
Photocatalytic NO removal: complete oxidation and reduction reaction for by-product inhibition and end-product recovery
Wen
Cui†
 ab,
Jiaqi
Wang†
a,
Yan
Li
a,
Pingqu
Liu
a and
Fan
Dong
*abc
ab,
Jiaqi
Wang†
a,
Yan
Li
a,
Pingqu
Liu
a and
Fan
Dong
*abc
aCollege of Resources and Environmental Engineering, Key Laboratory of Karst Georesources and Environment, Guizhou University, Guiyang 550025, China
bGuizhou Karst Environmental Ecosystems Observation and Research Station, Ministry of Education, Guiyang 550025, China
cResearch Center for Carbon-Neutral Environmental & Energy Technology, Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 611731, China. E-mail: dongfan@uestc.edu.cn
First published on 26th September 2024
Abstract
Nitrogen oxides (NOx, x = 1 and 2, the proportion of NO is about 95%), as one of the primary precursors for particulate matter and ozone, limit the continuous improvement of air quality. Photocatalytic NO purification technology has attracted significant attention, and much efforts have been devoted to realizing the complete photocatalytic oxidation and reduction of NO for toxic by-product inhibition and end-product recovery. This work presents a timely overview of the current research progress on the conversion of NO into nitrate/ammonia (NO3−/NH3), which can be further recycled and utilized. According to the principle of heterogeneous photocatalysis and considering the significance of the reaction microenvironment (surface active sites of photocatalysts, target pollutants and reaction media), this review systematically summarizes the progress on the strategies for controlling the surface structure of photocatalysts and reaction medium. Specifically, this critical overview is focused on various methods for the surface modification of photocatalysts, strategies to accelerate the mass transfer process of gaseous NO, and the effect of the additional introduction of a reductant/antioxidant in the reaction system. Furthermore, research trends and future prospects are discussed, aiming to provide insights into the breakthroughs and boost the development of photocatalytic NO removal technology.
 Wen Cui | Wen Cui, an associate professor and master's supervisor at the College of Resources and Environmental Engineering, Guizhou University, mainly engages in research on environmental and energy catalysis as well as atmospheric pollution control chemistry. Dr. Cui has led two projects funded by the National Natural Science Foundation of China, one sub-project of the National Key R&D Program, and one provincial or ministerial-level scientific research project. She is the author of 14 academic papers with an h-index of 37, and she has been selected as one of the top 2% of scientists worldwide in 2023. |
 Jiaqi Wang | Jiaqi Wang is pursuing her master's degree under the supervision of Wen Cui in the College of Resources and Environmental Engineering, Guizhou University. She is working on photocatalytic NO purification with biochar-based photocatalysts. |
 Yan Li | Yan Li is pursuing her master's degree in the College of Resources and Environmental Engineering, Guizhou University. Her research interests are developing integrated technology for low-energy air dehumidification and water production with solar thermal conversion. |
 Pingqu Liu | Pingqu Liu is an undergraduate student at the College of Environment Engineering, GuiZhou University, under the supervision of Wen Cui. His research interests are the development of supported photocatalysts. |
 Fan Dong | Prof. Fan Dong is a full professor at the Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China (UESTC). He serves as the director of the Research Center for Carbon-Neutral Environmental & Energy Technology at UESTC and Topic Editor of ACS ES&T Engineering. He has an h-index of 110 and total citations of more than 45 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. He was awarded National Natural Science Funds for Distinguished Youth Scientist of China (2022) and has been consistently recognized as a highly cited researcher by Clarivate Analytics (2018–2023). His research interests include environmental and energy catalysis, advanced
000. He was awarded National Natural Science Funds for Distinguished Youth Scientist of China (2022) and has been consistently recognized as a highly cited researcher by Clarivate Analytics (2018–2023). His research interests include environmental and energy catalysis, advanced
Environmental significanceNO is one of the primary precursors for the formation of particulate matter and ozone. Considering its insoluble characteristic and the intermediate chemical valence state of nitrogen in NO, it can be removed and even upcycled via the redox reaction. Photocatalysis, which makes full use of inexhaustible solar energy, can be an eco-friendly and cost-effective method to realize the complete oxidation and reduction of NO into nitrate/ammonia (NO3−/NH3), which can be further utilized. Herein, a comprehensive critical overview and perspectives, focusing on the complete photocatalytic oxidation and reduction of NO for toxic by-product inhibition and end-product recovery, are presented, which will hopefully inspire ongoing efforts to optimize the sustainable conversion process of NO and advance the practical application of photocatalytic NO removal technology. |
1. Introduction
Nitrogen oxides (NOx, x = 1 and 2, the proportion of NO is about 95%) are important precursors for the formation of particulate matter and ozone, which limit the continuous improvement of air quality.1–5 Thus, over the past decades, strategies ranging from clean combustion technology of source control to end-treatment technology of exhaust gas have been proposed to address this issue.6–8 Considering the characteristics of the insoluble and intermediate chemical valence state of nitrogen in NO, chemical methods via redox reaction can be a suitable end-treatment method; concretely, traditional treatment technologies such as selective catalytic reduction (SCR) and oxidation methods with strong oxidants have been widely applied.9–15 Nevertheless, in the context of carbon neutrality, seeking green technology is in line with the concept of sustainable development.16 Photocatalysis, which makes full use of accessible solar energy, is an eco-friendly and cost-effective method to address current environmental and energy crises.17,18 Correspondingly, photocatalytic NO purification technology has attracted significant interest, realizing remarkable progress.19–21 Recently, the complete photocatalytic oxidation/reduction reaction of NO has been proposed since the end-product (nitrate/ammonia) can be further recycled as nitrogen fertilizer or as the main raw material in the fertilizer and basic organic chemical industries, thus avoiding the formation of toxic by-products.22–26 However, realizing the efficient complete oxidation/reduction of NO into nitrate/ammonia is still a challenge to date.19,27–29Photocatalytic NO oxidation/reduction is a heterogeneous reaction, which is the reactant conversion process induced by surface/interface charge transfer between the reactants and photocatalyst, where the reaction microenvironment mainly consists of surface active sites on the photocatalyst, target pollutants and reaction media.30 In the gas–solid reaction, the photocatalyst is usually regarded as the core, where its surface structure determines the process of surface charge transfer and interfacial molecular transformation.31,32 Thus, the rational design of photocatalysts can be a fundamental way to regulate the surface charge arrangement and surface/interface charge transfer process, which is beneficial for optimizing the reaction pathway for toxic by-product inhibition, thus improving the selective catalytic conversion efficiency of reactants.30 In the gas–liquid–solid reaction, the reaction microenvironment is more complex, mainly reflecting the reaction medium.33 Besides the surface structure of the photocatalyst, the mass transfer process of gaseous reactants and the introduction of oxidants/reductants/sacrificing agents also need to be fully considered, which are the determinants in efficiently realizing selective catalytic conversion.
Photocatalytic NO oxidation is a reactive oxygen species-based (ROS-based) gas–solid reaction, and the diffusion of reactants (O2, H2O, NO) to the surface active sites of the photocatalyst for adsorption and activation is the premise of subsequent ROS formation and NO oxidation.30,34,35 Therefore, it is necessary to propose reasonable design and corresponding modification methods for the surface structure of photocatalysts, aiming to facilitate the adsorption and activation of the reactants and the separation of charge carriers for NO activation and the formation of ROS with deep oxidation ability to realize the efficient complete oxidation of NO.21,36–39 In the photocatalytic NO reduction process, the reaction microenvironment is more complex given that it generally proceeds in a gas–liquid–solid three-phase reaction system. The ultralow solubility of gaseous NO limits its transfer to the liquid phase for its subsequent conversion, and the reduction reaction is difficult to carry out spontaneously in an air environment.19,40 Therefore, in addition to optimizing the reactant activation process and charge carrier separation by precisely constructing the surface active sites in the photocatalyst, strategies for accelerating the mass transfer of gaseous NO and introducing applicable reductant/antioxidant into reaction system have also been proposed, aiming to achieve the efficient complete photocatalytic reduction reaction for NO upcycling.
In the past few decades, several excellent studies on photocatalytic NO removal have been published, and various critical reviews have emerged, summarizing the methods for the modification of the photocatalyst and the progress of typical photocatalysts for efficient N oxidation.19,41–47 However, photocatalytic NO complete oxidation/reduction with toxic by-product inhibition and end-product recovery has only recently attracted attention, and thus there is a lack of reviews focusing on this topic. Therefore, a comprehensive review is necessary, which can not only provide readers with a better understanding of the recent research progress in this field but also advance the development of photocatalytic NO removal technology. Herein, we attempt to present a critical review on photocatalytic NO removal, with the emphasis on summarizing the achieved progress in photocatalytic NO complete oxidation and reduction for toxic by-product inhibition and directed final product (NO3−/NH3) formation (Fig. 1). Some typical examples are also discussed in each section below, without bias towards studies from specific research groups. Meanwhile, the limitations and potential research directions to overcome the related challenges are also highlighted. It is hoped that this review will inspire ongoing efforts to precisely adjust the reaction microenvironment for efficient photocatalytic NO removal.
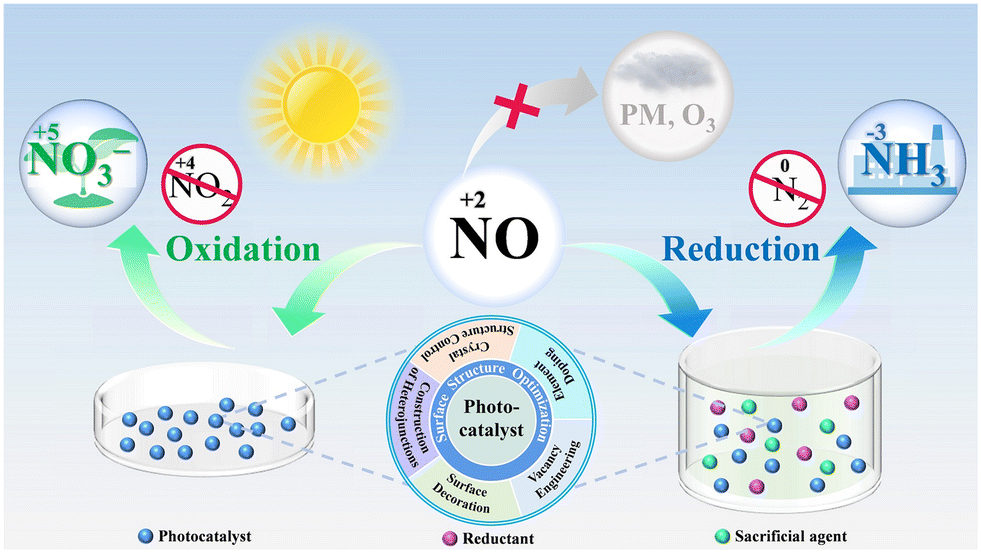 | ||
| Fig. 1 Illustration of the complete photocatalytic oxidation and reduction of NO for toxic by-product inhibition and directed final product (NO3−/NH3) formation. | ||
2. Photocatalytic complete NO oxidation
Although there are numerous studies on photocatalytic NO oxidation, the complete oxidation of NO without the formation of toxic intermediates has only attracted attention in recent years (Fig. 2a and b). The rational design of the surface structure of photocatalysts has been gradually proposed given that it is the fundamental way to optimize the reaction pathway and photocatalytic performance by regulating the surface charge transfer and interfacial molecular transformation. In general, the strategies for the surface modification of photocatalysts can be summarized as element doping, vacancy engineering, surface modification, construction of heterojunctions and crystal structure control, which can tune the surface properties and band structure of photocatalysts, and thus influence the adsorption/activation of reactants and subsequent photocatalytic NO oxidation reaction. Specifically, the surface structure optimization of photocatalysts can not only facilitate the adsorption and activation of O2/H2O to generate abundant ROS for complete NO oxidation and even induce the formation of ROS with direct complete oxidation ability (ROS-H) for the one-step oxidation of NO into NO3−, but also accelerate the adsorption and activation of NO for the convenient subsequent selective catalytic oxidation for the formation of nitrate (Fig. 2c). Generally, the whole reaction process and promotion mechanism, including the surface charge transfer, interfacial molecule conversion, ROS evolution, and NO oxidation, are revealed by experimental characterization and theoretical simulation.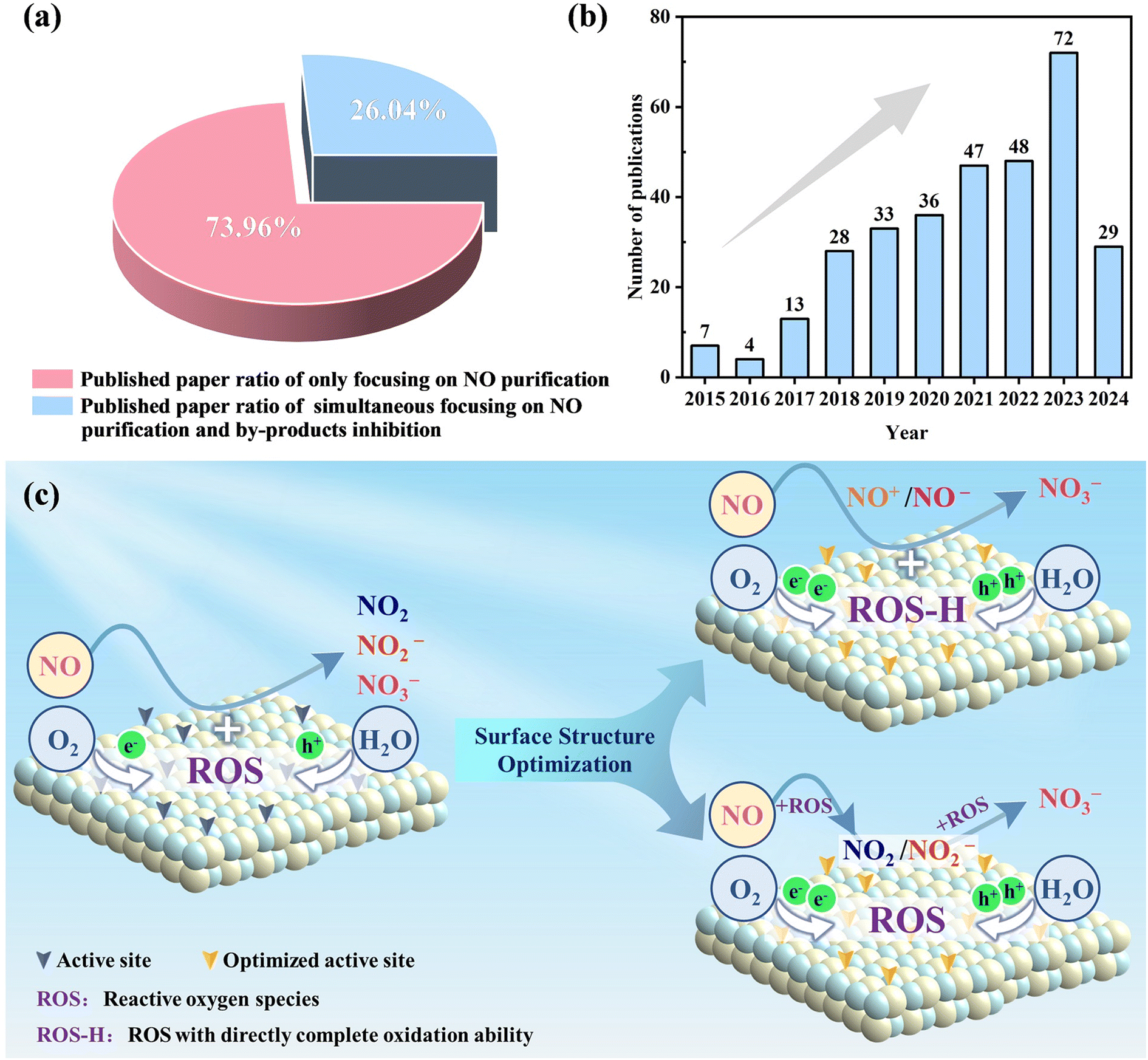 | ||
| Fig. 2 (a) Proportion of published papers only focusing on NO purification and simultaneously focusing on NO purification and by-product inhibition. (b) Increasing number of papers simultaneously focusing on NO purification and by-product inhibition with publication year. The search terms are as follows: “Photocataly* or photodegra* or photochemi* or visible* light or photoreactivity” (topic) and “NO or Nitric-Oxide$ or Nitrogen-Oxide$” (topic) and “oxidation or removal or conversion or degradation or remove or abatement” (topic). The document types are “Article” and the publication date is “All years (1985–2024)” via Web of Science Core Collection on 14/7/2024. (c) Illustration of the mechanism for the inhibition of by-product formation. | ||
2.1 Element doping
Element doping has been widely employed to redistribute the surface charge distribution and/or regulate the band structure of photocatalysts to optimize their photocatalytic performance.48–51 Generally, the introduction of heteroatoms can break the balance of atomic structure, and thus induce charge redistribution in the original photocatalyst, contributing to the formation of charge localization/delocalization centers due to the electronegativity difference between atoms. Yuan et al. constructed localized excess electron regions both in La-doped (BiO)2CO3 and La-doped ZnO, which was conductive to the adsorption and activation of O2, subsequently promoting the formation ROS for the efficient photocatalytic deep oxidation of NO.52,53The introduction of heteroatoms in graphitic carbon nitride (g-C3N4) has been widely reported for photocatalytic NO oxidation, which can achieve control of the interlayer or intralayer charge configuration of g-C3N4, promoting the activation of reactants and separation of charge carriers for the generation of ROS participating in the complete oxidation of NO. B-doped g-C3N4 enabled electron localization to accelerate the activation of the reactants and intermediates, and then facilitated photocatalytic NO oxidation reaction.54 Zhang et al. introduced Zn atoms into the g-C3N4 interlayer to form a Zn–N3 bridging structure (Zn-CN), which induced the adsorption of O2 and NO to fabricate the Zn–O2–NO structure, and then realized the direct formation of nitrate without NO2 generation (Fig. 3a).55 Besides, Sr-doped g-C3N4 with different doping modes was reported, and its corresponding functions were also elaborated (Fig. 3b).56 Sr-doping with simultaneous N atom replacement, cavity padding, and intercalation facilitated the activation of O2 to produce ˙O2−, endowing g-C3N4 with the ability to activate H2O2 for the generation of ˙OH and improving the transfer of photogenerated electrons, respectively. The different doping modes complement each other to realize efficient and deep NO removal.
 | ||
| Fig. 3 (a) NO oxidization process over g-C3N4 and Zn-CN.55 Reproduced from ref. 55 with permission from Elsevier, Copyright 2023. (b) Heat absorption and release of various possible structures of Sr multi-site doped g-C3N4.56 Reproduced from ref. 56 with permission from Elsevier, Copyright 2019. | ||
Meanwhile, the band structure of photocatalysts can be tuned by element doping, which is conductive to extending their light adsorption range and enhancing their redox ability. Besides, the introduction of heteroatoms can induce the formation of new intermediate energy levels, facilitating the separation, migration and transfer of photogenerated carriers to enhance the photocatalytic performance. For example, Guo et al. constructed N-doped BiOCl (N-BiOCl) with intermediate energy levels (Fig. 4a), which optimized the migration process of charge carriers to effectively generate ˙O2− for the direct conversion of NO into NO3−.57 The surface charge distribution and band structure of the photocatalyst could also be optimized simultaneously by element doping. Fe ions in Fe-doped TiO2 (Fe-TiO2) not only provided more active sites but also induced new states due to the Fe 3d orbit between the band gap of Fe-TiO2 (Fig. 4b and c), which accelerated the adsorption and activation of the reactants, enhanced the light absorption range and promoted the separation of charge carriers for the deep oxidation of NO (Fig. 4d–g).58 The B atom in B-doped Bi2O2CO3 not only acted as a channel of charge transfer to promote the activation of reactants, but also contributed to the formation of intermediate energy levels for the separation of electron–hole pairs, which induced the deep oxidation of NO.59
 | ||
| Fig. 4 (a) Total density of states of BiOCl and N-BiOCl.57 Reproduced from ref. 57 with permission from Elsevier, Copyright 2022. Density of states for (b) TiO2 and (c) Fe-TiO2. Charge density difference distribution in optimized (d) NO adsorbed on TiO2, (e) NO adsorbed on Fe-TiO2, (f) O2 adsorbed on TiO2 and (g) O2 adsorbed on Fe-TiO2. Light blue and yellow represent charge loss and charge accumulation, respectively. The calculated positive Bader charge (Δq) indicates the electron transfer from the catalyst surface to adsorbed molecule.58 Reproduced from ref. 58 with permission from Elsevier, Copyright 2020. | ||
There is the possibility of forming defects in the original material during the introduction of heteroatoms. The presence of vacancies also has an effect on the surface charge distribution and band structure of photocatalysts. Zhang et al. found that La3+ doping narrowed the band gap of Bi5O7I and induced the formation of oxygen vacancies (OVs).60 The narrow band structure improved the electronic excitation in the photocatalyst, and the presence of OVs facilitated the activation of O2 to generate ˙O2− for complete NO oxidation. Besides, the co-doping of atoms in photocatalysts has been proposed, where the heteroatoms play a synergistic role in elevating the photocatalytic NO oxidation performance. In Na–Ca co-doped g-C3N4, sodium doping promoted the separation of electron–hole pairs and strengthened the redox capacity, while calcium doping enhanced the chemisorption of NO2 to inhibit the emission of toxic by-products.61 Also, Lu et al. further prepared K–Ca co-doped g-C3N4 and found that K and Ca resulted in a better improvement than other elements in the alkali and alkaline-earth family.62 K–Ca co-doped g-C3N4 possessed both electrons with strong reducibility and holes with strong oxidizability to generate ˙O2− and ˙OH, participating in photocatalytic NO removal, which was also was conductive to the chemisorption and physisorption of NO and NO2 on the photocatalyst surface for their further complete oxidation into NO3−.
Supported single-atom catalysts cannot only maximize the atomic efficiency of metals but also provide an alternative strategy to tune the activity and selectivity of catalytic reactions, which have also been employed to optimize the reaction pathway of photocatalytic NO oxidation. Guo et al. proposed the anchoring of atomically dispersed Ag1 atoms on a {001} facet-exposed BiOCl nanosheet (Ag1/BiOCl), creating Ag single atom sites with a triangle two-coordinated Cl–Ag1–Cl local configuration (Fig. 5a and b), which achieved >90% NO conversion to the favorable NO3− with high selectivity (>97%) (Fig. 5c and d).63 The triangle Cl–Ag1–Cl sites not only promoted light adsorption and electron transfer kinetics in BiOCl but also facilitated end-on O2 adsorption, contributing to the one-electron molecular oxygen activation to form ˙O2−. The boosted generation of ˙O2− could selectively react with NO to form nitrate with strong bidentate adsorption on Cl–Ag1–Cl sites, which inhibited the dissociation of nitrate to produce unfavorable NO2 (Fig. 5e). Also, single-atomic Na (Na-SA) played a “three-in-one” role in carbon nitride (CN).64 Specifically, the introduction of Na-SA induced electron transfer from Na to the complex N2C of CN and the formation of a built-in electric field between layers, which increased the light harvesting ability, facilitated the adsorption of reactants and accelerated the separation of carriers, thus improving the NO removal rate and preventing the production of toxic by-products. Highly dispersed Ni-incorporated C3N5 was facilely prepared for efficient photocatalytic NO oxidation.65 The surface anchored Ni sites not only optimized the optical property but also promoted the separation of photogenerated carriers to facilitate the activation of O2 for ROS formation, and it was also demonstrated that ˙O2− mainly contributed to NO → NO2 conversion, while 1O2 and ˙OH could further convert NO2 into the harmless NO3−.
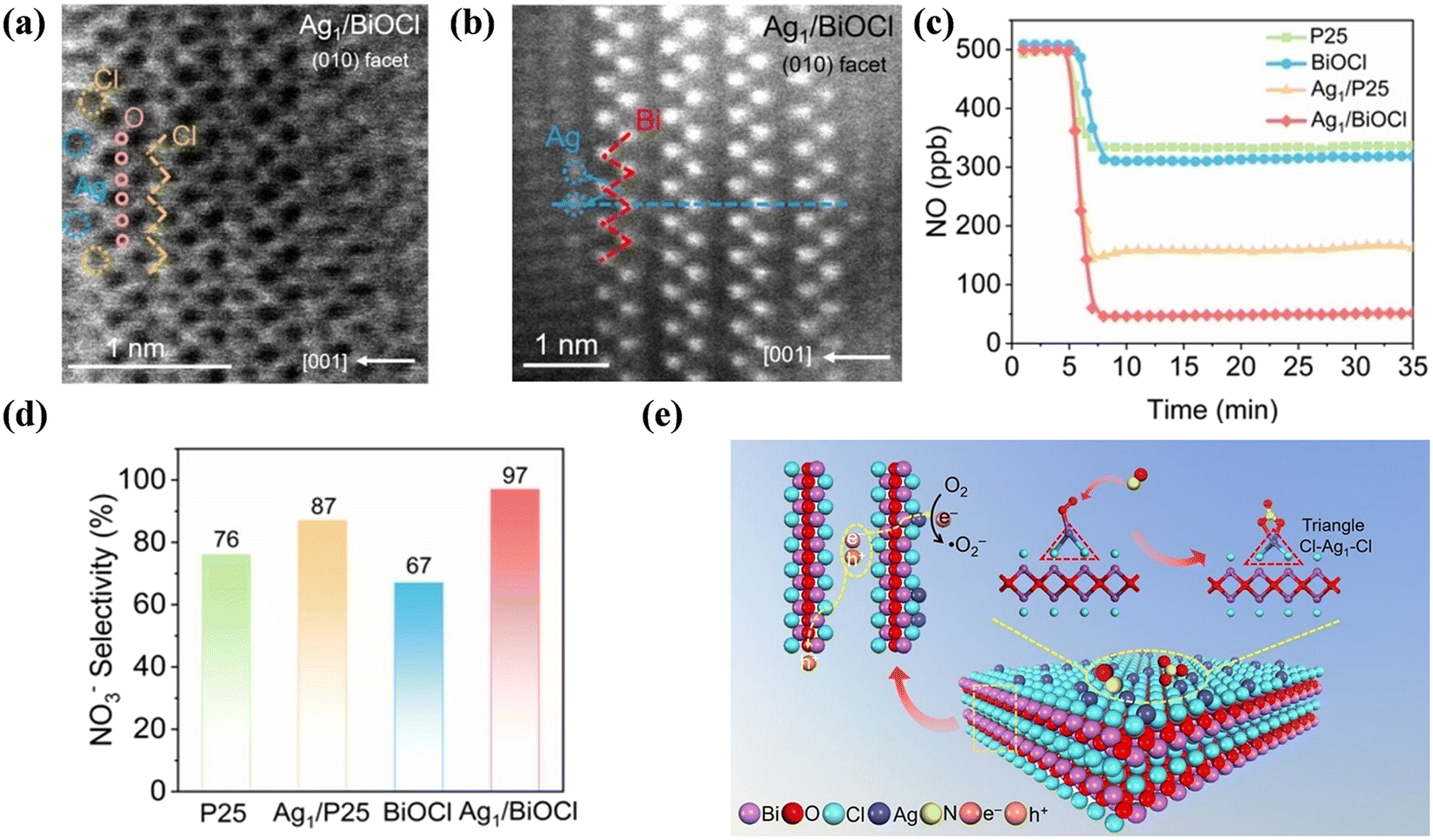 | ||
| Fig. 5 (a) Atomic-resolution ABF-STEM and (b) HADDF-STEM images of Ag1/BiOCl. (c) Photocatalytic NO oxidation of P25, BiOCl, Ag1/P25 and Ag1/BiOCl. (d) NO3− selectivity of Ag1/P25 and Ag1/BiOCl under visible light irradiation. (e) Schematic of photocatalytic NO oxidation at the Cl–Ag1–Cl sites under visible light.63 Reproduced from ref. 63 with permission from John Wiley and Sons, Copyright 2023. | ||
2.2 Vacancy engineering
Surface vacancy engineering is an effective means to tailor the local surface microstructure of photocatalysts, which can affect their surface properties and band structure. Defects are located in places where the perfect periodic arrangement of atoms in crystal materials is broken, thus inducing surface charge rearrangement.66 The construction of OVs is the most common approach to improve the photocatalytic NO oxidation performance. Generally, OVs can serve as charge regulators to redistribute the surface charge in photocatalysts. The confinement of electrons by OVs is beneficial for the adsorption and activation of O2 for generating ˙O2−, contributing to the direct oxidization of NO into nitrate without NO2 generation.28,67–70Besides, the introduction of OVs is an approach by which the optical property, charge separation, and surface structure of photocatalysts can be tuned.71–74 The OVs of blue TiO2 (TiO2-OV) with localized electrons not only facilitated the activation of O2 through the single-electron pathway to generate ˙O2− for the transformation of NO into nitrate, but also simultaneously induced photogenerated hole annihilation to inhibit the side-reaction between holes and NO for avoiding the formation of toxic by-products, which achieved the highly selective removal of NO (Fig. 6a–c).71 Besides facilitating the adsorption and activation of reactants, the OVs in Bi3TaO7 (OVs-BTO) induced the redistribution of local electrons to form a fast charge transfer channel between OVs and adjacent Ta atoms, increasing the transfer rate of photogenerated carriers, which was helpful to promote the deep oxidation of NO into nitrate (Fig. 6d).72 Zhang et al. synthesized g-C3N4 containing non-intrinsic OVs by oxygen pre-doping (Vo-CN) followed by elimination (Fig. 6e).73 The introduction of OVs induced the formation of a mid-gap state near the Fermi level to accelerate the separation of photogenerated carriers and provided favorable channels for carrier migration to accelerate O2 and NO adsorption, which promoted the generation of ROS, thus achieving efficient NO removal with high nitrate selectivity (Fig. 6f and g). Furthermore, the introduction of an intermediate energy level derived from the introduced vacancy can endow insulators with semiconductor-like photocatalytic activity, where BaSO4 with Ba-vacancy has already exhibited a photocatalytic NO oxidation performance.75 Ba-vacancy also induces the redistribution of surface charge to accelerate the activation of NO by donating electrons to electron-deficient areas, contributing to the conversion of NO into a higher valance state for further favorable photocatalytic oxidation.
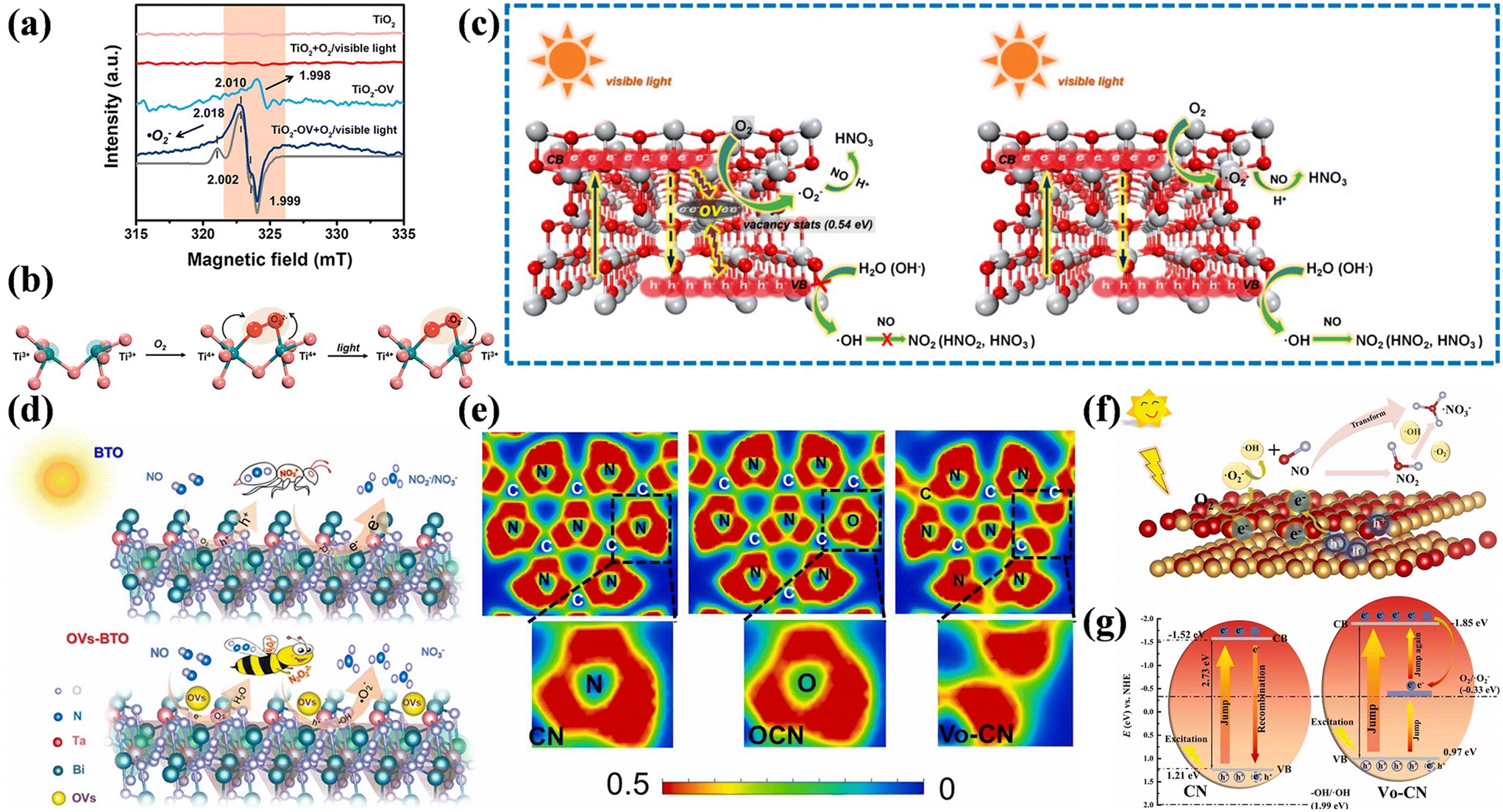 | ||
| Fig. 6 (a) EPR spectra for TiO2-OV and TiO2 under visible-light (420 nm) irradiation at 120 K. The corresponding simulated spectrum is indicated by the gray line. (b) Schematic of the interfacial charge-transfer path on TiO2-OV. The red and green ellipses represent the electron clouds, respectively. The oxygen vacancy with two localized electrons is essentially two unsaturated titanium ions (Ti3+). (c) Schematic drawing of the interfacial electron-transfer processes during photocatalytic NO oxidation on TiO2 and TiO2-OV.71 Reproduced from ref. 71 with permission from the American Chemical Society, Copyright 2019. (d) Mechanism of deep NO photo-oxidation by BTO and OVs-BTO.72 Reproduced from ref. 72 with permission from Elsevier, Copyright 2022. (e) Electronic location function of CN, OCN, and Vo-CN samples. (f) The pathway of photocatalytic NO oxidation on VO-CN. (g) Detailed photocatalytic reaction mechanism on the surface of VO-CN under visible light irradiation.73 Reproduced from ref. 73 with permission from Elsevier, Copyright 2024. | ||
Besides OVs, other types of defects have been fabricated to introduce extra active sites and optimize the band structure and surface electronic structure of photocatalysts, aiming to improve their performance. The metal-free layered conjugated semiconductor g-C3N4 has become one of the outstanding candidates for photocatalytic NO removal and different types of vacancies are easy to be constructed for further improving its photocatalytic performance.39,76–79 Carbon vacancy-modified C3N4 nanotubes were equipped with efficient performance for the selective oxidation of NO to nitrates, which was ascribed to the trapping of photo-induced electrons by carbon vacancies, and then reaction with surface-adsorbed O2 and H2O to form ˙O2− and ˙OH for NO complete oxidation, respectively (Fig. 7a).76 Li et al. prepared C3N4 with three-coordinate nitrogen vacancy (AC-CN4), where the introduction of nitrogen vacancy as the reactive sites either weakened the adsorption of intermediates/final products or facilitated the activation of O2 to generate 1O2, achieving an enhanced NO-oxidation performance and good reusability (Fig. 7b).77 Besides, the three-coordinate nitrogen vacancy in amorphous carbon nitride was beneficial to expand its visible light responsive range, decrease the activation barrier of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O triple bond, and boost the generation of 1O2 and ˙O2− for complete NO removal (Fig. 7c and d).78 The integration of ion doping and vacancy engineering has also been proposed to boost the photocatalytic activity of g-C3N4 towards NO removal.79 The introduction of N vacancies and K+ ions induced the formation of charge channels and achieved a preferred pathway (NO → NO+ → NO3−) for NO removal.
O triple bond, and boost the generation of 1O2 and ˙O2− for complete NO removal (Fig. 7c and d).78 The integration of ion doping and vacancy engineering has also been proposed to boost the photocatalytic activity of g-C3N4 towards NO removal.79 The introduction of N vacancies and K+ ions induced the formation of charge channels and achieved a preferred pathway (NO → NO+ → NO3−) for NO removal.
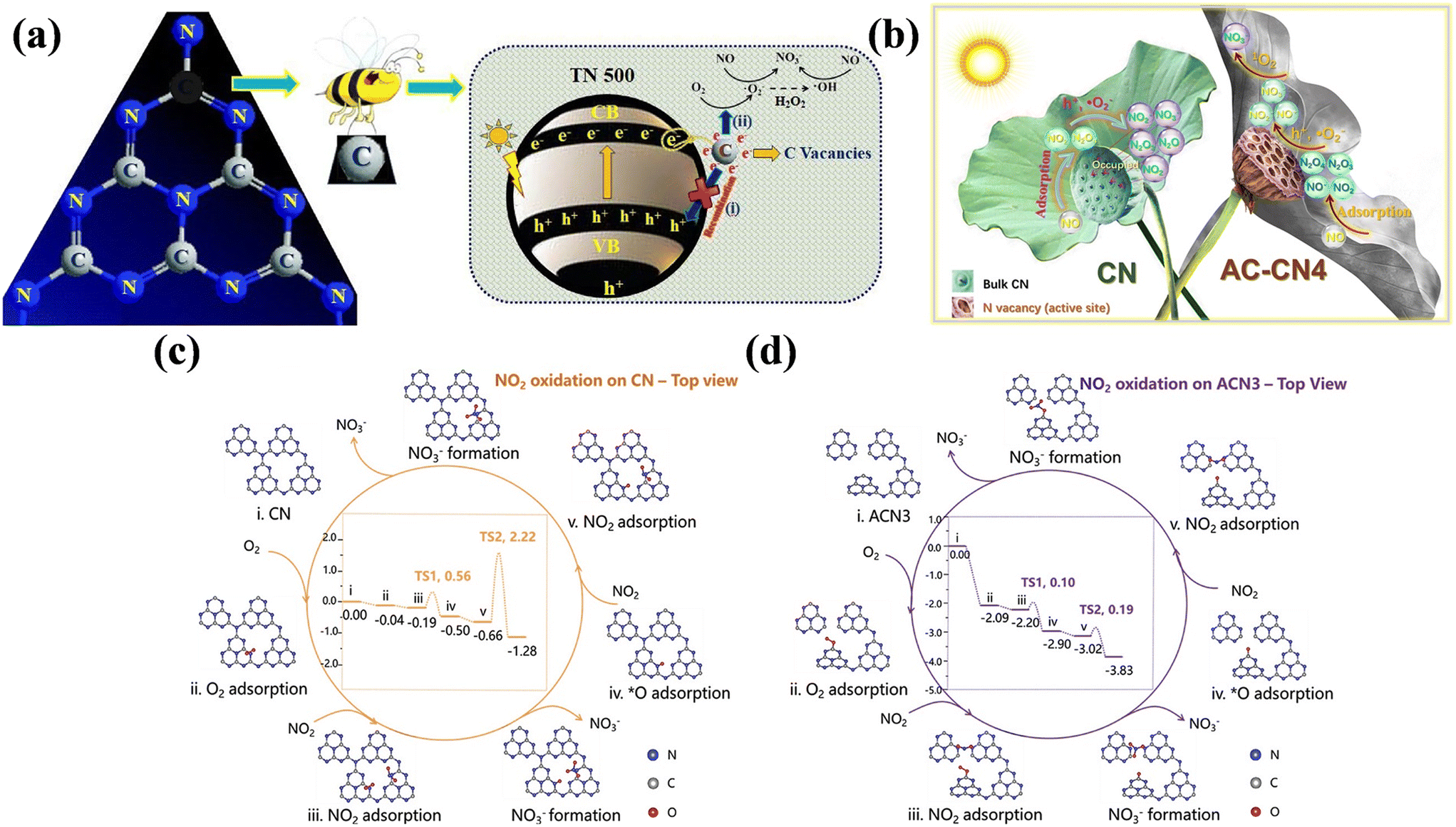 | ||
| Fig. 7 (a) Proposed photocatalytic oxidation mechanism of NO over C3N4 with carbon vacancies.76 Reproduced from ref. 76 with permission from Elsevier, Copyright 2020. (b) Reaction mechanism for NO oxidation of CN and AC-CN4.77 Reproduced from ref. 77 with permission from Elsevier, Copyright 2020. Calculated reaction pathways for NO2 oxidation on (c) CN and (d) amorphous carbon nitride with N3C-site vacancies (ACN3).78 Reproduced from ref. 78 with permission from John Wiley and Sons, Copyright 2021. | ||
It is worth mentioning that not all types of vacancies are beneficial to increase the photocatalytic performance of materials. Different types of defects including OVs clusters, surface OVs, and bulk OVs were introduced in TiO2.80 The surface OVs not only significantly promoted the adsorption of H2O and facilitated charge transfer to the adsorbed H2O, forming ˙OH, but also conducive to the adsorption and desorption of NO, contributing to the best photocatalytic NO removal activity. Amorphous TiO2 with OVs clusters had no photocatalytic ability, and bulk OVs neither helped the adsorption of H2O nor improved the charge transfer to H2O. Rao et al. constructed position-manipulated OVs (OVs1 and OVs2) on the surface of (BiO)2CO3 (BOC) to maximize the formation of ROS for photocatalytic NO deep oxidation (Fig. 8a and b).37 The introduction of OVs1 (OVs1-BOC) induced the formation of an intermediate energy level and promoted the formation of ˙O2− and ˙OH (Fig. 8c–e), and the light absorption edge of BOC was redshift after the construction of OVs2 (OVs2-BOC), and thus only ˙O2− could be generated. OVs1-BOC (50.0%) exhibited a higher photocatalytic NO removal efficiency than OVs2-BOC (41.6%) under identical conditions, but OVs2-BOC possessed more stable photocatalytic activity (Fig. 8f). Bi2Sn2O7−x with distinct Sn/Bi-adjacent OVs (VO1–Bi2Sn2O7−x/VO2–Bi2Sn2O7−x) was synthesized and exhibited different photocatalytic NO oxidation performances (Fig. 8g–i).81 VO1–Bi2Sn2O7−x served as the adsorption sites to boost the adsorption and activation of NO and facilitated electron–hole separation for efficient NO removal, whereas VO2–Bi2Sn2O7−x became the recombination centers of charge carriers, and thus led to a decrease in the photocatalytic performance (Fig. 8j–n). Therefore, the rational construction of surface vacancies is the key to maximizing the effect on elevating the photocatalytic performance.
 | ||
| Fig. 8 Crystal structure models of (a) OVs1-BOC and (b) OVs2-BOC. Comparison of DOS/PDOS between (c) BOC and OVs1-BOC and (d) BOC and OVs2-BOC. (e) DMPO spin-trapping ˙O2− and ˙OH ESR spectra of BOC, OVs1-BOC, and OVs2-BOC under visible light irradiation. (f) Formation and photocatalytic mechanism on BOC, OVs1-BOC, and OVs2-BOC.37 Reproduced from ref. 37 with permission from the American Chemical Society, Copyright 2021. Structure model of (g) pristine Bi2Sn2O7, (h) VO1–Bi2Sn2O7−x (Sn-adjacent), and (i) VO2–Bi2Sn2O7−x (Bi-adjacent) with oxygen vacancies obtained using theoretical calculations. (j) Photocurrent responses, (k) electrochemical impedance spectra, (l) steady-state photoluminescence, (m) surface photovoltage spectra, and (n) transient fluorescence decay spectra of Bi2Sn2O7, VO1–Bi2Sn2O7−x, and VO2–Bi2Sn2O7−x.81 Reproduced from ref. 81 with permission from the Royal Society of Chemistry 2021. | ||
Surface defects have been universally regarded as the vital active sites for highly efficient photocatalytic reaction, but predesigned surface defects undergo dynamic transformations in response to the inevitable structural changes in catalysts during the reaction upon exposure to light field/electric field/temperature, and thus the reconstructed defective surfaces may act as the real active sites for catalytic reactions. Recently, Dong's group devoted their efforts to elaborating the dynamic evolution of surface defects and revealing the true reaction mechanism with real-time monitoring via in situ electron paramagnetic resonance (EPR) technology, which is a powerful tool for the quantitative characterization of vacancies based on accurate single-electron detection (Fig. 9a and b).82,83 TiO2 and BiOCl with predesigned surface defects were employed as the model catalyst, and the formation and evolution of photoexcited dynamic OVs were verified by in situ EPR technology (Fig. 9c and d).82,83 The photoswitchable OVs were found to be the true active sites during the reaction, which facilitated the activation of reactants, and thus realized the efficient conversion of NO into nitrates.
 | ||
| Fig. 9 (a) Schematic of operando EPR measurements. (b) Picture of the custom-made in situ reactor chamber and illustration of the obtained EPR spectra during different stages. (c) Cycling process of photoexcited OV (PE-Vo) generation and self-recovery on TiO2 (PD-Vo: predesigned OVs).82 Reproduced from ref. 82 with permission from the American Chemical Society, Copyright 2023. (d) Schematic illustration of photocatalytic NO oxidation in which PE-OVs on BiOCl serve as real active sites under UV irradiation and the reversible creation of PE-OVs on BiOCl.83 Reproduced from ref. 83 with permission from the American Chemical Society, Copyright 2022. | ||
Surface vacancies can normally increase the photocatalytic activity, but there is still a possibility that unstable vacancies suffer from deactivation during continuous reaction. With regards to preventing the deactivation of OVs in Bi-based photocatalysts, additional modification of Bi nanoparticles may be an accessible strategy (Fig. 10a).84,85 The surface-deposited Bi acts the active sites to activate O2 and H2O, which avoids O2 and H2O filling into OVs, thus preventing the deactivation of OVs (Fig. 10b). Besides, Ran et al. proposed the UV-light-induced destruction of Bi–O and Sb–O bonds to introduce OVs in BiSbO4 (BiSbO4-OV) (Fig. 10c); meanwhile, the mechanisms for the inactivation and regeneration of OVs were clarified to prevent the deactivation of photocatalysts.86 Although oxygen in the air can fill the OVs during photocatalytic reaction to lead to the consumption of OVs, and thus the deactivation of BiSbO4-OV, the deactivated photocatalyst was regenerated by re-irradiation with UV-light to reintroduce OVs for inducing the photocatalyst to return to its initial state (Fig. 10d).
