
Pulsed laser deposition of delafossite oxide thin films on YSZ (001) substrates as solar water splitting photocathodes†
Chenyu
Zhou
 a,
Atiya
Banerjee
a,
Esteban Luis
Fornero
b,
Zhaoyi
Xi
ac,
Xiao
Tong
a,
Eli
Stavitski
d,
Xiaohui
Qu
*a,
Sara E.
Mason
*a,
Dario J.
Stacchiola
*a and
Mingzhao
Liu
*a
a,
Atiya
Banerjee
a,
Esteban Luis
Fornero
b,
Zhaoyi
Xi
ac,
Xiao
Tong
a,
Eli
Stavitski
d,
Xiaohui
Qu
*a,
Sara E.
Mason
*a,
Dario J.
Stacchiola
*a and
Mingzhao
Liu
*a
aCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, New York 11973, USA. E-mail: xiaqu@bnl.gov; smason@bnl.gov; djs@bnl.gov; mzliu@bnl.gov
bInstituto de Desarrollo Tecnológico para la Industria Química, UNL-CONICET, Güemes 3450, 3000 Santa Fe, Argentina
cDepartment of Materials Science and Chemical Engineering, Stony Brook University, Stony Brook, New York 11794, USA
dNational Synchrotron Light Source II, Brookhaven National Laboratory, Upton, New York 11973, USA
First published on 15th November 2024
Abstract
Development of solar energy converters with earth-abundant and environmentally friendly materials is one of the key routes explored towards a sustainable future. In this work, crystalline delafossite-phase CuAlO2 and CuFeO2 thin film solar water splitting photocathodes were fabricated using pulsed laser deposition. It was found that the desired delafossite phase was formed only after high temperature annealing in an oxygen-free atmosphere. The homogeneous delafossite bulk structure of the films was determined by correlating simulation results from first-principles calculations with synchrotron-based X-ray absorption near edge structure (XANES) spectroscopy. Both CuAlO2 and CuFeO2 photocathodes are active for solar water splitting, with the latter more efficient due to its narrower band gap and improved light absorption.
Environmental significanceThis study investigates CuAlO2 and CuFeO2 thin film photocathodes for photoelectrochemical (PEC) water splitting, a sustainable method for converting solar energy into clean fuels. By exploring pulsed laser deposition to synthesize the delafossite phase of these materials, terminated by active Cu(I) sites, we aim to simplify the catalyst design, eliminating the need for complex heterostructures with protective layers and cocatalysts. Our measurements show notable photoelectrochemical (PEC) activities for both CuAlO2 and CuFeO2. Through combined synthesis, characterization, electrochemical measurements, and modeling, this work addresses the need to advance Cu(I) electrocatalysts. Our findings advance PEC systems, promoting renewable energy storage and reducing carbon emissions. |
Introduction
In recent decades, Cu(I)-based delafossite oxide materials have emerged as prominent candidates in diverse technological applications, mainly embodying distinct yet related roles: transparent conducting oxides1–3 and solar energy converters such as photoelectrodes for solar cells,4–6 solar hydrogen evolution,7–11 and CO2 conversion.12,13 Their unique crystal structures, exceptional electronic properties, and promising electrochemical behavior have spurred extensive research efforts aimed at harnessing their potential for renewable energy, optoelectronics, and beyond. An important family of delafossite-type oxides are formulated as CuMO2 (M = Al, Fe, Cr, Ga, etc.) compounds, where Cu and M have oxidation states of +1 and +3, respectively.11 Characterized by a layered arrangement featuring linearly coordinated Cu+ cations and layers of MO6 octahedra that share edges, delafossite oxide typically adopts a rhombohedral R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m structure with trigonal symmetry (Fig. 1a).
m structure with trigonal symmetry (Fig. 1a).
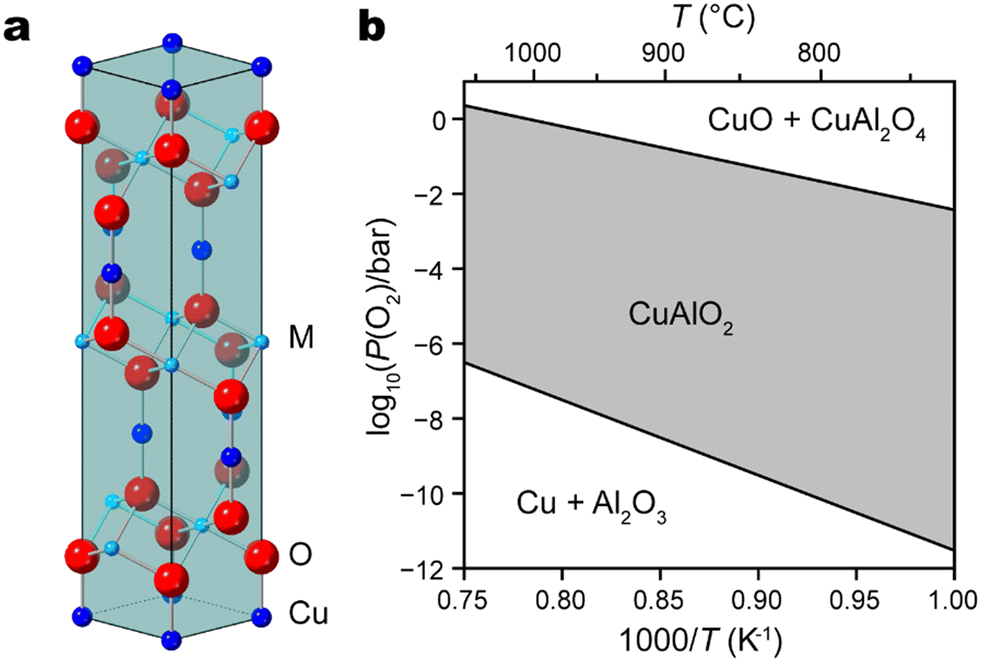 | ||
Fig. 1 (a) Crystal structure and unit cell of delafossite CuMO2 (Cu (blue), M = Al or Fe (cyan), O (red)). A few atoms outside of the unit cell are included for better view of the coordination shell around M atoms. (b) Ellingham diagram of the Cu–Al–O system, with the atomic ratio Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al = 1:1. :Al = 1:1. | ||
A wide range of deposition techniques have been reported to synthesize CuMO2 films, including sol–gel,14,15 hydrothermal,16–18 chemical spray pyrolysis,19 chemical vapor deposition (CVD),20,21 sputtering,22,23 and pulsed laser deposition (PLD).24–30 The last one is particularly attractive due to its capacity to transfer stoichiometry from target material to thin film. Prior research endeavors concerning PLD-grown delafossite materials are summarized in Table 1. In order to maintain Cu in its optimally active intermediate oxidation state of +1, the chemical potential of oxygen must be carefully controlled during delafossite synthesis and post-growth annealing at higher temperature is usually needed. As illustrated by the Ellingham diagram of CuAlO2, higher temperature will indeed stabilize the desired delafossite phase in an atmosphere of moderate oxygen partial pressure (Fig. 1b).
| Delafossite | Substrate | T (°C) | P O2 (mTorr) | Annealing | Crystal phase | Ref. |
|---|---|---|---|---|---|---|
| a Listed are the temperature and pressure conditions for the optimal outcome. b If the in-plane orientation of the thin film is tested to match rhombohedral crystal system, it is listed as epitaxial single-phase. If only the out-of-plane orientation is examined, it is listed as c-axis oriented single phase, otherwise. c Mixed with CuO and Al2O3 powders in closed crucible. Annealing is only for smoothing the surface of thin film. | ||||||
| CuAlO2 | SA (001) | 700 | 100 | — | c-Axis oriented single phase (00l)b | 1 |
| SA (001) | 100–200 | 100 | 1050 °C, air, 1.5 hc | c-Axis oriented single phase (00l) | 24 | |
| SA (001) | 500 | ∼20 | 900 °C, N2, 1 h | c-Axis oriented single phase (00l) | 25 | |
| SA (001) | 650 | 10 | 1100 °C, air, 30 min | c-Axis oriented single phase (00l) | 26 | |
| SA (001) | 700 | 100 | 1050 °C, air, 10 min | c-Axis oriented single phase (00l) | 27 | |
| CuFeO2 | SA (001) | 500 | 75 | 500 °C, O2, 10 min | Epitaxial single-phase (00l)b | 28 |
| SA (001) | 600 | 0.1 | — | c-Axis oriented single phase (00l) | 29 | |
| SA (001) | 600 | 0.1 | — | c-Axis oriented single phase (00l) | 30 | |
| AG | 750 | 1 | — | c-Axis oriented single phase (00l) | 31 | |
| SA (001) | 550–600 | 0.1 | — | Epitaxial single-phase (00l) | 32 | |
| SA (001) | 850 | 0.5 | — | Epitaxial single-phase (00l) | 33 | |
In addition, most delafossite growth efforts have been concentrated on substrates exhibiting trigonal or hexagonal symmetry, such as sapphire.1,24–30,32–34 Nonetheless, the insulating nature of sapphire poses challenges for applications necessitating high conductivity, such as photoelectrodes. Therefore, investigations in the realm of delafossite-based photovoltaic and photoelectrochemical research predominantly rely on fluorine-doped tin oxide (FTO) glass substrates, chosen for their transparency to light and efficient charge carrier transport.5,7,35–38 There are very few studies on other single crystalline substrate, such as yttrium-stabilized zirconia (YSZ), which has the advantage of being lattice-matched to indium tin oxide (ITO), a compelling alternative to FTO. Previously, we have demonstrated that epitaxial ITO layer can be grown on YSZ (001) for the fabrication of bismuth vanadate (BiVO4) photoanodes.39 However, to our knowledge, there is no previous studies on delafossite growth on this commonly available substrate.
In this work, we used PLD to fabricate CuAlO2 and CuFeO2 delafossite thin film photocathodes on ITO/YSZ (001) substrates and studied their solar water splitting activity in a photoelectrochemical cell as an example of their potential as energy converters. The structural properties of the deposited films were studied by thin film X-ray diffraction (XRD) and synchrotron-based X-ray absorption near edge structure (XANES) spectroscopy, demonstrating the growth of c-oriented single-phase delafossite thin films on YSZ (001) substrates. First-principles calculations of the XANES spectra confirmed that the thin films have atomic motifs matching the bulk structure.
Results and discussion
Delafossite thin films were prepared by pulsed laser deposition (PLD) on (001)-oriented yttrium-stabilized zirconia (YSZ) substrates, which were first coated with a thin layer of indium tin oxide (ITO) for electrical conductivity. According to its X-ray diffraction (XRD) pattern, the as-grown Cu–Al oxide thin film did not show any peaks corresponding to the delafossite phase, or any phases other than those in the ITO/YSZ substrate (Fig. 2a), which suggests that the as-grown Cu–Al oxide film was amorphous. Similarly, the XRD pattern of as-grown Cu–Fe oxide film did not contain any delafossite peaks, but showed the presence of the spinel phase of CuFe2O4 (Fig. 2b). According to the Ellingham diagram of the Cu–Fe–O system (ESI,† Fig. S1), the delafossite CuFeO2 is thermodynamically favored in a region with lower chemical potential of oxygen and higher temperature, while CuFe2O4 is stabilized in an oxygen-rich environment and at lower temperature.40 The observation suggests that the PLD chamber does not have an O2 partial pressure that is low enough to drive the CuFe2O4/CuFeO2 equilibrium in the favor of the latter.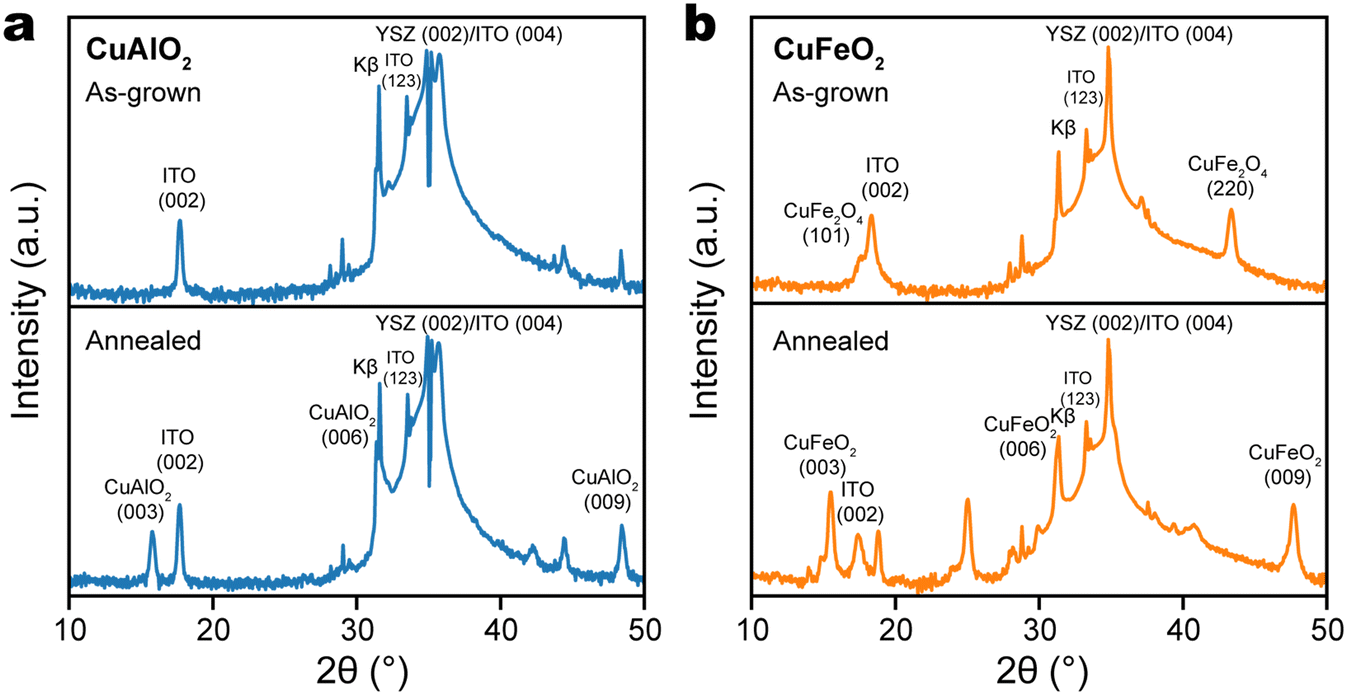 | ||
| Fig. 2 XRD patterns of (a) CuAlO2/ITO/YSZ and (b) CuFeO2/ITO/YSZ thin films, as-grown by PLD (top) and after annealing in N2 at 900 °C (bottom). | ||
To drive phase equilibrium toward the desired delafossite phase and to promote its crystallization, the thin film of Cu–Al and Cu–Fe oxides are processed by post-growth annealing at 900 °C in nitrogen. In the case of Cu–Al oxide, a single peak emerged at 15.78°, attributed to the CuAlO2 (003) plane, while the CuAlO2 (006) peak overlapped with the Kβ peak of YSZ (002) (Fig. 2a). This observation indicates the formation of a c-oriented single-phase CuAlO2 structure. In the case of Cu–Fe oxide, we observe the (003), (006), and (009) diffraction peaks of CuFeO2, respectively at 15.48°, 31.26°, and 47.68° (Fig. 2b). Because there was no epitaxial relationship between delafossite and the ITO/YSZ (001) substrate, the formation of delafossite phase required more strict control of conditions. We note that when the substrate is replaced with c-plane sapphire, c-axis oriented single-phase CuFeO2 was formed during PLD at the same deposition conditions (700 °C, 2 × 10−2 mbar N2 atmosphere), without the requirement of post-growth annealing (ESI,† Fig. S2a). Interestingly, the delafossite CuFeO2 phase can still form on sapphire even after a small amount of oxygen was introduced. As shown in ESI† Fig. S2b, both delafossite CuFeO2 and spinel CuFe2O4 phases can be identified from the XRD pattern of the Cu–Fe oxide film grown in 1 mTorr O2 (1.3 × 10−6 bar). This clearly reflects that a metastable thin film phase can be stabilized by reducing its surface energy through lattice matching to the substrate.
Although XRD confirmed the formation of delafossite phases of CuAlO2 and CuFeO2, in which Cu takes a formal oxidation number of +1, X-ray photoelectron spectroscopy (XPS) found that the surface of delafossite film was dominated by Cu +2 species. This was evident by the emergence of strong satellite peaks in the Cu 2p XPS spectra (Fig. 3a and c), which would be very weak if Cu had oxidation state of 0 or +1. Since XPS is a surface-sensitive technique, the observation suggested that the surface of delafossite films was oxidized upon air exposure. Chemically distinct surface Cu species are also indicated by the DFT optimized geometry, which is consistent with reported findings of Cu reconstructions at the (0001) surface of CuFeO2.41 The Al 2p peak of CuAlO2 was consistent with Al with oxidation state +3 and overlapped with the broader Cu 3p3/2 peak (Fig. 3b). The Fe 2p region of CuFeO2 must be fitted with two sets of 2p3/2–2p1/2 doublets, in addition to a pair of satellite peaks (Fig. 3d). However, this does not indicate the presence of Fe +2 species, since the lowest binding energy of Fe 2p3/2 remains at 710 eV, while Fe +2 would have Fe 2p3/2 binding energy at 709 eV. This is similar as the case of Fe2O3, which requires multiple sets of peaks to properly fit the Fe 2p features.42
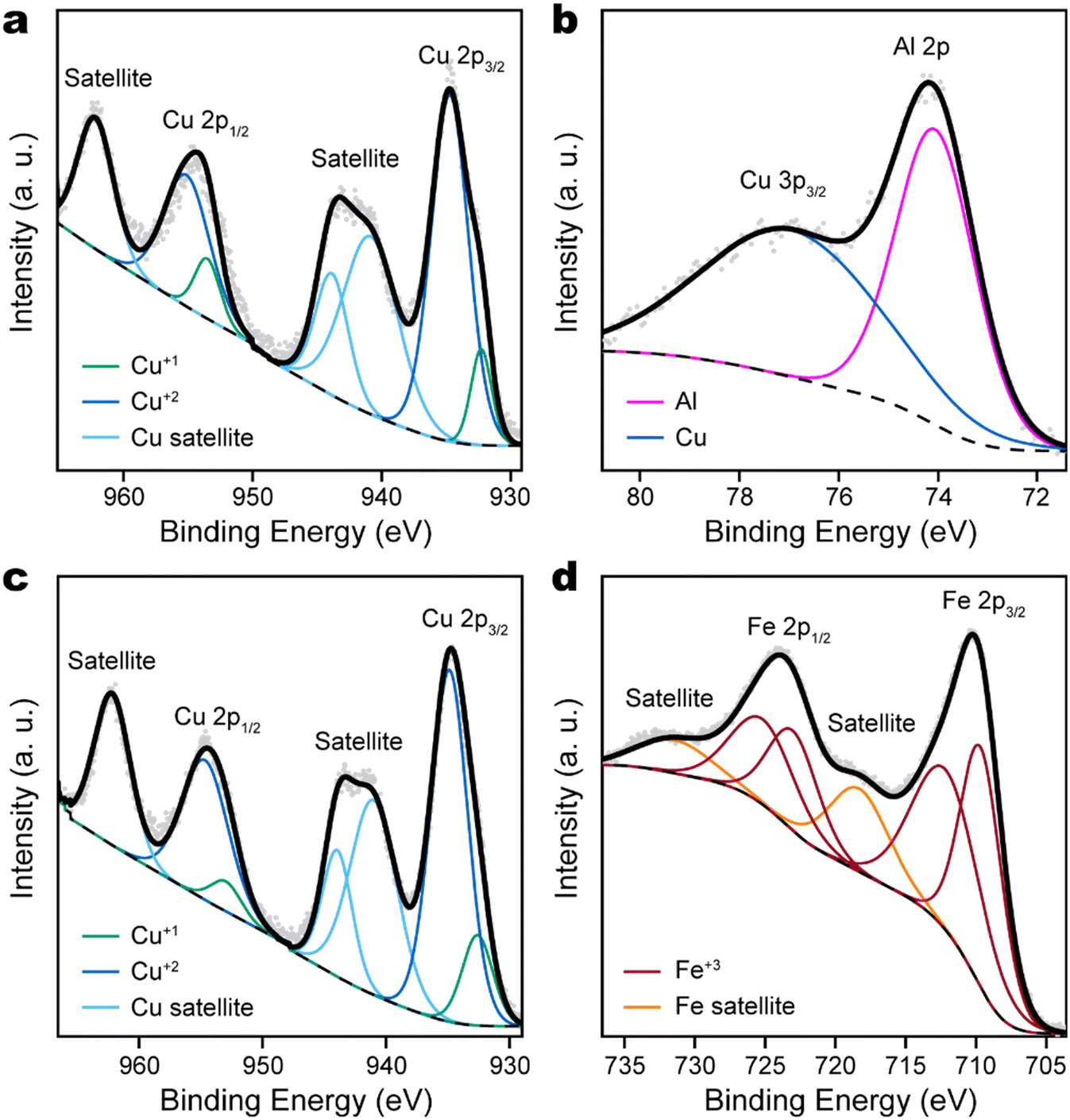 | ||
| Fig. 3 XPS spectra of CuAlO2 thin films, in (a) Cu 2p region and (b) Al 2p region, and of CuFeO2 thin films, in (c) Cu 2p region and (d) Fe 2p region, with corresponding spectral fitting. In each panel, the gray dots are experimental data, the solid black line is the fitted spectrum, and the dashed line is the fitted background. | ||
Nevertheless, X-ray absorption near edge structures (XANES) spectroscopy, a bulk-sensitive technique, confirmed that the bulk of the films remained delafossite despite the surface oxidation. A unique feature of delafossite lattice (A+B3+O2, space group Rm) is that the A+ cations are linearly coordinated and have a coordination number (C.N.) of 2, while the B3+ cations occupy octahedral sites (C.N. = 6). This contrasts with the caswellsilverite lattice of A+B3+O2, which is also in the space group Rm, but has A+ and B3+ cations both occupying octahedral sites. The linear coordination of Cu+ in our CuAlO2 and CuFeO2 films are confirmed by their XANES spectra at Cu K-edge (Fig. 4a). In both cases, the spectrum is characterized by a sharp shoulder feature that peaks at 8981 eV. This feature is consistent with the linear coordination of Cu+ cations and is similarly observed in Cu2O,43 in which each Cu+ ion is linearly coordinated with two O2− ions. According to Kau et al., this feature may be identified as the electric dipole-allowed 1s → 4px,y transition, which has a lower energy than the 1s → 4pz transition, due to the antibonding formation between Cu 4pz and ligands (z is along the O–Cu–O axis).44 In general, the sharp feature at 8981 eV would disappear, if the Cu+ cation is located at a higher C.N. site, e.g., trigonal or tetrahedral.43
 | ||
| Fig. 4 (a) Experimental Cu K-edge XANES spectra of delafossite CuAlO2 and CuFeO2 thin films. (b) Simulated Cu K-edge XANES spectra of delafossite CuAlO2 and CuFeO2, in which Cu takes a formal oxidation number of +1. (c) Simulated Cu K-edge XANES spectra of spinel CuAl2O4 and CuFe2O4, in which Cu takes a formal oxidation number of +2. | ||
The qualitative analysis of the Cu K-edge XANES is confirmed by first-principles computation. The structure of the thin delafossite film was relaxed using the Perdew–Burke–Ernzerhof (PBE) functional with a slab atomic model and the XANES were simulated with multiple scattering theory using the FDMNES program package.45 The simulated Cu+ K-edge spectra were quite similar between CuAlO2 and CuFeO2, with the characteristic pre-edge shoulder feature at 8981 eV observed clearly (Fig. 4b). Compared with the experimental spectra, the simulated delafossite spectra not only preserved the sharp shoulder features at 8991 eV, but also very well reproduced the shape of the lower energy (and stronger) white line peak at about 8994 eV. Given that XPS found surface Cu atoms were oxidized to an oxidation number of +2, we also simulated the XANES spectra of Cu +2 species following the same approach. For these simulations, spinel CuAl2O4 and CuFe2O4 were respectively chosen as model compounds for the surface oxidation of CuAlO2 and CuFeO2. Their lattice structures were taken from the Materials Project, mp-27719 for spinel CuAl2O4 and mp-770107 for CuFe2O4. As shown in Fig. 4c, the simulated Cu K-edge spectra of CuAl2O4 and CuFe2O4 showed little similarity with the experimental spectra. The pre-edge shoulder was much weaker than those experimentally observed for delafossite films and was moved to higher energy (8982 eV vs. 8981 eV). As such, we may conclude that the PLD-grown CuAlO2 and CuFeO2 films were dominated by the delafossite phase, despite the formation of small amount of Cu +2 species due to surface oxidation.
The photoelectrochemical (PEC) water reduction activities of both CuAlO2 and CuFeO2 thin films were evaluated using linear sweep voltammetry in a phosphate buffer solution at pH 7. Prior to the sweep, the dissolved O2 in the electrolyte solution was purged thoroughly by bubbling argon gas. This step is crucial to ensure that photoelectrons participate in water reduction rather than O2 reduction. In the PEC experiment, the illumination source is a Xenon arc lamp equipped with a filter to simulate AM 1.5 G solar radiation. As depicted in Fig. 5a and b, both CuAlO2 and CuFeO2 exhibit notable PEC activities upon illumination. However, CuFeO2 exhibits photocurrent density at the potential of hydrogen evolution (0 VRHE) that is one order of magnitude higher than that of CuAlO2. This stark difference can be attributed to the significantly narrower optical bandgap of CuFeO2 (2.0 eV vs. 3.5 eV),1,46 which results in more efficient absorption of incident light and charge carrier generation. Despite the chemical stability of CuFeO2, we note that its photocurrent density at 0 VRHE (0.25 mA cm−2) remains much lower than the theoretical limit of a semiconductor with 2 eV band gap (about 15 mA cm−2) and the highest photocurrent density achieved by Cu2O photocathode (about 10 mA cm−2).47,48 This suggests a low charge carrier separation efficiency that requires further study and optimization.
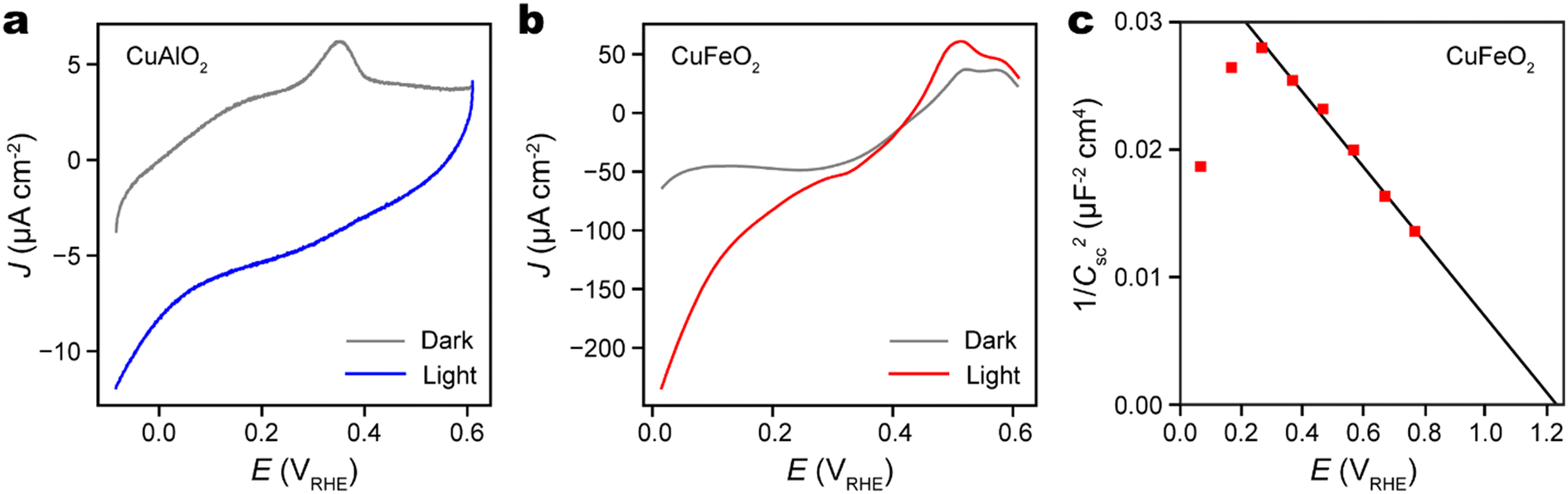 | ||
| Fig. 5 Photocurrent density (J) − potential (E) characteristics of (a) CuAlO2/ITO/YSZ and (b) CuFeO2/ITO/YSZ thin films. (c) Mott–Schottky plot of CuFeO2/ITO/YSZ thin film. The red squares are experimental data and the black line is the fitting for the linear range (0.4–0.8 VRHE). | ||
Mott–Schottky analysis, in which the space charge capacitance (Csc) is correlated with the electrode potential (E), was conducted to characterize the doping type and doping level of CuFeO2. The 1/C2sc − E plot had a uniform negative slope about −0.03 μF−2 cm4 V−1, which confirms the p-type doping of CuFeO2 (Fig. 5c). According to the Mott–Schottky relation, the slope is equal to −2(eεε0NA)−1, where e is the elementary charge, ε the semiconductor dielectric constant, ε0 the vacuum permittivity, and NA the acceptor density. Given that bulk CuFeO2 is ferroelectric with ε ∼ 104, we estimate that the CuFeO2 had an acceptor density of about 5 × 1017 cm−3.
In summary, we used PLD to fabricate delafossite CuAlO2 and CuFeO2 photocathodes on ITO-buffered YSZ (001) substrates and compared their solar water splitting activities using photoelectrochemistry measurements. Although the delafossite phase can be readily formed on a lattice-matching sapphire substrate, the stabilization of delafossite on ITO/YSZ was more challenging and achieved only after post-growth high temperature annealing. Cu K-edge XANES largely matched the expectation for Cu+ occupying a linearly coordinated center, through a careful comparison with first-principles calculation results. To date, all known electrocatalysts for CO2 reduction directly into valuable C2 or higher products contain copper as the active site.49–52 As a Cu-containing p-type oxide that has a visible light optical gap, the activity of CuFeO2 as a water splitting photocathode opens a door toward its application for the most desirable and challenging sustainable photoelectrochemical conversion reactions involving carbon dioxide.
Methods
Thin film synthesis
The ceramic targets for pulsed laser deposition (PLD) of Cu(I)-based delafossite were prepared via a solid-state sintering process. The CuO powder (Alfa Aesar) is mixed with the powder of trivalent metal oxide, either Al2O3 (Alfa Aesar) or Fe2O3 (Alfa Aesar), in a stoichiometric 1:1 ratio of the respective metal elements. The powder mixture was grounded in an agate mortar, pressed into pellets using a hydraulic pump at a pressure of 10 ton, and subsequently sintered in air at 800 °C, for 10 hours. During the PLD process, a laser fluence of 1.8 J cm−2 (KrF, 248 nm) and a repetition rate of 5 Hz were employed. Initially, a 50 nm thick layer of indium tin oxide (ITO) was deposited onto an yttrium-stabilized zirconia (YSZ) substrate at 600 °C in vacuum (with a base pressure of 6 × 10−7 mbar), as a conductive back contact for subsequent photoelectrochemical measurements. The deposition of Cu(I) delafossite took place at 700 °C in a nitrogen atmosphere with a pressure of 2 × 10−2 mbar. Following the deposition process, the system was gradually cooled down to room temperature under the same oxygen pressure, at a rate of 10 °C per minute. The deposited thin films were annealed in nitrogen at 900 °C for 1 h.
Materials characterization
The crystalline phases of thin film were characterized by X-ray diffraction (XRD, Rigaku Ultima III) using Cu Kα radiation (λ = 1.5418 Å). X-ray photoelectron spectroscopy (XPS) measurements are performed in a high vacuum (∼10−7 torr) using Al Kα (1486.6 eV) as the excitation source. X-ray absorption near edge structure (XANES) spectra at Cu K-edge are measured at the ISS beamline (8-ID)53 of NSLS-II, with the sample mounted in a similar fashion as for the XRF measurement. A fast scanning, liquid nitrogen cooled double crystal monochromator is used for continuous energy scans, with the energy range set to 8950–9200 eV. The X-ray absorption is determined by calculating the total fluorescence yield μ(E) = If/I0, where If is the fluorescence intensity and I0 the incident intensity. The intensity of the incident beam is measured by an ion chamber, and the X-ray fluorescence from the sample is captured by a PIPS (passivated implanted planar silicon) detector. The spectroscopic scans are repeated multiple times with the same settings, then binned and averaged to reduce the noise.Photoelectrochemical (PEC) measurements were carried out using a potentiostat (PAR VersaStat) in a three-electrode cell, with a delafossite thin film serving as the working electrode, Ag/AgCl as the reference electrode, and Pt wire as counter electrode. The simulated solar light was provided by a 150 W solar simulator equipped with an air mass 1.5 global (AM 1.5 G) filter (Newport) and the light power was calibrated to 1 Sun (100 mW cm−2) using a quartz-windowed Si solar cell (Newport). The electrolyte was a pH 7 phosphate buffer solution. Before the voltametric scan, the dissolved oxygen in the electrolyte solution was purged thoroughly by argon bubbling. The argon gas purging was maintained through the PEC experiment in a flow rate that forms minimal bubble.
First-principles calculations
Electronic structure calculations of CuMO2 (M = Al or Fe) were carried out using spin-polarized density functional theory as implemented in Quantum ESPRESSO54 using the generalized gradient approximation of Perdew, Burke, and Ernzerhof55 and ultrasoft pseudopotentials.56A plane-wave cutoff of 50 Ry and a charge density cutoff of 400 Ry were used, and all the atoms were fully relaxed during the geometry optimizations. The energy was sampled using k-point grids of 8 × 8 × 8. The optimized lattice constants a and c were 2.88 (3.07) and 17.13 (17.06) Å for CuAlO2 and CuFeO2, respectively.Data availability
All data supporting the findings of this study are presented in the main article and the ESI.†Conflicts of interest
All authors have given approval to the final version of the manuscript. The authors declare no competing financial interest.Acknowledgements
This research used Materials Synthesis & Characterization, Proximal Probes, and Theory & Computation facilities of the Center for Functional Nanomaterials (CFN), and the 8-ID (ISS) beamline of the National Synchrotron Light Source II (NSLS II), the U.S. Department of Energy Office of Science User Facilities, at Brookhaven National Laboratory under Contract No. DE-SC0012704.References
- H. Kawazoe, M. Yasukawa, H. Hyodo, M. Kurita, H. Yanagi and H. Hosono, Nature, 1997, 389, 939–942 CrossRef CAS.
- A. N. Banerjee and K. K. Chattopadhyay, Prog. Cryst. Growth Charact. Mater., 2005, 50, 52–105 CrossRef CAS.
- A. Stadler, Materials, 2012, 5, 661–683 CrossRef PubMed.
- M. Z. Yu, T. I. Draskovic and Y. Y. Wu, Phys. Chem. Chem. Phys., 2014, 16, 5026–5033 RSC.
- T. Zhu, et al. , J. Alloys Compd., 2016, 685, 836–840 CrossRef CAS.
- S. Akin, F. Sadegh, S. Turan and S. Sonmezoglu, ACS Appl. Mater. Interfaces, 2019, 11, 45142–45149 CrossRef CAS PubMed.
- C. G. Read, Y. Park and K. S. Choi, J. Phys. Chem. Lett., 2012, 3, 1872–1876 CrossRef CAS.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- Y. J. Jang and J. S. Lee, ChemSusChem, 2019, 12, 1835–1845 CrossRef CAS PubMed.
- C. L. Li, J. F. He, Y. Q. Xiao, Y. B. Li and J. J. Delaunay, Energy Environ. Sci., 2020, 13, 3269–3306 RSC.
- M. I. Diez-Garcia and R. Gomez, Sol. RRL, 2022, 6, 2100871 CrossRef CAS.
- C. Baiano, E. Schiavo, C. Gerbaldi, F. Bella, G. Meligrana, G. Talarico, P. Maddalena, M. Pavone and A. B. Munoz-Garcia, Mol. Catal., 2020, 496, 111181 CrossRef CAS.
- J. Gu, A. Wuttig, J. W. Krizan, Y. A. Hu, Z. M. Detweiler, R. J. Cava and A. B. Bocarsly, J. Phys. Chem. C, 2013, 117, 12415–12422 CrossRef CAS.
- N. Benreguia, A. Barnabe and M. Trari, J. Sol-Gel Sci. Technol., 2015, 75, 670–679 CrossRef CAS.
- M. S. Prevot, N. Guijarro and K. Sivula, ChemSusChem, 2015, 8, 1359–1367 CrossRef CAS PubMed.
- W. C. Sheets, E. Mugnier, A. Barnabe, T. J. Marks and K. R. Poeppelmeier, Chem. Mater., 2006, 18, 7–20 CrossRef CAS.
- D. H. Xiong, X. W. Zeng, W. J. Zhang, H. Wang, X. J. Zhao, W. Chen and Y. B. Cheng, Inorg. Chem., 2014, 53, 4106–4116 CrossRef CAS.
- M. Z. Yu, T. I. Draskovic and Y. Y. Wu, Inorg. Chem., 2014, 53, 5845–5851 CrossRef CAS.
- I. Garcia-Torregrosa, Y. G. Geertzema, A. S. M. Ismail, T. L. Lee, F. M. F. de Groot and B. M. Weckhuysen, ChemPhotoChem, 2019, 3, 1238–1245 CrossRef CAS.
- A. Yengantiwar, P. S. Shinde, S. L. Pan and A. Gupta, J. Electrochem. Soc., 2018, 165, H831–H837 CrossRef CAS.
- P. Piza-Ruiz, A. Saenz-Trevizo, Y. Verde-Gomez, P. Amezaga-Madrid and M. Miki-Yoshida, Ceram. Int., 2019, 45, 1156–1162 CrossRef CAS.
- N. Tsuboi, Y. Takahashi, S. Kobayashi, H. Shimizu, K. Kato and F. Kaneko, J. Phys. Chem. Solids, 2003, 64, 1671–1674 CrossRef CAS.
- A. Barnabe, E. Mugnier, L. Presmanes and P. Tailhades, Mater. Lett., 2006, 60, 3468–3470 CrossRef CAS.
- R. E. Stauber, J. D. Perkins, P. A. Parilla and D. S. Ginley, Electrochem. Solid-State Lett., 1999, 2, 654–656 CrossRef CAS.
- Z. H. Deng, X. D. Fang, R. H. Tao, W. W. Dong, D. Li and X. B. Zhu, J. Alloys Compd., 2008, 466, 408–411 CrossRef CAS.
- J. C. Lee, S. Y. Um, Y. W. Heo, J. H. Lee and J. J. Kim, J. Eur. Ceram. Soc., 2010, 30, 509–512 CrossRef CAS.
- M. Neumann-Spallart, S. P. Pai and R. Pinto, Thin Solid Films, 2007, 515, 8641–8644 CrossRef CAS.
- S. Z. Li, J. Liu, X. Z. Wang, B. W. Yan, H. Li and J. M. Liu, Phys. B, 2012, 407, 2412–2415 CrossRef CAS.
- R. A. Wheatley, S. Rojas, C. Oppolzer, T. Joshi, P. Borisov, D. Lederman and A. L. Cabrera, Thin Solid Films, 2017, 626, 110–116 CrossRef CAS.
- S. Vojkovic, J. Fernandez, S. Elgueta, F. E. Vega, S. D. Rojas, R. A. Wheatley, B. Seifert, S. Wallentowitz and A. L. Cabrera, SN Appl. Sci., 2019, 1, 1322 CrossRef.
- D. H. Choi, S. J. Moon, J. S. Hong, S. Y. An, I. B. Shim and C. S. Kim, Thin Solid Films, 2009, 517, 3987–3989 CrossRef CAS.
- T. Joshi, et al. , J. Appl. Phys., 2015, 117, 013908 CrossRef.
- S. J. Luo, et al. , J. Appl. Phys., 2020, 127, 065301 CrossRef CAS.
- K. Ueda, T. Hase, H. Yanagi, H. Kawazoe, H. Hosono, H. Ohta, M. Orita and M. Hirano, J. Appl. Phys., 2001, 89, 1790–1793 CrossRef CAS.
- S. Y. Choi, C. D. Kim, D. S. Han and H. Park, J. Mater. Chem. A, 2017, 5, 10165–10172 RSC.
- M. Miclau, N. Miclau, R. Banica and D. Ursu, Mater. Today: Proc., 2017, 4, 6975–6981 Search PubMed.
- M. S. Prevot, Y. Li, N. Guijarro and K. Sivula, J. Mater. Chem. A, 2016, 4, 3018–3026 RSC.
- M. S. Prevot, X. A. Jeanbourquin, W. S. Bouree, F. Abdi, D. Friedrich, R. van de Krol, N. Guijarro, F. Le Formal and K. Sivula, Chem. Mater., 2017, 29, 4952–4962 CrossRef CAS.
- W. Zhang, et al. , Chem. Mater., 2018, 30, 1677–1685 CrossRef CAS.
- T. Stöcker and R. Moos, Materials, 2018, 11, 1888 CrossRef.
- M. Ferri, J. D. Elliott, M. F. Camellone, S. Fabris and S. Piccinin, ACS Catal., 2021, 11(4), 1897–1910 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- A. A. Guda, et al. , npj Comput. Mater., 2021, 7, 203 CrossRef CAS.
- L. S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433–6442 CrossRef CAS.
- O. Bunău and Y. Joly, J. Phys.: Condens. Matter, 2009, 21, 345501 CrossRef.
- V. R. Galakhov, A. I. Poteryaev, E. Z. Kurmaev, V. I. Anisimov, S. Bartkowski, M. Neumann, Z. W. Lu, B. M. Klein and T.-R. Zhao, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 4584–4591 CrossRef CAS.
- J. Cheng, L. Wu and J. Luo, Chem. Phys. Rev., 2022, 3, 031306 CrossRef CAS.
- S. D. Tilley, ACS Energy Lett., 2023, 8, 2338–2344 CrossRef CAS.
- C. Long, et al. , Sci. Adv., 2023, 9, eadi6119 CrossRef CAS.
- W. Liu, et al. , Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed.
- X. Li, et al. , Chem, 2022, 8, 2148–2162 CAS.
- M. Jun, J. Kundu, D. H. Kim, M. Kim, D. Kim, K. Lee and S.-I. Choi, Adv. Mater., 2024, 36, 2313028 CrossRef CAS.
- D. Leshchev, M. Rakitin, B. Luvizotto, R. Kadyrov, B. Ravel, K. Attenkofer and E. Stavitski, J. Synchrotron Radiat., 2022, 29, 1095–1106 CrossRef CAS.
- P. Giannozzi, et al. , J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- K. F. Garrity, J. W. Bennett, K. M. Rabe and D. Vanderbilt, Comput. Mater. Sci., 2014, 81, 446–452 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4en00706a |
| This journal is © The Royal Society of Chemistry 2025 |