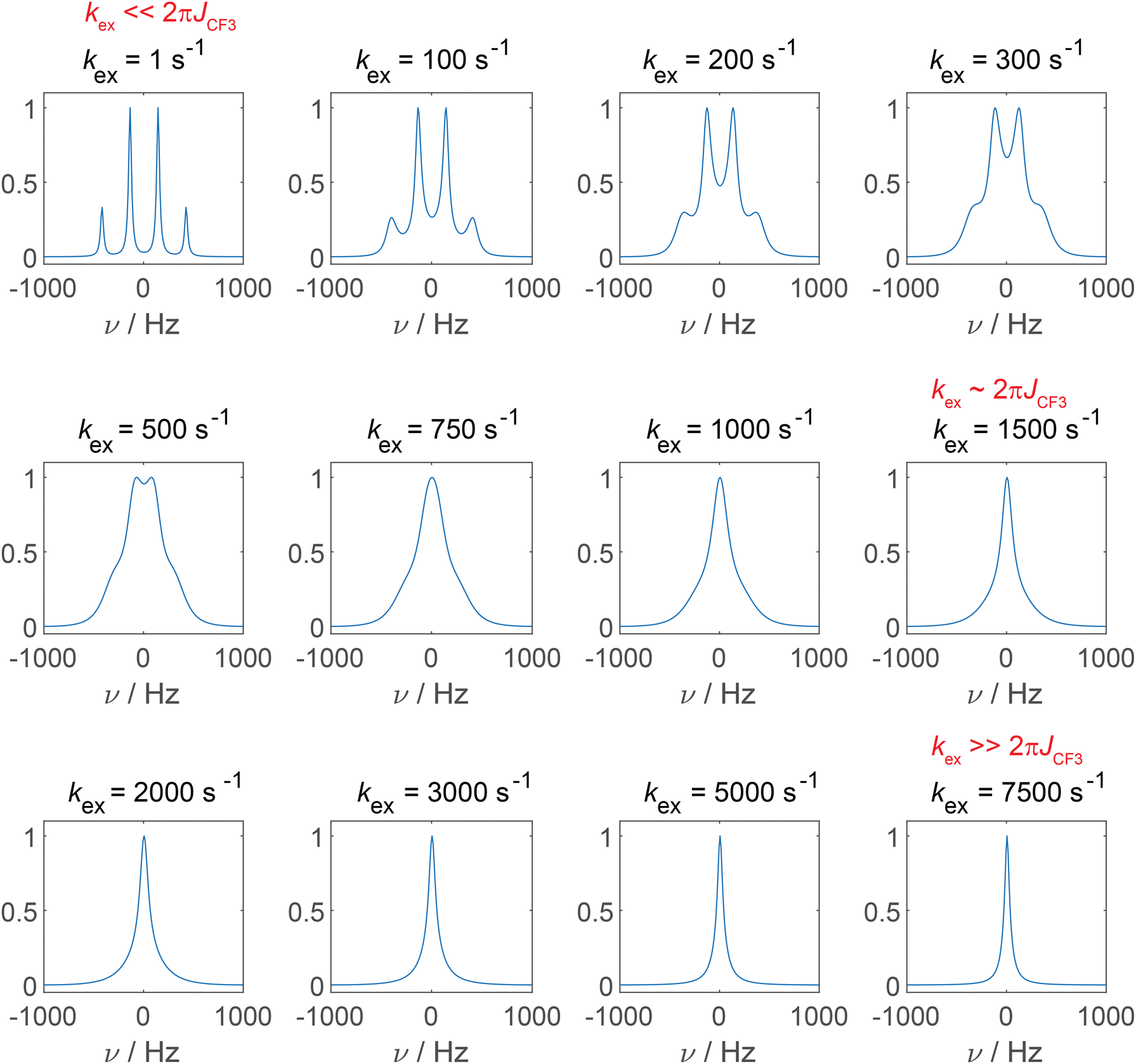 | ||
| Fig. 2 Simulation of the 13C NMR spectrum of a CF3 group affected by 19F SD assuming a 19F–13C J-coupling constant of 280 Hz and a linewidth of 10 Hz (FWHM = 1/(πT2)). The spectra have been simulated as a function of the SD rate constant kex. | ||
The SD rate constant scales down with increasing MAS frequency, since second-order terms in the effective Hamiltonian, which dominate the SD process, scale with the inverse of the spinning frequency.40 Therefore, we turned to MAS-dependent 13C CP-spectra of (S)-TFLA to investigate whether slowing down 19F SD has an effect on the CF3 multiplet line. As a matter of fact, 13C NMR spectra recorded at faster MAS frequencies would result in the expected CF3 line shape featuring the 1:3:3:1 quartet also for the enantiopure sample, assuming the main contribution to the observed line broadening comes from 19F SD. Fig. 3 shows the 13C-detected CP spectra of (S)-(a) and rac-TFLA (b) recorded at MAS frequencies ranging from 14.0 kHz to 60.0 kHz (spectra recorded in 3.2 mm and 1.3 mm rotors under high-power 1H SPINAL-64 decoupling39). For both substances, a narrowing of the multiplet lines and an increase in resolution of the quartet line shape is observed with increasing MAS frequency. In (S)-TFLA (Fig. 3a), the quartet becomes resolved at around 30.0 kHz MAS and above, while decreasing the MAS to 14.0 kHz produces a significantly broadened resonance as reported before. Similar observations can be drawn for the racemic sample (Fig. 3b), although the quartet becomes resolved at slightly lower MAS frequencies already. Fig. 3 also shows the simulations of the spectra (orange curves) based on a nonlinear least-square fit of the SD rate constant, kex, and the line width (FWHM = 1/(πT2)) according to the Bloch–McConnell equations (for more details see the Experimental Section). The comparison of the two sets of spectra and the SD rate constants taken from the simulations indicates that the SD rate is approximately a factor of three to four larger for (S)-TFLA compared to the racemic case (vide infra).
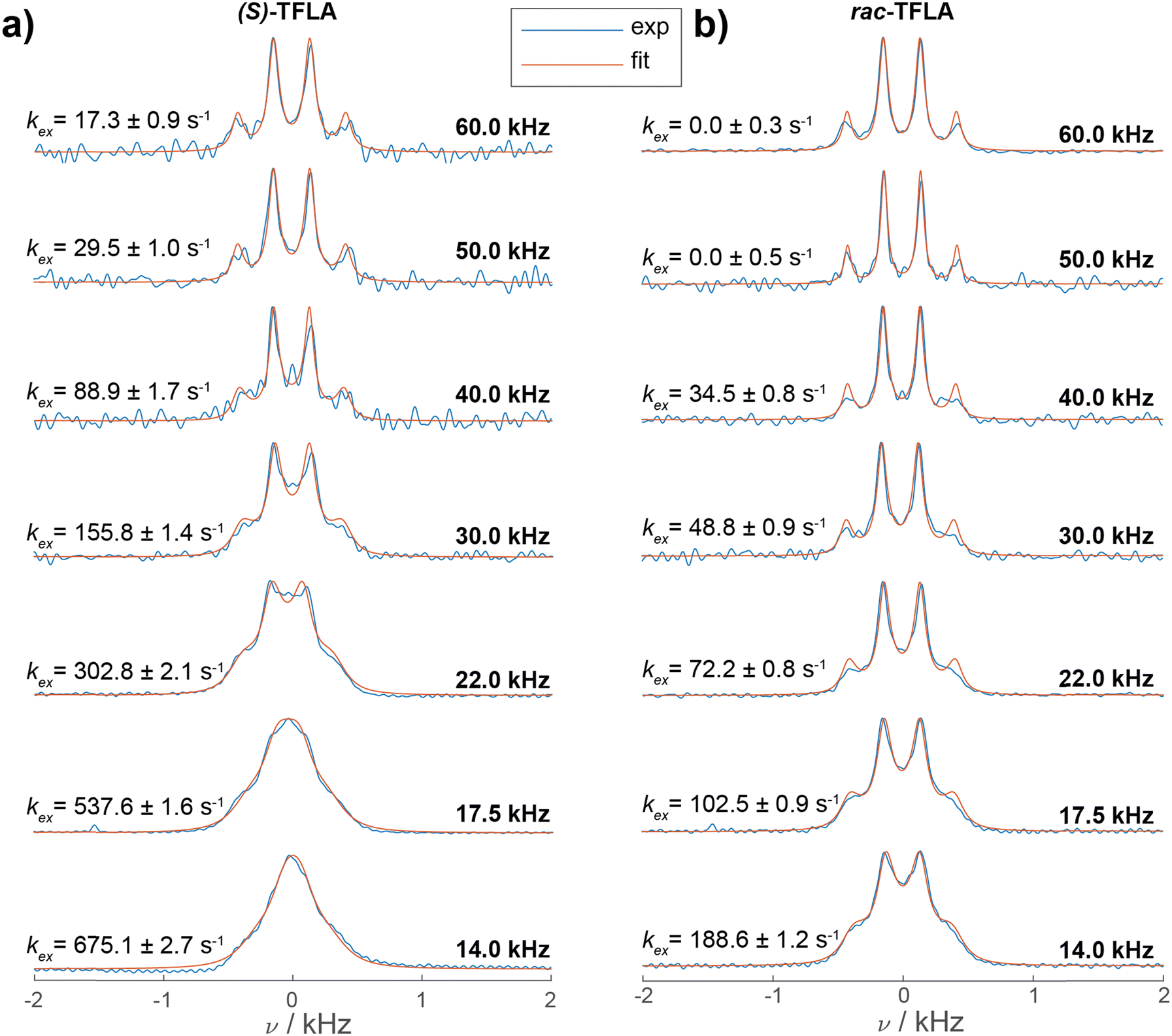 | ||
| Fig. 3 1H-13C CP spectra of (a) (S)- and (b) rac-TFLA recorded at various MAS frequencies and at 16.4 T (blue curves). The spectra measured at 14.0, 17.5 and 22.0 kHz MAS were recorded in a 3.2 mm rotor, the spectra at faster MAS in a 1.3 mm rotor. Displayed is a zoom in the 13C CF3 spectral region for the two compounds. For each spectrum the simulated spectrum resulting from Bloch–McConnell equations using as variable parameters the optimal values of T2 and kex obtained from a nonlinear least-square fit are shown (orange curves, for details regarding the error analysis see ESI†). | ||
Plotting the resulting fit of kex against the inverse of the MAS frequency in Fig. 4 reveals the expected linear decrease in kex for faster MAS frequencies as well as the mentioned factor of three to four difference in magnitude of kex between enantiopure and racemic TFLA. The fitted kex-values obtained from a nonlinear least-square minimization fit of the spectra at various MAS frequencies correspond to local minima, for which however we expect relatively large errors in the obtained rate constants. To determine the goodness-of-fit, we compared the 1D fit results to a 2D minimum χ2-estimation with variable T2- and kex-values (Fig. S2, ESI†) indeed showing that the minima reported in Fig. 3 are a good estimate for the rate constant.
 | ||
| Fig. 4 Plot of the dependence of the fitted kex-values (obtained from 1D nonlinear least-square fits of the experimental and simulated spectra) on the inverse of the MAS frequency, νMAS. The corresponding value of MAS frequency is reported on the x-axis on top for each datapoint. Dashed lines are the linear regressions of the datapoints for both enantiopure and racemic TFLA samples. The R2-values resulting from the linear regressions are reported in the legend. | ||
The 19F SD can alternatively be slowed down by applying off-resonance radio frequency (rf) irradiation on the 19F nuclei, thereby scaling the homonuclear 19F dipolar interaction and, thus, the SD rate constants as (P2(cosθ))2. Note that this will also scale the heteronuclear J-coupling, but with a different scaling factor, namely with cosθ. Such experiments have been performed for solid adamantane before.27 Fig. 5a shows a series of 19F–13C CP spectra of (S)- and rac-TFLA recorded at 15.0 kHz MAS and with variation of the angle of the fluorine effective field during data acquisition with respect to the static magnetic field direction, θ, as shown schematically in Fig. 5b. The angle θ was varied between 20° and 54.7° (magic angle), with the 90° case representing the control experiment. At the magic angle, 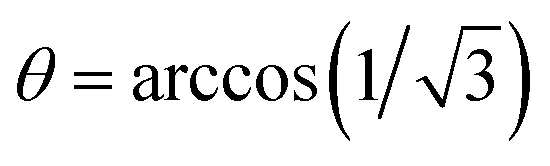 , the homonuclear 19F flip–flop term in the effective NMR-Hamiltonian vanishes and the contributions of 19F SD to the line shape should, therefore, be eliminated. In such a scenario, the expected quartet line shape should be observed. As can be concluded from the spectra, there is a clear deviation from both a symmetric quartet line shape when setting θ to the magic angle and a single decoupled line for θ = 90°, which is caused by second-order contributions between a rather large 19F chemical-shielding anisotropy (CSA) and the heteronuclear dipolar coupling under CW irradiation.41,42
, the homonuclear 19F flip–flop term in the effective NMR-Hamiltonian vanishes and the contributions of 19F SD to the line shape should, therefore, be eliminated. In such a scenario, the expected quartet line shape should be observed. As can be concluded from the spectra, there is a clear deviation from both a symmetric quartet line shape when setting θ to the magic angle and a single decoupled line for θ = 90°, which is caused by second-order contributions between a rather large 19F chemical-shielding anisotropy (CSA) and the heteronuclear dipolar coupling under CW irradiation.41,42
 | ||
| Fig. 5 (a) 19F–13C CP spectra of the CF3 spectral region for racemic (dashed red curve) and (S)-TFLA (straight black curve) recorded at 15.0 kHz MAS under off-resonance 19F decoupling. The angle θ of the effective 19F field with respect to B0 during CW decoupling varies as shown in the figure from 90° to 20°. (b) schematic representation for the calculations of the effective field during CW decoupling. (c) Numerical simulations of off-resonance 19F decoupled spectra as a function of θ and with kex = 160 s−1 representing rac-TFLA and kex = 670 s−1 representing (S)-TFLA. The simulations are based on the following parameters: J(19F–13C) = −280 Hz, J(19F–19F) =100 Hz, δ(19F–13C)/2π = 7870 Hz, δ(19F–19F)/2π = 10700 Hz, δσ{19F}/2π = 26352 Hz with all tensors oriented along the rotation axis of the CF3 group. The MAS frequency was set to 15.0 kHz, B1 to 90 kHz, and the offsets values as indicated in the figure. | ||
To examine whether the experimentally observed line shapes under off-resonance CW irradiation are compatible with the determined parameters, numerical simulations of the CF3 group including SD have been performed. The rf-field amplitude and the irradiation offsets were set to the same values as in the experimental measurements of Fig. 5a using an unscaled exchange-rate constant of kex = 160 s−1 for rac-TFLA and a value of 670 s−1 for (S)-TLFA as determined above. The agreement between the simulations and the experimental data is not perfect, but the general features of the line shapes are reproduced quite well. Furthermore, as can be observed by comparing the rac- and (S)-TFLA spectra in Fig. 5a and c, the CF3 line shapes for the two compounds become very similar if the SD is quenched near an effective field along the magic angle indeed supporting the assumption of different SD rate constants for the two compounds.
We initially assumed that the different SD rate constants are caused by differences in the homonuclear 19F–19F dipolar-coupling network in the two TFLA-phases, which might also explain the differences in their 19F static spin echo lineshapes (Fig. S3, see Fig. S4 for the corresponding MAS spectra, ESI†). We thus calculated the square root of the sum of squared dipolar couplings,43 dRSSi, using the published and refined crystal structures§ of rac- and (S)-TFLA, respectively20 (for more details see Experimental section). The resulting dRSSi taking only intermolecular CF3 group contacts into account and calculated between the centers of mass of the three fluorine nuclei in the CF3 groups are reported in Table S1 (ESI†) and amount to 11.4 kHz for rac-TFLA and 11.6 kHz for (S)-TFLA. The dRSSi-values have also been calculated taking an explicit averaging over all possible combinations on the circle into account and assuming “uncorrelated” rotations of the involved CF3 groups (10.6 kHz for rac- and 9.7 kHz for (S)-TFLA). In both cases, the dRSSi-values do not reflect the expected difference by a factor of  as predicted from the experimentally observed ratio of three of the SD rate constants. We also note that reducing particle size by means of milling the sample does not affect the line shape, e.g., due to reduced longitudinal relaxation times upon milling samples.44
as predicted from the experimentally observed ratio of three of the SD rate constants. We also note that reducing particle size by means of milling the sample does not affect the line shape, e.g., due to reduced longitudinal relaxation times upon milling samples.44
Although the detailed reason for the different SD rate constants remains currently unclear, we note that the spatial orientations of the two CF3 groups in the dimers differ (Fig. 1b). While the CF3 groups in (S)-TFLA are oriented face-to-face with an inversion centre between them (e.g., the 19F CSA- and the 19F–19F dipolar-coupling tensor are co-linear), they are stacked in rac-TFLA (non-linear tensor orientations). The importance of such geometric effects for instance on 31P{19F} REDOR dephasing curves involving CF3 groups has been reported.¶ 45 The differences in the SD rate constants might be caused by dynamics. Similar 13C and 19F T1 relaxation times for the CF3 groups in rac- and (S)-TFLA point to fast rotations of these groups with correlation times in the order of 10–100 ps for both samples. The above-mentioned face-to-face orientation of the CF3-groups in (S)-TFLA might however lead to faster SD if a correlated (cooperative) rotation of such groups exist, as for instance reported for the CF3-group in 3-(trifluoromethyl)phenanthrene.46,47 We have currently no direct experimental evidence for this hypothesis and will further explore this effect in our laboratories.
Conclusions
We show that 13C-detected solid-state NMR spectra of molecules containing CF3 groups can be affected by 19F spin diffusion leading to line broadening and sometimes even precluding the observation of the expected quartet multiplet at slow to moderate MAS frequencies in spectra without 19F decoupling. While in the case of small 19F spin-diffusion rate constants, the expected quartet line shape can be observed, this is not the case for faster rate constants. Our study on the small organic molecule TFLA crystallizing as enantiopure and racemic phases illustrates the high sensitivity of solid-state NMR to small differences in the 19F spin-diffusion rate constants by using the CF3 group as a highly sensitive marker. We hypothesize that these differences could be the result of correlated or uncorrelated motion of the two face-to-face oriented CF3 groups.Author contributions
E. B. and I. D. A. S. recorded the solid-state NMR experiments. C. Q. prepared and provided the TFLA samples. F. M. performed the PXRD structure refinement for (S)-TFLA. E. B. and M. E. performed the Bloch–McConnell and the numerical simulations. E. B., M. E. and T. W. carried out the analysis and interpretation of the data and wrote an initial version of the manuscript. All co-authors contributed to the writing of the final version of the manuscript. M. E. and T. W. designed the research, which was supervised by M. Z., C. B., M. E. and T. W.Data availability
The scripts and data used in this manuscript are available through a public Github repository hosted at https://github.com/ebartalucci/CF3_self_decoupling.git.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. W. acknowledges support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, project number 455240421 and Heisenberg fellowship, project number 455238107) and the Max Planck Society. M. Z., C. B. and T. W. appreciate funding by the DFG under Germany's Excellence Strategy – Exzellenzcluster 2186 “The Fuel Science Center” (ID 390919832). We acknowledge M.Sc. Florian Schreiner and Prof. Dr Michael Ryan Hansen (University of Münster. Germany) for enabling additional solid-state NMR experiments shown in Fig. S6d (ESI†), Dr Alexander A. Malär (Fraunhofer Headquarters, Germany) for providing a MATLAB script to analyse relaxation data, Dr Alexis Bordet (MPI CEC) for providing the SILP sample, B.Sc. Tomasz Wesolowski for contributions in the early phase of this project, Prof. Dr Hellmut Eckert (University of São Paulo, Brazil) for helpful discussions and also the support of Dr Kathrin Aebischer (ETH Zürich, Switzerland) for useful discussions on off-resonance decoupling and for providing the MATLAB function to process NMR spectra. Open Access funding provided by the Max Planck Society.References
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- G. Shabir, A. Saeed, W. Zahid, F. Naseer, Z. Riaz, N. Khalil, M. Muneeba and F. Albericio, Pharmaceuticals, 2023, 16, 1162 CrossRef CAS PubMed.
- A. S. Nair, A. K. Singh, A. Kumar, S. Kumar, S. Sukumaran, V. P. Koyiparambath, L. K. Pappachen, T. M. Rangarajan, H. Kim and B. Mathew, Processes, 2022, 10, 2054 CrossRef CAS.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed.
- D. O’Hagan, J. Fluorine Chem., 2010, 131, 1071–1081 CrossRef.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- Q. Wang, Y. Bian, G. Dhawan, W. Zhang, A. E. Sorochinsky, A. Makarem, V. A. Soloshonok and J. Han, Chin. Chem. Lett., 2024, 35, 109780 CrossRef CAS.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- H. L. Yale, J. Med. Pharm. Chem., 1959, 1, 121–133 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- C.-C. Tseng, G. Baillie, G. Donvito, M. A. Mustafa, S. E. Juola, C. Zanato, C. Massarenti, S. Dall’Angelo, W. T. A. Harrison, A. H. Lichtman, R. A. Ross, M. Zanda and I. R. Greig, J. Med. Chem., 2019, 62, 5049–5062 CrossRef CAS PubMed.
- B. E. Smart, J. Fluorine Chem., 2001, 109, 3–11 CrossRef CAS.
- A. Abula, Z. Xu, Z. Zhu, C. Peng, Z. Chen, W. Zhu and H. A. Aisa, J. Chem. Inf. Model., 2020, 60, 6242–6250 CrossRef CAS PubMed.
- J. Han, A. Wzorek, K. D. Klika and V. A. Soloshonok, in Frontiers of Organofluorine Chemistry, ed. I. Ojima, World Scientific, 2020, pp. 283–340 Search PubMed.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- C. Esterhuysen, A. Heßelmann and T. Clark, ChemPhysChem, 2017, 18, 772–784 CrossRef CAS PubMed.
- M. Wang, N. Garrison, P. M. Nguyen, A. Prasad, Y. Wang, H.-K. Kwon, G. Kim, M. A. Siegler and T. Lectka, J. Org. Chem., 2024, 89, 9681–9685 CrossRef CAS PubMed.
- V. A. Soloshonok, H. Ueki, M. Yasumoto, S. Mekala, J. S. Hirschi and D. A. Singleton, J. Am. Chem. Soc., 2007, 129, 12112–12113 CrossRef CAS PubMed.
- J. Han, O. Kitagawa, A. Wzorek, K. D. Klika and V. A. Soloshonok, Chem. Sci., 2018, 9, 1718–1739 RSC.
- R. Tonner, V. A. Soloshonok and P. Schwerdtfeger, Phys. Chem. Chem. Phys., 2011, 13, 811–817 RSC.
- M. A. Suhm and M. Albrecht, Phys. Chem. Chem. Phys., 2011, 13, 4159–4160 RSC.
- F. Puccetti, T. Rinesch, S. Suljić, K. Rahimi, A. Herrmann and C. Bolm, Chemistry, 2023, 9, 1318–1332 CrossRef CAS.
- C. Quaranta, I. D. A. A. Silva, S. Moos, E. Bartalucci, L. Hendrickx, B. M. D. Fahl, C. Pasqualini, F. Puccetti, M. Zobel, C. Bolm and T. Wiegand, Angew. Chem., Int. Ed., 2024, 63, e202410801 CAS.
- N. Bloembergen, Physica, 1949, 15, 386–426 CrossRef CAS.
- M. Ernst, A. Verhoeven and B. H. Meier, J. Magn. Reson., 1998, 130, 176–185 CrossRef CAS PubMed.
- G. Antonioli and P. Hodgkinson, J. Magn. Reson., 2004, 168, 124–131 CrossRef CAS PubMed.
- G. Sinning, M. Mehring and A. Pines, Chem. Phys. Lett., 1976, 43, 382–386 CrossRef CAS.
- M. Mehring and G. Sinning, Phys. Rev. B: Solid State, 1977, 15, 2519–2532 CrossRef CAS.
- M. Mehring, G. Sinning and A. Pines, J. Phys. B, 1976, 24, 73–76 CAS.
- L. B. Alemany, L. Zhang, L. Zeng, C. L. Edwards and A. R. Barron, Chem. Mater., 2007, 19, 735–744 CrossRef CAS.
- H. M. McConnell, J. Chem. Phys., 1958, 28, 430–431 CrossRef CAS.
- J. Tang and A. Pines, J. Chem. Phys., 1980, 72, 3290–3297 CrossRef CAS.
- J.-N. Dumez, P. Håkansson, S. Mamone, B. Meier, G. Stevanato, J. T. Hill-Cousins, S. S. Roy, R. C. D. Brown, G. Pileio and M. H. Levitt, J. Chem. Phys., 2015, 142, 044506 CrossRef PubMed.
- M. Negroni, D. Guarin, K. Che, L. M. Epasto, E. Turhan, A. Selimović, F. Kozak, S. Cousin, D. Abergel, G. Bodenhausen and D. Kurzbach, J. Phys. Chem. B, 2022, 126, 4599–4610 CrossRef CAS PubMed.
- D. Suter and R. R. Ernst, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 5608–5627 CrossRef CAS PubMed.
- K. Morimoto, J. Phys. Soc. Jpn., 1981, 50, 2404–2412 CrossRef CAS.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed.
- M. Chávez, T. Wiegand, A. A. Malär, B. H. Meier and M. Ernst, Magn. Reson., 2021, 2, 499–509 CrossRef PubMed.
- M. Ernst, S. Bush, A. C. Kolbert and A. Pines, J. Chem. Phys., 1996, 105, 3387–3397 CrossRef.
- M. Ernst, J. Magn. Reson., 2003, 162, 1–34 CrossRef CAS PubMed.
- V. E. Zorin, S. P. Brown and P. Hodgkinson, J. Chem. Phys., 2006, 125, 144508 CrossRef PubMed.
- K. E. Dempah, J. W. Lubach and E. J. Munson, Mol. Pharmaceutics, 2017, 14, 856–865 CrossRef CAS PubMed.
- E. A. Louie, P. Chirakul, V. Raghunathan, S. T. Sigurdsson and G. P. Drobny, J. Magn. Reson., 2006, 178, 11–24 CrossRef CAS PubMed.
- X. Wang, F. B. Mallory, C. W. Mallory, P. A. Beckmann, A. L. Rheingold and M. M. Francl, J. Phys. Chem. A, 2006, 110, 3954–3960 CrossRef CAS PubMed.
- P. A. Beckmann, J. Rosenberg, K. Nordstrom, C. W. Mallory and F. B. Mallory, J. Phys. Chem. A, 2006, 110, 3947–3953 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00275c |
| ‡ Present address: NMR facility, University of Missouri, 601 S. College Avenue, MO 65211, USA. |
| § We note here that the (S)-TFLA crystal structure has been refined based on the deposited structure (CSD number: 666327, ref. 20) (see ESI,† Fig. S5), since the experimentally observed PXRD pattern does not match with the predicted one (for details see ref. 25). |
| ¶ As a final remark, we point out that we investigated two additional samples containing a CF3 group, namely a supported-ionic liquid phase (SILP) containing bistriflimide as an anion, and the organic molecule 4-(trifluoromethyl)benzene-1-carboximidamide hydrochloride hydrate (data discussed in ESI,† Fig. S6). For both cases, the CF3 groups spectra show a resolved quartet, in agreement with the slow exchange-regime scenario of Fig. 2. |
| This journal is © the Owner Societies 2025 |