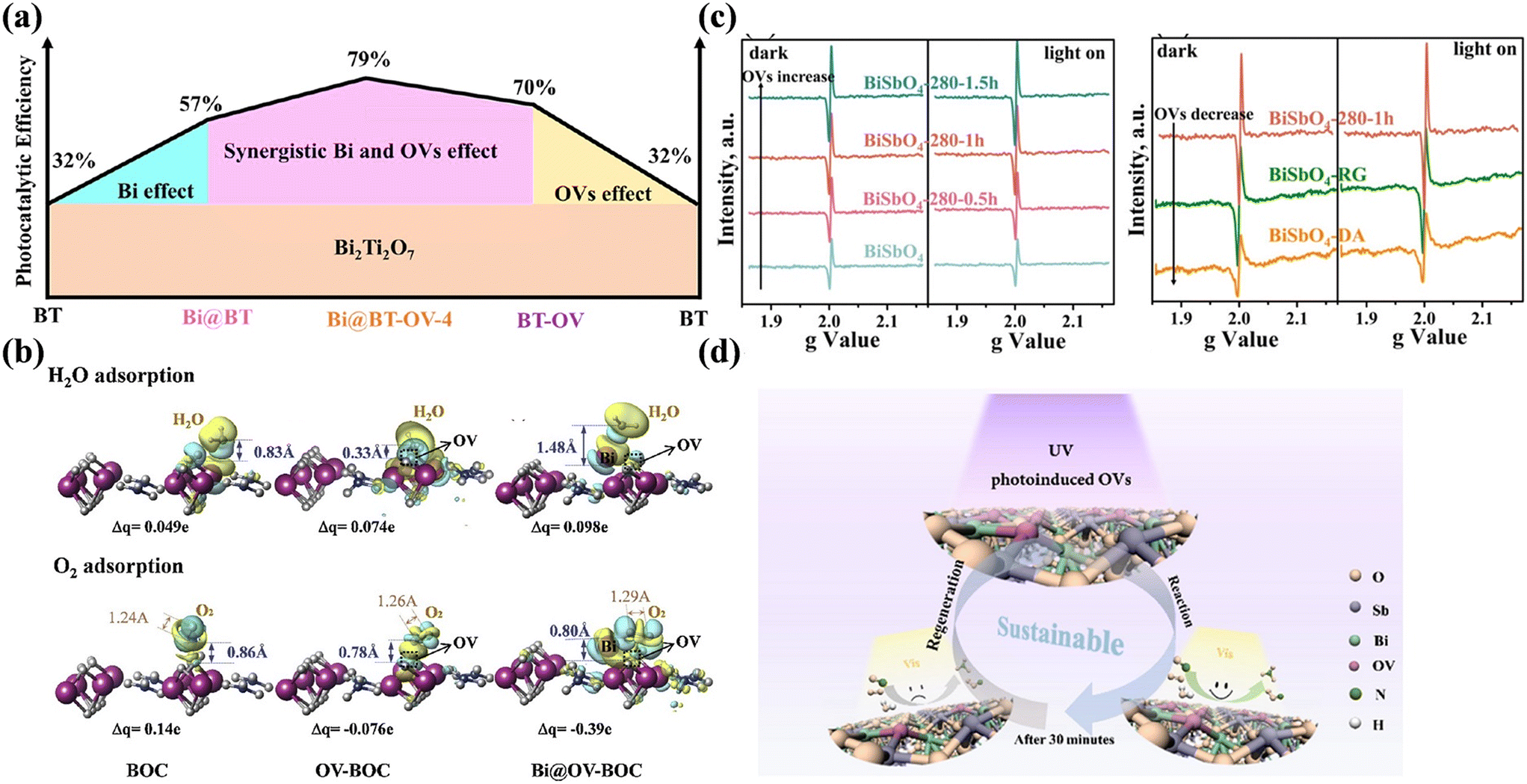 | ||
| Fig. 10 (a) Schematic of the co-effect of OVs and Bi on the photocatalytic performance of Bi-deposited porous Bi2Ti2O7 possessing rich OVs.84 Reproduced from ref. 84 with permission from Elsevier, Copyright 2022. (b) Charge difference distribution of optimized H2O and O2 adsorption on Bi2O2CO3 (BOC), Bi2O2CO3 nanosheets with OVs (OV-BOC) and Bi metal nanoparticle-decorated Bi2O2CO3 nanosheets with OVs (Bi@OV-BOC).85 Reproduced from ref. 85 with permission from Elsevier, Copyright 2020. (c) Solid-state EPR spectra of BiSbO4-OV samples. (d) Formation, inactivation and regeneration process of OVs on BiSbO4 and photocatalytic NO oxidation mechanism.86 Reproduced from ref. 86 with permission from the American Chemical Society, Copyright 2019. | ||
2.3 Surface decoration
Surface decoration is another approach to adjust the surface structure of photocatalysts by introducing a small number of metal/non-metal nanoparticles or surface functional groups on their surface, constructing micro-sized heterogeneous structures to tune their optical property, charge separation, and surface reaction for improving their photocatalytic performance. The surface deposition of noble metal nanoparticles is an attractive strategy to fully utilize the localized surface plasmonic resonance (LSPR) effect of noble metals for extending the spectral response and facilitating the separation of charge carriers in photocatalysts. Au, Ag, and Pt nanoparticles usually present strong plasmon resonance absorption at a specific wavelength under visible light irradiation.87–91 For example, Guo et al. deposited Au nanoparticles on Bi5Ti3FeO15 (Au/Bi5Ti3FeO15) to improve its light absorption and inhibit the recombination of photogenerated carriers by the synergistic LSPR effect and interface effect (Fig. 11a).88 According to density functional theory (DFT) simulation, the ohmic contact, which was formed at the interfaces between Bi5Ti3FeO15 and Au, contributed to the electron transfer and reactant activation for the generation of ˙O2− and ˙OH, realizing efficient and stable photocatalytic NO removal. Similarly, zero-dimensional Pt quantum dot-decorated La2Ti2O7 nanosheets exhibited outstanding efficiency for electron–hole separation and carrier transfer, which was beneficial for ROS formation for efficient NO removal with extremely low NO2 production.89 | ||
| Fig. 11 (a) Schematic of photocatalytic NO removal by Au/Bi5FeTi3O15.88 Reproduced from ref. 88 with permission from Springer Nature, Copyright 2022. (b) Schematic of charge separation on the integrated effect of the Schottky junction and LSPR effect and corresponding photocatalytic reaction on Ag nanoparticle-decorated SrTiO3.92 Reproduced from ref. 92 with permission from the American Chemical Society, Copyright 2022. | ||
Besides, the introduction of plasmonic metals can not only induce the LSPR effect but also construct a Schottky junction to synergistically promote the separation and transfer of carriers.92,93 Ag nanoparticle-decorated SrTiO3 showed a six-fold increment in NO conversion rate and significant decline in toxic NO2 simultaneously.92 The elevated photocatalytic performance could be attributed to the following: (1) the LSPR effect of Ag nanoparticles promoted the light absorption and charge transfer in the photocatalyst, (2) the formation of a Schottky junction facilitated the charge separation and induced electron transfer from the silver particles to SrTiO3 to enhance the lifetime of excitons, and (3) the formation of an Ag–O bond between Ag nanoparticles and SrTiO3 increased the charge density of adjacent Ti, which offered a favorable channel for the adsorption and activation of reactants for the subsequent formation of ROS and complete oxidation of NO (Fig. 11b).
The introduced metal nanoparticles can also serve as active sites to promote the adsorption and activation of reactants, contributing to the optimization of the reaction pathway for the highly efficient complete photocatalytic oxidation of NO. g-C3N4 with monodisperse Au nanoparticle decoration (Au@CN) exhibited highly enhanced NO oxidation activity and superior NO2 inhibition ability.94 Besides the accelerated adsorption and activation of reactants and separation efficiency of carriers achieved by the modification of Au nanoparticles, the unique electronic structure of Au@CN tended to promote nitrate formation with a low rate-determining barrier, and thus largely suppressed the formation of the toxic intermediate NO2 (Fig. 12a–d). Furthermore, Li et al. designed Pd nanoparticle-decorated g-C3N4 (PdCN) by DFT simulation firstly, and it was found that the PdCN was equipped with a lower activation barrier in the rate-determining step and higher selectivity for the final product (nitrate) instead of the toxic intermediate (NO2) (Fig. 12e and f).95 PdCN was also fabricated and exhibited outstanding photocatalytic NO complete oxidation (Fig. 12g and h), which is consistent with the theoretical prediction. Therefore, the rational utilization of theoretical prediction can provide a quick and efficient method to screen and design efficient photocatalysts.
 | ||
| Fig. 12 Calculated reaction pathway: calculated climbing image nudged elastic band (CI-NEB) reaction pathway (a and b) and structures of initial states, transitional states and final states (c and d) for NO photo-oxidation by ˙O2− on CN and Au@CN.94 Reproduced from ref. 94 with permission from Elsevier, Copyright 2019. Calculated reaction pathway for NO conversion by ˙O2− and optimized structures of adsorbed NO and NO2 on (e) CN and (f) PdCN. (g) NO conversion and (h) NO2 production on CN and PdCN.95 Reproduced from ref. 95 with permission from Elsevier, Copyright 2020. | ||
Non-noble plasmonic metal-based photocatalysts have been identified as alternatives to noble metal-based photocatalysts due to their advantages such as earth-abundance and cost effectiveness.96 Metal Bi, which is equipped with relatively small effective mass for the conduction of electrons, long mean free path, and high charge carrier mobility, exhibits interesting electronic properties. Correspondingly, metal Bi possesses a distinctive LSPR-response covering the entire UV-vis-NIR spectral region, and thus can directly drive the photocatalytic redox reaction. Dong et al. firstly found that Bi nanoparticles with a 100–200 nm size exhibited notable photocatalytic NO removal efficiency under 280 nm light irradiation, which was ascribed to the UV-mediated LSPR effect of Bi nanoparticles (Fig. 13a).97 Meanwhile, significant research has been directed to the rational design of Bi-decorated nanomaterials for photocatalytic NO removal, such as SiO2 nanoparticle-modified Bi microspheres,98 bismuth sphere-assembled graphene oxide,99 and Bi-decorated amorphous bismuth oxide.100 Although various Bi-decorated hybrid structures have been proposed in the earlier study, complete photocatalytic NO oxidation has only gradually attracted attention in recent years. Zhang et al. transformed Bi spheres with a surface amorphous Bi2O3 layer to a Bi@Bi2O2CO3 core–shell photocatalyst (Bi@C-BOC), realizing an increase in photocatalytic NO removal efficiency and the inhibition of NO2 formation (Fig. 13b).101 The crystalline Bi2O2CO3 layer was beneficial for the transfer of hot electrons on plasmonic Bi to Bi2O2CO3, contributing to the formation of ROS for the complete oxidation of NO (Fig. 13c).
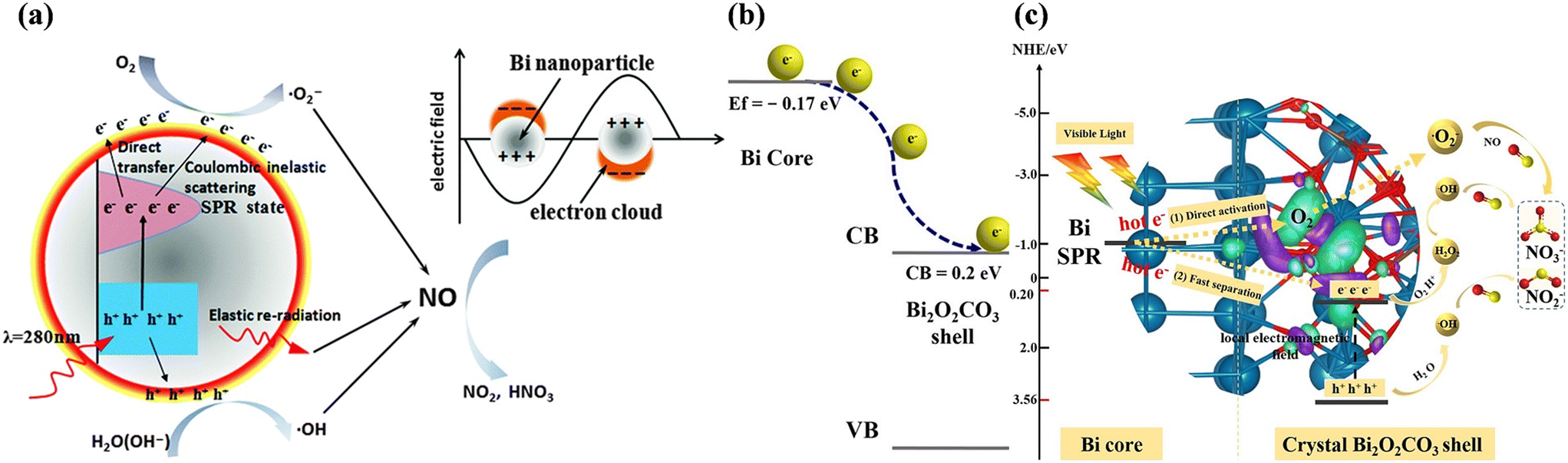 | ||
| Fig. 13 (a) Schematic of the plasmonic photocatalysis mechanism of Bi nanoparticles.97 Reproduced from ref. 97 with permission from the Royal Society of Chemistry, Copyright 2014. (b) Schematic of directional photogenerated electron transfer processes over Bi@C-BOC. (c) Proposed photocatalytic mechanism for enhancing the photocatalytic activity over Bi@C-BOC under visible light irradiation.101 Reproduced with permission from ref. 101. Reproduced from ref. 101 with permission from Elsevier, Copyright 2021. | ||
Besides, the integration of the LSPR effect and defect engineering has attracted widespread attention on Bi-decorated photocatalysts, which can synergistically promote the adsorption of reactants and the separation of carriers to increase the photocatalytic NO removal efficiency.102–106 Bi/BiOBr nanoflowers with OVs (BOB-25) were fabricated to achieve an enhanced photocatalytic performance and decreased NO2 generation.102 Concretely, NO tended to donate outer layer electrons to OVs to form NO+, and also additional active sites were introduced to promote the adsorption and activation of the reactants (Fig. 14a). Besides, plasmonic Bi with a larger work function than BiOBr served as an electron sink and the impurity level was formed because of the presence of OVs, and thus a directional transfer channel for electrons was formed, which inhibited the recombination of photoinduced carriers (Fig. 14b). Furthermore, Bi/Bi2O2−xCO3 nanosheets with surface OVs promoted the generation of H2O2 by capturing electrons from the defect states of Bi2O2−xCO3via the two-electron reduction of O2, and also the surface OVs in Bi–O layers provided a channel for electron transfer between Bi and Bi2O2−xCO3 to increase the charge separation efficiency, which induced the highly efficient complete oxidation of NO (Fig. 14c and d).103 A unique electron transfer covalent loop ([Bi2O2]2+ → Bi-metal → O2−) was formed in Bi-metal@Bi2O2[BO2(OH)] with OVs, which could guide the directional transfer of carriers to improve the separation efficiency of charge carriers and the yield of ROS (Fig. 14e).104 Simultaneously, the Bi metal functioned as an electron donor to activate NO, and thus induce a new reaction path of NO → NO− → NO3−, inhibiting the generation of toxic intermediates (Fig. 14f).
 | ||
| Fig. 14 (a) Proposed NO adsorption and photocatalytic conversion routes over BOB-25. (b) Plausible diagram for ROS generation under visible light over BOB and BOB-25.102 Reproduced from ref. 102 with permission from Elsevier, Copyright 2023. (c) Possible mechanisms of H2O2 formation over Bi/Bi2O2−xCO3 nanosheet. (d) Schematic of charge transfer in the Bi/Bi2O2−xCO3 system and the possible mechanism of photocatalysis.103 Reproduced from ref. 103 with permission from Elsevier, Copyright 2019. (e) Charge density difference in Bi-metal@Bi2O2[BO2(OH)] with OVs: electron accumulation is labeled in blue and depletion in yellow, and the isosurfaces are set to 0.002 eV Å−3. (f) Principal photocatalytic oxidation reaction mechanism of NO on Bi-metal@Bi2O2[BO2(OH)].104 Reproduced from ref. 104 with permission from Elsevier, Copyright 2022. | ||
The surface decoration of functional groups also has been proposed to improve the photocatalytic performance. The introduction of hydroxyl functional groups, which are equipped with the ability to extend the light absorption, provide extra active sites, and promote the adsorption and activation of reactants, is one of most widely used strategies.107,108 Yang et al. found that the intercalation of –OH group in BiOI could enhance visible light absorption, accelerate the carrier transfer, and reduce the energy barriers for the conversion of NO2 into NO2− and NO3−.108 Besides, cyano/hydroxyl group co-functionalized g-C3N4 realized efficient photocatalytic NO conversion with an extremely low NO2 production ratio (4.8%).109 The insertion of cyano groups induced the optimization of the band structure to promote the formation of ˙O2− in g-C3N4, and the intermediate NO2, which tended to adsorb on the surface of hydroxyl groups instead of being released, could be complete oxidized by changing the conversion pathway from NO → NO2 to NO → NO2 → NO3−.
2.4 Construction of heterojunctions
The construction of heterojunctions is the most commonly used strategy to accelerate the spatial separation of photogenerated electron–hole pairs for higher photocatalytic activity.110,111 One of mainstream heterojunctions, type II heterojunctions, have been widely constructed for photocatalytic NO purification.112–117 Meanwhile, quantum dots (QDs), which are equipped with excellent visible light absorption, multi-exciton effect and adjustable energy bands, are introduced to fabricate type II heterojunctions for efficient NO removal.118–120 For example, BaSnO3 loaded with CdS QDs (CdS/BaSnO3) exhibited efficient photocatalytic NO removal efficiency with suppressed NO2 formation (Fig. 15a).118 The introduction of CdS QDs not only improved the light absorption range and induced the generation of ˙O2−, but also could provide new active sites for the adsorption and activation of NO. Type II heterojunctions with 2D/2D structures have also attracted attention due to their advantages of enlarged interface area and intimate contact to regulate carriers for boosting the photocatalytic performance. The BiOBr–g-C3N4 heterojunction with surface OVs formed a spatial conductive network framework, which greatly boosted the separation efficiency of electron–hole pairs, and thus facilitated the photocatalytic NO oxidation performance.119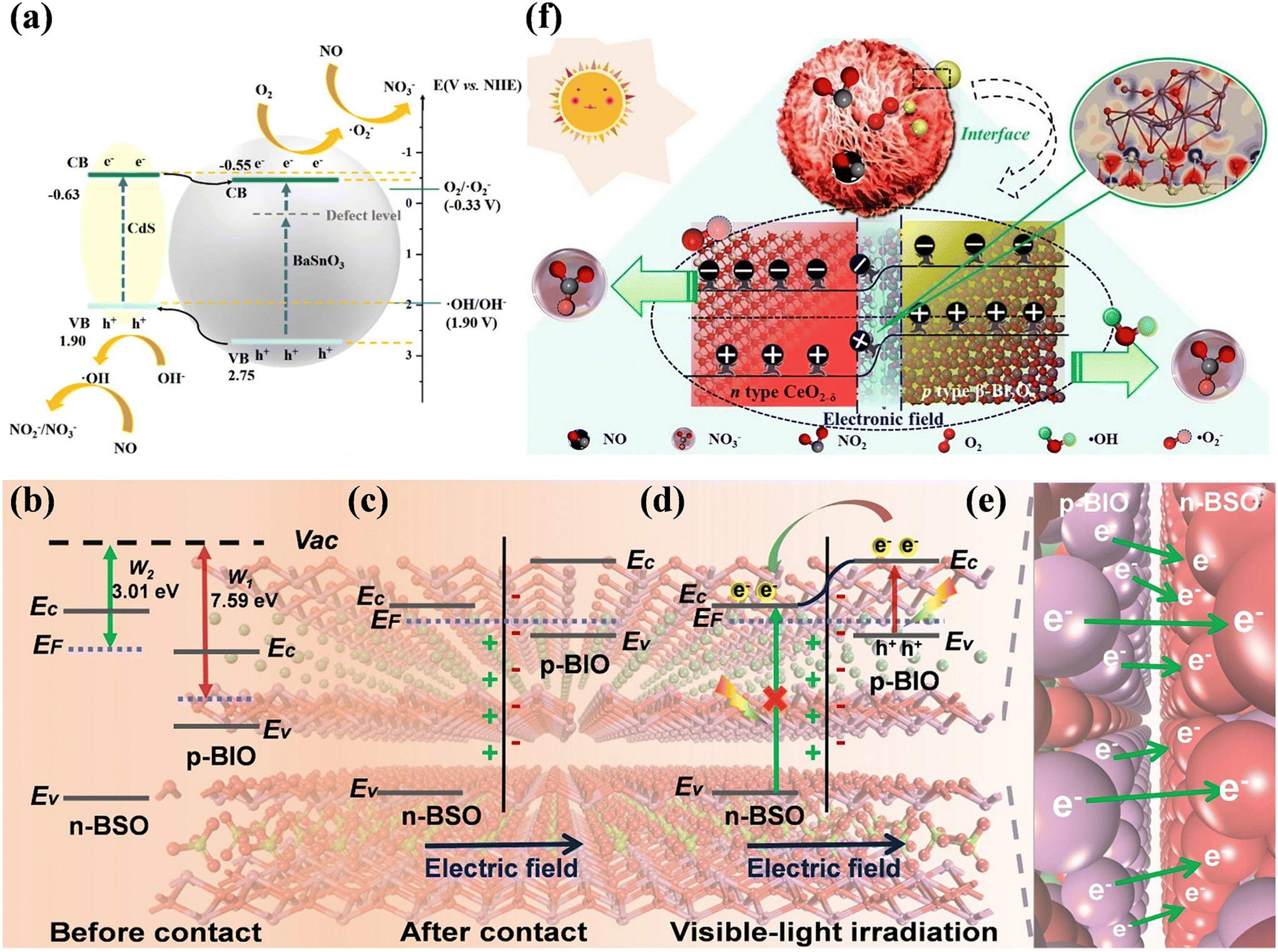 | ||
| Fig. 15 (a) Proposed process of NO + O2 reaction over CdS/BaSnO3 under visible light irradiation.118 Reproduced from ref. 118 with permission from Elsevier, Copyright 2023. Schematic of the BiOI/Bi2O2SO4 p–n heterojunction: (b) before contact, (c) after contact, (d) under visible-light irradiation, and (e) visual representation of electron migration process from p-BIO to n-BSO (the background image is the atomic structure of the BiOI/Bi2O2SO4 p–n heterojunction).121 Reproduced from ref. 121 with permission from Elsevier, Copyright 2021. (f) Schematic of photocatalytic NO removal over 4% β-Bi2O3/CeO2−δ under visible light irradiation.122 Reproduced from ref. 122 with permission from Elsevier, Copyright 2021. | ||
Based on type II heterojunctions, the p–n heterojunction has been proposed, which can accelerate the electron–hole migration across the heterojunction by providing an additional internal electric field (IEF) for the optimization of photocatalytic performance.110,111 Generally, combining theoretical calculations and experimental characterization, the directional transfer of charge under the influence of IEF was confirmed.121–123 Geng et al. constructed the BiOI/Bi2O2SO4 p–n heterojunction, and thus a directional charge transfer channel induced by the IEF was established to allow photogenerated electrons to migrate from BiOI to Bi2O2SO4 (Fig. 15b–e).121 The boosted charge separation and transfer contributed to an electron-rich interfacial environment, promoting the reactant activation and ROS generation, and thus promoting the photocatalytic complete oxidation of NO. Besides, the synergistic effects of OVs and p–n heterojunctions was demonstrated, and the well-designed β-Bi2O3/CeO2−δ p–n heterojunction with OVs exhibited efficient deep oxidation of NO.122 Concretely, the light responsive range was broadened to the visible region and the spatial charge transfer was achieved by the IEF, and also the OVs could further promote the migration of charge carriers at the interface, which contributed to the production of ˙OH and ˙O2− participating in NO removal (Fig. 15f).
Another mainstream Z-scheme heterojunction has been widely acknowledged, aiming to not only accelerate the separation of carriers but also maximize the redox potential of the heterojunction system.124,125 For example, the covalent organic skeleton (COF)/TiO2 Z scheme heterojunction was constructed to achieve the highly selective oxidation of NO (Fig. 16a).126 The introduced COF improved the light capture capability and separation of photogenerated carriers, and the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– structure of COF with accumulated photogenerated electrons was identified as the activation sites of O2 to form ˙O2−; meanwhile, the photogenerated holes were inhibited to react with adsorbed water to generate ˙OH, further hindering the critical path of NO oxidation to produce toxic by-products NO2 (Fig. 16b–f). Noteworthily, other COF/MOx (M = Zn, Zr, Sn, Ce, and Nb) catalysts also exhibited superior selectivity and activity, meaning that this scheme is credited with universality. Besides, layered 2D/2D W18O49/g-C3N4 heterostructures were equipped with well-developed interfaces, thus showing extended light absorption from the visible light to NIR region and improved photocatalytic NO oxidation performances.127 The combination of a Z-scheme heterojunction and OVs also could synergistically promote the deep oxidation of NO.128–130 Besides, Lu et al. reported the preparation of the ternary Z-scheme photocatalyst SnO2/NCDs/ZnSn(OH)6 (NCDs = N-doped carbon quantum dots), which possessed strong redox ability for NO complete oxidation.131 The NCDs, as an electron transport bridge, improved both the broad-spectrum light-harvesting ability and the rapid separation of photoinduced electrons, contributing to the formation of ROS for NO removal (Fig. 16g).
N– structure of COF with accumulated photogenerated electrons was identified as the activation sites of O2 to form ˙O2−; meanwhile, the photogenerated holes were inhibited to react with adsorbed water to generate ˙OH, further hindering the critical path of NO oxidation to produce toxic by-products NO2 (Fig. 16b–f). Noteworthily, other COF/MOx (M = Zn, Zr, Sn, Ce, and Nb) catalysts also exhibited superior selectivity and activity, meaning that this scheme is credited with universality. Besides, layered 2D/2D W18O49/g-C3N4 heterostructures were equipped with well-developed interfaces, thus showing extended light absorption from the visible light to NIR region and improved photocatalytic NO oxidation performances.127 The combination of a Z-scheme heterojunction and OVs also could synergistically promote the deep oxidation of NO.128–130 Besides, Lu et al. reported the preparation of the ternary Z-scheme photocatalyst SnO2/NCDs/ZnSn(OH)6 (NCDs = N-doped carbon quantum dots), which possessed strong redox ability for NO complete oxidation.131 The NCDs, as an electron transport bridge, improved both the broad-spectrum light-harvesting ability and the rapid separation of photoinduced electrons, contributing to the formation of ROS for NO removal (Fig. 16g).
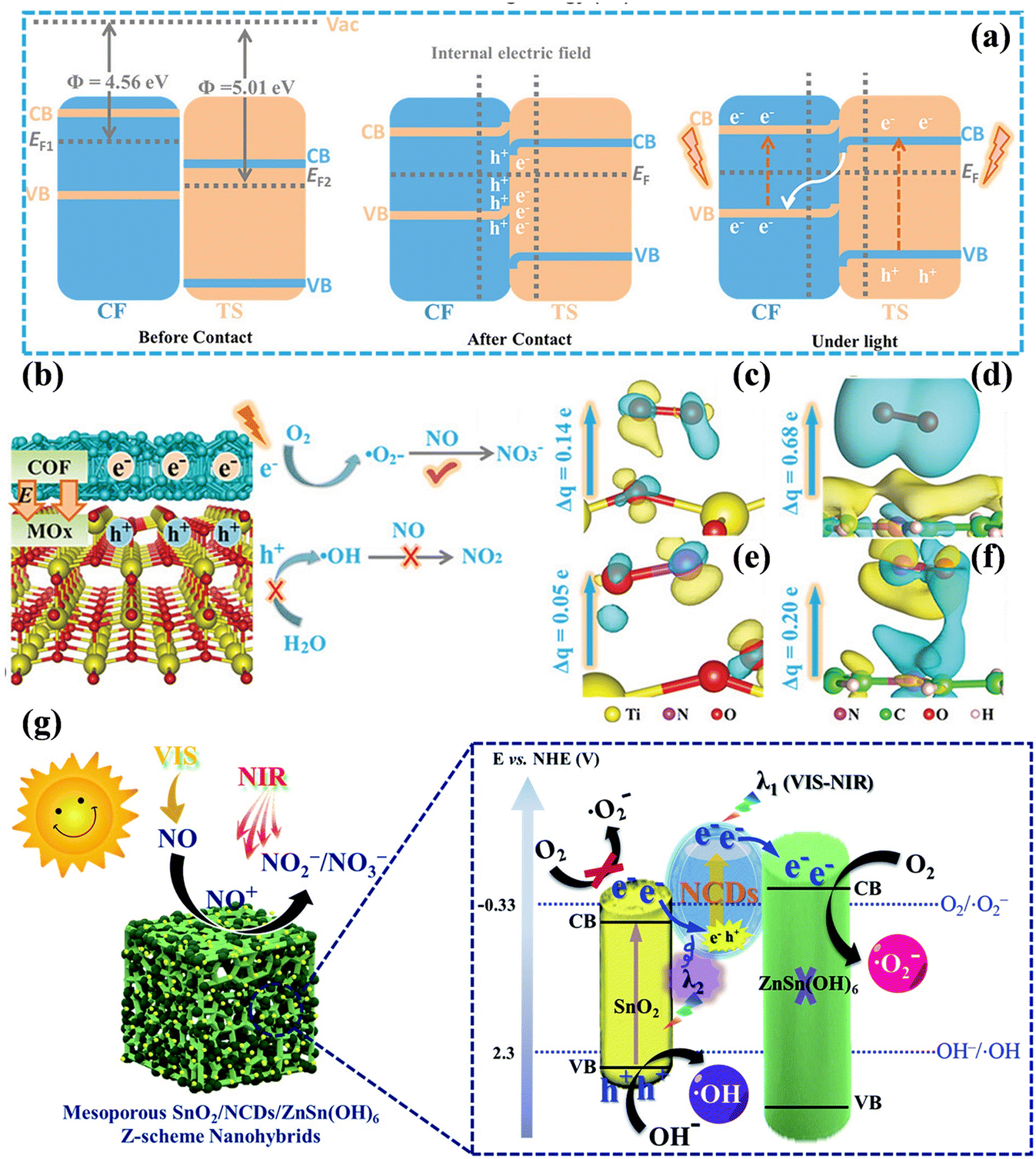 | ||
| Fig. 16 (a) Photoexcited carrier transfer process in COF and TiO2. (b) Mechanism of photocatalytic NO oxidation on COF/MOx. Charge difference distribution of adsorbed O2 and NO on (c and d) TiO2 and (e and f) COF, respectively.126 Reproduced from ref. 126 with permission from John Wiley and Sons, Copyright 2023. (g) Pathway of photocatalytic NO transformation and the reaction mechanism on the surface of the SnO2/NCDs/ZnSn(OH)6 Z-scheme nanohybrid under vis-NIR irradiation.131 Reproduced from ref. 131 with permission from the Royal Society of Chemistry, Copyright 2019. | ||
The S-scheme heterojunction, which is composed of reduction photocatalysts (RP) and oxidation photocatalysts (OP) with staggered band structures, has been proposed given that the pointless photogenerated charge carriers are recombined and a strong redox potential is introduced.132–134 Recently, the S-scheme heterojunction was also employed in photocatalytic NO removal.135–137 Moreover, Lv et al. developed a synergistic strategy to improve the photocatalytic NO oxidation efficiency with doping elements, building an S-scheme heterojunction and introducing OVs (0.1BTO/LTO-OV), which realized highly efficient NO removal with NO2 inhibition (Fig. 17a).135 Specifically, the doped electronic states and oxygen defect states narrowed the band gap to extend the light absorption range, and the S-scheme heterojunction formed between Bi-doped La2Ti2O7 and La-doped Bi4Ti3O12 enhanced the separation efficiency of photogenerated carriers and improved the transfer rate of electrons across the interface, which was conductive to the activation of the reactants and ROS formation for photocatalytic NO purification (Fig. 17b). A ternary g-C3N4/TiO2/Ti3C2 MXene photocatalyst was constructed, which possessed an interconnected nanosheet structure to increase the light absorption and provide a reaction interface between the reactants and photocatalysts.136 Simultaneously, the separation of photogenerated carriers was promoted by the formed S-scheme heterojunction and the Ti3C2 tightly bonded with TiO2 could accelerate the transfer of photogenerated holes, resulting in a high NO removal efficiency and low NO2 generation (Fig. 17c).
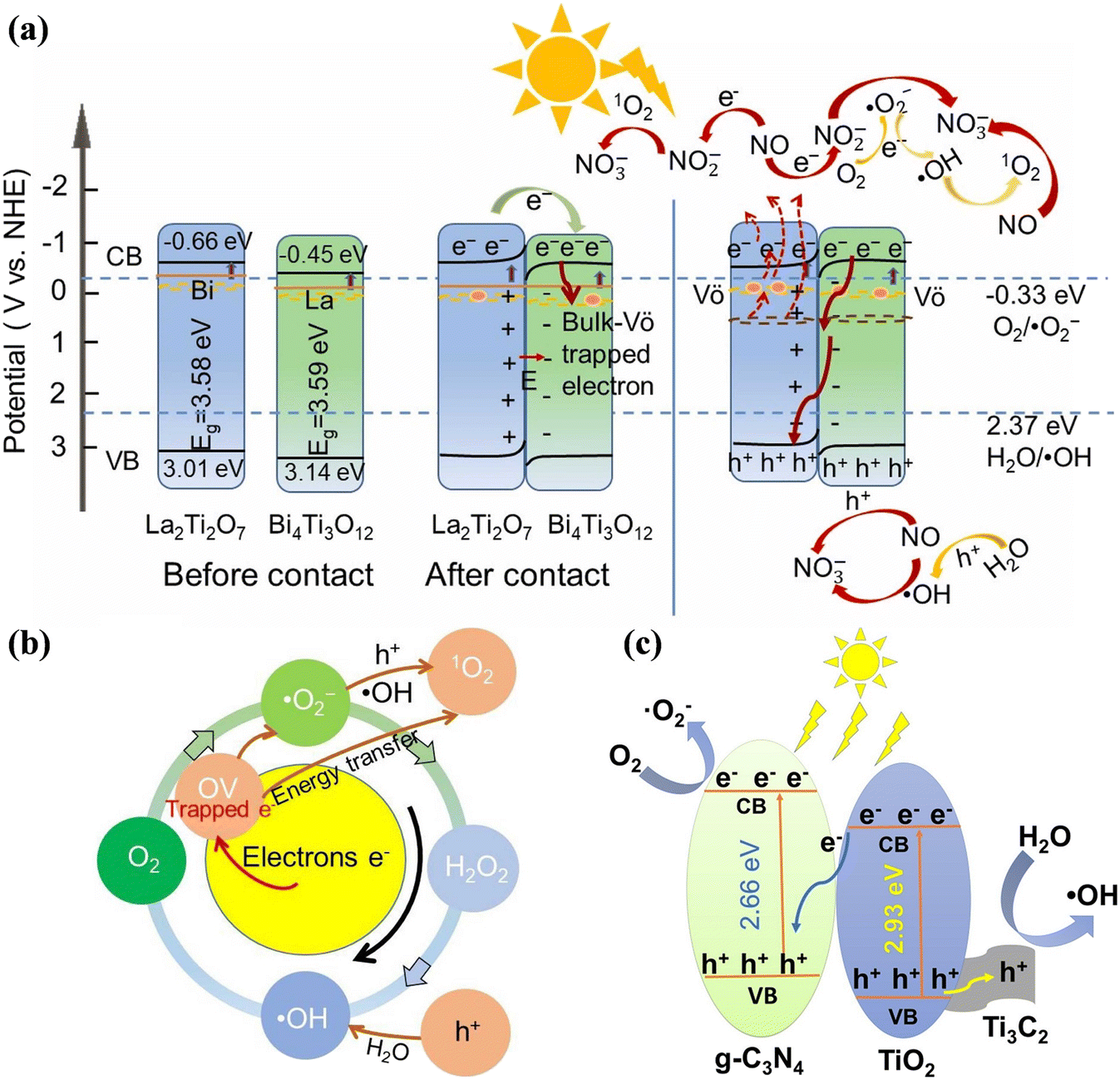 | ||
| Fig. 17 (a) Schematic of charge transfer and radical generation over 0.1BTO/LTO-OV S-scheme heterojunction for NO removal by different routes. (b) Schematic generation of reactive oxygen species of 0.1BTO/LTO-OV.135 Reproduced from ref. 135 with permission from Elsevier, Copyright 2024. (c) Schematic of the energy band structure and electron–hole separation of g-C3N4/TiO2/Ti3C2.136 Reproduced from ref. 136 with permission from Elsevier, Copyright 2021. | ||
Furthermore, insulator–semiconductor heterostructures, as a new family of photocatalysts, have been investigated because earth-abundant insulators possess the advantages of low cost, easy availability and good stability. Dong's group constructed a series of insulator-based heterojunctions for photocatalytic NO oxidation, challenging the traditional opinion that free electrons cannot be transferred to insulators. For example, the SrCO3–BiOI core–shell structure exhibited an enhanced visible light absorbance between 400–600 nm and could efficiently convert NO into harmless nitrate via the NO → NO+ and NO2+ → nitrate or nitrite routes (Fig. 18a).138 As confirmed by DFT simulation and experimental characterization, the covalent interaction between the O 2p orbital of insulator (SrCO3, n-type) and the Bi 6p orbital of the photocatalyst (BiOI, p-type) provided an electron transfer channel between SrCO3 and BiOI by the formation of a p–n heterostructure, allowing photogenerated electrons to transfer from the photocatalyst to the conduction band of the insulator, thus promoting the formation of ˙OH and ˙O2− for photocatalytic NO oxidation (Fig. 18b and c). Similarly, the photogenerated electrons from the semiconductor (BiOI) could directly transfer to the insulator (BaCO3) through the electron delivery channel (Bi atoms → adjacent carbonate layer) due to the potential difference between the Bi layer of BiOI and the carbonate layer of BaCO3 under visible light irradiation, contributing to the utilization of free electrons on BaCO3 to produce ROS participating in deep oxidation of NO (Fig. 18d–f).139 Besides, alkaline earth metal sulfate-based heterostructures also could induce electron transfer from the semiconductor to insulator, and thus accelerate the separation of charge carriers for efficient NO removal.140,141
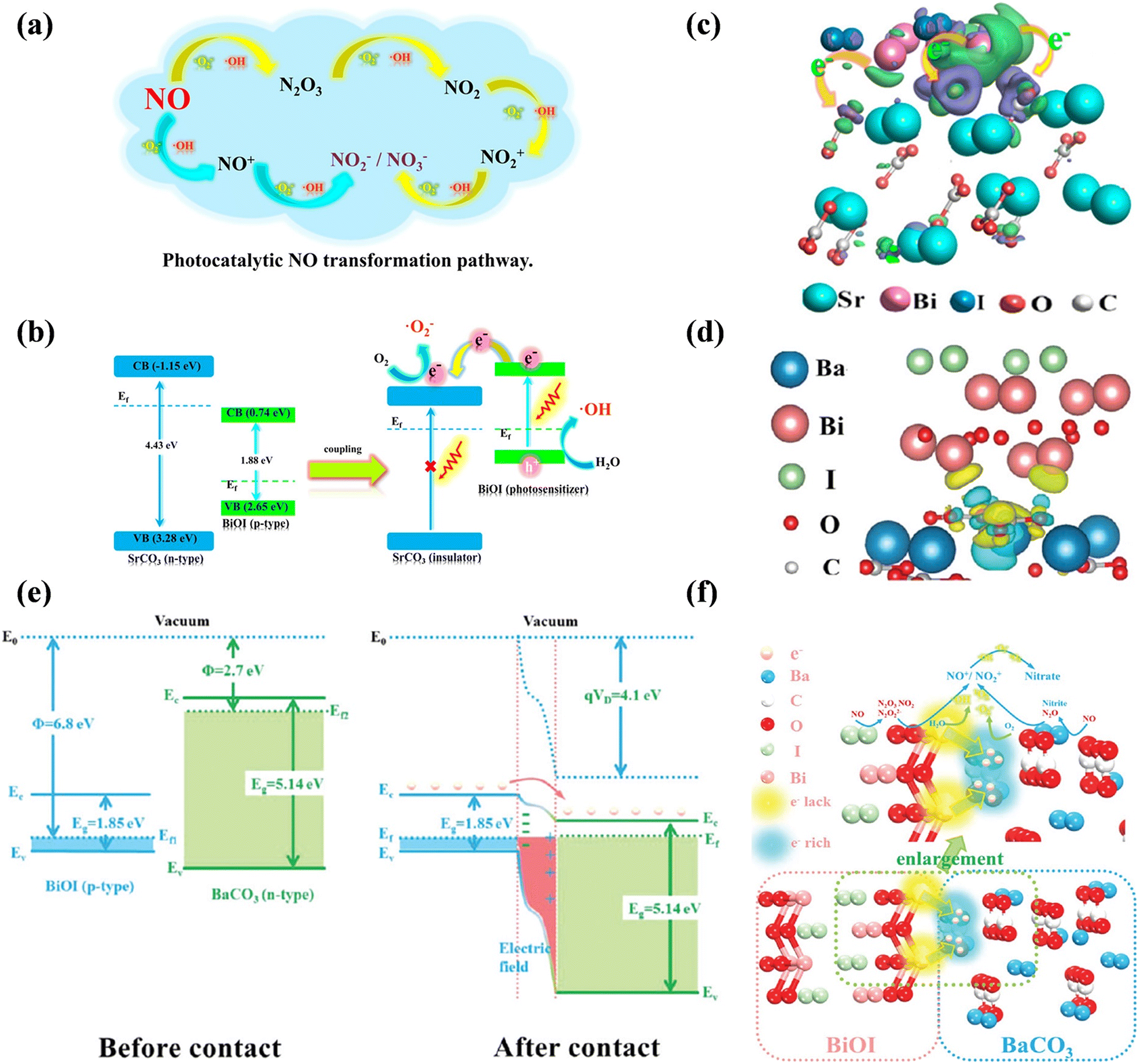 | ||
| Fig. 18 (a) Photocatalytic NO transformation pathway and (b) visible light photocatalysis mechanism of the insulator-based SrCO3–BiOI core–shell heterojunction. (c) Charge difference distribution between BiOI and SrCO3: charge accumulation is given in purple and depletion in green, and the isosurface is set to 0.003 eV Å−3.138 Reproduced from ref. 138 with permission from the American Chemical Society, Copyright 2018. (d) Charge difference distribution between BiOI and BaCO3: charge accumulation is shown in blue and depletion in yellow, and the isosurface is set to 0.002 eV Å−3. (e) Schematic of the band configuration and the charge separation at the interface of BaCO3/BiOI heterojunctions under visible light irradiation; Φ is the work function, VD is the contact potential, E0 is the vacuum level, Ec is the bottom of the conduction band, Ev is the top of the valence band, Eg is the band gap, Ef1 and Ef2 are the Fermi levels of BiOI and BaCO3, respectively, and Ef is the Fermi level of the BaCO3/BiOI heterojunction. (f) Proposed schematic diagram of the separation and transfer of photogenerated carriers and the photocatalytic process over the insulator–semiconductor heterojunction.139 Reproduced from ref. 139 with permission from the Royal Society of Chemistry, Copyright 2018. | ||
2.5 Crystal structure control
Photocatalysts with different exposed crystal structures often show different catalytic activities given that physicochemical properties are related to the surface atomic arrangement and coordination. For example, Wang et al. developed ZrO2 with monoclinic (Zr-M) and tetragonal (Zr-T) phases and found that monoclinic ZrO2 exhibited an outstanding NO conversion efficiency and extremely low NO2 selectivity.27 There is abundant Lewis acid sites exposed on monoclinic ZrO2, which served as electron acceptors and induced itinerant electron redistribution, charge carrier transfer, and further oxidation of ˙O2− for the directed formation of 1O2, realizing the selective oxidation of NO into NO3− (Fig. 19a and b). Also, the photocatalytic NO oxidation activity of TiO2 strongly depended on the percentage of anatase phase, and in comparison with rutile and mixed-phase TiO2, the removal efficiency was enhanced with an increase in the anatase content, while the selectivity for NO2 formation exhibited the opposite trend.142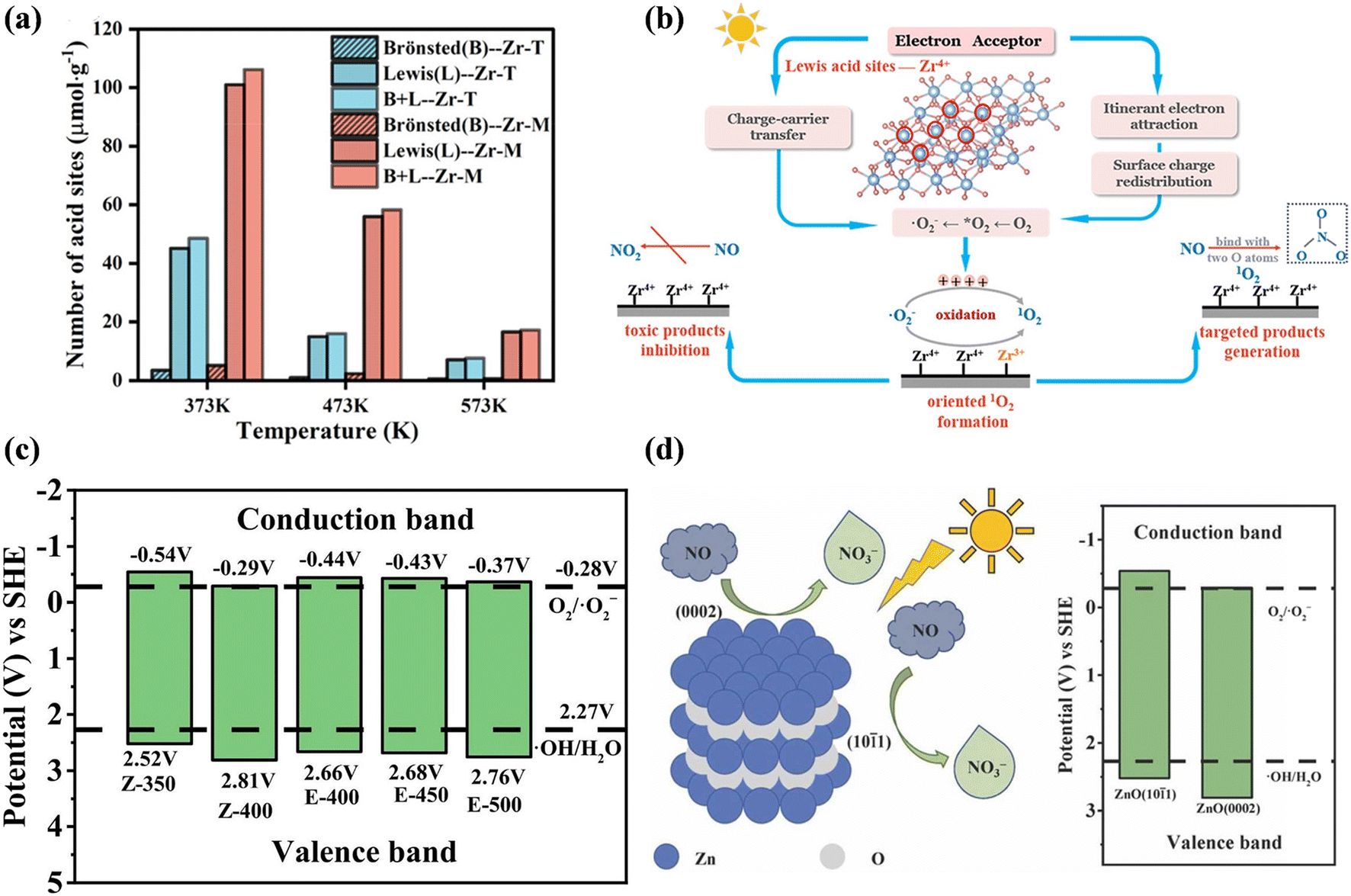 | ||
| Fig. 19 (a) Number of acid sites on Zr-T and Zr-M. (b) Evolution process of ROS and photocatalytic NO oxidation process over Zr-M.27 Reproduced from ref. 27 with permission from the American Chemical Society, Copyright 2023. (c) Energy level diagram arrangement of ZnO prepared from different precursors and calcination temperatures. (d) Photocatalytic mechanism of ZnO with mixed exposure of {101(−)1} and {0002} planes.143 Reproduced from ref. 143 with permission from Elsevier, Copyright 2022. | ||
Numerous facet-dependent photocatalysts also have been designed to optimize the band structure and surface electronic structure, which can adjust the adsorption and activation of reactants on the active sites and the behavior of carrier dynamics for achieving photocatalytic NO deep oxidation. The ZnO crystal with mixed exposure of {101(−)1} and {0002} planes exhibited a superior photocatalytic performance to that with exposed {0002} facets for NO removal. The nanocrystals with predominant {0002} crystal plane led to a shrinkage in band structure, and thus the low valence band position and poor affinity for NO decreased the NO removal efficiency and selectivity for NO3− production (Fig. 19c and d).143 SrBi2Ta2O9 with a higher {200} crystal plane exposure ratio showed higher photocatalytic NO deep oxidation activity compared to {001}-BiO and {001}-TaO facets.144 The promoted conversion of NO into NO3− was attributed to the greater exposure of {200} planes, which (1) facilitated the adsorption of NO; (2) endowed electron-enriching ability between the two types of the {001} layers to induce polarized electro-field for charge separation; and (3) provided more active sites for the formation for ˙O2−.
Crystal structure control of Bi-based photocatalysts has been widely employed, and photocatalytic NO deep oxidation has also been reported.145–149 The different exposed facets of Bi-based photocatalysts can provide various activate sites for the adsorption of reactants, which have an effect on ROS formation and the subsequent NO oxidation reaction. Bi2O2CO3 with {001} facets (001-BOC) exhibited much higher photocatalytic activity than that with {110} facets (110-BOC) given that the different adsorption and activation patterns directed the conversion pathway of NO deep oxidation.145 In combination with time-dependent in situ DRIFTS spectra and DFT calculation results, 001-BOC enabled the transformation of NO into NO− or cis-N2O22−, while 110-BOC induced NO to be activated into NO+ or N2O3 during the adsorption and activation process, which is conductive to the subsequent oxidation of the intermediates to the final products (NO3−) and the desorption of NO3− on 001-BOC (Fig. 20a). Li et al. fabricated BiOCl with tailored crystal planes, where efficient ROS generation and smoother NO conversion on the {010} facet-exposed BiOCl promoted the complete oxidation of NO.146 Specifically, in comparison with the {001} facet-exposed BiOCl (BOC-001), the alternating distribution of [Bi2O2]2+ and Cl− on the {010} facet of BiOCl (BOC-010) not only promoted H2O2 dissociation, which is the rate-determining step of ˙O2− generation (˙OH → H2O2 → ˙O2−), but also decreased the activation energies of NO2 to suppress NO2 accumulation on the photocatalyst (Fig. 20b–d).
 | ||
| Fig. 20 (a) DFT-calculated adsorption energy and bond length of several major intermediate adsorption products on 110-BOC and 001-BOC; all lengths are given in Å.145 Reproduced from ref. 145 with permission from the Royal Society of Chemistry, Copyright 2019. Calculated reaction pathways and activation energies (Ea) for (b) ˙OH and (c) ˙O2− induced NO conversion on BOC-001 and BOC-010. (d) Calculated reaction pathways and activation energies (Ea) for ROS generation and transformation on BOC-001 and BOC-010.146 Reproduced from ref. 146 with permission from Elsevier, Copyright 2019. | ||
Besides, the geometric structures of OVs on different crystal faces of BiOCl are totally different, affecting the transfer and transformation of surface charge to adjust the photocatalytic NO oxidation reaction.150 BiOCl with exposed {010} facets 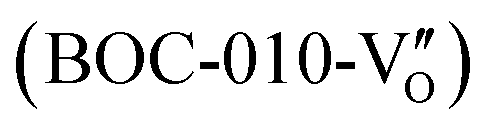 displayed an open channel structure with O, Bi, and Cl atoms exposed, and OVs could localize two electrons to form an archetypal F center
displayed an open channel structure with O, Bi, and Cl atoms exposed, and OVs could localize two electrons to form an archetypal F center 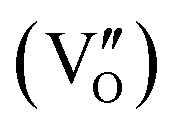 . BiOCl with exposed {001} facets
. BiOCl with exposed {001} facets 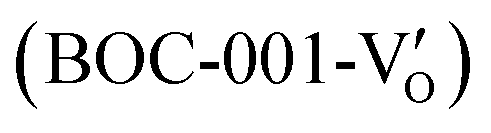 possessed a closely packed O-exposed structure, and OV was a single electron-trapped center
possessed a closely packed O-exposed structure, and OV was a single electron-trapped center 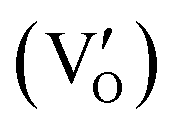 . During the photocatalytic NO oxidation process,
. During the photocatalytic NO oxidation process,  induced two-electron charging
induced two-electron charging 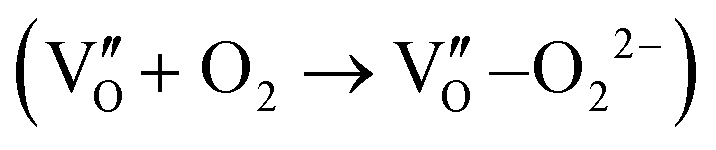 and subsequent one-electron decharging
and subsequent one-electron decharging 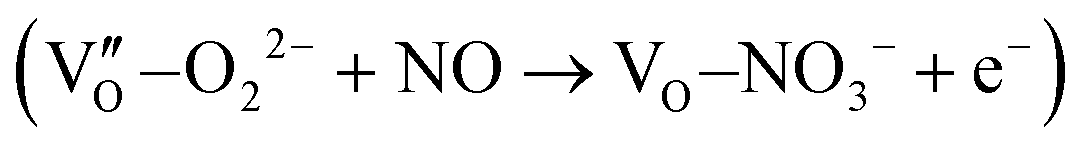 , and the back-donated electron was retrapped by VO to produce a new single-electron-trapped
, and the back-donated electron was retrapped by VO to produce a new single-electron-trapped 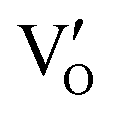 for a second round of selective NO oxidation
for a second round of selective NO oxidation 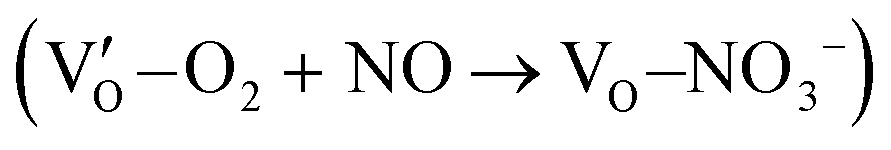 (Fig. 21b). Noteworthily,
(Fig. 21b). Noteworthily, 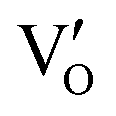 could directly oxidized NO by O22− into more stable bidentate nitrate in an exothermic process with a small energy barrier, but the
could directly oxidized NO by O22− into more stable bidentate nitrate in an exothermic process with a small energy barrier, but the 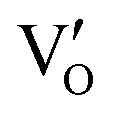 -mediated NO oxidation product possessed a remarkable geometric difference as monodentate nitrate (Fig. 21a). Besides, one
-mediated NO oxidation product possessed a remarkable geometric difference as monodentate nitrate (Fig. 21a). Besides, one 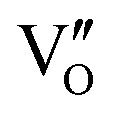 could oxidize two molecules of NO, resulting in a remarkably higher atomic vacancy utilization efficiency (Fig. 21c and d). The interfacial charging–decharging strategy by the synergistic effect of crystal structure control and vacancy engineering realized highly efficient NO complete oxidation.
could oxidize two molecules of NO, resulting in a remarkably higher atomic vacancy utilization efficiency (Fig. 21c and d). The interfacial charging–decharging strategy by the synergistic effect of crystal structure control and vacancy engineering realized highly efficient NO complete oxidation.
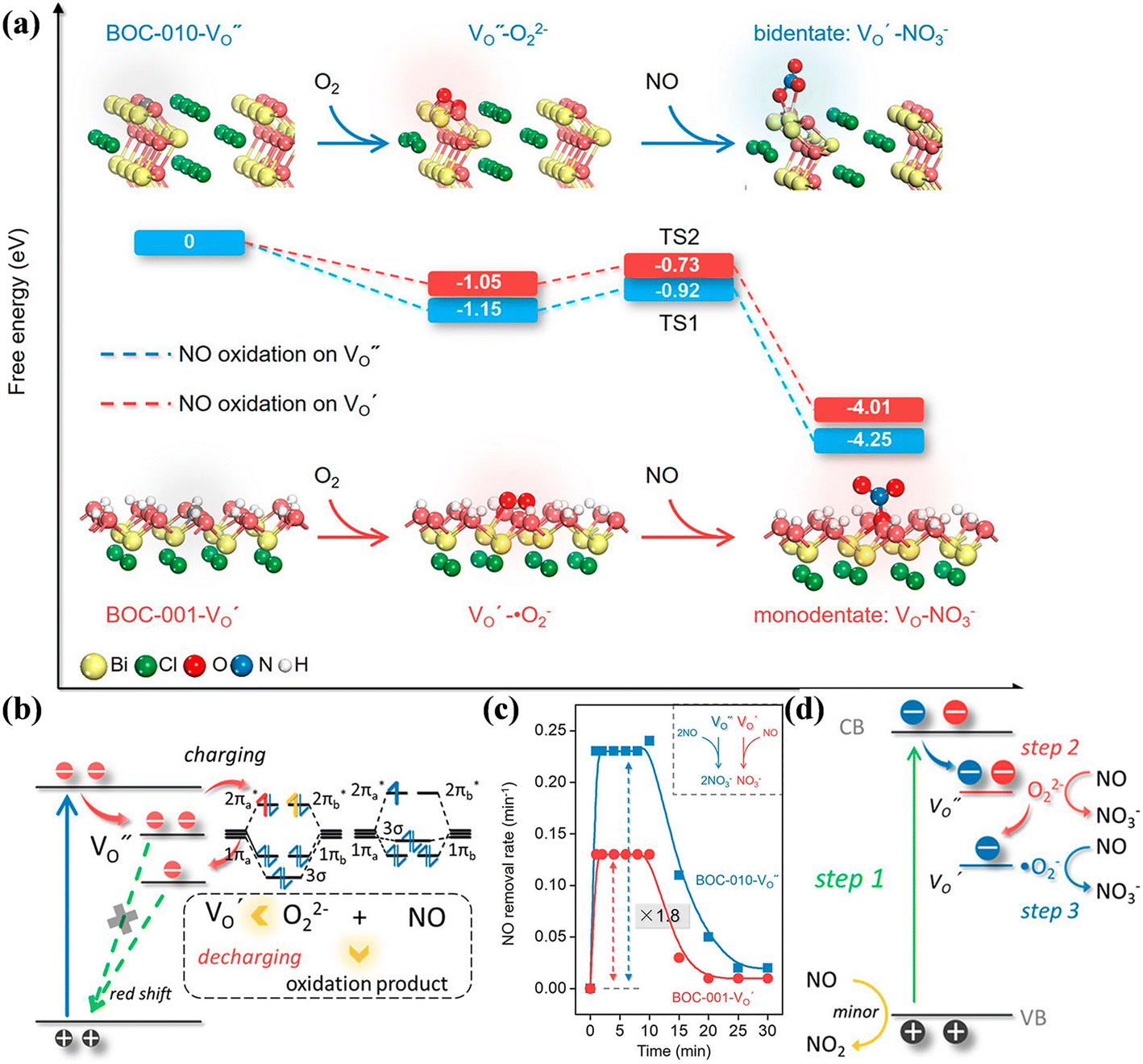 | ||
Fig. 21 (a) Free energy change during O22−- and ˙O2−-mediated NO oxidation on the VO of BOC-010 and BOC-001, respectively. (b) Schematic illustration of the charging–decharging scheme on the 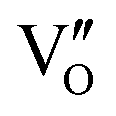 of BOC-010. (c) Corresponding NO oxidation rate dynamics and corresponding diagram of reaction process (inset). (d) Schematic illustration of NO removal and corresponding electronic excitation process on of BOC-010. (c) Corresponding NO oxidation rate dynamics and corresponding diagram of reaction process (inset). (d) Schematic illustration of NO removal and corresponding electronic excitation process on 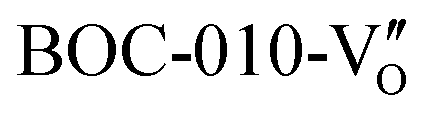 .150 Reproduced from ref. 150 with permission from the American Chemical Society, Copyright 2019. .150 Reproduced from ref. 150 with permission from the American Chemical Society, Copyright 2019. | ||
3. Photocatalytic complete NO reduction
To date, SCR technology is still the primary reduction technology for NO abatement, but the high energy consumption and massive demand of reductant during the treatment process decrease the environmental benefit and economic value of SCR technology.151–153 Thus, an alternative NO reduction reaction route by electrocatalysis has been proposed to achieve NO upcycling under ambient conditions and with water-supplied proton as the reductant, attracting wide attention from researchers.154–157 In recent years, the complete photocatalytic reduction of NO into NH3 has emerged. Although photocatalytic NO reduction technology is in its infancy, it possesses the potential to become one of the promising strategies to address the problems faced by both NO removal and NH3 synthesis due to the conversion of an air pollutant into valuable substances by directly utilizing the renewable solar energy as the energy source.19,29,158Goldstein et al. firstly demonstrated the potential feasibility of photocatalytic reduction NO into NH3 (Fig. 22a).159 TiO2 loaded with 10–130 electrons per particle, which was constructed by using γ-irradiation of acidic TiO2 colloid solutions containing 2-propanol, was studied for NO reduction under anaerobic conditions. There was competition among the reaction paths to produce NH3, N2O and N2, where the complete reduction of NO into NH3 proceeded via five consecutive one-electron transfer reactions. Inspired by the electrocatalytic NO reduction process, a 0D/3D g-C3N4 QD/three-dimensional macroporous and mesoporous (3DOMM) TiO2−x electrode was developed and its performances in both electrocatalytic and photocatalytic reduction of NO to NH3 were investigated (Fig. 22b).160 The g-C3N4 QD/3DOMM-TiO2−x electrode exhibited a much higher NH3 yield of 3841.5 μg h−1 mg−1 during the electrocatalytic test, and the photocatalytic NO reduction reaction was carried out in a single-compartment cell using g-C3N4 QD/3DOMM-TiO2−x as the photoelectrode. The NH3 yield in the photocatalytic test was estimated to be 95.07 μg h−1 mg−1, which was attributed to the fact that g-C3N4 QDs/3DOMM-TiO2−x possessed the following features: (1) a large specific surface area to provide surface active sites for the adsorption of the reactants, (2) macroporous and mesoporous channels to accelerate the mass and charge transfer, and (3) S-scheme heterostructure to facilitate charge separation. However, although the photocatalytic NO reduction route was initially developed, the photocatalytic efficiency and product selectivity still need to be further optimized.
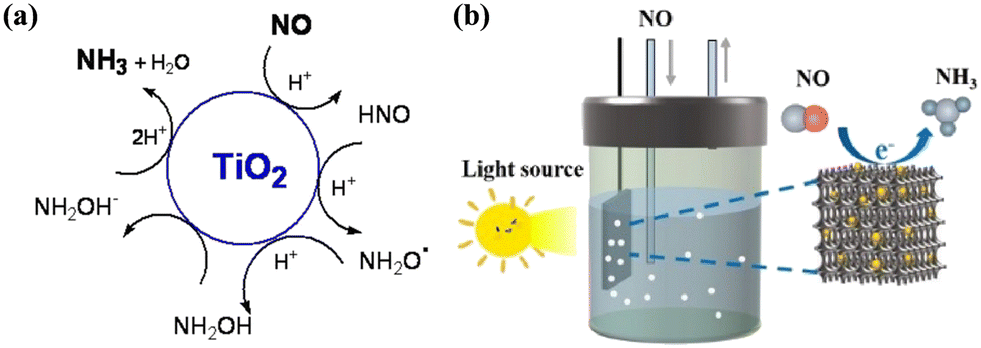 | ||
| Fig. 22 (a) Proposed mechanism of NO reduction to NH3 by TiO2 electrons in colloid solution via consecutive one-electron transfer steps.159 Reproduced from ref. 159 with permission from the American Chemical Society, Copyright 2015. (b) Schematic diagram of photocatalytic NO reduction to NH3 on g-C3N4 QD/3DOMM-TiO2−x electrode.160 Reproduced from ref. 160 with permission from Elsevier, Copyright 2023. | ||
It is worth noting that the ultralow solubility of gaseous NO limits its transfer to liquid-phase reaction systems for its subsequent conversion and upcycling, leading to an unsatisfactory performance. Therefore, integrated processes combining continuous chemical absorption and electrocatalytic/biological reduction have emerged for NO removal from flue gas.161–163 Recently, Li et al. developed an on-site coupling system by combining continuous chemical absorption and photocatalytic reduction reaction to increase the solubility of NO for facilitating subsequent NO upcycling, achieving outstanding conversion efficiency of NO (89.05% ± 0.71%) and production selectivity of NH3 (95.58% ± 0.95%) (Fig. 23a).29 In the synchronous NO absorption and conversion system, an Fe(II) ethylenediaminetetraacetic acid complex [Fe(II)EDTA] was used as the absorbent to generate Fe(II)EDTA–NO, and the Au nanoparticle-decorated TiO2 photocatalyst was fabricated by an operando deposition method during the photocatalytic reaction. Also, formaldehyde (HCHO) was introduced as an antioxidant to avoid the transformation of Fe(II) into Fe(III) and a quenching agent of photogenerated holes, which realized continuous NO reduction and Fe(II)EDTA regeneration.
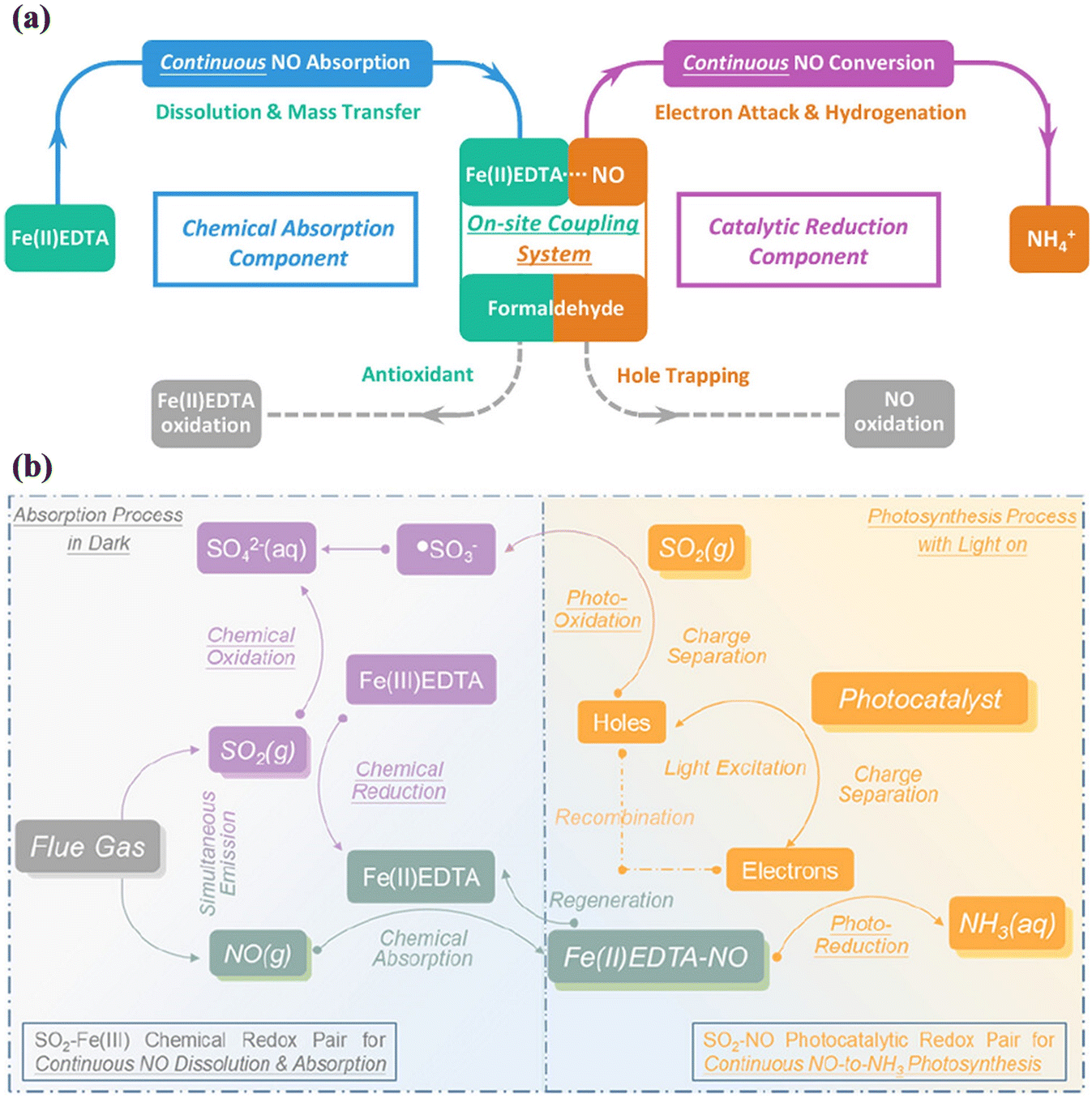 | ||
| Fig. 23 (a) Illustration of the on-site coupling system of continuous chemical absorption and catalytic reduction of NO.29 Reproduced from ref. 29 with permission from the American Chemical Society, Copyright 2023. (b) Illustration for the coupled continuous absorption and conversion process of NO assisted by SO2 poison.158 Reproduced from ref. 158 with permission from the American Chemical Society, Copyright 2023. | ||
Generally, the NO released by anthropogenic sources is produced from the process of fossil fuel combustion, together with multicomponent air pollutants.164,165 Sulfur dioxide (SO2) is recognized as the main coexisting species with NO, and thus the potential impact of SO2 on NOx purification should be seriously considered. Correspondingly, the simultaneous removal and utilization of NOx and SO2 in the flue gas has attracted interest from researchers.166,167 A redox pair of SO2–NO, namely the synergetic SO2 oxidation and NO reduction driven by absorption–photocatalysis system, was proposed, using Fe(II)EDTA as the absorbent and commercial TiO2 as the photocatalyst (Fig. 23b).158 The constructed redox pairs of SO2–Fe(III) hindered the side reaction of Fe(II)-to-Fe(III) oxidation to guarantee the sufficient concentration of Fe(II)EDTA for continuous NO absorption in the absorption process, and with regard to the photocatalytic reaction process, the redox pair of SO2–NO facilitated the separation of photogenerated charge carriers by h+-induced SO2 oxidation reaction and e−-promoted NO reduction reaction. This contributed to ultrahigh selectivity for both NO-to-NH3 upcycling and SO2-to-SO42− purification under ambient conditions.
4. Conclusions and future outlooks
This review presented a timely overview of NO removal by complete photocatalytic oxidation and reduction reaction for toxic by-product inhibition and directed final product formation. The corresponding strategies for the optimization of the reaction microenvironment were systematically summarized, mainly reflecting how to reasonably adjust the surface structure of photocatalysts and the reaction medium. Specifically, this critical review focused on various methods for the surface modification of photocatalysts, strategies to accelerate the mass transfer of gaseous NO, and the influence of the additional introduction of reductant/antioxidant to the reaction system. In general, significant research progress in photocatalytic NO removal with toxic by-product inhibition and end-product recovery has been achieved, but there are still many critical issues, which are provided below together with our perspectives to advance photocatalytic NO removal technology.(1) Photocatalysts are not indestructible and their deactivation becomes noticeable at certain timescales, which can affect the reaction efficiency and even hinder their commercial viability. However, deactivation studies have not mirrored the recent advances in improving the stability of photocatalysts and exploring available regeneration methods, which deserves more attention and effort for advancing practical applications.
(2) In actual scenarios where NO pollution exists, multicomponent pollutants inevitably coexist, while most research only focused on the single NO pollutant. The interaction between different pollutants should be considered, including the synergistic/antagonistic effect and corresponding mechanism, which can lay the foundation for the application of photocatalytic technology in real situations.
(3) In the current design route of photocatalysts, powder materials are the most common form. However, powdery photocatalysts suffer from loss under the action of air/water flow during the reaction process, which not only decreases the reaction efficiency and increases the running cost, but also leads to secondary pollution. In this case, supporting photocatalysts by loading them on a carrier material may be a feasible strategy, which can induce the powder to be firmly supported, and also provide a good reaction site.
(4) With regard to the ultralow solubility of gaseous NO, integrated processes combining chemical absorption and reduction have been proposed to promote the absorption of NO for the convenience of subsequent photocatalytic reduction process. In the process of practical application, the maturity, economy and operability of the coupling technologies should be the primary consideration.
(5) In the current research on NO reduction reaction, it is preferred to pursue the excellent synthesis rate of NH3; however, continuous and efficient NO conversion should be the most important target given that it is the primary obstacle in the further development of photocatalytic NO purification and upcycling technology. It is suggested that the rational design of the reaction system (including photocatalyst, reaction medium, and reactor) will facilitate the synergistic promotion of NO conversion efficiency, ammonia selectivity, ammonia yield, ammonia recovery and long-term stability.
(6) It is important and urgent to establish in situ characterization methods with high spatial and temporal resolution to dynamically monitor the reaction process, and thus reveal the true active sites and conversion pathway of reactants, which can essentially guide the precise design of photocatalysts and the reaction pathway.
(7) Theoretical simulation methods have been widely employed to clarify the crystal structure of photocatalysts and reaction pathways, but it is worth noting that the theoretical simulation should be based on experimental results. Thus, optimizing the matching degree between theoretical simulation methods and experiments should be seriously considered.
Data availability
The data used to support the findings of this review are available from the corresponding author upon request.Author contributions
Wen Cui: conceptualization, writing – original draft, writing – review & editing, project administration, and funding acquisition. Jiaqi Wang: data curation, writing – original draft, writing – review & editing, formal analysis. Yan Li and Pingqu Liu: data curation, and writing – review & editing. Fan Dong: conceptualization, writing – review & editing, project administration, and funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant numbers: 22366008], the Guizhou Provincial Basic Research Program (Natural Science) [grant numbers: ZK (2023)045], the Special Research Fund of Natural Science (Special Post) of Guizhou University [grant numbers: (2021)36].References
- B. W. Chu, Q. X. Ma, J. Liu, J. Z. Ma, P. Zhang, T. Z. Chen, Q. C. Feng, C. Y. Wang, N. Yang, H. N. Ma, J. J. Ma, A. G. Russell and H. He, Air Pollutant Correlations in China: Secondary Air Pollutant Responses to NOx and SO2 Control, Environ. Sci. Technol. Lett., 2020, 7, 695–700 CrossRef CAS.
- K. Li, D. J. Jacob, H. Liao, L. Shen, Q. Zhang and K. H. Bates, Anthropogenic Drivers of 2013-2017 Trends in Summer Surface Ozone in China, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 422–427 CrossRef CAS.
- S. C. Anenberg, J. Miller, R. Minjares, L. Du, D. K. Henze, F. Lacey, C. S. Malley, L. Emberson, V. Franco, Z. Klimont and C. Heyes, Impacts and Mitigation of Excess Diesel-Related NOx Emissions in 11 Major Vehicle Markets, Nature, 2017, 545, 467–471 CrossRef CAS.
- T. Sha, X. Y. Ma, H. X. Zhang, N. Janechek, Y. Y. Wang, Y. Wang, L. Castro García, G. D. Jenerette and J. Wang, Impacts of Soil NOx Emission on O3 Air Quality in Rural California, Environ. Sci. Technol., 2021, 55, 7113–7122 CrossRef CAS PubMed.
- E. Y. Pfannerstill, C. Arata, Q. D. Zhu, B. C. Schulze, R. Ward, R. Woods, C. Harkins, R. H. Schwantes, J. H. Seinfeld, A. Bucholtz, R. C. Cohen and A. H. Goldstein, Temperature-Dependent Emissions Dominate Aerosol and Ozone Formation in Los Angeles, Science, 2024, 384, 1324–1329 CrossRef CAS PubMed.
- T. V. W. Janssens and P. N. R. Vennestrøm, A Molecular Dance to Cleaner Air, Science, 2017, 357, 866–867 CrossRef CAS PubMed.
- A. Islam, S. H. Teo, C. H. Ng, Y. H. Taufiq-Yap, S. Y. T. Choong and M. R. Awual, Progress in Recent Sustainable Materials for Greenhouse Gas (NOx and SOx) Emission Mitigation, Prog. Mater. Sci., 2022, 132, 101033–101080 CrossRef.
- Z. F. Hao, G. Q. Liu, P. F. Wang, W. Y. Zhang, W. M. Sun, L. R. Zheng, S. J. Guo and S. H. Zhan, In situ Visualizing Reveals Potential Drive of Lattice Expansion on Defective Support toward Efficient Removal of Nitrogen Oxides, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2311180121 CrossRef CAS PubMed.
- M. P. Heap, S. L. Chen, J. C. Kramlich, J. M. Mccarthy and D. W. Pershing, An Advanced Selective Reduction Process for NOx Control, Nature, 1988, 335, 620–622 CrossRef CAS.
- D. D. Chen, A. Khetan, H. Lei, V. Rizzotto, J. Y. Yang, J. X. Jiang, Q. M. Sun, B. X. Peng, P. R. Chen, R. Palkovits, D. Q. Ye and U. Simon, Copper Site Motion Promotes Catalytic NOx Reduction under Zeolite Confinement, Environ. Sci. Technol., 2023, 57, 16121–16130 CrossRef CAS PubMed.
- R. P. Miao, D. W. Chen, Z. Y. Guo, Y. Y. Zhou, C. Chen and S. Y. Wang, Recent Advances in Electrocatalytic Upgrading of Nitric Oxide and Beyond, Appl. Catal., B, 2023, 344, 123662–123680 CrossRef.
- L. J. Yan, H. F. Zhu, X. Y. Liu, D. C. Peng, J. Zhang, D. O. Cheng, A. L. Chen and D. S. Zhang, Synergistic Catalytic Removal of NOx and n-Butylamine via Spatially Separated Cooperative Sites, Environ. Sci. Technol., 2024, 58, 11781–11790 CrossRef CAS PubMed.
- M. Y. Bian, K. J. Liu, D. Y. Zheng, X. Y. Han, X. Yang, Y. F. Fang, C. H. Liu, J. W. Zhao, Y. B. Zhang and X. G. Yang, Metal-Free β Zeolite used as an Efficient NH3-SCR Catalyst can Achieve Complete Immunity to SO2: Unique Design Strategy of Sulfur-Resistant Catalyst, Chem. Eng. J., 2024, 481, 148563 CrossRef CAS.
- D. Y. Zheng, K. J. Liu, Z. S. Zhang, Q. Fu, M. Y. Bian, X. Y. Han, X. Shen, X. H. Chen, H. J. Xie, X. Wang, X. G. Yang, Y. B. Zhang and S. Y. Song, Essential Features of Weak Current for Excellent Enhancement of NOx Reduction over Monoatomic V-based Catalyst, Nat. Commun., 2024, 15, 6688 CrossRef CAS PubMed.
- X. Yang, K. J. Liu, X. Y. Han, J. H. Xu, M. Y. Bian, D. Y. Zheng, H. J. Xie, Y. B. Zhang and X. G. Yang, Transformation of Waste Battery Cathode Material LiMn2O4 into Efficient Ultra-Low Temperature NH3-SCR Catalyst: Proton Exchange Synergistic Vanadium Modification, J. Hazard. Mater., 2023, 459, 132209 CrossRef CAS PubMed.
- I. Ioannou, Á. Galán-Martín, J. Pérez-Ramírez and G. Guillén-Gosálbez, Trade-offs between Sustainable Development Goals in Carbon Capture and Utilisation, Energy Environ. Sci., 2022, 16, 113–124 RSC.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Photocatalytic Solar Hydrogen Production from Water on a 100-m2 Scale, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- F. He, W. Jeon and W. Choi, Photocatalytic Air Purification Mimicking the Self-Cleaning Process of the Atmosphere, Nat. Commun., 2021, 12, 2528–2531 CrossRef CAS PubMed.
- T. Xue, J. Li, L. C. Chen, K. L. Li, Y. Hua, Y. Yang and F. Dong, Photocatalytic NOx Removal and Recovery: Progress, Challenges and Future Perspectives, Chem. Sci., 2024, 15, 9026–9046 RSC.
- W. R. Dai, S. Zhang, H. Shang, S. N. Xiao, Z. L. Tian, W. B. Fan, X. L. Chen, S. K. Wang, W. Chen and D. Q. Zhang, Breaking the Selectivity Barrier: Reactive Oxygen Species Control in Photocatalytic Nitric Oxide Conversion, Adv. Funct. Mater., 2024, 34, 2309426–2309439 CrossRef CAS.
- H. Shang, H. B. Jia, W. B. Zhang, S. J. Li, Q. Wang, Q. Y. Yang, C. Zhang, Y. X. Shi, Y. J. Wang, P. P. Li, Y. C. He, S. N. Xiao, D. Wang and D. Q. Zhang, Surface Hydrogen Bond-Induced Oxygen Vacancies of TiO2 for Two-Electron Molecular Oxygen Activation and Efficient NO Oxidation, Environ. Sci. Technol., 2023, 57, 20400–20409 CrossRef CAS.
- Y. H. Shi, Z. F. Zhao, D. Yang, J. D. Tan, X. Xin, Y. Q. Liu and Z. Y. Jiang, Engineering Photocatalytic Ammonia Synthesis, Chem. Soc. Rev., 2023, 52, 6938–6956 RSC.
- H. M. Liu, L. C. Bai, A. Bergmann, B. R. Cuenya and J. S. Luo, Electrocatalytic Reduction of Nitrogen Oxide Species to Ammonia, Chem, 2024 DOI:10.1016/j.chempr.2024.07.006.
- K.-H. Liu, M. H. Liu, Z. W. Lin, Z.-F. Wang, B. Q. Chen, C. Liu, A. Guo, M. Konishi, S. Yanagisawa, G. Wagner and J. Sheen, NIN-Like Protein 7 Transcription Factor is a Plant Nitrate Sensor, Science, 2022, 377, 1419–1425 CrossRef CAS PubMed.
- S. J. Li, H. Shang, Y. Tao, P. P. Li, H. H. Pan, Q. Wang, S. Zhang, H. B. Jia, H. N. Zhang, J. Z. Cao, B. X. Zhang, R. Zhang, G. S. Li, Y. R. Zhang, D. Q. Zhang and H. X. Li, Hydroxyl Radical-Mediated Efficient Photoelectrocatalytic NO Oxidation with Simultaneous Nitrate Storage Using A Flow Photoanode Reactor, Angew. Chem., Int. Ed., 2023, 62, e202305538 CrossRef CAS.
- R. Abualia, K. Ötvös, O. Novák, E. Bouguyon, K. Domanegg, A. Krapp, P. Nacry, A. Gojon, B. Lacombe and E. Benková, Molecular Framework Integrating Nitrate Sensing in Root and Auxin-Guided Shoot Adaptive Responses, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2122460119 CrossRef CAS.
- S. X. Wang, W. Cui, B. Lei, X. A. Dong, Y. Tang and F. Dong, Targeted NO Oxidation and Synchronous NO2 Inhibition via Oriented 1O2 Formation Based on Lewis Acid Site Adjustment, Environ. Sci. Technol., 2023, 57, 12890–12900 CrossRef CAS PubMed.
- H. Li, H. J. Zhu, Y. B. Shi, H. Shang, L. Z. Zhang and J. Wang, Vacancy-Rich and Porous NiFe-Layered Double Hydroxide Ultrathin Nanosheets for Efficient Photocatalytic NO Oxidation and Storage, Environ. Sci. Technol., 2022, 56, 1771–1779 CrossRef CAS.
- J. Y. Li, J. L. Wang, S. J. Shen, R. M. Chen, M. Liu and F. Dong, Beyond Purification: Highly Efficient and Selective Conversion of NO into Ammonia by Coupling Continuous Absorption and Photoreduction under Ambient Conditions, Environ. Sci. Technol., 2023, 57, 5445–5452 CrossRef CAS PubMed.
- W. Cui, J. Y. Li and F. Dong, Optimizing the Gas–Solid Photocatalytic Reactions for Air Purification, ACS ES&T Eng., 2022, 2, 1103–1115 Search PubMed.
- J. M. Luo, S. Q. Zhang, M. Sun, L. X. Yang, S. L. Luo and J. C. Crittenden, A Critical Review on Energy Conversion and Environmental Remediation of Photocatalysts with Remodeling Crystal Lattice, Surface, and Interface, ACS Nano, 2019, 13, 9811–9840 CrossRef CAS.
- K. Khan, L. F. Xu, M. Shi, J. S. Qu, X. P. Tao, Z. C. Feng, C. Li and R. Li, Surface Assembly of Cobalt Species for Simultaneous Acceleration of Interfacial Charge Separation and Catalytic Reactions on Cd0.9Zn0.1S Photocatalyst, Chin. J. Catal., 2021, 42, 1004–1012 CrossRef CAS.
- J. J. Lv, R. N. Yin, L. M. Zhou, J. Li, R. Kikas, T. Xu, Z. J. Wang, H. L. Jin, X. Wang and S. Wang, Microenvironment Engineering for the Electrocatalytic CO2 Reduction Reaction, Angew. Chem., Int. Ed., 2022, 61, e202207252–e202207275 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Generation and Detection of Reactive Oxygen Species in Photocatalysis, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS.
- Y. B. Shi, Z. P. Yang, L. J. Shi, H. Li, X. P. Liu, X. Zhang, J. D. Cheng, C. Liang, S. Y. Cao, F. R. Guo, X. Liu, Z. H. Ai and L. Z. Zhang, Surface Boronizing Can Weaken the Excitonic Effects of BiOBr Nanosheets for Efficient O2 Activation and Selective NO Oxidation under Visible Light Irradiation, Environ. Sci. Technol., 2022, 56, 14478–14486 CrossRef CAS PubMed.
- Q. Li, J. J. Zhao, H. Shang, Z. Ma, H. Y. Cao, Y. Zhou, G. S. Li, D. Q. Zhang and H. X. Li, Singlet Oxygen and Mobile Hydroxyl Radicals Co-Operating on Gas–Solid Catalytic Reaction Interfaces for Deeply Oxidizing NOx, Environ. Sci. Technol., 2022, 56, 5830–5839 CrossRef CAS.
- F. Rao, G. Q. Zhu, W. B. Zhang, Y. H. Xu, B. W. Cao, X. J. Shi, J. Z. Gao, Y. H. Huang, Y. Huang and M. Hojamberdiev, Maximizing the Formation of Reactive Oxygen Species for Deep Oxidation of NO via Manipulating the Oxygen-Vacancy Defect Position on (BiO)2CO3, ACS Catal., 2021, 11, 7735–7749 CrossRef CAS.
- X. J. Song, W. J. Jiang, Z. H. Cai, X. Y. Yue, X. Chen, W. X. Dai and X. Z. Fu, Visible Light-Driven Deep Oxidation of NO and its Durability over Fe Doped BaSnO3: The NO+ Intermediates Mechanism and the Storage Capacity of Ba Ions, Chem. Eng. J., 2022, 444, 136709–136721 CrossRef CAS.
- J. Z. Liao, W. Cui, J. Y. Li, J. P. Sheng, H. Wang, X. A. Dong, P. Chen, G. M. Jiang, Z. M. Wang and F. Dong, Nitrogen Defect Structure and NO+ Intermediate Promoted Photocatalytic NO Removal on H2 Treated g-C3N4, Chem. Eng. J., 2020, 379, 122282–122289 CrossRef CAS.
- R. Chen, T. S. Zhang, Y. Q. Guo, J. W. Wang, J. X. Wei and Q. J. Yu, Recent Advances in Simultaneous Removal of SO2 and NOx from Exhaust Gases: Removal Process, Mechanism and Kinetics, Chem. Eng. J., 2021, 420, 127588 CrossRef CAS.
- N. Li, C. Y. Wang, K. Zhang, H. Q. Lv, M. Z. Yuan and D. W. Bahnemann, Progress and Prospects of Photocatalytic Conversion of Low-Concentration NOx, Chin. J. Catal., 2022, 43, 2363–2387 CrossRef CAS.
- X. Lu, L. Wang, Z. L. Li, Z. M. Wang, Y. M. Gan and R. Hailili, 2D Bismuth-Based Nanomaterials for Photocatalytic Nitrogen Oxide Removal: Progress and Prospects, ACS Sustainable Chem. Eng., 2024, 12, 11444–11466 CrossRef CAS.
- Y. F. Lu, M. J. Chen, L. Jiang, J. J. Cao, H. W. Li, S. C. Lee and Y. Huang, Oxygen Vacancy Engineering of Photocatalytic Nanomaterials for Enrichment, Activation, and Efficient Removal of Nitrogen Oxides with High Selectivity: A Review, Environ. Chem. Lett., 2022, 20, 3905–3925 CrossRef CAS.
- C. Ma, J. Wei, K. Jiang, J. Chen, Z. Yang, X. Yang, G. Yu, C. Zhang and X. Li, Typical Layered Structure Bismuth-Based Photocatalysts for Photocatalytic Nitrogen Oxides Oxidation, Sci. Total Environ., 2023, 855, 158644 CrossRef CAS PubMed.
- H. Shang, H. B. Jia, P. P. Li, H. Li, W. B. Zhang, S. J. Li, Q. Wang, S. N. Xiao, D. Wang, G. S. Li and D. Q. Zhang, Highly Selective and Efficient Photocatalytic NO Removal: Charge Carrier Kinetics and Interface Molecular Process, Nano Res., 2023, 17, 1003–1026 CrossRef.
- Z. Y. Wang, X. J. Shi, M. J. Chen, J. J. Cao, W. K. Ho, S. C. Lee, C. Y. Wang and Y. Huang, Polymeric Carbon Nitride-Based Photocatalysts for the Removal of Nitrogen Oxides: A Review, Environ. Chem. Lett., 2023, 21, 2913–2952 CrossRef CAS.
- Y. Yang, X. Z. Zheng, W. Ren, J. F. Liu, X. L. Fu, S. G. Meng, S. F. Chen and C. Cai, Recent Advances in Special Morphologic Photocatalysts for NOx Removal, Front. Environ. Sci. Eng., 2022, 16, 137 CrossRef CAS.
- R. Asahi, T. Morikawa, H. Irie and T. Ohwaki, Nitrogen-Doped Titanium Dioxide as Visible-Light-Sensitive Photocatalyst: Designs, Developments, and Prospects, Chem. Rev., 2014, 114, 9824–9852 CrossRef CAS PubMed.
- L. B. Jiang, X. Z. Yuan, Y. Pan, J. Liang, G. M. Zeng, Z. B. Wu and H. Wang, Doping of Graphitic Carbon Nitride for Photocatalysis: A Reveiw, Appl. Catal., B, 2017, 217, 388–406 CrossRef CAS.
- W. Wang, M. O. Tade and Z. P. Shao, Nitrogen-Doped Simple and Complex Oxides for Photocatalysis: A Review, Prog. Mater. Sci., 2018, 92, 33–63 CrossRef CAS.
- S. Q. Zhang, X. S. Yi, G. H. Hu, M. X. Chen, H. Shen, B. Li, L. X. Yang, W. L. Dai, J. P. Zou and S. L. Luo, Configuration Regulation of Active Sites by Accurate Doping Inducing Self-Adapting Defect for Enhanced Photocatalytic Applications: A Review, Coord. Chem. Rev., 2022, 478, 214970–214997 CrossRef.
- C. W. Yuan, R. M. Chen, J. D. Wang, H. Z. Wu, J. P. Sheng, F. Dong and Y. J. Sun, La-doping Induced Localized Excess Electrons on (BiO)2CO3 for Efficient Photocatalytic NO Removal and Toxic Intermediates Suppression, J. Hazard. Mater., 2020, 400, 123174–123180 CrossRef CAS.
- C. W. Yuan, W. Cui, Y. J. Sun, J. D. Wang, R. M. Chen, J. Zhang, Y. X. Zhang and F. Dong, Inhibition of the Toxic Byproduct during Photocatalytic NO Oxidation via La Doping in ZnO, Chin. Chem. Lett., 2020, 31, 751–754 CrossRef CAS.
- J. R. Li, M. X. Ran, P. Chen, W. Cui, J. Y. Li, Y. J. Sun, G. M. Jiang, Y. Zhou and F. Dong, Controlling the Secondary Pollutant on B-Doped g-C3N4 During Photocatalytic NO removal: A Combined DRIFTS and DFT Investigation, Catal. Sci. Technol., 2019, 9, 4531–4537 RSC.
- R. Y. Zhang, Y. H. Cao, D. E. Doronkin, M. Z. Ma, F. Dong and Y. Zhou, Single-Atom Dispersed Zn-N3 Active Sites Bridging the Interlayer of g-C3N4 to Tune NO Oxidation Pathway for the Inhibition of Toxic By-Product Generation, Chem. Eng. J., 2023, 454, 140084–140092 CrossRef CAS.
- M. Zhou, G. H. Dong, F. K. Yu and Y. Huang, The Deep Oxidation of NO was Realized by Sr Multi-Site Doped g-C3N4 via Photocatalytic Method, Appl. Catal., B, 2019, 256, 117825–117835 CrossRef CAS.
- J. X. Guo, T. Shen, J. Liang, S. L. He, Y. Jing, Y. H. Chu and W. L. Cen, Study on the Effective Removal of NO by Light-Driven N-BiOCl, Fuel, 2022, 329, 125422–125435 CrossRef CAS.
- G. Cheng, X. Liu, X. J. Song, X. Chen, W. X. Dai, R. S. Yuan and X. Z. Fu, Visible-Light-Driven Deep Oxidation of NO over Fe Doped TiO2 Catalyst: Synergic Effect of Fe and Oxygen Vacancies, Appl. Catal., B, 2020, 277, 119196–119212 CrossRef CAS.
- J. Zhang, W. Cui, P. Chen, K. L. Li, H. Wu and F. Dong, B Doped Bi2O2CO3 Hierarchical Microspheres: Enhanced Photocatalytic Performance and Reaction Mechanism for NO Removal, Catal. Today, 2021, 380, 230–236 CrossRef CAS.
- J. Y. Zhang, G. Q. Zhu, S. P. Li, F. Rao, H. Qadeer-Ul, J. Z. Gao, Y. Huang and M. Hojamberdiev, Novel Au/La-Bi5O7I Microspheres with Efficient Visible-Light Photocatalytic Activity for NO Removal: Synergistic Effect of Au Nanoparticles, La Doping, and Oxygen Vacancy, ACS Appl. Mater. Interfaces, 2019, 11, 37822–37832 CrossRef CAS.
- Z. Z. Lu, S. Q. Li and J. Y. Xiao, Synergetic Effect of Na-Ca for Enhanced Photocatalytic Performance in NOx Degradation by g-C3N4, Catal. Lett., 2021, 151, 370–381 CrossRef CAS.
- Z. Z. Lu, S. Q. Li and J. Y. Xiao, K-Ca Synergetic Modified g-C3N4 for Efficient Photocatalytic NO Removal with Low-NO2-Emission, Catal. Lett., 2022, 153, 2558–2570 CrossRef.
- F. R. Guo, C. L. Mao, C. Liang, P. Xing, L. H. Yu, Y. B. Shi, S. Y. Cao, F. Y. Wang, X. Liu, Z. H. Ai and L. Z. Zhang, Triangle Cl-Ag1-Cl Sites for Superior Photocatalytic Molecular Oxygen Activation and NO Oxidation of BiOCl, Angew. Chem., Int. Ed., 2023, 62, e202314243–e202314250 CrossRef CAS.
- X. F. Li, Z. Hu, Q. Li, M. Lei, J. J. Fan, S. A. C. Carabineiro, Y. Liu and K. L. Lv, Three in One: Atomically Dispersed Na Boosting the Photoreactivity of Carbon Nitride towards NO Oxidation, Chem. Commun., 2020, 56, 14195–14198 RSC.
- L. X. Han, J. M. Huang, J. Zhan, X. H. Zhang, S. Y. Wang and H. Chen, Construction of Highly Dispersed Ni Sites on N-rich Carbon Nitride for Enhanced Photocatalytic NO Removal, Adv. Sustainable Syst., 2022, 7, 220009–220017 Search PubMed.
- S. Bai, N. Zhang, C. Gao and Y. J. Xiong, Defect Engineering in Photocatalytic Materials, Nano Energy, 2018, 53, 296–336 CrossRef CAS.
- P. Y. Dong, C. X. Wang, J. J. Tan, Y. Wang, X. G. Xi and J. L. Zhang, Solar-Driven Photocatalytic Removal of NO over A Concrete Paving Eco-Block Containing Black TiO2, J. Mater. Chem. A, 2023, 11, 25429–25440 RSC.
- Z. Y. Zhao, Y. H. Cao, F. Dong, F. Wu, B. X. Li, Q. Zhang and Y. Zhou, The Activation of Oxygen through Oxygen Vacancies in BiOCl/PPy to Inhibit Toxic Intermediates and Enhance the Activity of Photocatalytic Nitric Oxide Removal, Nanoscale, 2019, 11, 6360–6367 RSC.
- J. Z. Liao, K. L. Li, H. Ma, F. Dong, X. L. Zeng and Y. J. Sun, Oxygen Vacancies on the BiOCl Surface Promoted Photocatalytic Complete NO Oxidation via Superoxide Radicals, Chin. Chem. Lett., 2020, 31, 2737–2741 CrossRef CAS.
- Z. Hu, K. N. Li, X. F. Wu, N. Wang, X. F. Li, Q. Li, L. Li and K. L. Lv, Dramatic Promotion of Visible-Light Photoreactivity of TiO2 Hollow Microspheres towards NO Oxidation by Introduction of Oxygen Vacancy, Appl. Catal., B, 2019, 256, 117860–117869 CrossRef CAS.
- H. Shang, M. Q. Li, H. Li, S. Huang, C. L. Mao, Z. H. Ai and L. Z. Zhang, Oxygen Vacancies Promoted the Selective Photocatalytic Removal of NO with Blue TiO2 via Simultaneous Molecular Oxygen Activation and Photogenerated Hole Annihilation, Environ. Sci. Technol., 2019, 53, 6444–6453 CrossRef CAS.
- L. Liu, P. Ouyang, Y. H. Li, Y. Y. Duan, F. Dong and K. L. Lv, Insight into the Mechanism of Deep NO Photo-Oxidation by Bismuth Tantalate with Oxygen Vacancies, J. Hazard. Mater., 2022, 439, 129637–129650 CrossRef CAS PubMed.
- C. L. Zhang, Y. K. Xu, H. J. Bai, D. F. Li, L. Wei, C. L. Feng, Y. H. Huang, Z. S. Wang, X. Y. Li, X. D. Cui, C. G. Hu and F. Wang, Boosting Visible-Light Photocatalytic NO Removal by Non-Intrinsic Oxygen Vacancies in Graphitic Carbon Nitride, Nano Energy, 2024, 121, 109197–109207 CrossRef CAS.
- H. J. Liu, P. Chen, X. Y. Yuan, Y. X. Zhang, H. W. Huang, L. A. Wang and F. Dong, Pivotal Roles of Artificial Oxygen Vacancies in Enhancing Photocatalytic Activity and Selectivity on Bi2O2CO3 Nanosheets, Chin. J. Catal., 2019, 40, 620–630 CrossRef CAS.
- W. Cui, L. C. Chen, J. Y. Li, Y. Zhou, Y. J. Sun, G. M. Jiang, S. C. Lee and F. Dong, Ba-Vacancy Induces Semiconductor-Like Photocatalysis on Insulator BaSO4, Appl. Catal., B, 2019, 253, 293–299 CrossRef CAS.
- Y. H. Li, M. L. Gu, T. Shi, W. Cui, X. M. Zhang, F. Dong, J. S. Cheng, J. J. Fan and K. L. Lv, Carbon Vacancy in C3N4 Nanotube: Electronic Structure, Photocatalysis Mechanism and Highly Enhanced Activity, Appl. Catal., B, 2020, 262, 118281–118291 CrossRef CAS.
- Y. H. Li, M. L. Gu, M. Zhang, X. M. Zhang, K. L. Lv, Y. Q. Liu, W. Ho and F. Dong, C3N4 with Engineered Three Coordinated (N3C) Nitrogen Vacancy Boosts the Production of 1O2 for Efficient and Stable NO Photo-Oxidation, Chem. Eng. J., 2020, 389, 124421–124436 CrossRef CAS.
- Y. Y. Duan, Y. Wang, L. Y. Gan, J. Z. Meng, Y. J. Feng, K. W. Wang, K. Zhou, C. Wang, X. D. Han and X. Y. Zhou, Amorphous Carbon Nitride with Three Coordinate Nitrogen (N3c) Vacancies for Exceptional NOx Abatement in Visible Light, Adv. Energy Mater., 2021, 11, 2004001–2004010 CrossRef CAS.
- Y. Xia, H. Yang, W. K. Ho, B. C. Zhu and J. G. Yu, Promoting the Photocatalytic NO Oxidation Activity of Hierarchical Porous g-C3N4 by Introduction of Nitrogen Vacancies and Charge Channels, Appl. Catal., B, 2024, 344, 123604–123613 CrossRef CAS.
- X. L. Shen, G. H. Dong, L. Wang, L. Q. Ye and J. W. Sun, Enhancing Photocatalytic Activity of NO Removal through an in situ Control of Oxygen Vacancies in Growth of TiO2, Adv. Mater. Interfaces, 2019, 6, 1901032–1901041 CrossRef CAS.
- Y. F. Lu, M. J. Chen, T. T. Huang, Y. Huang, J. J. Cao, H. W. Li, W. K. Ho and S. C. Lee, Oxygen Vacancy-Dependent Photocatalytic Activity of Well-Defined Bi2Sn2O7-x Hollow Nanocubes for NOx Removal, Environ. Sci.: Nano, 2021, 8, 1927–1933 RSC.
- Y. He, J. P. Sheng, Q. Ren, Y. J. Sun, W. C. Hao and F. Dong, Operando Identification of Dynamic Photoexcited Oxygen Vacancies as True Catalytic Active Sites, ACS Catal., 2022, 13, 191–203 CrossRef.
- Q. Ren, Y. He, H. Wang, Y. J. Sun and F. Dong, Photo-Switchable Oxygen Vacancy as the Dynamic Active Site in the Photocatalytic NO Oxidation Reaction, ACS Catal., 2022, 12, 14015–14025 CrossRef CAS.
- Q. H. Zhu, R. Hailili, Y. Xin, Y. T. Zhou, Y. Huang, X. Z. Pang, K. Zhang, P. K. J. Robertson, D. W. Bahnemann and C. Y. Wang, Efficient Full Spectrum Responsive Photocatalytic NO Conversion at Bi2Ti2O7: Co-Effect of Plasmonic Bi and Oxygen Vacancies, Appl. Catal., B, 2022, 319, 121888–121897 CrossRef CAS.
- P. Chen, H. J. Liu, Y. J. Sun, J. Y. Li, W. Cui, L. A. Wang, W. D. Zhang, X. Y. Yuan, Z. M. Wang, Y. X. Zhang and F. Dong, Bi Metal Prevents the Deactivation of Oxygen Vacancies in Bi2O2CO3 for Stable and Efficient Photocatalytic NO Abatement, Appl. Catal., B, 2020, 264, 118545–118555 CrossRef CAS.
- M. X. Ran, H. Wang, W. Cui, J. Y. Li, P. Chen, Y. J. Sun, J. P. Sheng, Y. Zhou, Y. X. Zhang and F. Dong, Light-Induced Generation and Regeneration of Oxygen Vacancies in BiSbO4 for Sustainable Visible Light Photocatalysis, ACS Appl. Mater. Interfaces, 2019, 11, 47984–47991 CrossRef CAS.
- W. H. Li and N. Yang, Green Fabrication of AuNPs/CMTKP/g-C3N4 Nanocomposites with Enhanced Photocatalytic Activity for the Removal of Nitric Oxide under Visible-Light Irradiation, J. Cleaner Prod., 2020, 256, 120257 CrossRef CAS.
- Y. Y. Guo, Y. C. Ou, F. Rao, W. B. Zhang, J. Sun, Y. N. Yang, X. Yang, C. Wang and J. J. Wang, Au Nanoparticles Loaded Bi5Ti3FeO15 Ferroelectric Nanosheets with Enhanced Photocatalytic Activity for NO Removal, Catal. Lett., 2022, 153, 1861–1870 CrossRef.
- L. Lv, H. D. Yang, Q. W. Chen, H. Q. Fan and J. P. Zhou, La2Ti2O7 Nanosheets Modified by Pt Quantum Dots for Efficient NO Removal Avoiding NO2 Secondary Pollutant, Environ. Res., 2023, 223, 115441–115450 CrossRef CAS.
- D. P. Bui, M. T. Nguyen, H. H. Tran, S. J. You, Y. F. Wang and P. V. Viet, Green Synthesis of Ag@SnO2 Nanocomposites for Enhancing Photocatalysis of Nitrogen Monoxide Removal under Solar Light Irradiation, Catal. Commun., 2020, 136, 105902–105905 CrossRef CAS.
- V. V. Pham, T. Q. Nguyen, H. V. Le and T. M. Cao, Visible Light Photocatalytic NOx Removal with Suppressed Poisonous NO2 Byproduct Generation over Simply Synthesized Triangular Silver Nanoparticles Coupled with Tin dioxide, Nanoscale Adv., 2024, 6, 2380–2389 RSC.
- Z. Chen, H. B. Yin, R. Wang, Y. Peng, C. F. You and J. H. Li, Efficient Electron Transfer by Plasmonic Silver in SrTiO3 for Low-Concentration Photocatalytic NO Oxidation, Environ. Sci. Technol., 2022, 56, 3604–3612 CrossRef CAS PubMed.
- L. Lv, Q. U. Hassan, Q. W. Chen, H. Q. Fan and J. P. Zhou, Photodegradation of NO with Minimal NO2 Secondary Pollution by La2Ti2O7 Nanosheets Loaded with Ag Particles, Appl. Surf. Sci., 2023, 619, 156657–156665 CrossRef CAS.
- K. L. Li, W. Cui, J. Y. Li, Y. J. Sun, Y. H. Chu, G. M. Jiang, Y. Zhou, Y. X. Zhang and F. Dong, Tuning the Reaction Pathway of Photocatalytic NO Oxidation Process to Control the Secondary Pollution on Monodisperse Au Nanoparticles@g-C3N4, Chem. Eng. J., 2019, 378, 122184–122192 CrossRef CAS.
- K. L. Li, Y. He, P. Chen, H. Wang, J. P. Sheng, W. Cui, G. Leng, Y. H. Chu, Z. M. Wang and F. Dong, Theoretical Design and Experimental Investigation on Highly Selective Pd Particles Decorated C3N4 for Safe Photocatalytic NO Purification, J. Hazard. Mater., 2020, 392, 122357–122365 CrossRef CAS PubMed.
- M. Sayed, J. G. Yu, G. Liu and M. Jaroniec, Non-Noble Plasmonic Metal-Based Photocatalysts, Chem. Rev., 2022, 122, 10484–10537 CrossRef CAS.
- F. Dong, T. Xiong, Y. J. Sun, Z. W. Zhao, Y. Zhou, X. Feng and Z. B. Wu, A Semimetal Bismuth Element as A Direct Plasmonic Photocatalyst, Chem. Commun., 2014, 50, 10386–10389 RSC.
- Z. L. Ni, W. D. Zhang, G. M. Jiang, X. P. Wang, Z. Z. Lu, Y. J. Sun, X. W. Li, Y. X. Zhang and F. Dong, Enhanced Plasmonic Photocatalysis by SiO2@Bi Microspheres with Hot-Electron Transportation Channels via Bi-O-Si Linkages, Chin. J. Catal., 2017, 38, 1174–1183 CrossRef CAS.
- X. W. Li, W. D. Zhang, W. Cui, Y. J. Sun, G. M. Jiang, Y. X. Zhang, H. W. Huang and F. Dong, Bismuth Spheres Assembled on Graphene Oxide: Directional Charge Transfer Enhances Plasmonic Photocatalysis and in situ DRIFTS Studies, Appl. Catal., B, 2018, 221, 482–489 CrossRef CAS.
- X. W. Li, Y. J. Sun, T. Xiong, G. M. Jiang, Y. X. Zhang, Z. B. Wu and F. Dong, Activation of Amorphous Bismuth Oxide via Plasmonic Bi Metal for Efficient Visible-Light Photocatalysis, J. Catal., 2017, 352, 102–112 CrossRef CAS.
- P. Zhang, Y. F. Rao, Y. Huang, M. J. Chen, T. T. Huang, W. K. Ho, S. C. Lee, J. B. Zhong and J. J. Cao, Transformation of Amorphous Bi2O3 to Crystal Bi2O2CO3 on Bi Nanospheres Surface for Photocatalytic NOx Oxidation: Intensified Hot-Electron Transfer and Reactive Oxygen Species Generation, Chem. Eng. J., 2021, 420, 129814–129824 CrossRef CAS.
- Y. Xin, Q. H. Zhu, T. Gao, X. M. Li, W. Zhang, H. Wang, D. H. Ji, Y. Huang, M. Padervand, F. Yu and C. Y. Wang, Photocatalytic NO Removal over Defective Bi/BiOBr Nanoflowers: The Inhibition of Toxic NO2 Intermediate via High Humidity, Appl. Catal., B, 2023, 324, 122238–122247 CrossRef.
- Y. F. Lu, Y. Huang, Y. F. Zhang, T. T. Huang, H. W. Li, J. J. Cao and W. Ho, Effects of H2O2 Generation over Visible Light-Responsive Bi/Bi2O2-xCO3 Nanosheets on Their Photocatalytic NOx Removal Performance, Chem. Eng. J., 2019, 363, 374–382 CrossRef CAS.
- X. Zhou, J. Zhang, X. M. Wang, T. Q. Tan, R. M. Fang, S. Chen and F. Dong, Efficient NO Removal and Photocatalysis Mechanism over Bi-Metal@Bi2O2[BO2(OH)] with Oxygen Vacancies, J. Hazard. Mater., 2022, 436, 129271–129280 CrossRef CAS.
- X. M. Li, Q. B. Dong, F. Li, Q. H. Zhu, Q. Y. Tian, L. Tian, Y. Y. Zhu, B. Pan, M. Padervand and C. Y. Wang, Defective Bi@BiOBr/C Microrods Derived from Bi-MOF for Efficient Photocatalytic NO Abatement: Directional Regulation of Interfacial Charge Transfer via Carbon–Loading, Appl. Catal., B, 2024, 340, 123238–123249 CrossRef CAS.
- N. Li, W. W. Zhao, J. T. Zhang, X. H. Liu, Y. Q. Gao and L. Ge, Plasmonic Bi-Modified Bi2Sn2O7 Nanosheets for Efficient Photocatalytic NO Removal, Catalysts, 2024, 14, 275–287 CrossRef CAS.
- A. Martinez-Oviedo, Y. K. Kshetri, B. Joshi and S. W. Lee, Surface Modification of Blue TiO2 with Silane Coupling Agent for NOx Abatement, Prog. Nat. Sci.: Mater. Int., 2021, 31, 230–238 CrossRef CAS.
- W. P. Yang, Q. Ren, F. Y. Zhong, Y. X. Wang, J. L. Wang, R. M. Chen, J. Y. Li and F. Dong, Promotion Mechanism of -OH Group Intercalation for NOx Purification on BiOI Photocatalyst, Nanoscale, 2021, 13, 20601–20608 RSC.
- H. Wang, X. Q. Xu, A. Labidi, H. T. Ren, A. A. Allam, A. Rady, Y. Huang, S. W. Wei, M. Padervand, S. Ghasemi and C. Y. Wang, Cyano/Hydroxyl Groups Co-Functionalized g-C3N4 for Photocatalytic NO Removal: A Synergistic Strategy towards Inhibition of Toxic Intermediate NO2, Catalysts, 2023, 13, 1433–1450 CrossRef CAS.
- H. L. Wang, L. S. Zhang, Z. G. Chen, J. Q. Hu, S. J. Li, Z. H. Wang, J. S. Liu and X. C. Wang, Semiconductor Heterojunction Photocatalysts: Design, Construction, and Photocatalytic Performances, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- H. J. Li, Y. Zhou, W. G. Tu, J. H. Ye and Z. G. Zou, State-of-the-Art Progress in Diverse Heterostructured Photocatalysts toward Promoting Photocatalytic Performance, Adv. Funct. Mater., 2015, 25, 998–1013 CrossRef CAS.
- B. K. Xie, D. Y. Chen, N. J. Li, Q. F. Xu, H. Li, J. H. He and J. M. Lu, Fabrication of an FAPbBr3/g-C3N4 Heterojunction to Enhance NO Removal Efficiency under Visible-Light Irradiation, Chem. Eng. J., 2022, 430, 132968–132977 CrossRef CAS.
- V. V. Pham, T. H. T. Vinh, N. T. N. Dung and C. M. Thi, Facile Ball-Milling Synthesis of TiO2 Modified ZnO for Efficient Photocatalytic Removal of Atmospheric Nitric Oxide Gas under Solar Light Irradiation, Chem. Phys. Lett., 2021, 775, 138642–138648 CrossRef.
- E. Ebrahimi, M. Irfan, F. Shabani, Y. Kocak, B. Karakurt, E. Erdem, H. V. Demir and E. Ozensoy, Core-Crown Quantum Nanoplatelets with Favorable Type-II Heterojunctions Boost Charge Separation and Photocatalytic NO Oxidation on TiO2, ChemCatChem, 2020, 12, 6329–6343 CrossRef CAS.
- F. Chang, Z. X. Wei, J. Y. Wang, S. S. Zhao and D. G. Liu, Ultra-Stable Type-II Heterojunctions Bi4O5I2/FeVO4 of Reinforced Photocatalytic NOx Removal Abilities in Visible Light, Mater. Chem. Phys., 2022, 291, 126729–126738 CrossRef CAS.
- T. H. Huy, B. D. Phat, F. Kang, Y. F. Wang, S. H. Liu, C. M. Thi, S. J. You, G. M. Chang and P. V. Viet, SnO2/TiO2 Nanotube Heterojunction: The first Investigation of NO Degradation by Visible Light-Driven Photocatalysis, Chemosphere, 2019, 215, 323–332 CrossRef CAS.
- S. M. Hossain, H. Park, H. J. Kang, J. S. Mun, L. Tijing, I. Rhee, J. H. Kim, Y. S. Jun and H. K. Shon, Facile Synthesis and Characterization of Anatase TiO2/g-CN Composites for Enhanced Photoactivity under UV-Visible Spectrum, Chemosphere, 2021, 262, 128004–128016 CrossRef CAS PubMed.
- X. J. Song, A. Q. Li, Z. H. Cai, X. Chen, W. X. Dai and X. Z. Fu, Synthesis of CdS QDs/BaSnO3 Nanocomposites via Electrostatic Self-Assembly for Visible Light Photocatalytic Deep Oxidation of NO, Appl. Surf. Sci., 2023, 623, 157004–157012 CrossRef CAS.
- D. N. Liu, D. Y. Chen, N. J. Li, Q. F. Xu, H. Li, J. H. He and J. M. Lu, Surface Engineering of g-C3N4 by Stacked BiOBr Sheets Rich in Oxygen Vacancies for Boosting Photocatalytic Performance, Angew. Chem., Int. Ed., 2020, 59, 4519–4524 CrossRef CAS.
- Y. Z. Zou, Y. Xie, S. Yu, L. C. Chen, W. Cui, F. Dong and Y. Zhou, SnO2 Quantum Dots Anchored on g-C3N4 for Enhanced Visible-Light Photocatalytic Removal of NO and Toxic NO2 Inhibition, Appl. Surf. Sci., 2019, 496, 143630–143637 CrossRef CAS.
- Q. Geng, H. T. Xie, H. Ye, Y. J. Sun, X. F. Hou, Z. M. Wang and D. Fan, Atomic Interfacial Structure and Charge Transfer Mechanism on in-situ Formed BiOI/Bi2O2SO4 p-n Heterojunctions with Highly Promoted Photocatalysis, Appl. Catal., B, 2021, 297, 120492–120502 CrossRef CAS.
- J. L. Nie, G. Q. Zhu, W. B. Zhang, J. Z. Gao, P. Zhong, X. T. Xie, Y. Huang and M. Hojamberdiev, Oxygen Vacancy Defects-Boosted Deep Oxidation of NO by β-Bi2O3/CeO2-δ p-n Heterojunction Photocatalyst in situ Synthesized from Bi/Ce(CO3) (OH) Precursor, Chem. Eng. J., 2021, 424, 130327–130337 CrossRef CAS.
- H. Z. Wu, C. W. Yuan, R. M. Chen, J. D. Wang, F. Dong, J. Y. Li and Y. J. Sun, Mechanisms of Interfacial Charge Transfer and Photocatalytic NO Oxidation on BiOBr/SnO2 p-n Heterojunctions, ACS Appl. Mater. Interfaces, 2020, 12, 43741–43749 CrossRef CAS.
- Q. L. Xu, L. Y. Zhang, J. G. Yu, S. Wageh, A. A. Al-Ghamdi and M. Jaroniec, Direct Z-Scheme Photocatalysts: Principles, Synthesis, and Applications, Mater. Today, 2018, 21, 1042–1063 CrossRef CAS.
- X. Li, C. Garlisi, Q. S. Guan, S. Anwer, K. Al-Ali, G. Palmisano and L. X. Zheng, A Review of Material Aspects in Developing Direct Z-Scheme Photocatalysts, Mater. Today, 2021, 47, 75–107 CrossRef CAS.
- Y. J. Zhang, Z. Hu, H. Zhang, H. Li and S. Yang, Uncovering Original Z Scheme Heterojunctions of COF/MOx (M = Ti, Zn, Zr, Sn, Ce, and Nb) with Ascendant Photocatalytic Selectivity for Virtually 99.9% NO-to-NO3− Oxidation, Adv. Funct. Mater., 2023, 33, 23003851–23003860 Search PubMed.
- X. Zhang, H. S. Chen, S. P. Jiang and P. Yang, W18O49/Crystalline g-C3N4 Layered Heterostructures with Full Solar Energy Harvesting towards Efficient H2O2 Generation and NO Conversion, Nano Energy, 2024, 120, 109160–109172 CrossRef CAS.
- Y. Jing, A. D. Fan, J. X. Guo, T. Shen, S. D. Yuan and Y. H. Chu, Synthesis of An Ultrathin MnO2 Nanosheet-Coated Bi2WO6 Nanosheet as a Heterojunction Photocatalyst with Enhanced Photocatalytic Activity, Chem. Eng. J., 2022, 429, 132193–132207 CrossRef CAS.
- T. Shen, X. K. Shi, J. X. Guo, J. Li and S. D. Yuan, Photocatalytic Removal of NO by Light-Driven Mn3O4/BiOCl Heterojunction Photocatalyst: Optimization and Mechanism, Chem. Eng. J., 2021, 408, 128014–128017 CrossRef CAS.
- F. Chang, C. Yang, X. M. Wang, S. S. Zhao, J. L. Wang, W. P. Yang, F. Dong, G. Q. Zhu and Y. Kong, Mechanical Ball-Milling Preparation and Superior Photocatalytic NO Elimination of Z-scheme Bi12SiO20-Based Heterojunctions with Surface Oxygen Vacancies, J. Cleaner Prod., 2022, 380, 135167–135179 CrossRef CAS.
- Y. F. Lu, Y. Huang, J. J. Cao, H. W. Li, W. K. Ho and S. C. Lee, Constructing Z-Scheme SnO2/N-Doped Carbon Quantum Dots/ZnSn(OH)6 Nanohybrids with High Redox Ability for NOx Removal under VIS-NIR Light, J. Mater. Chem. A, 2019, 7, 15782–15793 RSC.
- T. F. Qahtan, T. O. Owolabi, O. E. Olubi and A. Hazam, Emerging Tandem S-Scheme Heterojunction Photocatalysts, Coord. Chem. Rev., 2024, 514, 215839–215859 CrossRef CAS.
- B. C. Zhu, J. Sun, Y. Y. Zhao, L. Y. Zhang and J. G. Yu, Construction of 2D S-Scheme Heterojunction Photocatalyst, Adv. Mater., 2023, 36, 2310600–2310624 CrossRef PubMed.
- Q. L. Xu, L. Y. Zhang, B. Cheng, J. J. Fan and J. G. Yu, S-Scheme Heterojunction Photocatalyst, Chem, 2020, 6, 1543–1559 CAS.
- L. Lv, L. Lei, Q. W. Chen, C. L. Yin, H. Q. Fan and J. P. Zhou, Oxygen Vacancies-Modified S-scheme Heterojunction of Bi-doped La2Ti2O7 and La-Doped Bi4Ti3O12 to Improve the NO Gas Removal Avoiding NO2 Product, Appl. Catal., B, 2024, 343, 123464–123478 CrossRef CAS.
- X. S. Hu, Y. W. Wang, Z. Ling, H. R. Song, Y. Cai, Z. Li, D. Y. Zu and C. P. Li, Ternary g-C3N4/TiO2/Ti3C2 MXene S-scheme Heterojunction Photocatalysts for NOx Removal under Visible Light, Appl. Surf. Sci., 2021, 556, 149817–149823 CrossRef CAS.
- Z. Y. Meng, Y. F. Ma, B. F. Chen, Y. H. Li, H. Ma, B. C. Zhu and F. Dong, One-Step in-situ Construction Engineering of ZnO-Zn2SnO4 Heterojunction for Deeply Photocatalytic Oxidation of Nitric Oxide, J. Colloid Interface Sci., 2024, 664, 433–443 CrossRef CAS.
- H. Wang, Y. J. Sun, G. M. Jiang, Y. X. Zhang, H. W. Huang, Z. B. Wu, S. C. Lee and F. Dong, Unraveling the Mechanisms of Visible Light Photocatalytic NO Purification on Earth-Abundant Insulator-Based Core-Shell Heterojunctions, Environ. Sci. Technol., 2018, 52, 1479–1487 CrossRef CAS PubMed.
- H. Wang, Y. J. Sun, W. J. He, Y. Zhou, S. C. Lee and F. Dong, Visible Light Induced Electron Transfer from a Semiconductor to An Insulator Enables Efficient Photocatalytic Activity on Insulator-Based Heterojunctions, Nanoscale, 2018, 10, 15513–15520 RSC.
- G. H. Dong, L. L. Zhao, X. X. Wu, M. S. Zhu and F. Wang, Photocatalysis Removing of NO based on Modified Carbon Nitride: The Effect of Celestite Mineral Particles, Appl. Catal., B, 2019, 245, 459–468 CrossRef CAS.
- H. Wang, W. Cui, X. A. Dong, J. Y. Li, Q. S. Chen, Z. M. Wang, Y. J. Sun, J. P. Sheng, Y. Zhou, Y. X. Zhang and F. Dong, Interfacial Activation of Reactants and Intermediates on CaSO4 Insulator-Based Heterostructure for Efficient Photocatalytic NO Removal, Chem. Eng. J., 2020, 390, 124609–124616 CrossRef CAS.
- A. J. Wang, Q. Q. Wu, C. Han, H. Yang and X. X. Xue, Significant Influences of Crystal Structures on Photocatalytic Removal of NOx by TiO2, J. Photochem. Photobiol., A, 2021, 407, 113020–113028 CrossRef CAS.
- Y. S. Wan, J. B. Li, J. P. Ni, C. Wang, C. S. Ni and H. Chen, Crystal-Facet and Microstructure Engineering in ZnO for Photocatalytic NO Oxidation, J. Hazard. Mater., 2022, 435, 129073–129082 CrossRef CAS PubMed.
- N. Li, Q. H. Zhu, G. M. Liu, Q. Zhao, H. Q. Lv, M. Z. Yuan, Q. G. Meng, Y. T. Zhou, J. K. Xu and C. Y. Wang, Modulation of Photocatalytic Activity of SrBi2Ta2O9 Nanosheets in NO Removal by Tuning Facets Exposure, J. Mater. Sci. Technol., 2022, 122, 91–100 CrossRef CAS.
- P. Chen, Y. J. Sun, H. J. Liu, Y. Zhou, G. M. Jiang, S. C. Lee, Y. X. Zhang and F. Dong, Facet-Dependent Photocatalytic NO Conversion Pathways Predetermined by Adsorption Activation Patterns, Nanoscale, 2019, 11, 2366–2373 RSC.
- J. Y. Li, R. M. Chen, W. L. Cen, P. Yan, K. L. Li, P. Wang, S. Shu, Y. H. Chu and F. Dong, Quantifying the Activation Energies of ROS-Induced NOx Conversion: Suppressed Toxic Intermediates Generation and Clarified Reaction Mechanism, Chem. Eng. J., 2019, 375, 122026–122033 CrossRef CAS.
- T. Cao, W. C. Huo, Z. Y. Guo, C. Jing, Y. X. Chen, Y. X. Zhang and Z. Zhou, Constructing Defective (BiO)2CO3 with Different Dominated Facets for Efficiently Photocatalytic NO Oxidization and in situ Reaction Pathway Study, Appl. Surf. Sci., 2019, 498, 143848–143854 CrossRef CAS.
- J. R. Li, W. D. Zhang, M. X. Ran, Y. J. Sun, H. W. Huang and F. Dong, Synergistic Integration of Bi Metal and Phosphate Defects on Hexagonal and Monoclinic BiPO4: Enhanced Photocatalysis and Reaction Mechanism, Appl. Catal., B, 2019, 243, 313–321 CrossRef CAS.
- B. Lei, W. Cui, J. P. Sheng, H. Wang, P. Chen, J. Y. Li, Y. J. Sun and F. Dong, Synergistic Effects of Crystal Structure and Oxygen Vacancy on Bi2O3 Polymorphs: Intermediates Activation, Photocatalytic Reaction Efficiency, and Conversion Pathway, Sci. Bull., 2020, 65, 467–476 CrossRef CAS.
- H. Li, H. Shang, Y. Li, X. Cao, Z. Yang, Z. Ai and L. Zhang, Interfacial Charging-Decharging Strategy for Efficient and Selective Aerobic NO Oxidation on Oxygen Vacancy, Environ. Sci. Technol., 2019, 53, 6964–6971 CrossRef CAS.
- L. P. Han, S. X. Cai, M. Gao, J.-Y. Hasegawa, P. L. Wang, J. P. Zhang, L. Y. Shi and D. S. Zhang, Selective Catalytic Reduction of NOx with NH3 by Using Novel Catalysts: State of the Art and Future Prospects, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS PubMed.
- R. D. Zhang, N. Liu, Z. G. Lei and B. H. Chen, Selective Transformation of Various Nitrogen-Containing Exhaust Gases toward N2 over Zeolite Catalysts, Chem. Rev., 2016, 116, 3658–3721 CrossRef CAS.
- Y. J. Pu, X. Y. Xie, W. J. Jiang, L. Yang, X. Jiang and L. Yao, Low-temperature Selective Catalytic Reduction of NOx with NH3 over Zeolite Catalysts: A Review, Chin. Chem. Lett., 2020, 31, 2549–2555 CrossRef CAS.
- D. Kim, D. Shin, J. Heo, H. Lim, J.-A. Lim, H. M. Jeong, B.-S. Kim, I. Heo, I. Oh, B. Lee, M. Sharma, H. Lim, H. Kim and Y. Kwon, Unveiling Electrode–Electrolyte Design-Based NO Reduction for NH3 Synthesis, ACS Energy Lett., 2020, 5, 3647–3656 CrossRef CAS.
- J. Liang, P. Y. Liu, Q. Y. Li, T. S. Li, L. C. Yue, Y. S. Luo, Q. Liu, N. Li, B. Tang, A. A. Alshehri, I. Shakir, P. O. Agboola, C. H. Sun and X. P. Sun, Amorphous Boron Carbide on Titanium Dioxide Nanobelt Arrays for High-Efficiency Electrocatalytic NO Reduction to NH3, Angew. Chem., Int. Ed., 2022, 61, e202202087 CrossRef CAS PubMed.
- J. Long, S. M. Chen, Y. L. Zhang, C. X. Guo, X. Y. Fu, D. H. Deng and J. P. Xiao, Direct Electrochemical Ammonia Synthesis from Nitric Oxide, Angew. Chem., Int. Ed., 2020, 59, 9711–9718 CrossRef CAS PubMed.
- Y. M. Zang, Q. Wu, S. H. Wang, B. B. Huang, Y. Dai and Y. D. Ma, High-Throughput Screening of Efficient Biatom Catalysts Based on Monolayer Carbon Nitride for the Nitric Oxide Reduction Reaction, J. Phys. Chem. Lett., 2022, 13, 527–535 CrossRef CAS.
- R. M. Chen, J. Y. Li, J. L. Wang, W. P. Yang, S. J. Shen and F. Dong, Continuous NO Upcycling into Ammonia Promoted by SO2 in Flue Gas: Poison Can Be a Gift, Environ. Sci. Technol., 2023, 57, 12127–12134 CrossRef CAS PubMed.
- S. Goldstein, D. Behar, T. Rajh and J. Rabani, Nitric Oxide Reduction to Ammonia by TiO2 Electrons in Colloid Solution via Consecutive One-Electron Transfer Steps, J. Phys. Chem. A, 2015, 119, 2760–2769 CrossRef CAS.
- L. Chen, D. C. Shen, B. J. Li, Z. T. Xiao, W. T. Sun, X. Liu, J. J. Ma, C. H. Li and W. T. Wang, The Efficient Electrocatalytic and Photocatalytic Reduction of Nitric Oxide into Ammonia over 0D/3D g-C3N4 Quantum Dots/3DOMM-TiO2-x Heterojunction, Ceram. Int., 2023, 49, 23129–23139 CrossRef CAS.
- E. K. Pham and S.-G. Chang, Removal of NO from Flue Gases by Absorption to An Iron(ii) Thiochelate Complex and Subsequent Reduction to Ammonia, Nature, 1994, 369, 139–141 CrossRef CAS.
- S. H. Zhang, H. Chen, Y. F. Xia, N. Liu, B. H. Lu and W. Li, Current Advances of Integrated Processes Combining Chemical Absorption and Biological Reduction for NOx removal from Flue Gas, Appl. Microbiol. Biotechnol., 2014, 98, 8497–8512 CrossRef CAS PubMed.
- W. Li, C. Z. Wu, S. H. Zhang, K. Shao and Y. Shi, Evaluation of Microbial Reduction of Fe(III)EDTA in a Chemical Absorption-Biological Reduction Integrated NOx Removal System, Environ. Sci. Technol., 2007, 41, 639–644 CrossRef CAS PubMed.
- X. Y. Liu, P. L. Wang, Y. J. Shen, L. R. Zheng, L. P. Han, J. Deng, J. P. Zhang, A. Y. Wang, W. Ren, F. Gao and D. S. Zhang, Boosting SO2-Resistant NOx Reduction by Modulating Electronic Interaction of Short-Range Fe–O Coordination over Fe2O3/TiO2 Catalysts, Environ. Sci. Technol., 2022, 56, 11646–11656 CrossRef CAS PubMed.
- Z. P. Si, Y. J. Shen, J. B. He, T. T. Yan, J. P. Zhang, J. Deng and D. S. Zhang, SO2-Induced Alkali Resistance of FeVO4/TiO2 Catalysts for NOx Reduction, Environ. Sci. Technol., 2022, 56, 605–613 CrossRef CAS.
- X. R. Qi, L. P. Han, J. Deng, T. W. Lan, F. L. Wang, L. Y. Shi and D. S. Zhang, SO2-Tolerant Catalytic Reduction of NOx via Tailoring Electron Transfer between Surface Iron Sulfate and Subsurface Ceria, Environ. Sci. Technol., 2022, 56, 5840–5848 CrossRef CAS PubMed.
- T. Y. Liu and J. P. D. Abbatt, Oxidation of Sulfur Dioxide by Nitrogen Dioxide Accelerated at the Interface of Deliquesced Aerosol Particles, Nat. Chem., 2021, 13, 1173–1177 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |