
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modular perovskite-BiVO4 artificial leaves towards syngas synthesis on a m2 scale†
Virgil
Andrei‡
 ab,
Yu-Hsien
Chiang‡
b,
Motiar
Rahaman‡
a,
Miguel
Anaya‡
bc,
Taeheon
Kang
c,
Edoardo
Ruggeri
b,
Samuel D.
Stranks
*bc and
Erwin
Reisner
*a
ab,
Yu-Hsien
Chiang‡
b,
Motiar
Rahaman‡
a,
Miguel
Anaya‡
bc,
Taeheon
Kang
c,
Edoardo
Ruggeri
b,
Samuel D.
Stranks
*bc and
Erwin
Reisner
*a
aYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk
bCavendish Laboratory, University of Cambridge, J J Thomson Avenue, Cambridge CB3 0HE, UK. E-mail: sds65@cam.ac.uk
cDepartment of Chemical Engineering & Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge CB3 0AS, UK
First published on 13th January 2025
Abstract
Metal halide perovskite-based artificial leaves have emerged in recent years as a promising design towards direct solar fuel synthesis. However, the complexity of these layered devices and reliance on solution-based techniques hinders the scalability and performance of existing prototypes. Here, we utilise vacuum processing of the perovskite light absorbers, as an industrially compatible method to produce large-scale devices. Accordingly, we fabricate fully evaporated 10 cm2 PV devices sustaining a 1 V photovoltage, which allow perovskite-BiVO4 tandem photoelectrochemical devices with a selective Cu92In8 alloy catalyst to sustain unassisted water and CO2 splitting over 36 hours. To demonstrate the modularity of this approach, we designed a 0.7 × 0.5 m2 “artificial tree” reactor containing a 10 × 10 array of artificial leaves, which was benchmarked during the 3-days final of the EIC Horizon Prize “Fuel from the Sun”, through an outdoor demonstration at the Joint Research Center of the European Commission in Ispra, Italy. Such real-world tests reveal key insights into practical operation that are not encountered during standardised laboratory experiments, but are crucial for upscaling of emerging solar fuel technologies.
Broader contextSyngas, a mixture of H2 and CO, is a key industrial intermediate in the production of liquid fuels through the Fischer–Tropsch process. Hence, solar-driven CO2-to-syngas conversion is a key strategic goal towards securing a sustainable carbon economy. This is reflected through support from policymakers including the U.S. Department of Energy and the European Commission. To this end, the €5 million EIC Horizon Prize “Fuel from the Sun” was launched in 2017 to incentivise research on direct light-driven fuel production beyond water splitting, and accelerate the translation of these technologies from laboratory prototypes towards real-world applications. This report describes the insights gained by a University of Cambridge team throughout the competition, from advances in device and catalyst design for unassisted syngas synthesis, to the practical engineering challenges encountered during outdoor testing of a 0.35 m2 solar fuel reactor. |
Introduction
Metal halide perovskite semiconductors have emerged as excellent materials for light harvesting applications, with photovoltaic (PV) energy conversion efficiencies of lab-based cells increasing rapidly from 3.8% in 20091 to 26.7% today.2,3 Single and multijunction perovskite PV modules can exhibit a very small energy payback time of 0.09 years and a greenhouse gas emission factor as low as 13.4 g CO2 equivalent per kWh, which compare favourably to other technologies including market-leading silicon cells.4 Perovskite-based technologies can therefore play a key part in achieving society's decarbonisation goals.Accordingly, these materials have attracted similar interest from the solar fuels community,5 with PV-electrolysers,6,7 photoelectrochemical (PEC) systems8 and nanoparticle photocatalysts9,10 developed for proton11–22 and CO2 reduction,23–28 O2 evolution,11,13,16,19,29–37 NH3 production,38 organic transformations39–43 or waste/pollutant degradation and valorisation.12,39,44,45 Among those, compact artificial leaves have emerged as promising alternatives for unassisted water splitting and CO2 conversion, as they bridge the gap between the performance of bulky PV-electrolysis systems and the separation and recovery challenges of photocatalytic powder suspensions.46 Despite the rich product scope, most prototypes are only reported on a <1 cm2 scale, as such layered devices rely on multiple deposition techniques involving solution processing and annealing steps. These deposition methods lead to inhomogeneities on areas beyond a few cm2, which can induce pinholes affecting the dark current and photocurrent.46,47 These film defects increase the likelihood of device shorting and device degradation under operation.
Thermal evaporation can provide key advantages for perovskite PV fabrication over solution methods, as solvent-free steps avoid potentially hazardous and expensive solvents (e.g., dimethylformamide), which can degrade underlying layers during processing.48,49 Moreover, vacuum processing ensures a homogeneous large-area deposition on a broad variety of conductive substrates with active areas spanning from <10 mm2 multiple pixel devices to >10 × 10 cm2 PV modules, which is key for both single and multijunction applications.48,49
Here, we utilise vacuum deposition as a scalable technique for the fabrication of 10 cm2 artificial leaves (Fig. 1). Fully evaporated perovskite PV devices can be interfaced to selective Pt and Cu92In8 electrocatalysts for H2 evolution or CO2 reduction, enabling unassisted water and CO2 splitting in a tandem configuration with a BiVO4 photoanode for O2 evolution. The vacuum deposition allows us to fabricate >100 individual artificial leaves, which are used to demonstrate the modularity of PEC artificial leaves under outdoor operation. To this end, we develop a series of small- to large-scale PEC reactors approaching a m2 size, which reveal practical insights into the effect of atmospheric conditions (temperature, pressure) and light intensity during real-world outdoor tests.
 | ||
| Fig. 1 Vacuum deposition of 10 cm2 perovskite solar cells for PEC artificial leaves. (a) Architecture of a perovskite-BiVO4 artificial leaf embedding a fully evaporated PV device. (b) Reverse scan J–V curves of a representative large-area device under 1 sun (orange, AM 1.5G) illumination and no irradiation (blue). Left inset: photograph of the PV device with a 4.3 × 2.2 cm2 active area. Right inset: schematic depiction of the multiple-source evaporation chamber. | ||
Results and discussion
Perovskite PV device fabrication
Devices with an ITO|spiro-TTB|perovskite|C60|BCP|Cu architecture were fully evaporated to ensure film uniformity and compatibility with multiple layers in the stack (Fig. 1 and Fig. S1, ESI†). A FA0.7Cs0.3Pb(I0.9Br0.1)3 perovskite layer with a 1.62 eV band gap was deposited by co-evaporation from three PbI2, CsBr and FAI sources (FA: formamidinium).48,49 2,2′,7,7′-Tetra(N,N-di-p-tolyl)amino-9,9-spirobifluorene (spiro-TTB) and C60|BCP charge transport layers ensured effective separation of holes and electrons, respectively. We note that we could not achieve sufficient uniformity of layers over large area with leading solution-processed hole transport layers such as phosphonic-based self-assembled monolayers,50 specifically motivating the choice of evaporated spiro-TTB.51 The deposition of a crystalline perovskite film onto ITO|spiro-TTB was confirmed by X-ray diffraction (XRD) (Fig. S2, ESI†). Device performance was first optimized on 8-pixel devices with individual active areas of 0.12 cm2, which were deposited onto 2.54 × 2.54 cm2 ITO substrates. These displayed an average open-circuit voltage (VOC) of 0.97 ± 0.05 V, short circuit current density (JSC) of −18.8 ± 2.8 mA cm−2, a fill factor (FF) of 58.4 ± 8.1% and PV cell efficiency (PCE) of 10.8 ± 2.6% (see Fig. S3 and S4, ESI,† for representative JV curves and histograms).Custom-made masks enabled the simultaneous fabrication of up to four devices with a 4.3 × 2.2 cm2 photoactive area onto 5 × 3 cm2 ITO substrates (Fig. S1, ESI†). A representative 10 cm2 device sustained a VOC of 1.04 V and a moderately high JSC of −17.2 mA cm−2, similar to those of smaller 8-pixel devices. A significantly lower FF of 28.7% could be traced back to the sheet resistance of conductive ITO glass (15 Ω sq−1), which is known to induce resistive losses over larger areas, especially at absolute currents beyond 100 mA. This, combined with parasitic contact effects and dark currents, results in a linear, ohmic photocurrent behaviour with applied voltage.47,52,53 Still, the device attained a PCE of 5.3% (Fig. 1b), representing a four-fold improvement over the efficiency of our solution-processed devices.47
Cu92In8 alloy catalyst development
In parallel with PV fabrication, a dendritic CuxIny electrocatalyst was synthesized following a template-assisted galvanostatic electrodeposition method.54 A reported procedure was adapted for catalyst deposition on 10 cm2 Cu foils,54 whereas the alloy composition was adjusted to favour long-term CO2 reduction (see Methods and Fig. S5, ESI†). Scanning electron microscopy (SEM) and high-angle annular dark field (HAADF) images revealed a hierarchical catalyst morphology, with nanoporous dendrites forming macropores with a 33 ± 3.3 μm pore size, whereas scanning transmission electron microscopy (STEM) elemental mapping confirmed the homogeneous distribution of Cu and In on the dendrite surface (Fig. 2c, d and Fig. S6, ESI†). Energy-dispersive X-ray (EDX) spectroscopy indicated an alloy composition of 92 wt% Cu and 8 wt% In (Cu92In8), whereas XRD confirmed the presence of CuIn alloy phases, along with some residual Cu and Cu2O phases. Accordingly, the 10 cm2 catalyst preserved the morphology and elemental distribution of previously reported 0.84 cm2 Cu96In4 samples.54 | ||
| Fig. 2 Characterisation of the 10 cm2 Cu92In8 catalyst. (a) and (b) Long-term electrochemical CO2 reduction test. (a) Current trace during a 36 h CPE experiment at −0.4 V vs. RHE. Inset shows a higher cathodic current density at the beginning of the experiment due to electrochemical reduction of the surface oxide layer that originated due to aerial oxidation. The oxide reduction process was completed within 3 min as the transient reached a steady state. (b) Amounts and FYs of gas products from electrolysis. CPE is performed in 0.5 M KHCO3, under CO2, pH 7.4. (c) SEM image of the porous Cu92In8 catalyst. (d) STEM mapping supports the bimetallic nature of the catalyst dendrites. | ||
A long-term controlled potential electrolysis (CPE) experiment was carried out at −0.4 V against the reversible hydrogen electrode (RHE) for 36 h, resembling the daytime operation of a PEC device during a 3-day test (Fig. 2a and b). The experiment was performed in a 3-electrode, one-compartment setup with a 10 cm2 Cu92In8 catalyst, a Ag/AgCl reference and graphite foil counter electrode. A steady current density of −0.36 ± 0.03 mA cm−2 was sustained during CPE (Fig. 2a). A higher cathodic current was observed over the first ∼3 min due to electrochemical reduction of the residual surface oxides (see inset of Fig. 2a). 164.4 μmol cm−2 CO and 80.9 μmol cm−2 H2 were obtained over the 36 h CPE test, amounting to faradaic yields (FYs) of 68.4% and 33.7% for CO and H2, respectively. A steady CO production was observed with time, whereas the H2 evolution rate increased after the first 15 h. This can be traced back to an in situ reorganisation of the alloy phases, which is known to expose the Cu phase leading to higher H2 evolution activity at the applied electrochemical conditions.54 A cyclic voltammetry (CV) scan performed after long-term CPE indicated an early onset potential at around −0.3 V vs. RHE for aqueous CO2 reduction (Fig. S7, ESI†), which suited our standalone artificial leaves for unassisted syngas (CO + H2) production.
Perovskite-BiVO4 artificial leaves
The perovskite PV devices were next interfaced to Pt or Cu92In8 catalysts for proton or CO2 reduction using graphite epoxy (GE) paste, which acted as a conductive encapsulant (see Methods).55 The resulting photocathodes (abbreviated PVK|GE|Pt or PVK|GE|Cu92In8) displayed early onset potentials of 1.0 and 0.8 V vs. RHE, respectively, resulting in a good overlap with CV curves of BiVO4 photoanodes (Fig. 3a and Fig. S8b, ESI†). Photocurrents under 1 sun irradiation remained similar when light was filtered through BiVO4, which confirms the limiting effect of sheet resistance on perovskite photocathodes. Dark currents of PVK|GE|Cu92In8 photocathodes (Fig. 3a) could be traced back to a combination of surface Cu reoxidation at positive applied potentials, current leakage through the PV device and transient capacitive effects of the porous catalyst, which were diminished in case of planar PVK|GE|Pt electrodes (Fig. S8b, ESI†). Further controls indicated that only negligible photocurrents below −5 μA cm−2 are observed in the absence of a catalyst, i.e., GE has no intrinsic catalytic activity (Fig. S8a, ESI†).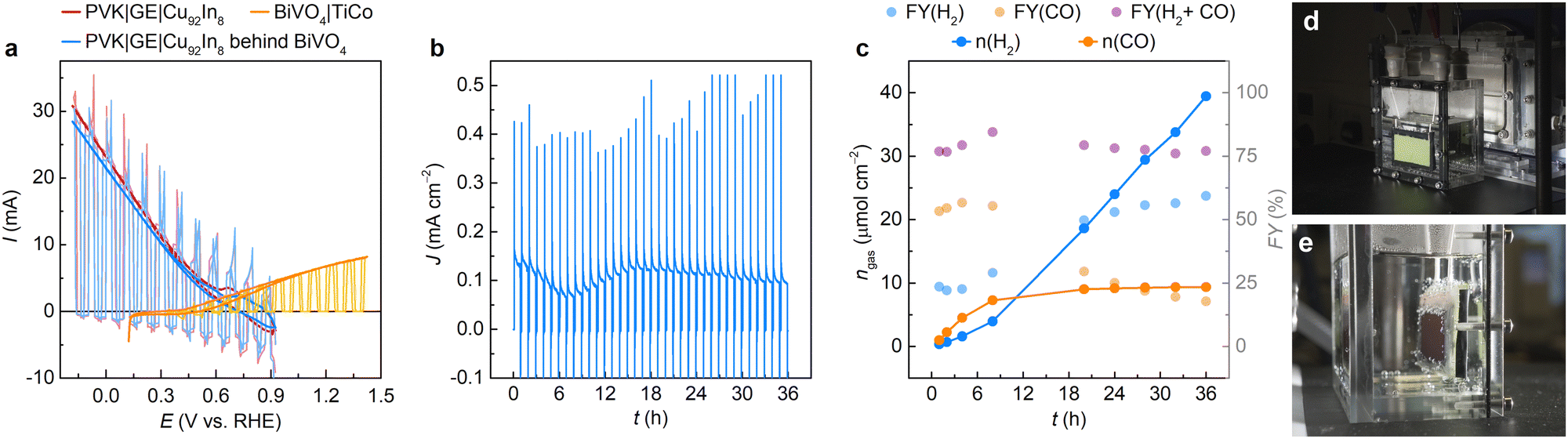 | ||
| Fig. 3 Performance of a 10 cm2 BiVO4 – PVK|GE|Cu92In8 artificial leaf coupling unassisted aqueous syngas production to O2 evolution. (a) CVs of individual perovskite and BiVO4 photoelectrodes. The sign of photocathode traces is reversed to illustrate photocurrent overlap. (b) CPE trace recorded at 0 V applied bias under 1 sun irradiation (AM 1.5G, 100 mW cm−2), in a 0.5 M KHCO3 solution, pH 7.4, under CO2. Light was chopped in 50 min on, 10 min off cycles to aid benchmarking against reported systems. (c) Corresponding faradaic yields and product amounts as a function of time. (d) and (e) Photographs of the 10 cm2 tandem device during operation in a custom-built PEC reactor. | ||
The perovskite photocathodes were attached to BiVO4 photoanodes in a back-to-back, tandem configuration (Fig. 1a and Fig. S9, ESI†). The PEC performance of these 10 cm2 artificial leaves was tested in a 2-electrode, one-compartment setup, using a custom-built Perspex (acrylic glass) reactor with an inner volume of 8 × 8 × 4 cm3 (Fig. 3 and Fig. S10, ESI†).
