
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzymatic synthesis of semi-IPNs within hydrogel-based microfluidics†
Chen
Jiao
 ab,
Dietmar
Appelhans
a,
Brigitte
Voit
ab,
Nico
Bruns
c and
Jens
Gaitzsch
*a
ab,
Dietmar
Appelhans
a,
Brigitte
Voit
ab,
Nico
Bruns
c and
Jens
Gaitzsch
*a
aLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Straße 6, 01069 Dresden, Germany. E-mail: gaitzsch@ipfdd.de
bTechnische Universität Dresden, Faculty of Chemistry and Food Chemistry, Organic Chemistry of Polymers, 01069 Dresden, Germany
cTechnical University of Darmstadt, Department of Chemistry and Centre for Synthetic Biology, Peter-Grünberg-Straße 4, 64287 Darmstadt, Germany
First published on 26th December 2024
Abstract
With the goal of achieving environmentally friendly polymer synthesis strategies, enzyme-promoted polymerisation has gradually attracted people's attention. The development of hydrogel-based microfluidics provides a new carrier system for enzymatic catalysis. Here, we report a new technique for enzyme-promoted free radical polymerisation, supported on hydrogel microdots (μHDs) within a microfluidic chip. Free radical polymerisation initiated by free horseradish peroxidase (HRP) in vials confirmed the formation of poly(N-isopropyl acrylamide) (PNiPAAm), achieving high molecular weight (500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) in 5 min. For polymerisation in microfluidics, disulphide-bearing μHDs were mounted on a PDMS-on-glass chip. Utilising a disulphide-thiol exchange reaction, modified HRP was then captured “from the flow” through the chip, which was confirmed by fluorescence microscopy. Various polymerisation parameters were studied in the microfluidic chip, and the successful polymer formation was confirmed by copolymerisation with a fluorescent comonomer. The physical entanglement fixed the formed polymer on the μHDs, forming a structure similar to a semi-interpenetrating network (semi-IPN). Thus, this technique provides a new direct approach to achieving semi-IPNs within microfluidic chips, showcasing the versatility in which microfluidic systems can be utilised.
000 Da) in 5 min. For polymerisation in microfluidics, disulphide-bearing μHDs were mounted on a PDMS-on-glass chip. Utilising a disulphide-thiol exchange reaction, modified HRP was then captured “from the flow” through the chip, which was confirmed by fluorescence microscopy. Various polymerisation parameters were studied in the microfluidic chip, and the successful polymer formation was confirmed by copolymerisation with a fluorescent comonomer. The physical entanglement fixed the formed polymer on the μHDs, forming a structure similar to a semi-interpenetrating network (semi-IPN). Thus, this technique provides a new direct approach to achieving semi-IPNs within microfluidic chips, showcasing the versatility in which microfluidic systems can be utilised.
Introduction
Over the years, conventional free radical polymerisation often relies on harsh reaction conditions, transition metal catalysts, etc., that raise environmental concerns.1–3 A promising way to tackle these concerns and to provide a more environmentally friendly and sustainable synthesis is the use of biocatalysts. The emergence of enzyme-promoted polymerisation demonstrates the potential to reduce resource consumption and waste generation by offering a greener approach to polymer synthesis. Enzymes, as natural catalysts, offer a way to make high-quality polymers with precise control over structure, properties, and function.4–7 An area that has grown considerably over the past two decades is the enzyme-promoted free-radical polymerisation of vinyl monomers.8–11Horseradish peroxidase (HRP) is an enzyme that possesses the remarkable ability to facilitate the oxidation of numerous organic compounds, including phenols and anilines, by utilising hydroperoxides such as hydrogen peroxide (H2O2) and creating radicals.12 Just like all peroxidases, HRP acts as an “electron relay” that connects a two-electron transfer step (reduction of H2O2 to water) to two subsequent single-electron transfer steps (Fig. S1†). The catalytic cycle of the enzyme relies on the conversion of its’ iron-containing heme to different oxidative states at its active site, namely HRP-I and HRP-II.4,6,13 In brief, the water molecule that is bound to the native HRP is first replaced by H2O2, and when the O–O bond of the H2O2 is broken in a heterolytic manner, HRP-I is generated. HRP-I is converted back into its’ initial state in two steps by abstracting a hydrogen atom from a reducing substrate in each step, leading to the generation of two radical species.14 If these radicals are to be utilised for the purpose of initiating a radical polymerisation, β-diketones are often used as reducing substrates.15 Among various β-diketones, acetylacetone (ACAC) was found to be an effective mediator, leading, e.g., to high polymerisation yield and molecular weight of polyacrylamide obtained this way.15,16 However, it is important to note that an insufficient concentration of ACAC can lead to HRP deactivation by H2O2 in the initial stages of polymerisation.16 In addition, the option of the heme unit also affecting the radical polymerisation itself has to be taken into account. As a result, the ternary initiation system of HRP, H2O2 and ACAC has to be at the correct ratio as it can otherwise undergo a transformation into an ‘enzymatically promoted redox system’.16 Furthermore, enzyme-catalyzed polymerization enables the manipulation of polymer properties by adjusting the molecular weight or its distribution.4,5,7
Integrating this system into microfluidics would enable a combination with the modern fields of flow chemistry and microreactors.6 Microfluidic systems are one of the rapidly developing technologies especially for biomedical applications.17,18 Due to the miniaturisation, it is superior to conventional fluidic systems as transport pathways of heat and mass are shortened, leading to reduced operating time, liquid volume, and reagent consumption.19–24 Since they can be operated easily at room temperature, microfluidic systems are also ideal to host enzymatic (cascade) reactions.25
Typically, enzymes are immobilised in inorganic, organic or polymeric materials by binding them to the surface of supports or by entrapment. It is a way to enhance their long-term stability and activity as well as facilitate the separation of reaction products.26–28 Using hydrogels to immobilise enzymes is a feasible approach to combine them with microfluidics because of their high compatibility with enzymes and ease of photolithographic patterning. The integration of a large number of hydrogel microdots (μHDs) into a microfluidic system enables an increase in specific surface area and thus maximizes enzyme–substrate interactions.29,30 These compartments allow for the physical entrapment of proteins, such as enzymes,31 and our group has reported the capture and release of specific proteins and molecules through disulphide bonds and host–guest interactions in microfluidics.32,33 Compartmentalized enzymatic cascade reactions were achieved by immobilizing enzymes on multifunctional μHDs within PDMS-on-glass chips.25,29,30 The PDMS-on-glass chip is a transparent and stable conventional chip that enables rapid prototyping and production using standard techniques.30,31,34–36 A core part of these works are double cross-linked hydrogels that contain a permanent cross-linker (often bisacrylamide (BIS)) and a reversible cross-linker (often bis-acrylylcystamine (BAC)) to include redox chemistry by breaking and reforming disulphide bonds in BAC. Together, the microfluidic platform and the double cross-linked hydrogels lay the foundation for polymerisations-on-a-chip, i.e., capturing enzymes for conducting enzyme-mediated radical polymerisations within microfluidics, which is the main topic of this paper (Fig. 1).
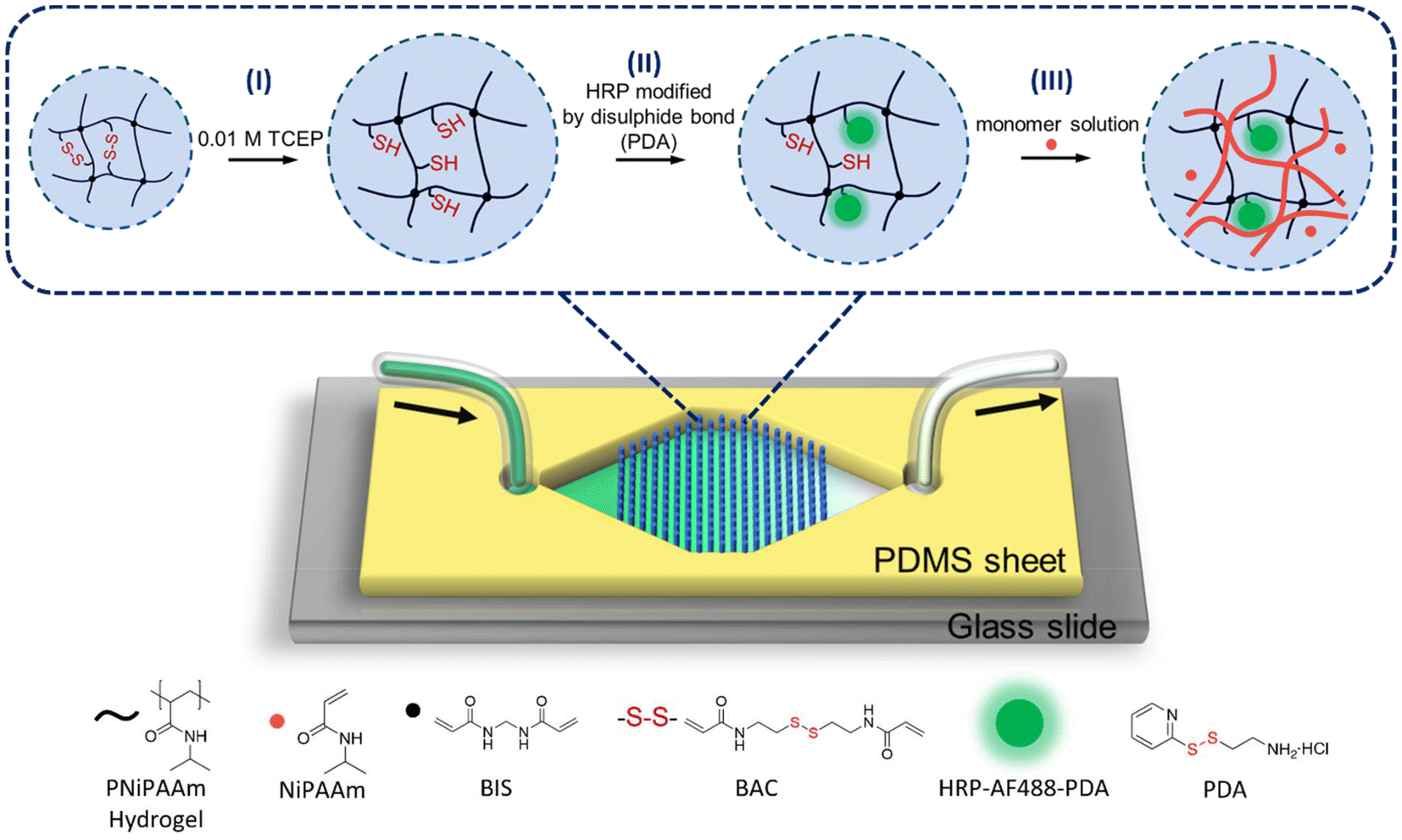 | ||
| Fig. 1 Schematic drawing of the microfluidic chip design and the free radical polymerisation initiated by enzyme HRP captured in hydrogel microdots within a microfluidic device. (I) Hydrogel microdots were prepared by photopatterning. Then, reducing agent TCEP was added to cleave disulphide bonds. (II) HRP was captured via a thiol-disulphide exchange reaction. The HRP was modified with fluorescence marker AF488 for tracking and 2-(2-pyridyldithio)-ethylamine (PDA) for introducing disulphide bonds. (III) The monomer solution (red dots) was perfused to achieve enzyme-promoted free radical polymerisation (formation of red polymer chains). | ||
Here, we utilise our simple and rapid method to capture HRP in microfluidic reactors in order to initiate polymerisations with the ternary system HRP, H2O2, and ACAC (Fig. 1). HRP was modified with 2-(2-pyridyldithio)-ethylamine (PDA) as a linker that contains a disulphide bond. Integration of PNiPAAm-BAC-BIS μHDs into microfluidic devices was achieved by effective in situ photopolymerisation, and HRP was then captured “on the flow” utilising the reducing agent tris(2-carboxyethyl)phosphine (TCEP). Finally, the reaction mixture containing H2O2, ACAC, and the monomer was perfused, and the monomer was polymerised by the enzyme within the μHDs. A polymer could be formed and, much to our surprise, instead of being able to collect a soluble polymer in the outflow, semi-IPNs formed on the chip. We thus discovered that this microfluidic reactor design even offers the potential to synthesise special polymer architectures.
Results and discussion
In-vial HRP initiated free radical polymerisation
In conjunction with the reported enzyme-promoted polymerisation systems, various parameters such as enzyme selection, monomer concentration, and reaction time should be considered and potentially optimised before transitioning to microfluidic chips.4,5,7,10,37 Reaction conditions were hence first established by performing polymerisations in solution with glass vials as reaction vessels. HRP was selected as an efficient enzyme to initiate radical polymerisations. To gain a better understanding of the polymerisation process, NiPAAm was chosen as a well-studied model monomer.38–40 Any repulsive forces between the monomer, the formed polymer, and the existing μHDs could potentially hinder the polymerisation and were avoided using NiPAAM as the μHDs consist of cross-linked PNiPAAm. Using this polymer then allowed for exploiting its LCST around 35 °C for rapid identification.41Following recently published procedures, we started our investigation using a similar monomer concentration range (2 wt%), and the molar ratio of monomer to ternary initiation system NiPAAm/HRP/ACAC/H2O2 was set to 1:10−5:0.05:0.002 (Fig. 2a and Table S1†).4,7,16,37 Enzyme-promoted polymerisation was carried out in deionised water at room temperature for 1 h. To verify the feasibility of the initiation system and the influence of oxygen from air on the reaction, several control experiments have been performed (Fig. S2†). The polymerisation of monomer NiPAAm under different compositions and argon-purging conditions was measured (Fig. S2c†). After reaction times of 1 min and 60 min, the reaction mixtures have been exposed to a hot water bath (Fig. S2a and b,† respectively). A cloudy solution meant that the temperature-responsive PNiPAAM precipitated and, hence, that the polymerisation was successful. Four samples were prepared in this series. Sample no. 1 was synthesised under argon in the presence of the monomer and the ternary initiation system, and lead to successful polymerisation (Fig. 2b). Samples no. 2, 3 and 4 were conducted without purging with argon, without HRP, and without H2O2 (Fig. S2c†), respectively. Surprisingly, the corresponding NiPAAm in sample no. 2 also polymerised well within 5 min in the open vessel, although not as fast as sample no. 1. It indicated that the oxygen did not completely consume all active radicals and there were still enough active radicals to allow for a polymerisation. This was an important basis for the subsequent polymerisation reaction in the PDMS-based microfluidic chip, which is known to be gas-transmissive.42 Moreover, air-contact of the reagent solutions during the transfer to the chip and the polymerisation process on the chip is unavoidable. Samples no. 3 and 4, lacking HRP and H2O2, respectively, did not show any polymerisation after 60 min of reaction, as shown by the lack of a precipitate after heating (Fig. S2b†).43 As per these observations, no polymer could be obtained when either component of the ternary initiation system was absent from the reaction medium. Thus, the presence of HRP, H2O2, and the appropriate mediator is crucial for a successful polymerisation.
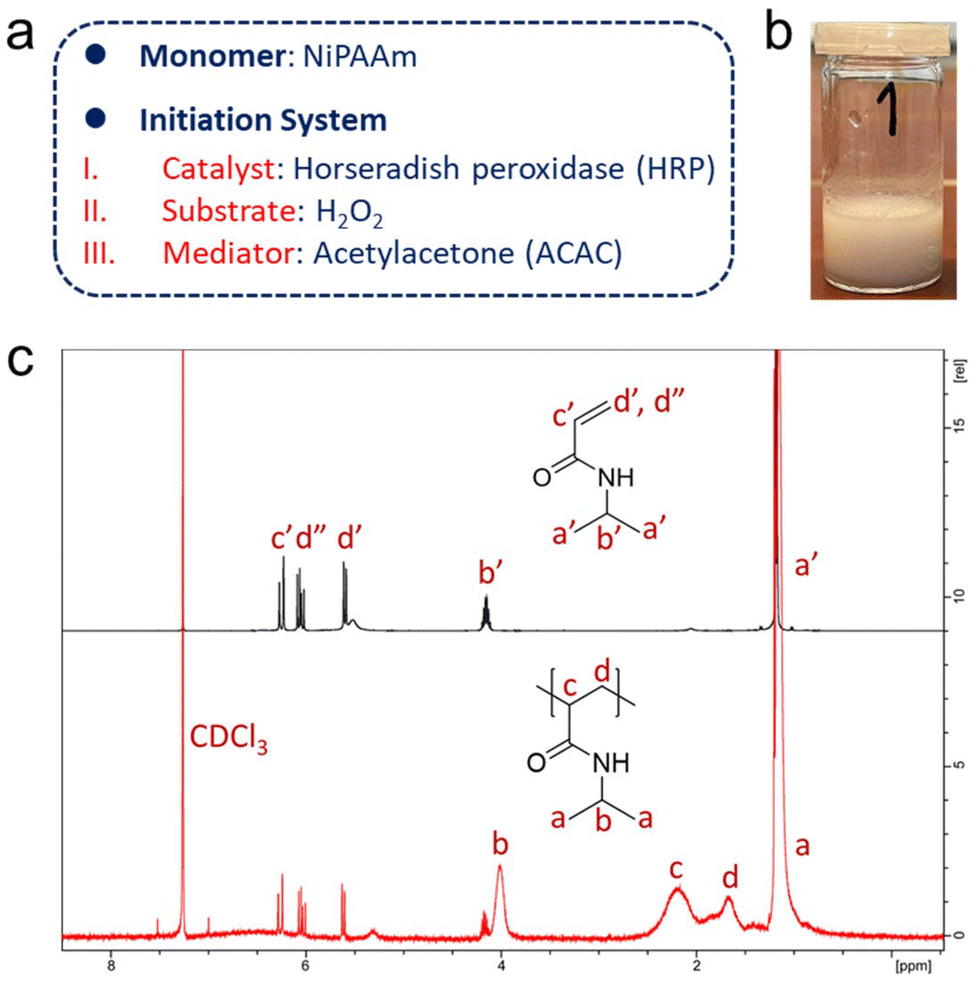 | ||
| Fig. 2 (a) Selection of monomers and composition of the ternary initiation system. (b) Product of the free radical polymerisation of NiPAAm initiated by HRP in solution, using a glass vial as reaction vessel. The photo depicts a polymerised product in water after being heated in a water bath of about 50 °C. “1” refers to sample no. 1, for which the free radical polymerisation of NiPAAm was allowed to run for 1 min (part from Fig. S2†). (c) 1H NMR spectra (CDCl3) of NiPAAm and of PNiPAAm (freeze-dried, unpurified) obtained from a polymerisation initiated by free HRP in vials (reaction time: 60 min). Signals a′–d′ and d′′ are from the monomer NiPAAm, and signals a–d are from the polymer PNiPAAm. | ||
1H NMR spectroscopy was also used to confirm the production of polymers. It should be noted that the samples were directly freeze-dried after 1 h of polymerisation in vials and used for NMR measurements without further purification. All characteristic signals of the polymer PNiPAAm could be clearly assigned in the NMR spectrum of the mixture.44 Broad peaks with chemical shifts of δ = 1.93–2.57 (signal c in Fig. 2c) and 1.49–1.83 (signal d in Fig. 2c) appeared, which were attributed to the protons of the main chain of PNiPAAm. Residual monomer could be identified by the signals of the protons of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds in the monomers with chemical shifts of δ = 6.27, 6.05, and 5.60 (signals c′, d′′, and d′ in Fig. 2c, respectively). The octet and doublet signals, assigned to the protons on the isopropyl group in the monomer with chemical shifts of δ = 4.15 and 1.18 (signals b′ and a′ in Fig. 2c), shifted and became broad in the spectrum of the reaction product, also indicative of polymer formation. An accurate conversion could not be calculated due to the very low feed and high monomer loss during lyophilisation as a workup. Concentrations of other components like HRP and ACAC of the ternary initiation system were too small to be detected in the 1H NMR spectrum.
C double bonds in the monomers with chemical shifts of δ = 6.27, 6.05, and 5.60 (signals c′, d′′, and d′ in Fig. 2c, respectively). The octet and doublet signals, assigned to the protons on the isopropyl group in the monomer with chemical shifts of δ = 4.15 and 1.18 (signals b′ and a′ in Fig. 2c), shifted and became broad in the spectrum of the reaction product, also indicative of polymer formation. An accurate conversion could not be calculated due to the very low feed and high monomer loss during lyophilisation as a workup. Concentrations of other components like HRP and ACAC of the ternary initiation system were too small to be detected in the 1H NMR spectrum.
In order to determine the appropriate polymerisation time, the reaction was followed by size exclusion chromatography (SEC) (Fig. S3 and Table S2†) with samples taken after 5, 10, 15, 20, 40, and 60 min. All polymerisations yielded polymers with a number average molecular weight (Mn) of around 500000 g mol−1 with similar dispersity of around 1.3, independent of the reaction time, and the SEC traces did not show obvious trends. It should be noted that the dispersity and Mn could be underestimated following the upper exclusion volume of the used columns (700000 g mol−1), as well as the potential loss of unreacted monomers and low-molecular-weight oligomers by vacuum freeze-drying during work-up. Bearing in mind the goal of this research, a polymerisation on a microfluidic chip, these results had to be interpreted in this context. According to our previous reports, the standard residence time in our established single-chamber chip is 4.7 min at a flow rate: 5 μL min−1, which is hence within the time required for polymerisation.29,30 Therefore, the feasibility of the ternary initiation system was demonstrated in bulk solution, and the fast reaction rates should allow for a polymerisation in the microfluidic reactor.
Modifications of HRP
In order for HRP to be effectively captured by μHDs within a microfluidic chip, it is necessary to modify the enzyme. The modification process is similar to our previously published procedure,33 using the fluorescence marker Alexa Fluor 488 (AF488) for imaging purposes and 2-(2-pyridyldithio)-ethylamine (PDA) for the introduction of disulphide bonds (as shown in Fig. 3a).33,45 In brief, HRP was first treated with the AF488 N-hydroxysuccinimide (NHS) ester, which reacted with the amine groups of surface-exposed lysines on the HRP. Next, PDA was used to modify carboxylic acid groups of the HRP-AF488 conjugate via the activation with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDAC) and NHS.46 The resulting modified enzyme was then analysed by matrix-assisted laser desorption ionisation with time-of-flight detection mass spectrometry (MALDI-TOF MS) (Fig. 3b) to determine the average number of AF488 and PDA groups per HRP molecule.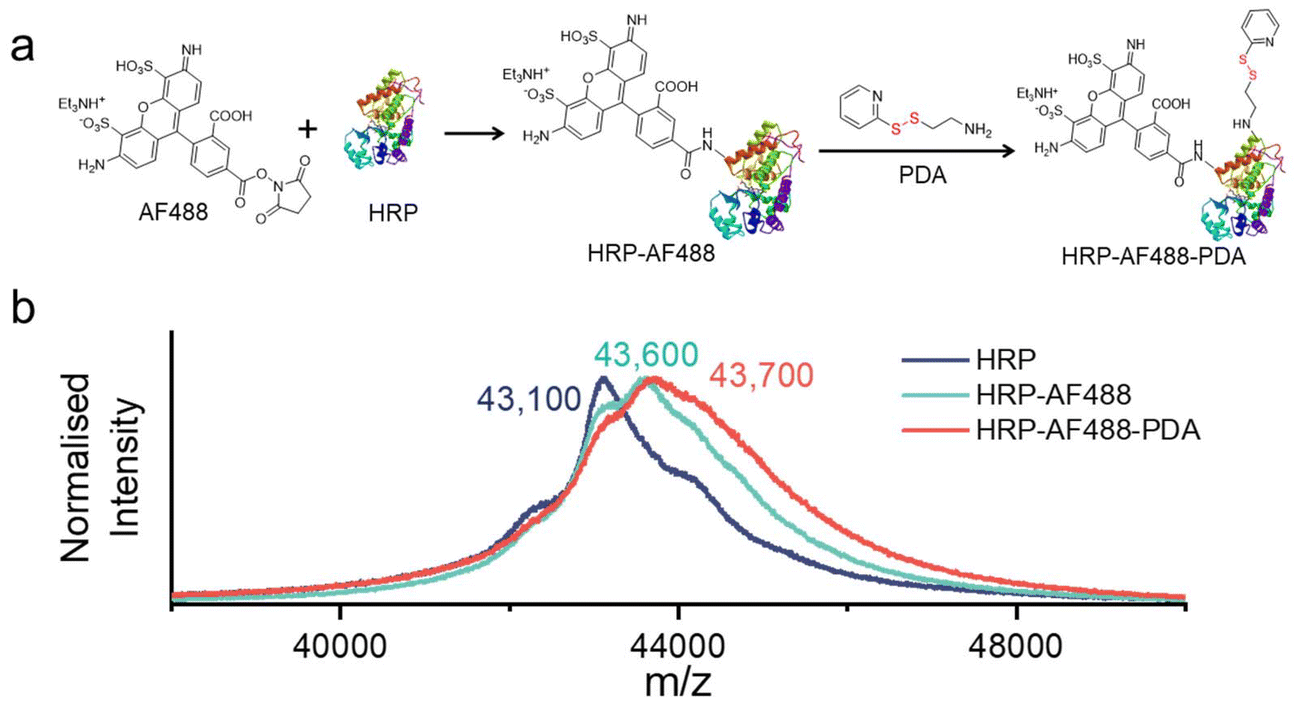 | ||
| Fig. 3 (a) Reaction scheme of the modification of HRP with the fluorescent dye AF488 and disulphide bonds (PDA). (b) MALDI-TOF MS spectra in the [M + X]+ (X: Na or H) range of HRP, HRP-AF488, and HRP-AF488-PDA. | ||
The molecular weights of HRP-AF488 and HRP-AF488-PDA were significantly higher than the unmodified HRP (with respect to the most dominant isoenzyme, Fig. 3b, green and blue lines). HRP-AF488 showed an increase in molecular weight compared to unmodified HRP of Δm/z ≈ 450 to 500 Da, which was less than the molar mass of AF488 (733 g mol−1). Hence, not every HRP was modified by AF488, but on average, 0.6 AF488 molecules were bound per enzyme, i.e., 60% of the HRP molecules were modified with the dye. The shape of the MALDI-TOF MS spectrum of HRP-AF488 supported the partial modification, as it overlaps with the mass spectrum of unmodified HRP in the lower range (until around 43000 m/z, i.e., near the HRP maximum) but then deviates from the HRP mass in the region beyond the maximum intensity. For HRP-AF488-PDA, the molecular weight gain compared to the starting material (Δm/z ≈ 110 to 120 Da) was again less than the molar mass of PDA (186 g mol−1), suggesting a yield of 65%. This means that each successfully synthesised HRP-AF488-PDA and residual HRP-PDA carried only one PDA group per enzyme. The various shoulders in the MALDI-TOF MS spectrum of HRP-AF488-PDA suggest the presence of some unmodified HRP (first shoulder), HRP-PDA (plateau afterwards), HRP-AF488 (maximum intensity) and HRP-AF488-PDA (shoulder following maximum) (Fig. 3b, red line). Still, one can conclude that the disulphide bonds for linkage to the μHDs and the dye AF488 were successfully introduced into HRP, laying the foundation for subsequent microfluidic tests. The activity of the modified enzyme had to be tested and hence the polymerisation initiated by free HRP-AF488-PDA was attempted. Formation of a white precipitate upon heating the reaction mixture after 5 min demonstrated that the modified HRP was still able to promote the polymerisation (Fig. S9†).
Capture of HRP-AF488-PDA by μHDs in microfluidics
Our versatile and established microfluidic platform, a reactor with 227 BIS/BAC double cross-linked micrometer-sized hydrogel dots (μHDs) integrated into a PDMS-on-glass chip, was used for HRP capture.32,33 The process of HRP capture by disulphide exchange on the μHDs in microfluidics was divided into four steps (Fig. 4a, mechanism in Fig. 4b). The first step involved perfusing 0.01 M TCEP in deionised water at a flow rate of 5 μL min−1 for 60 min to break the disulphide bonds in BAC. In the second step, the chip was washed with deionised water to remove residual TCEP. A 50 μM aqueous solution of HRP-AF488-PDA was then perfused in the third step at a flow rate of 5 μL min−1 for 60 min to capture the enzyme. Step four was a crucial second washing step to remove any unreacted material from the chip, and completed the procedure. Long-term perfusion and low flow rates ensured sufficient thiol-disulphide exchange between μHDs and HRP-AF488-PDA to capture the enzyme on the μHDs.35 The μHDs that captured HRP-AF488-PDA showed strong green fluorescence in confocal laser fluorescence microscopy (Fig. 4c), which corresponds to the black areas (μHDs) in the bright-field micrograph (Fig. S4†).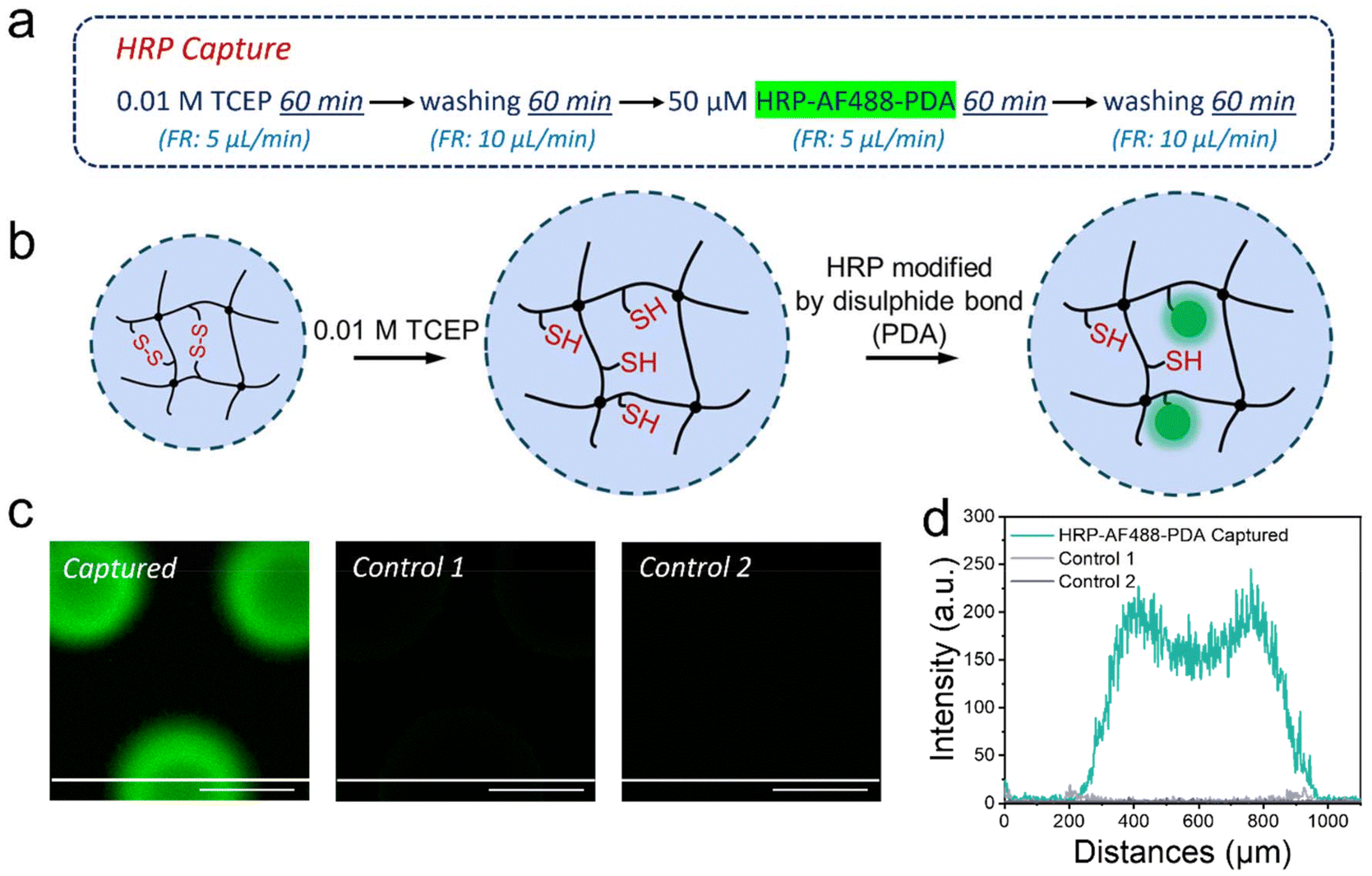 | ||
| Fig. 4 (a) The four perfusion steps to capture the modified HRP (HRP-AF488-PDA as highlighted) in the microfluidic chip. (b) Schematic drawing of the HRP-AF488-PDA capture by redox-responsive double cross-linked μHDs. (c) Fluorescence micrographs of μHDs that captured HRP-AF488-PDA and of two control samples, with a scale bar of 500 μm and a white line indicating the position of the cross-section analysis. Control 1 was produced with HRP that lacked PDA modification and in control 2, the μHDs were not reduced with TCEP. Both controls did not result in capture of proteins in the microfluidic device. (d) Cross-sectional fluorescence intensity values of a HRP-AF488-PDA-modified μHD and of μHD in the two controls, analysed from the white line shown in the micrographs of part c. | ||
Two controls were used to prove enzyme capture, and the perfusion order for both is shown in Fig. S5.† The first control did see TCEP reduction prior to the enzyme capture, but HRP-AF488 without PDA attached was administered. The enzyme now lacked the prerequisite for disulphide exchange, and hence, extremely low fluorescence intensity (Fig. 4c) was detected after the perfusion cycle. This was negligible compared to the successful modification, as shown by cross-sectional analysis (Fig. 4d). In the second control, no TCEP was perfused to break disulphide bonds in the hydrogels. As a result, no free thiol groups were available for the exchange with the disulphide bonds on HRP-AF488-PDA. Again, the extremely weak fluorescence intensity detected after this control cycle indicated that no HRP-AF488-PDA was captured (Fig. 4c), which was again proven by the cross-section analysis (Fig. 4d). Together with the controls, it was hence proven that the discussed 4-step procedure (Fig. 4a) resulted in HRP-AF488-PDA being captured in the μHDs on the microfluidic chip.
The concentration of HRP is of great significance to the successful operation of the ternary initiation system as it has to be high enough to enable the initiation of the polymerisation. It is, hence, necessary to quantify the HRP-AF488-PDA that was captured by the μHDs (capture efficiency) and the concentration in the chip. Fluorometric quantification was chosen for being fast, convenient, and of high sensitivity. The disassembled PDMS-on-glass chip was immersed directly in excess TCEP aqueous solution. As the reducing agent broke the disulphide bonds between HRP and the μHDs, the captured HRP was released from the μHDs. Long-term rinsing and high concentration of the reducing agent were applied to ensure the complete release of the captured HRP-AF488-PDA. The fluorescence spectra in Fig. S6† show the emission of the sample under the excitation at a wavelength of 499 nm, where the emission peak at 520 nm (AF488 emission) was used to determine the concentration of the enzyme. According to the calibration curve of HRP-AF488-PDA (Fig. S7a†), the concentration of HRP-AF488-PDA in the eluent was 0.027 μmol L−1. Since the volume of the rinsing solution to collect the released enzyme was 36 mL (see ESI 1.9†), the amount of captured HRP-AF488-PDA was 0.00097 μmol. According to the perfusion time (60 min), flow rate (5 μL min−1), and concentration (50 μmol L−1) of HRP-AF488-PDA, the amount of perfused enzyme was 0.015 μmol. Based on that, the capture efficiency was at least 6.5%. Since approximately 35% of HRP was not modified with AF488, as measured by MALDI-TOF, the actual efficiency is likely to be up to 9%. In addition, the concentration of captured HRP-AF488-PDA in the microfluidic chip was calculated to be at least 36.1 μmol L−1 or up to 49 μmol L−1 if unmodified HRP is accounted for. For comparison, the concentration of HRP for initiating the polymerisations in solution within glass vials was 1.77 μmol L−1, which is 1/25 of the captured amount. The amount of captured HRP-AF488-PDA was thus high enough for the intended polymerisation.
HRP-promoted radical polymerisation in microfluidics
The successful HRP-initiated radical polymerisation of PNiPAAm in solution and the capture of HRP by μHDs in the microfluidic chip laid a solid foundation for conducting enzymatic polymerisations in the microfluidic reactors. The polymerisation, i.e., perfusion of the monomer solution, was conducted directly after step four of capturing HRP. All chemicals in the monomer solution (NiPAAm, ACAC, and H2O2) had the same concentration as for the vial tests (see Table S1†). Although contact with air was unavoidable during the transfer to the microfluidic chip, the monomer solutions were purged with argon beforehand to reduce the interference of oxygen.As already noted, a flow rate of 5 μL min−1 was suitable as it would provide sufficient residence time (i.e., reaction time) in the chip and was hence chosen as a starting point (Measurement I in Table S3†). In an initial trial with continuous perfusion, the effluent was collected directly to monitor polymer formation. When heated above the cloud point temperature of PNiPAAm, the effluent was still clear and transparent without any precipitation, hence containing no polymer (Fig. S8†). A lower flow rate of 3 μL min−1 was also investigated under the same conditions (Measurement II in Table S3†) and gave no polymer as well. Capping of free thiol groups on the μHDs with maleimide was considered next, as thiol groups are known to cause chain transfer reactions in radical polymerisations which could lead to μHDs with covalently attached polymer chains. Thus, a 0.1 M aqueous maleimide solution was perfused through the microfluidic chip to cap free thiols before the polymerisation mixture was perfused (Measurement III in Table S3†). Again, no polymer was found in the effluent, indicating that chain transfer to free thiols within the hydrogel was not the cause that no polymer was rinsed off the microfluidic chip. Raising the monomer concentration to 1.0 M did also not result in release of polymer (Measurement IV in Table S3†). As it was not clear what hindered the presence of polymer in the effluent, static incubation was considered next in order to ensure the required interactions of all ingredients to allow for a polymerisation (Measurement V in Table S3†). After the 1 M monomer solution was pumped into the chip, the flow was stopped for 2 h, after which the chip was rinsed with deionised water at a flow rate of 5 μL min−1. Again, the effluent did not contain polymer. As the polymer might have been attached to the enzyme, the following step was to perfuse 0.01 M TCEP aqueous solution after 1 h of static incubation to release the enzyme and possibly the formed polymer (Measurement VI in Table S3†). This also resulted in no detectable polymer in the effluent.
All of these experiments could mean that enzymatic polymerisation did not occur within the microfluidic chip, or that the polymer was formed, but could not leave the chip. The latter would be the result from a formed semi-IPN of enzymatically synthesized PNiPAAm within the μHDs. It would mean that the polymerisation of the perfused NiPAAm was achieved in the microreactor, but that the resulting polymer was immobilised on the μHDs due to physical entanglements. Following this notion, a fluorescent comonomer was used as a tracer to demonstrate just that. 2.5 × 10−5 mol% (relative to NiPAAm) of Rhodamine B acrylate (RhBA) (Fig. 5b) was hence added to the protocol of Measurement I in Table S3† while all other components remained unchanged (Table S4†). Following the reaction, fluorescence confocal microscopy confirmed the presence of AF488- (green, from HRP) and RhB- (red, from the synthesised copolymer) labelled molecules immobilised in μHDs (Fig. 5c). In the combined micrograph it can be observed that the RhB-labelled polymers endowed μHDs with a larger diameter than what is observed with the AF488-labelled enzyme, indicating a corona of a poly(NiPAAm-co-RhBA) around the initial μHD network. Since the red fluorescence may result from captured, unreacted RhB-labelled monomer rather than the polymer, a control experiment was performed. It was done with the same sequence of steps, but without capturing HRP (sequence in Fig. S10†). The μHDs showed a detectable, but extremely low fluorescence intensity (Fig. S11†), which might be caused by some unspecific interactions between the μHDs and the RhB-labelled monomers. Together with this control, it was now clear that HRP catalysed the initiation of the radical polymerisation of NiPAAm and RhBA within the μHDs and that the enzymatically synthesized poly(NiPAAm-co-RhBA) formed a semi-IPN within the μHDs as well as a polymer corona around the μHDs. As a result, they could not exit the microfluidic chip through perfusion. As stated earlier, NiPAAm was initially chosen to minimize possible repulsive interactions between the μHDs and the formed polymer, both of which are based on PNiPAAm. Being based on the same polymer now made entanglements of the formed polymer chains and the network of the μHDs more likely and may have promoted the formation of these PNiPAAm-in-PNiPAAm semi-IPNs.
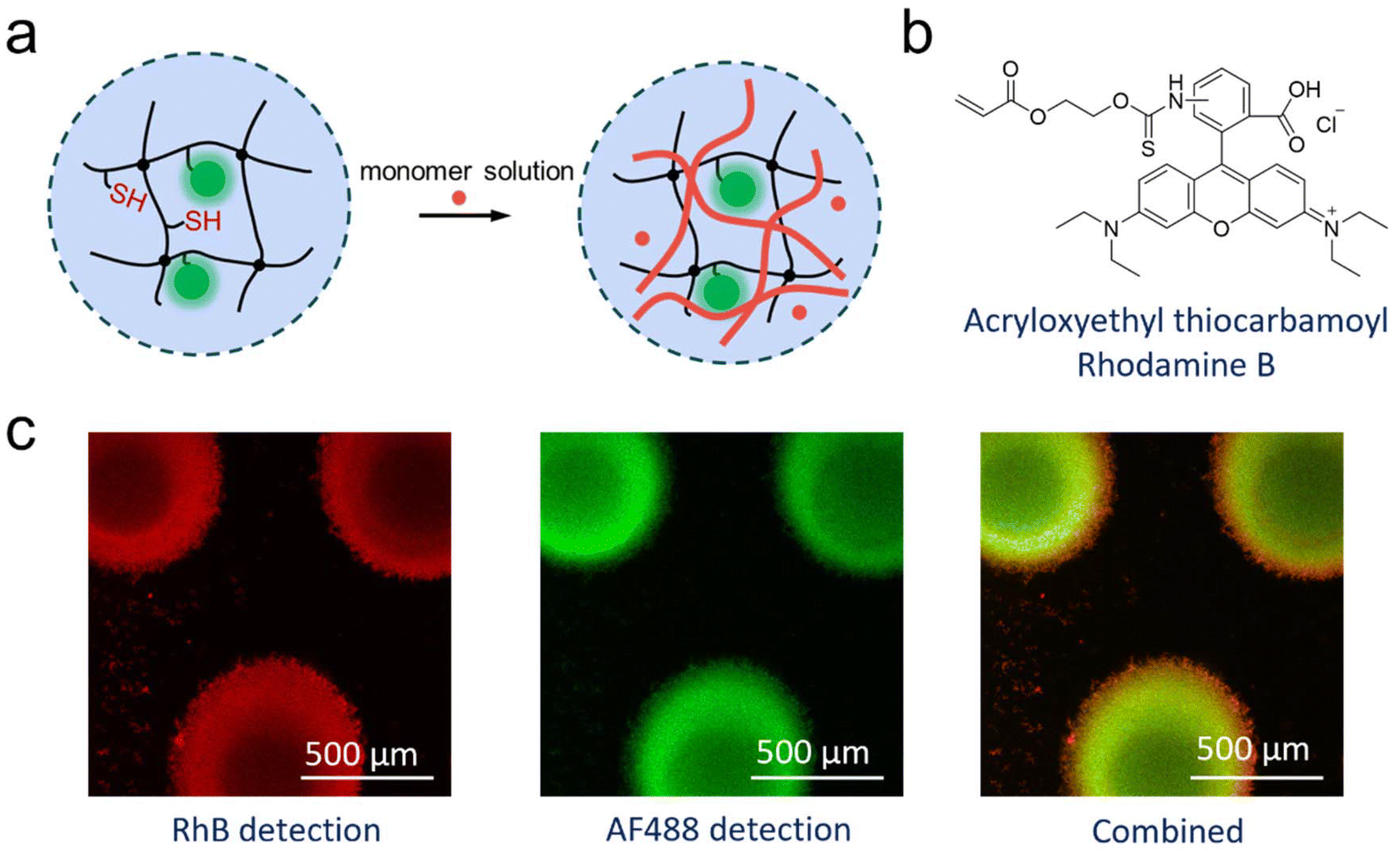 | ||
| Fig. 5 (a) Schematic drawing of the HRP-initiated radical polymerisation within HRP-containing μHDs in microfluidic chips. The black network represents the PNiPAAm μHDs, the green dots represent fluorescently labelled HRP, and the red part represents the perfused reaction mixture as well as the formed polymer. (b) Chemical structure of RhBA. (c) Fluorescence micrographs of μHDs after the enzymatic copolymerisation of NiPAAm and RhBA. The images reveal immobilised HRP-AF488 within the μHDs (shown by the green emission of AF488), and interpenetrating and/or entangled poly(NiPAAm-co-RhBA) in and on μHDs (shown by the red emission of RhB). | ||
After the formation of the semi-IPN, the enzyme had fulfilled its purpose and could be released from the chip. On this basis, the reducing agent TCEP was perfused to release only the captured HRP from μHDs. Fluorescence micrographs were utilised to document the release of both, HRP-AF488-PDA and the produced polymer, over time (Fig. 6a). The residual fluorescence intensity in the images was monitored for 120 min to characterise the release ratio of the enzyme and the formed copolymer (Fig. 6b). In the initial 60 min period, the fluorescence intensity of HRP-AF488-PDA declined rapidly before reaching a plateau at around 120 min (traced by AF488), indicating a final release of 91%. In sharp contrast, the produced polymer showed almost no release in the first 20 min, after which the release slowly increased to 27% after 120 min (traced by RhB). The overlaid fluorescence micrographs show almost exclusively red fluorescence from RhB after 120 min and hence, the polymerised PNiPAAm remained within the μHDs in the form of semi-IPN after release of the enzyme. Only small amounts of the polymer were therefore attached to the enzyme or otherwise able to leave the semi-IPN during the flow, possible following comparatively little entanglements within the μHDs. The rest of the produced polymer remained on the μHDs. Even though the enzyme was captured closer to the surface of the μHDs (green corona in Fig. 6a), the semi-IPN was formed almost homogeneously over the whole μHDs. Thus, our approach unexpectedly provided a new method to realise semi-IPNs through enzymatic polymerisation in flow on a microfluidic chip. Unlike bulk synthesis, microfluidics enable IPN formation in a dynamic flow and support in situ polymerization at precise locations. The compact, portable design of microfluidic chips allows for the production of micro-scale semi-IPNs tailored for micro-scale applications.
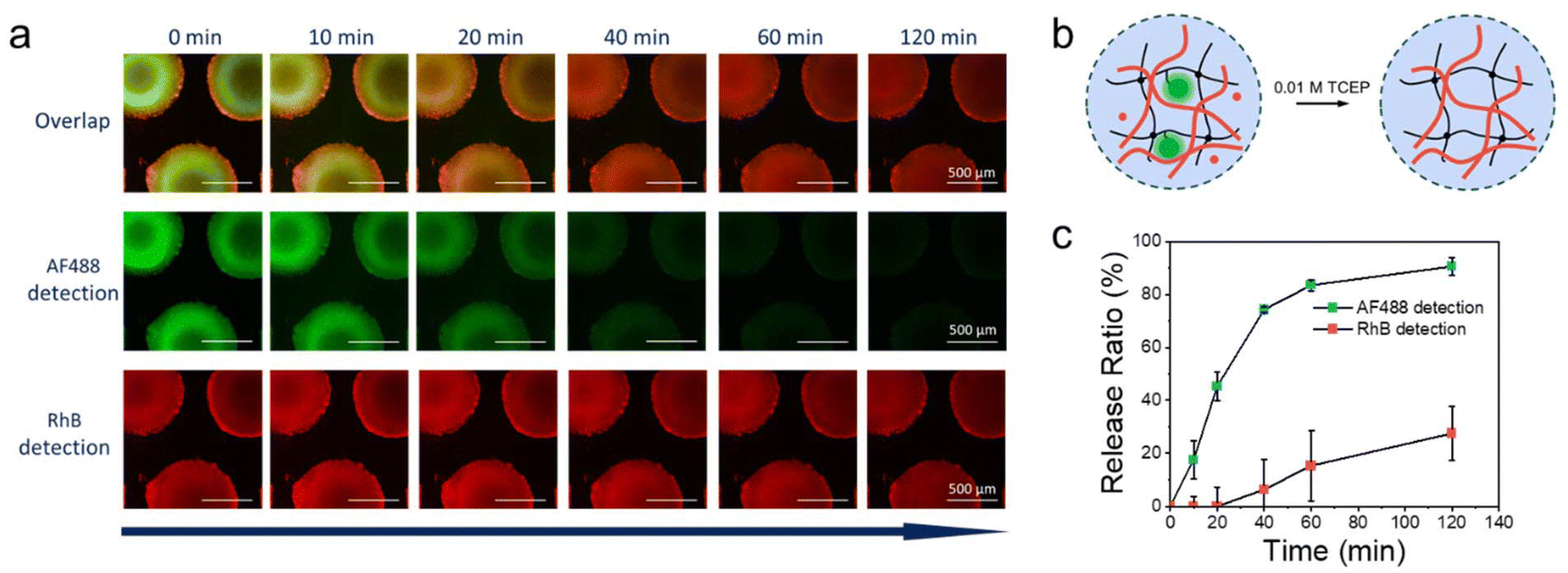 | ||
| Fig. 6 (a) Time series of fluorescence microscopy images of HRP- and poly(NiPAAm-co-RhBA)-containing μHDs under TCEP reduction, with the scale bar of 500 μm. (b) Schematic drawing of the release of HRP and of enzymatically synthesized poly(NiPAAm-co-RhBA) from μHDs by TCEP. (c) Release of HRP-AF488-PDA and of poly(NiPAAm-co-RhBA) by TCEP reduction as measured by the relative reduction in fluorescence intensity of the microscopy images shown in (a). | ||
Conclusion
This work successfully combined enzyme-promoted radical polymerisation with hydrogel-based microfluidic reactors. Following vigorous tests of HRP as an enzyme for NiPAAm polymerisation, it was then successfully modified to be loaded on the redox-responsive μHDs on the microfluidic chip by a disulphide-thiol exchange reaction. Even though the free modified enzyme did allow for a radical polymerisation, no free polymer could be retrieved from the effluent of the microfluidic chip. Instead, this setup led to the formation of PNiPAAm-in-PNiPAAm semi-IPNs. Physical entanglement with the network forming the μHDs during the formation of the semi-IPNs resulted in the immobilisation of the formed polymer within and on the surface of the μHDs. These semi-IPNs formed by polymerisation during flow remained on the chip, also after the enzyme was released, indicating relatively strong physical entanglements. Although this led to difficulties in collecting and analysing the formed polymers, it provided additional proof of the formation of the semi-IPN. It opens the door to explore this new approach to achieve semi-IPNs by enzymatic polymerisations in microfluidic flow reactors. Other monomer/μHD combinations as well as the manipulation of polymer molecular weight/properties by different enzymes will be looked at in future research. Our results demonstrate that this novel design, with its low solvent consumption and minimal use of chemicals, enables the synthesis of unique polymer structures within the microfluidic reaction chamber under dynamic flow conditions. As an early-stage fundamental study, the semi-IPN polymers prepared at the microscale complement traditional bulk semi-IPNs and hold potential value for biomedical applications, such as tissue engineering and drug delivery.Experimental section
Materials, synthesis and characterisations of polymers, preparation of hydrogel arrays, microfluidic testing and additional experimental data are supplied in the ESI.†Author contributions
The manuscript was written by contributions from all authors and all authors have approved the final version of the manuscript.Chen Jiao: methodology, validation, formal analysis, investigation, data curation, writing – original draft. Dietmar Appelhans: conceptualisation, writing – review and editing, supervision, funding acquisition. Brigitte Voit: conceptualisation, writing – review and editing, supervision, project administration, funding acquisition. Nico Bruns: conceptualisation, writing – review and editing. Jens Gaitzsch: conceptualisation, data curation, writing – review and editing, supervision, project administration.
Data availability
Experimental data is available in the ESI.† Additional data for this article, including [NMR spectra, UV-Vis spectra, computational data] are available at Zenodo at https://doi.org/10.5281/zenodo.14046444.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge the financial support by the China Scholarship Council (CSC, for C.J.) and Deutsche Forschungsgemeinschaft (Graduiertenkolleg “Hydrogelbasierte Mikrosysteme”, DFG-GRK 1865). The authors thank Alissa Seifert for GPC measurements and Dr Silvia Moreno for discussions on enzyme modification and fluorescence quantification. The authors thank the workshop of Leibniz Institute of Polymer Research (IPF) for the production of POM moulds, Mohammed Hadi Shahadha (Dresden University of Technology, Dresden, Germany) for manufacturing microfluidic chip moulds and Dr Franziska Obst (Dresden University of Technology, Dresden, Germany) as well as Gerald Hielscher (Dresden University of Technology, Dresden, Germany) for manufacturing photomasks.References
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 3087–3092 Search PubMed.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309–15314 CrossRef CAS.
- T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087–1097 RSC.
- M. Daoud Attieh, Y. Zhao, A. Elkak, A. Falcimaigne-Cordin and K. Haupt, Angew. Chem., Int. Ed., 2017, 56, 3339–3343 CrossRef CAS PubMed.
- C. Fodor, B. Gajewska, O. Rifaie-Graham, E. A. Apebende, J. Pollard and N. Bruns, Polym. Chem., 2016, 7, 6617–6625 RSC.
- F. Hollmann and I. W. C. E. Arends, Polymers, 2012, 4, 759–793 CrossRef.
- S. J. Sigg, F. Seidi, K. Renggli, T. B. Silva, G. Kali and N. Bruns, Macromol. Rapid Commun., 2011, 32, 1710–1715 CrossRef CAS PubMed.
- S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353 CrossRef CAS.
- A. Singh and D. L. Kaplan, J. Polym. Environ., 2002, 10, 85–91 CrossRef CAS.
- K. J. Rodriguez, M. M. Pellizzoni, R. J. Chadwick, C. Guo and N. Bruns, Methods Enzymol., 2019, 627, 249–262 CAS.
- S. Shoda, H. Uyama, J. Kadokawa, S. Kimura and S. Kobayashi, Chem. Rev., 2016, 116, 2307–2413 CrossRef CAS PubMed.
- Y. Huang, J. Ren and X. Qu, Chem. Rev., 2019, 119, 4357–4412 CrossRef CAS PubMed.
- T. Su, D. Zhang, Z. Tang, Q. Wu and Q. Wang, Chem. Commun., 2013, 49, 8033–8035 RSC.
- R. S. Premachandran, S. Banerjee, X. K. Wu, V. T. John, G. L. McPherson, J. Akkara, M. Ayyagari and D. Kaplan, Macromolecules, 1996, 29, 6452–6460 CrossRef CAS.
- D. Teixeira, T. Lalot, M. Brigodiot and E. Maréchal, Macromolecules, 1998, 32, 70–72 CrossRef.
- A. Durand, T. Lalot, M. Brigodiot and E. Maréchal, Polymer, 2001, 42, 5515–5521 CrossRef CAS.
- D. Mark, S. Haeberle, G. Roth, F. von Stetten and R. Zengerle, Chem. Soc. Rev., 2010, 39, 1153–1182 RSC.
- E. K. Sackmann, A. L. Fulton and D. J. Beebe, Nature, 2014, 507, 181–189 CrossRef CAS PubMed.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS.
- L. Shang, Y. Cheng and Y. Zhao, Chem. Rev., 2017, 117, 7964–8040 CrossRef CAS.
- Y. Liu, L. Sun, H. Zhang, L. Shang and Y. Zhao, Chem. Rev., 2021, 121, 7468–7529 CrossRef CAS PubMed.
- W. Li, L. Zhang, X. Ge, B. Xu, W. Zhang, L. Qu, C. H. Choi, J. Xu, A. Zhang, H. Lee and D. A. Weitz, Chem. Soc. Rev., 2018, 47, 5646–5683 RSC.
- T. M. Choi, G. H. Lee, Y. S. Kim, J. G. Park, H. Hwang and S. H. Kim, Adv. Mater., 2019, 31, e1900693 CrossRef.
- H. Wang, Y. Liu, Z. Chen, L. Sun and Y. Zhao, Sci. Adv., 2020, 6, eaay1438 CrossRef CAS PubMed.
- A. Koball, F. Obst, J. Gaitzsch, B. Voit and D. Appelhans, Small Methods, 2024, 2400282, DOI:10.1002/smtd.202400282.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- D. Brady and J. Jordaan, Biotechnol. Lett., 2009, 31, 1639–1650 CrossRef CAS.
- C. Garcia-Galan, Á. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- F. Obst, D. Simon, P. J. Mehner, J. W. Neubauer, A. Beck, O. Stroyuk, A. Richter, B. Voit and D. Appelhans, React. Chem. Eng., 2019, 4, 2141–2155 RSC.
- F. Obst, A. Beck, C. Bishayee, P. J. Mehner, A. Richter, B. Voit and D. Appelhans, Micromachines, 2020, 11, 167 CrossRef.
- F. Obst, M. Mertz, P. J. Mehner, A. Beck, K. Castiglione, A. Richter, B. Voit and D. Appelhans, ACS Appl. Mater. Interfaces, 2021, 13, 49433–49444 CrossRef CAS.
- C. Jiao, N. Liubimtsev, Z. Zagradska-Paromova, D. Appelhans, J. Gaitzsch and B. Voit, Macromol. Rapid Commun., 2023, 44, 2200869 CrossRef CAS.
- C. Jiao, F. Obst, M. Geisler, Y. Che, A. Richter, D. Appelhans, J. Gaitzsch and B. Voit, Polymers, 2022, 14, 267 CrossRef CAS.
- A. Beck, F. Obst, M. Busek, S. Grunzner, P. J. Mehner, G. Paschew, D. Appelhans, B. Voit and A. Richter, Micromachines, 2020, 11, 167 CrossRef PubMed.
- S. F. Chong, R. Chandrawati, B. Stadler, J. Park, J. Cho, Y. Wang, Z. Jia, V. Bulmus, T. P. Davis, A. N. Zelikin and F. Caruso, Small, 2009, 5, 2601–2610 CrossRef CAS PubMed.
- A. Beck, P. J. Mehner, A. Voigt, F. Obst, U. Marschner and A. Richter, Adv. Mater. Technol., 2022, 7, 2200185 Search PubMed.
- M. V. Dinu, M. Spulber, K. Renggli, D. Wu, C. A. Monnier, A. Petri-Fink and N. Bruns, Macromol. Rapid Commun., 2015, 36, 507–514 CrossRef CAS PubMed.
- E. Gil and S. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- S. Lanzalaco and E. Armelin, Gels, 2017, 3, 36 CrossRef PubMed.
- X. Z. Zhang, D. Q. Wu and C. C. Chu, Biomaterials, 2004, 25, 3793–3805 CrossRef CAS PubMed.
- Z. Osváth and B. Iván, Macromol. Chem. Phys., 2017, 218, 1600470 CrossRef.
- B. Wilks and M. E. Rezac, J. Appl. Polym. Sci., 2002, 85, 2436–2444 CrossRef CAS.
- H. Wei, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, Prog. Polym. Sci., 2009, 34, 893–910 CrossRef CAS.
- S.-W. Kuo, J.-L. Hong, Y.-C. Huang, J.-K. Chen, S.-K. Fan, F.-H. Ko, C.-W. Chu and F.-C. Chang, J. Nanomater., 2012, 2012, 749732 CrossRef.
- C. Battistella and H. A. Klok, Biomacromolecules, 2017, 18, 1855–1865 CrossRef CAS PubMed.
- X. Huang, M. Li, D. C. Green, D. S. Williams, A. J. Patil and S. Mann, Nat. Commun., 2013, 4, 2239 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01259c |
| This journal is © The Royal Society of Chemistry 2025 |