A BiVO4 – PVK|GE|Pt PEC tandem could attain a reproducible onset voltage of −0.8 V and photocurrents of ∼0.6 mA cm−2 at 0 V applied bias, displaying a minimal photocurrent decay and no visible photocathode degradation during a 10 h indoor stability test (Fig. S8, S11, S12 and Movie S1, ESI†). The H2 FY amounted to 99.2% after 10 h, corresponding to a 0.69% solar-to-fuel conversion efficiency (STF). Accordingly, the evaporated perovskite device structure and hydrophobic graphite paste coating resulted in a threefold improvement in performance and stability over our previously reported 10 cm2 perovskite-BiVO4 devices for overall water splitting, which could only sustain photocurrents of 0.2 mA cm−2 at 0 V applied bias, with an onset voltage of −0.5 V and complete photocurrent degradation within 3 h.47
10 cm2 BiVO4 – PVK|GE|Cu92In8 tandems were also benchmarked under 1 sun illumination for 36 h, comparable to daylight availability over a 3-day outdoor test (Fig. 3 and Fig. S13, S14 and Movie S1, ESI†). CV scans of tandem devices displayed an onset voltage of −0.5 V, indicating that artificial leaves can operate in the absence of external bias (Fig. S13 and S14a, ESI†). Accordingly, a PEC tandem sustained a steady-state photocurrent density of around 0.1 mA cm−2 during long-term CPE at 0 V applied bias (Fig. 3b and Fig. S13, ESI†), representing a tenfold increase in stability over earlier, Field's metal encapsulated devices.47 The artificial leaf produced 88.6 μmol CO and 373.1 μmol H2 over 36 h (see amounts normalised per area in Fig. 3c), corresponding to faradaic yields of 17.7% (CO) and 59.3% (H2), with a CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 ratio of 0.24:1. Accordingly, the 10 cm2 device attained STF values of 0.098% (H2) and 0.032% (CO), which compare favourably to small-scale artificial leaves23,54,56 with active areas <1 cm2 (Table S1, ESI†). This performance was reproducible, with a second prototype yielding 79.6 μmol CO and 215.6 μmol H2 over 36 h, with FYs of 22.3% (CO) and 60.3% (H2), a CO:H2 ratio of 0.37:1, and STF values of 0.062% (H2) and 0.025% (CO) (Fig. S14b and c, ESI†).
:H2 ratio of 0.24:1. Accordingly, the 10 cm2 device attained STF values of 0.098% (H2) and 0.032% (CO), which compare favourably to small-scale artificial leaves23,54,56 with active areas <1 cm2 (Table S1, ESI†). This performance was reproducible, with a second prototype yielding 79.6 μmol CO and 215.6 μmol H2 over 36 h, with FYs of 22.3% (CO) and 60.3% (H2), a CO:H2 ratio of 0.37:1, and STF values of 0.062% (H2) and 0.025% (CO) (Fig. S14b and c, ESI†).
Scalability towards a m2 scale
One intrinsic advantage of standalone PEC artificial leaves is their modularity. Since each unit can operate independently from each other, multiple smaller leaves can outperform a larger device of the same area, without suffering from resistive losses or mass transport effects.56 Moreover, the degradation of one smaller device (e.g., from moisture infiltration, or light-induced interfacial degradation) does not affect the performance of the entire device array, as in the case of a single larger-area device.56–58 This gives PEC artificial leaves another advantage over PV modules, where the shorting of one PV cell can affect the performance of the entire assembly, necessitating expensive solutions such as by-pass diodes.To test this design principle on a larger scale, we developed modular PEC reactors approaching a m2 scale for the EIC Horizon Prize “Fuel from the Sun: Artificial Photosynthesis” from the European Commission (Fig. 4, Fig. S15, S16 and Discussion S1, ESI†).59 To this end, >100 BiVO4 – PVK|GE|Cu92In8 artificial leaves were assembled over a period of 2 months by a team of 5 postgraduate students and postdoctoral researchers, while a 0.7 × 0.5 (0.35) m2 “artificial tree” reactor was constructed by the Mechanical Workshop (Yusuf Hamied Department of Chemistry, University of Cambridge), to house a 10 × 10 array of 10 cm2 PEC devices (Fig. 4a, d and Fig. S17, ESI†). The large reactor was engineered pursuing versatility, low cost and ease of fabrication, making this design applicable to a wide scope of photo(electro)catalytic systems within the broader artificial photosynthesis community. Accordingly, the reactor was assembled from UV-vis-transparent Perspex panels, which were processed using an automated laser cutter machine. The latter allowed the rapid patterning of sample holder frames, which can be swapped for devices of different sizes, without needing adjustments to the main reactor body (Fig. 4a). Based on multi-physics modelling and simulation studies,60,61 small openings were allowed in these sample holders, to permit syngas collection in the headspace and avoid pH gradient build-up between the cathodic and anodic sides of the reactor (Fig. 4d).
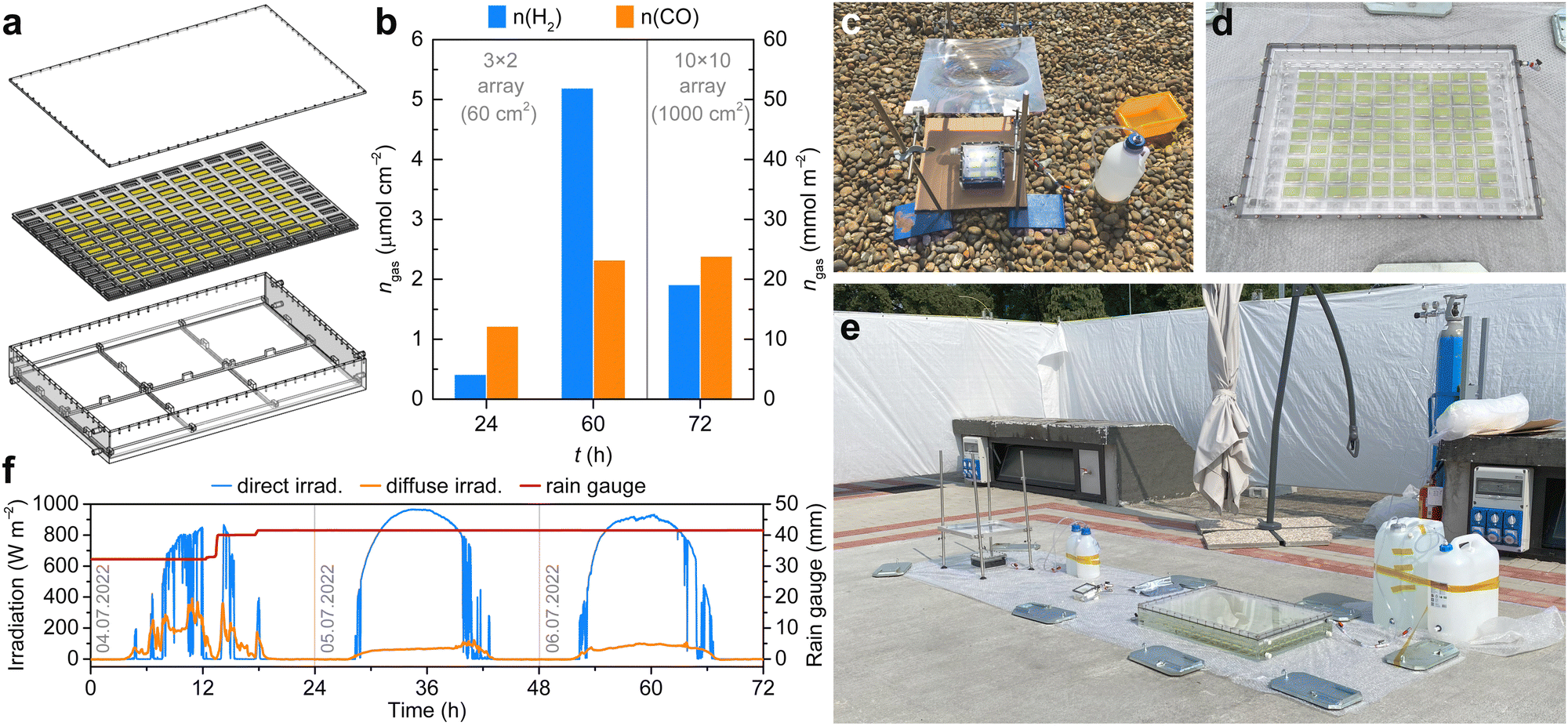 | ||
| Fig. 4 Outdoor testing of modular PEC reactors. (a) 3D design of the 0.7 × 0.5 m2 Perspex reactor. (b) Product amounts obtained during 2.5- and 3-day tests with the medium- and large-scale reactors, respectively. (c) Medium (3 × 2 array) reactor with a Fresnel lens, at the start of the 2.5-day test (11:00, 21st June 2022 – Maxwell Centre Cambridge, United Kingdom). (d) Large-scale reactor with a 10 × 10 array of 10 cm2 artificial leaves. (e) Testing pad at the start of the 3-day outdoor test (09:00, 4th July 2022 – JRC Ispra, Italy). Left: Medium-scale reactor with Fresnel lens, connected to 2.5 L gas and liquid collection tanks. Middle: Small-scale reactor connected to a gas sampling bag. Right: Large-scale reactor connected to gas (25 L) and liquid (20 L) collection tanks. Tests were performed under solar irradiation, at ambient temperature, in a CO2-purged 0.5 M KHCO3 solution. (f) Weather conditions during the 3-day outdoor test. A thunderstorm yielded a quarter of the yearly precipitations in Ispra over the first day. | ||
A medium-scale test was first performed on the rooftop of the Maxwell Centre in Cambridge, to assess the design of our overall PEC platform (including gas collection and light concentration modules) under real-world operating conditions (Fig. 4b, c and Fig. S18, ESI†). To this end, a 16 × 16 cm2 reactor46 was fitted with a custom-made 3D-printed sample holder accommodating a 3 × 2 array of 10 cm2 artificial leaves. In this setup, the reactor was connected to a 2.5 L external gas tank via tubing, which was fitted with on–off and one-way valves. The gas tank was filled with 1 M KOH solution, and connected to a second empty 2.5 L tank to collect spill-over KOH solution, which was meant to prevent overpressure in the PEC reactor. Accordingly, the excess product and CO2 mixture would flow into the gas tank, where CO2 would be captured in form of carbonate, leaving syngas and O2 to displace the KOH solution (Fig. S19 and Movie S2, ESI†). The synthesis of concentrated syngas was essential to obtain a combustible, non-diluted gaseous fuel − a key evaluation criterium of the Horizon competition (Discussion S1, ESI†).
The outdoor test in Cambridge began at noon on 21st June 2022 with clear sky (Fig. 4c). A 39.5 × 39.5 cm2 static Fresnel lens was dismounted within the first hour and not used thereafter in this test, as a drift of the focal point caused sample shadowing, which emphasizes the importance of light tracking for solar concentration. Due to leakage of KOH from the gas tank, the on–off valve was also switched off after 2 hours. Nevertheless, 1.22 μmol cm−2 of CO and 0.41 μmol cm−2 H2 were retained in the PEC reactor headspace after ∼24 h. In contrast, 2.32 μmol cm−2 of CO and 5.19 μmol cm−2 of H2 were detected after ∼60 h (i.e., by the end of 23rd June 2022, Fig. 4b). This gradual decrease in the CO:H2 ratio was in line with the catalyst behaviour during earlier laboratory experiments (Fig. 2).
A full-scale test of our different setups was assembled a week later on the 5 × 5 m2 testing pad provided as one of three teams59,62,63 shortlisted for the EIC Horizon Prize final (Joint Research Center of the European Commission, Ispra, Italy, Fig. 4e). Accordingly, the single-sample reactor was connected to a gas bag for product collection, while the medium (3 × 2 array) and large (10 × 10 array) reactors were connected to 1 M KOH-filled gas tanks, as described earlier (Fig. S20, ESI†). The reactors were filled with 0.5 M KHCO3 and purged with pure CO2 for over 30 min on the day of the competition start.
While in theory this design would effectively achieve product separation from the gas mixture, in practice the system was affected by atmospheric conditions, which are not routinely considered under standardised laboratory or even controlled outdoor conditions, as well as leakage through the valves. Accordingly, overcast weather including a thunderstorm on the first day of the competition (4th July 2022) resulted in a severely decreased light flux (Fig. 4f and Fig. S21, ESI†). Most photogenerated charges were likely employed to reduce the surface Cu oxide, instead of producing syngas, meaning that no overpressure was produced in the reactors throughout the day. Therefore, underpressure caused by the temperature drop during the night and the failure of the one-way valves led to KOH being drawn from the gas tank into the main reactors. The basic pH led to a further depletion of CO2 and hence underpressure (see inwards-curved reactor window in Fig. S22e, ESI†), which could only partly be compensated by further CO2 purging.
More importantly, the basic pH solution resulted in a dissolution of the BiVO4 photoanodes, which are only stable under near-neutral pH. This gradual process is accelerated under operation, when charges pass through the irradiated electrodes.23 This exposed the underlying perovskite light absorbers, indicating degraded devices (metallic Cu reflection) among healthy devices (dark layers) (Fig. S22, ESI†). As a result, only 0.64 vol% CO and 0.80 vol% H2 were detected in the large reactor headspace (0.43 vol% CO and 0.58 vol% H2 in the respective gas tank), as certified by the official test report of the competition (Data S1, ESI†). This corresponds to absolute product amounts of 2.39 μmol cm−2 of CO and 1.91 μmol cm−2 of H2 in the reactor headspace, comparable to those of the earlier rooftop test (Fig. 4b). Despite purging, CO2 content was only 1.61 vol%, whereas N2, O2 and Ar amounted to 97 vol% of the headspace composition (Data S1, ESI†). This indicates air infiltration due to underpressure in the reactor, which may have also led to the competing O2 reduction reaction. For comparison, the 10 × 10 array reactor was expected to produce 8.9 mmol CO and 37.3 μmol H2 (corresponding to around 220 mL CO and 910 mL H2) during the 3-day outdoor test, as extrapolated from laboratory tests of individual artificial leaves (Fig. 3). While these results indicate that further system engineering is necessary, they also provide some general guidelines for PEC reactor design (Text Box 1). In practice, leakage can be suppressed using industrial-grade reactors, connectors and tubing,62 whereas flow systems and gas separating membranes can be implemented for product collection.64 However, these components increase system complexity and require external electricity inputs, which contribute towards the overall energy balance of the process.
Ultimately, the Horizon Prize provided a timely opportunity to compare alternative artificial photosynthesis technologies at different stages of development, from SrTiO3 photocatalytic sheets62 and tandem silicon-perovskite PV-electrolysers63 feeding solar hydrogen to thermo/biocatalytic reactors for methane production, to our integrated artificial leaf panels for direct CO2-to-syngas conversion. Such initiatives will further incentivise efforts to transition these technologies from laboratory prototypes towards real-world applications.
Text box 1. Practical aspects in the design of PEC systems.• Reactor engineering must match the simplicity of the device design. Yet, solar tracking remains essential for concentrated light applications.• Thorough medium-scale prototyping, characterisation and troubleshooting (TRL 3–5) is necessary before assembling large-scale systems (TRL 6 and beyond).65,66 • Benchmarking must include or simulate atmospheric conditions including temperature and pressure changes, lack of sufficient sunlight and diurnal cycle, or component malfunction. • Manual sample fabrication67 and storage are challenging for systems meant to operate for several days. |
Conclusions
Our results demonstrate that thermal evaporation can contribute significantly towards solvent-free, scalable and reproducible manufacturing of perovskite-based artificial leaves. Accordingly, 10 cm2 perovskite-BiVO4 tandem PEC devices could demonstrate syngas production coupled to O2 evolution over 36 h, whereas water splitting tandems surpassed both efficiency and stability of previously reported solution-processed devices. Moreover, vacuum deposition enabled us to test the modularity of artificial leaves towards a 1 m2 scale under harsh outdoor environments. While further progress is required on the reactor engineering side, such deposition techniques are suitable for the adoption of perovskite light absorbers in production chains and multijunction device applications, which are less compatible with solution methods.Methods
Materials
FAI (GreatCell Solar), PbI2 (TCI, >98%), CsBr (Sigma), C60 (Creaphys), BCP (Ossila), Bi(NO3)3·5H2O (98%, Sigma-Aldrich), NaI (≥99%, laboratory reagent grade, Fischer), p-benzoquinone (≥98%, Sigma-Aldrich), ethanol (absolute, VWR), vanadyl acetylacetonate (VO(acac)2; 98.0%, Sigma-Aldrich), graphite (powder, <20 mm, synthetic, Sigma-Aldrich), H3BO3 (BioReagent, ≥99.5%, Sigma-Aldrich), K2SO4 (≥99%, ACS reagent, Sigma-Aldrich), CuSO4·5H2O (99.995%, Sigma-Aldrich), In2(SO4)3·xH2O (99.99%, Sigma-Aldrich), H2SO4 (Suprapur 96%, Sigma-Aldrich), Cu foil (99.9%, Alfa Aesar), and KHCO3 (≥99.5%, BioUltra, Sigma-Aldrich) were used without further purification.Vacuum-processed perovskite PV devices
ITO coated glass substrates (15 Ω sq−1, Kintech) were employed for small-scale device fabrication (0.12 cm2 active area) and 9.46 cm2 devices. The substrates were cleaned with soap, deionized water, acetone and isopropanol in a sonication bath for 15 min at each step. Then, clean substrates were treated by UV-Ozone for 15 min, and transferred to a CreaPhys organic evaporator inside an MBraun N2 glovebox (O2 and H2O levels below 0.5 ppm). A spiro-TTB hole extraction layer (25 nm for small-scale and 60 nm for 9.46 cm2 device) was evaporated on the conductive substrates, followed by mixed cation lead mixed halide perovskite deposition. The substrates are transferred to a CreaPhys PEROvap evaporator inside an MBraun N2 glovebox (O2 and H2O levels below 0.5 ppm), where the perovskite deposition is conducted. The chamber was pumped down to a pressure of 1–3 × 10−6 mbar for the deposition. We employed a specifically designed cooling system that maintains the evaporator walls, source shutters and shields at −20 °C throughout the entire process. This functionality minimises re-evaporation of the precursors and cross-contamination between sources, ensuring fine control over the evaporation rates and high reproducibility. To this end, FAI, PbI2 and CsBr were filled into three different crucibles, using fresh FAI and PbI2 powders for every deposition. The tooling factor of each chemical was calibrated by checking the film thicknesses by profilometry inside the N2-filled glovebox (DEKTAK XT profilometer). Three quartz crystal microbalances (QCMs) mounted on top of the vapor sources allowed us to monitor the deposition rate of each source and control the composition. The substrate temperature was maintained at around 18 °C. The distance between evaporator sources and substrate holder is around 35 cm.48 The perovskite films were post-annealed at 120 °C for 20 min. After cooling down to room temperature, 25 nm of C60 and 8 nm of BCP were sequentially evaporated on the perovskite film for small-scale devices, whereas 40 nm of C60 and 15 nm of BCP were deposited on 9.46 cm2 samples. A 100 nm layer of Cu was lastly evaporated in the same perovskite deposition system.Synthesis of the CO2 reduction catalyst
The Cu92In8 alloy catalyst was synthesised on 2.3 × 6 cm2 Cu foil substrates via a template-assisted electrodeposition method following previously reported protocols.54,68 Prior to electrodeposition, the Cu foil substrates were electropolished in 50% ortho-phosphoric acid by applying 2.0 V for 120 s, rinsed in Milli-Q water and dried under air. The active area for electrodeposition (2.3 × 4.3 cm2) was set by masking the substrate with Kapton tape, which is inert and stable under acidic medium. The bimetallic catalyst electrodeposition was conducted in a clean glass beaker containing copper sulfate and indium sulfate precursor salts (total 0.05 M) in 1.5 M sulfuric acid solution (pH ∼ 0.5). A 3-electrode set-up was used with the electropolished Cu foil as working electrode, a Ag/AgCl (double junction, saturated NaCl, Metrohm, Switzerland) electrode as reference, and another 42 cm2 Cu foil as counter electrode (Fig. S5, ESI†). For the galvanostatic deposition process, a current density of j = −0.45 A cm−2 was applied for 100 s to prepare the large-scale catalyst. After electrodeposition, the catalyst was cleaned by dipping into Milli-Q water for 120 s and dried under N2 stream at room temperature.BiVO4/TiCo photoanodes
10 cm2 BiVO4 electrodes were prepared by adapting previously reported methods.46,47 Unpatterned 3 × 5 cm2 FTO substrates were first cleaned in Piranha solution. An electrodeposition solution was prepared by adding 200 mL of a 0.02 M Bi(NO3)3·5H2O, 0.4 M NaI aqueous solution to 90 mL of a 0.3 M benzoquinone solution in ethanol. BiOI was electrodeposited onto FTO substrates by applying −0.3 V vs. Ag/AgCl for 5 s and −0.1 V vs. Ag/AgCl for 180 s, with graphite foil as the counter electrode. To allow rapid electrode fabrication, FTO substrates were directly clamped to the working electrode and fully immersed in the electrodeposition solution, resulting in a near-complete BiOI coverage. 40 μL cm−2 of a 0.4 M VO(acac)2 (acac: acetylacetonate) solution was dropcast onto BiOI samples and annealed at 723 K for 1 h with a 1 K min−1 ramp rate. The resulting electrodes were stirred in 0.2 M NaOH solution for 0.5–2 h to wash away excess V2O5 from the BiVO4 film. A TiCoOx (TiCo) O2 evolution catalyst was deposited on BiVO4 by spin coating 10 μL cm−2 of a 4.8 mg mL−1 [Ti4O(OEt)15(CoCl)] solution in toluene at 2000 rpm for 10 s under air. A 0.5 × 5 cm2 stripe of BiVO4 was scratched away using a razor blade, to expose the underlying FTO for photoanode wiring. The photoactive area of ∼10 cm2 was set by masking the BiVO4 near the electrode edges with Araldite 5-Minute Rapid two component epoxy, which also insulated the electrical contact.Perovskite-BiVO4 artificial leaves
Perovskite photocathodes were first encapsulated using a conductive GE paste, which provides increased moisture protection and enables a direct catalyst adhesion without additional binders. The paste was prepared by mixing graphite powder with Araldite Standard two component epoxy in a 3:4 mass ratio. The GE paste was evenly spread over the Cu contact layer of the PV device, before attaching the Cu92In8 electrocatalyst. The GE was allowed to settle for 24 hours, before sealing the edges and the exposed wiring with Araldite 5-Minute Rapid two component epoxy.55,69 Alternatively, a 5 nm thin Pt film was sputtered onto hardened, exposed graphite epoxy as H2 evolution catalyst. The same Araldite Rapid epoxy was used to attach the perovskite and BiVO4 photoelectrodes in a back-to-back, tandem PEC configuration. Perovskite-BiVO4 artificial leaves (as those shown in Fig. 3) were connected to potentiostats using PVC insulated wires (Alpha Wire 3050 WH001). In case of standalone devices for outdoor tests, both photoelectrodes were directly connected using a thin strip of 3M conductive Cu tape. Its edges were coated with RS PRO Ag conductive paste to improve electrical contact with ITO or FTO, respectively.
Photovoltaic characterization
We performed current density–voltage (J–V) curve measurements of 9.46 cm2 devices with illumination from a Xe lamp (Abet Sun 2000 Solar Simulator, AAB class) with 1 sun intensity (100 mW cm−2, AM 1.5G), calibrated with a reference Si diode. The device performance was recorded with a Keithley 2636A source meter, controlled by a LabVIEW program. The J–V curve is obtained by scanning from 1.2 to −0.1 V (reverse scan) and from −0.1 to 1.2 V (forward scan) with a step size of 20 mV and a delay time of 100 ms. J–V curves of small-scale devices were measured in air using a custom-built Cicci setup, with a forward/reverse scan rate of 200 mV s−1. All solar cells were measured under the standard 1 sun AM 1.5G spectrum using a Sunbrick Base-UV large area AAA LED solar simulator (G2V), with a spectral mismatch of <5%. The system was calibrated separately using both a silicon KG5 filter reference cell (RERA Solutions, model number: RK5N3199) and an Avantes (AvaSpec-ULS2048CL-EVO-FCPC) spectroradiometer. Computer numerical controlled (CNC) metal masks with a circular aperture area of 0.118 cm2 were used. Devices were mounted in a custom designed holder (Cicci) with a thermoelectric cell base to maintain the temperature at 25 °C during measurements.Photoelectrochemical characterization
A LOT-QD LS0816-H large-area solar simulator was calibrated to 1 sun (100 mW cm−2) using a certified Newport 843-R optical power meter. PEC experiments were conducted using Ivium CompactStat and Biologic VSP potentiostats, without stirring, at room temperature, in custom-made Perspex (polymethyl methacrylate) reactors designed in SolidWorks. Tests were conducted in 0.1 M H3BO3, 0.1 M K2SO4, under N2 (adjusted to pH 8.5) for water splitting, and in 0.5 M KHCO3, under CO2 (pH 7.4) for CO2 reduction. 3-Electrode experiments were performed with a Ag/AgCl/NaCl(sat) (Basi MW-2030) reference and Pt mesh counter electrode, whereas potentials were converted to the RHE scale following the equation: E (V vs. RHE) = E (V vs. Ag/AgCl) + 0.059 V × pH + 0.197 V (at 298 K). All CV scans were recorded at a 10 mV s−1 scan rate unless otherwise mentioned. The edges of BiVO4 photoanodes were only covered by black tape during laboratory benchmarking to prevent diffuse irradiation from reaching the perovskite light absorber. CO and H2 amounts were quantified using a Shimadzu GC-2010 Plus gas chromatograph by manual sampling from the reactor headspace. Product amounts obtained in laboratory experiments were corrected for leakage using a previously reported model,56 whereas dissolved gases were accounted for using Henry's law.47 No leakage correction could be applied to outdoor experiments due to the small number of data points and external factors (variable light irradiation and atmospheric conditions, solution and gas leakage).Materials characterization
A Bruker D8 ADVANCE system with a Cu focus X-ray tube (Ka: 1.54 Å) and 40 kV operation voltage was used to obtain XRD patterns. During the measurement, the samples were kept in air. The scan range for 2θ was from 5° to 55° with a step size of 0.01° and a dwell time of 0.15 s per step. SEM and EDX analysis were performed using a TESCAN MIRA3 field emission gun-scanning electron microscope equipped with an Oxford Instruments Aztec Energy X-maxN 80 EDX system. A Thermo Scientific Talos F200X G2 TEM (operating voltage 200 kV) was used for HAADF-STEM images and EDX maps. 300-mesh Ni grids with holey carbon film were used for TEM sample preparation. Powder XRD (PXRD) spectra of the catalyst were recorded by a Panalytical X’Pert Pro (K-α Cu radiation) diffractometer using a 1° min−1 scan rate.Author contributions
V. A., Y.-H. C., M. R., M. A., S. D. S. and Er. R. designed the project. Y.-H. C. optimised the vacuum-deposited solar cells. Y.-H. C., M. A. and Ed. R. fabricated large area devices in 2022, and T. K. prepared devices for characterisation afterwards. Y.-H. C. and T. K. characterised the perovskite materials and solar cells. V. A. developed the perovskite encapsulation, and helped design the large-scale Perspex PEC reactors. M. R. designed, synthesized, and characterized the CO2 reduction catalyst. V. A., M. R., Ed. R. fabricated and characterized the perovskite-BiVO4 artificial leaves. V. A., Y.-H. C., M. R., M. A., S. D. S. and Er. R. set up and conducted the outdoors experiments in Ispra, Italy. V. A. drafted the manuscript, all authors contributed to the discussion and completion of the manuscript. S. D. S. and Er. R. supervised the work.Data availability
The raw data that support the findings of this study are available from the University of Cambridge data repository, https://doi.org/10.17863/CAM.115131.Conflicts of interest
S. D. S. is a co-founder of Swift Solar, Inc., developing perovskite PV modules.Acknowledgements
This work was supported by the Winton Programme for the Physics of Sustainability and St John's College (Title A research fellowship to V. A.); the Taiwan Cambridge Trust and Rank Prize fund (to Y.-H. C.); a Marie Sklodowska-Curie Individual European Fellowship (SolarFUEL, GAN 839763, to M. R.); a SNSF EPM Fellowship (P2BEP2_184483, to M. R.); a Leverhulme Trust and Royal Academy of Engineering Research Fellowship and the Marie Skłodowska-Curie actions under EU Horizon 2020 (GAN 841386, to M. A.); an ERC Starting Grant ‘HYPERION’ (756962, to S. D. S.), the Royal Society and Tata Group (UF150033, URF\R\221026 and RF\ERE\221004, to S. D. S.); UK Research and Innovation (ERC Advanced grant EP/X030563/1 to E. R.), the UK Department of Science, Innovation and Technology and the Royal Academy of Engineering Chair in Emerging Technologies programme (CIET-2324-83 to E. R.) as well as the Yusuf Hamied Department of Chemistry, the School of Technology and Maxwell Centre, University of Cambridge (fabrication costs and travel). Part of this work was undertaken using equipment facilities and a grant provided by the Henry Royce Institute, via the grant Henry Royce Institute, Cambridge Equipment EP/P024947/1 with additional funding from the “Centre for Advanced Materials for Integrated Energy Systems (CAM-IES)” (EP/P007767/1). We thank Matthew Woolley and Simon Dowe (Mechanical Workshop, Yusuf Hamied Department of Chemistry, University of Cambridge) for aid in constructing the large-scale reactors. We thank Sebastian D. Pike and Dominic S. Wright (University of Cambridge) for a sample of the TiCoOx precursor. For the purpose of open access, the authors have applied a Creative Commons Attribution (CC BY) license to any Author Accepted Manuscript version arising from this submission.References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051, DOI:10.1021/ja809598r.
- M. A. Green, E. D. Dunlop, M. Yoshita, N. Kopidakis, K. Bothe, G. Siefer, D. Hinken, M. Rauer, J. Hohl-Ebinger and X. Hao, Prog. Photovoltaics Res. Appl., 2024, 32, 425–441, DOI:10.1002/pip.3831.
- National Renewable Energy Laboratory, Best Research-Cell Efficiency Chart, available at: https://www.nrel.gov/pv/cell-efficiency.html, accessed 17 November 2024.
- X. Tian, S. D. Stranks and F. You, Nat. Sustain., 2021, 4, 821–829, DOI:10.1038/s41893-021-00737-z.
- A. M. K. Fehr, T. G. Deutsch, F. M. Toma, M. S. Wong and A. D. Mohite, ACS Energy Lett., 2023, 8, 4976–4983, DOI:10.1021/acsenergylett.3c01865.
- J. Gao, F. Sahli, C. Liu, D. Ren, X. Guo, J. Werner, Q. Jeangros, S. M. Zakeeruddin, C. Ballif, M. Grätzel and J. Luo, Joule, 2019, 3, 2930–2941, DOI:10.1016/j.joule.2019.10.002.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596, DOI:10.1126/science.1258307.
- J. Chen, C. Dong, H. Idriss, O. F. Mohammed and O. M. Bakr, Adv. Energy Mater., 2019, 10, 1902433, DOI:10.1002/aenm.201902433.
- H. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123, DOI:10.1021/acsenergylett.0c00058.
- S. Peng, Z. Yang, M. Sun, L. Yu and Y. Li, Adv. Mater., 2023, 35, 2304711, DOI:10.1002/adma.202304711.
- H. Choi, S. Seo, C. J. Yoon, J.-B. Ahn, C.-S. Kim, Y. Jung, Y. Kim, F. M. Toma, H. Kim and S. Lee, Adv. Sci., 2023, 10, 2303106, DOI:10.1002/advs.202303106.
- Y. Choi, R. Mehrotra, S.-H. Lee, T. V. T. Nguyen, I. Lee, J. Kim, H.-Y. Yang, H. Oh, H. Kim, J.-W. Lee, Y. H. Kim, S.-Y. Jang, J.-W. Jang and J. Ryu, Nat. Commun., 2022, 13, 5709, DOI:10.1038/s41467-022-33435-1.
- A. M. K. Fehr, A. Agrawal, F. Mandani, C. L. Conrad, Q. Jiang, S. Y. Park, O. Alley, B. Li, S. Sidhik, I. Metcalf, C. Botello, J. L. Young, J. Even, J. C. Blancon, T. G. Deutsch, K. Zhu, S. Albrecht, F. M. Toma, M. Wong and A. D. Mohite, Nat. Commun., 2023, 14, 3797, DOI:10.1038/s41467-023-39290-y.
- I. S. Kim, M. J. Pellin and A. B. F. Martinson, ACS Energy Lett., 2019, 4, 293–298, DOI:10.1021/acsenergylett.8b01661.
- J.-H. Kim, S. Seo, J.-H. Lee, H. Choi, S. Kim, G. Piao, Y. R. Kim, B. Park, J. Lee, Y. Jung, H. Park, S. Lee and K. Lee, Adv. Funct. Mater., 2021, 31, 2008277, DOI:10.1002/adfm.202008277.
- R. Rhee, T. G. Kim, G. Y. Jang, G. Bae, J. H. Lee, S. Lee, S. Kim, S. Jeon and J. H. Park, Carbon Energy, 2023, 5, e232, DOI:10.1002/cey2.232.
- Z. Song, C. Li, L. Chen, K. Dolia, S. Fu, N. Sun, Y. Li, K. Wyatt, J. L. Young, T. G. Deutsch and Y. Yan, ACS Energy Lett., 2023, 8, 2611–2619, DOI:10.1021/acsenergylett.3c00654.
- Z. Xu, L. Chen, C. J. Brabec and F. Guo, Small Methods, 2023, 7, 2300619, DOI:10.1002/smtd.202300619.
- H. Chen, M. Zhang, T. Tran-Phu, R. Bo, L. Shi, I. Di Bernardo, J. Bing, J. Pan, S. Singh, J. Lipton-Duffin, T. Wu, R. Amal, S. Huang, A. W. Y. Ho-Baillie and A. Tricoli, Adv. Funct. Mater., 2020, 31, 2008245, DOI:10.1002/adfm.202008245.
- S. Park, W. J. Chang, C. W. Lee, S. Park, H.-Y. Ahn and K. T. Nam, Nat. Energy, 2016, 2, 16185, DOI:10.1038/nenergy.2016.185.
- H. Wang, X. Wang, R. Chen, H. Zhang, X. Wang, J. Wang, J. Zhang, L. Mu, K. Wu, F. Fan, X. Zong and C. Li, ACS Energy Lett., 2019, 4, 40–47, DOI:10.1021/acsenergylett.8b01830.
- H. Zhang, Z. Yang, W. Yu, H. Wang, W. Ma, X. Zong and C. Li, Adv. Energy Mater., 2018, 8, 1800795, DOI:10.1002/aenm.201800795.
- V. Andrei, B. Reuillard and E. Reisner, Nat. Mater., 2020, 19, 189–194, DOI:10.1038/s41563-019-0501-6.
- J. Chen, J. Yin, X. Zheng, H. Ait Ahsaine, Y. Zhou, C. Dong, O. F. Mohammed, K. Takanabe and O. M. Bakr, ACS Energy Lett., 2019, 4, 1279–1286, DOI:10.1021/acsenergylett.9b00751.
- H. Zhang, Y. Chen, H. Wang, H. Wang, W. Ma, X. Zong and C. Li, Adv. Energy Mater., 2020, 10, 2002105, DOI:10.1002/aenm.202002105.
- S. K. Kuk, Y. Ham, K. Gopinath, P. Boonmongkolras, Y. Lee, Y. W. Lee, S. Kondaveeti, C. Ahn, B. Shin, J.-K. Lee, S. Jeon and C. B. Park, Adv. Energy Mater., 2019, 9, 1900029, DOI:10.1002/aenm.201900029.
- Y.-F. Xu, M.-Z. Yang, B.-X. Chen, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663, DOI:10.1021/jacs.7b00489.
- Y. Jiang, J.-F. Liao, H.-Y. Chen, H.-H. Zhang, J.-Y. Li, X.-D. Wang and D.-B. Kuang, Chem, 2020, 6, 766–780, DOI:10.1016/j.chempr.2020.01.005.
- M. Daboczi, J. Cui, F. Temerov and S. Eslava, Adv. Mater., 2023, 35, 2304350, DOI:10.1002/adma.202304350.
- D. Hansora, J. W. Yoo, R. Mehrotra, W. J. Byun, D. Lim, Y. K. Kim, E. Noh, H. Lim, J.-W. Jang, S. I. Seok and J. S. Lee, Nat. Energy, 2024, 9, 272–284, DOI:10.1038/s41560-023-01438-x.
- T. G. Kim, J. H. Lee, G. Hyun, S. Kim, D. H. Chun, S. Lee, G. Bae, H.-S. Oh, S. Jeon and J. H. Park, ACS Energy Lett., 2022, 7, 320–327, DOI:10.1021/acsenergylett.1c02326.
- H. Choi, Y. Y. Kim, S. Seo, Y. Jung, S.-M. Yoo, C. S. Moon, N. J. Jeon, S. Lee, K. Lee, F. M. Toma, J. Seo and S. Lee, Adv. Energy Mater., 2023, 13, 2300951, DOI:10.1002/aenm.202300951.
- J. Yun, H. Lee, Y. S. Park, W. Jeong, C.-S. Jeong, J. Lee, J. Lee, J. Tan, S. Ma, G. Jang, C. U. Lee, S. Moon, H. Im, S. Lee, D.-Y. Yee, J.-H. Kim and J. Moon, Adv. Energy Mater., 2023, 13, 2301693, DOI:10.1002/aenm.202301693.
- S. K. Karuturi, H. Shen, A. Sharma, F. J. Beck, P. Varadhan, T. Duong, P. R. Narangari, D. Zhang, Y. Wan, J.-H. He, H. H. Tan, C. Jagadish and K. Catchpole, Adv. Energy Mater., 2020, 10, 2000772, DOI:10.1002/aenm.202000772.
- W. Yang, J. Park, H.-C. Kwon, O. S. Hutter, L. J. Phillips, J. Tan, H. Lee, J. Lee, S. D. Tilley, J. D. Major and J. Moon, Energy Environ. Sci., 2020, 13, 4362–4370, 10.1039/D0EE02959A.
- I. Poli, U. Hintermair, M. Regue, S. Kumar, E. V. Sackville, J. Baker, T. M. Watson, S. Eslava and P. J. Cameron, Nat. Commun., 2019, 10, 2097, DOI:10.1038/s41467-019-10124-0.
- S. Nam, C. T. K. Mai and I. Oh, ACS Appl. Mater. Interfaces, 2018, 10, 14659–14664, DOI:10.1021/acsami.8b00686.
- A. Tayyebi, R. Mehrotra, M. A. Mubarok, J. Kim, M. Zafari, M. Tayebi, D. Oh, S.-H. Lee, J. E. Matthews, S.-W. Lee, T. J. Shin, G. Lee, T. F. Jaramillo, S.-Y. Jang and J.-W. Jang, Nat. Catal., 2024, 7, 510–521, DOI:10.1038/s41929-024-01133-4.
- S. Bhattacharjee, V. Andrei, C. Pornrungroj, M. Rahaman, C. M. Pichler and E. Reisner, Adv. Funct. Mater., 2022, 32, 2109313, DOI:10.1002/adfm.202109313.
- X.-D. Wang, Y.-H. Huang, J.-F. Liao, Z.-F. Wei, W.-G. Li, Y.-F. Xu, H.-Y. Chen and D.-B. Kuang, Nat. Commun., 2021, 12, 1202, DOI:10.1038/s41467-021-21487-8.
- H. Huang, H. Yuan, J. Zhao, G. Solís-Fernández, C. Zhou, J. W. Seo, J. Hendrix, E. Debroye, J. A. Steele, J. Hofkens, J. Long and M. B. J. Roeffaers, ACS Energy Lett., 2019, 4, 203–208, DOI:10.1021/acsenergylett.8b01698.
- I. Rosa-Pardo, D. Zhu, A. Cortés-Villena, M. Prato, L. de Trizio, L. Manna, R. E. Galian and J. Pérez-Prieto, ACS Energy Lett., 2023, 8, 2789–2798, DOI:10.1021/acsenergylett.3c00771.
- X. Zhu, Y. Lin, J. San Martin, Y. Sun, D. Zhu and Y. Yan, Nat. Commun., 2019, 10, 2843, DOI:10.1038/s41467-019-10634-x.
- D. Wang, S. H. Lee, S. Han, J. Kim, N. V. T. Trang, K. Kim, E.-G. Choi, P. Boonmongkolras, Y. W. Lee, B. Shin, Y. H. Kim and C. B. Park, Green Chem., 2020, 22, 5151–5160, 10.1039/D0GC01521K.
- D. Cardenas-Morcoso, A. F. Gualdrón-Reyes, A. B. Ferreira Vitoreti, M. García-Tecedor, S. J. Yoon, M. S. de la Fuente, I. Mora-Seró and S. Gimenez, J. Phys. Chem. Lett., 2019, 10, 630–636, DOI:10.1021/acs.jpclett.8b03849.
- V. Andrei, G. M. Ucoski, C. Pornrungroj, C. Uswachoke, Q. Wang, D. S. Achilleos, H. Kasap, K. P. Sokol, R. A. Jagt, H. Lu, T. Lawson, A. Wagner, S. D. Pike, D. S. Wright, R. L. Z. Hoye, J. L. MacManus-Driscoll, H. J. Joyce, R. H. Friend and E. Reisner, Nature, 2022, 608, 518–522, DOI:10.1038/s41586-022-04978-6.
- V. Andrei, R. L. Z. Hoye, M. Crespo-Quesada, M. Bajada, S. Ahmad, M. de Volder, R. Friend and E. Reisner, Adv. Energy Mater., 2018, 8, 1801403, DOI:10.1002/aenm.201801403.
- Y.-H. Chiang, M. Anaya and S. D. Stranks, ACS Energy Lett., 2020, 5, 2498–2504, DOI:10.1021/acsenergylett.0c00839.
- Y.-H. Chiang, K. Frohna, H. Salway, A. Abfalterer, L. Pan, B. Roose, M. Anaya and S. D. Stranks, ACS Energy Lett., 2023, 8, 2728–2737, DOI:10.1021/acsenergylett.3c00564.
- A. Al-Ashouri, A. Magomedov, M. Roß, M. Jošt, M. Talaikis, G. Chistiakova, T. Bertram, J. A. Márquez, E. Köhnen, E. Kasparavičius, S. Levcenco, L. Gil-Escrig, C. J. Hages, R. Schlatmann, B. Rech, T. Malinauskas, T. Unold, C. A. Kaufmann, L. Korte, G. Niaura, V. Getautis and S. Albrecht, Energy Environ. Sci., 2019, 12, 3356–3369, 10.1039/C9EE02268F.
- F. Sahli, J. Werner, B. A. Kamino, M. Bräuninger, R. Monnard, B. Paviet-Salomon, L. Barraud, L. Ding, J. J. Diaz Leon, D. Sacchetto, G. Cattaneo, M. Despeisse, M. Boccard, S. Nicolay, Q. Jeangros, B. Niesen and C. Ballif, Nat. Mater., 2018, 17, 820–826, DOI:10.1038/s41563-018-0115-4.
- H. Lu, V. Andrei, K. J. Jenkinson, A. Regoutz, N. Li, C. E. Creissen, A. E. H. Wheatley, H. Hao, E. Reisner, D. S. Wright and S. D. Pike, Adv. Mater., 2018, 30, 1804033, DOI:10.1002/adma.201804033.
- I. Y. Ahmet, Y. Ma, J.-W. Jang, T. Henschel, B. Stannowski, T. Lopes, A. Vilanova, A. Mendes, F. F. Abdi and R. van de Krol, Sustainable Energy Fuels, 2019, 3, 2366–2379, 10.1039/C9SE00246D.
- M. Rahaman, V. Andrei, C. Pornrungroj, D. Wright, J. J. Baumberg and E. Reisner, Energy Environ. Sci., 2020, 13, 3536–3543, 10.1039/D0EE01279C.
- C. Pornrungroj, V. Andrei, M. Rahaman, C. Uswachoke, H. J. Joyce, D. S. Wright and E. Reisner, Adv. Funct. Mater., 2021, 31, 2008182, DOI:10.1002/adfm.202008182.
- V. Andrei, R. A. Jagt, M. Rahaman, L. Lari, V. K. Lazarov, J. L. MacManus-Driscoll, R. L. Z. Hoye and E. Reisner, Nat. Mater., 2022, 21, 864–868, DOI:10.1038/s41563-022-01262-w.
- F. Toor, T. G. Deutsch, J. W. Pankow, W. Nemeth, A. J. Nozik and H. M. Branz, J. Phys. Chem. C, 2012, 116, 19262–19267 Search PubMed.
- P. Buabthong, J. M. Evans, K. Z. Rinaldi, K. M. Kennedy, H. J. Fu, Z. P. Ifkovits, T.-J. Kuo, B. S. Brunschwig and N. S. Lewis, ACS Energy Lett., 2021, 6, 3709–3714, DOI:10.1021/acsenergylett.1c01174.
- European Commission, Directorate-General for Research and Innovation, Artificial photosynthesis: fuel from the sun: EIC Horizon Prize: the chancellor masters and scholars of the University of Cambridge, Publications Office of the European Union, 2022 DOI:10.2777/682437.
- A. Hankin, F. E. Bedoya-Lora, C. K. Ong, J. C. Alexander, F. Petter and G. H. Kelsall, Energy Environ. Sci., 2017, 10, 346–360, 10.1039/C6EE03036J.
- M. R. Singh, C. Xiang and N. S. Lewis, Sustainable Energy Fuels, 2017, 1, 458–466, 10.1039/C7SE00062F.
- T. Yamada, H. Nishiyama, H. Akatsuka, S. Nishimae, Y. Ishii, T. Hisatomi and K. Domen, ACS Eng. Au, 2023, 3, 352–363, DOI:10.1021/acsengineeringau.3c00034.
- A. R. A. Maragno, G. Cwicklinski, M. Matheron, R. Vanoorenberghe, J.-M. Borgard, A. Morozan, J. Fize, M. Pellat, C. Cavazza, V. Artero and S. Charton, Joule, 2024, 8, 2325–2341, DOI:10.1016/j.joule.2024.05.012.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307, DOI:10.1038/s41586-021-03907-3.
- C. G. Manning, Technology Readiness Levels, available at: https://www.nasa.gov/directorates/somd/space-communications-navigation-program/technology-readiness-levels/, accessed 1 February 2024.
- European Space Agency, Technology Readiness Levels (TRL), available at: https://www.esa.int/Enabling_Support/Space_Engineering_Technology/Shaping_the_Future/Technology_Readiness_Levels_TRL, accessed 1 February 2024.
- K. P. Sokol and V. Andrei, Nat. Rev. Mater., 2022, 7, 251–253, DOI:10.1038/s41578-022-00432-1.
- H.-C. Shin and M. Liu, Chem. Mater., 2004, 16, 5460–5464, DOI:10.1021/cm048887b.
- V. Andrei, K. Bethke and K. Rademann, Phys. Chem. Chem. Phys., 2016, 18, 10700–10707, 10.1039/C5CP06828B.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee05780e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |