
Precisely controlled yet dynamically exchanged micelles via the self-assembly of amphiphilic acrylate random copolymers in water†
Hiroyuki Kono ,
Makoto Ouchi and
Takaya Terashima*
,
Makoto Ouchi and
Takaya Terashima*
Department of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: terashima.takaya.2e@kyoto-u.ac.jp
First published on 20th December 2024
Abstract
Herein, we investigated the self-assembly of amphiphilic acrylate random copolymers bearing hydrophilic poly(ethylene glycol) chains and hydrophobic dodecyl groups into micelles in water. The random copolymers formed precise yet dynamic micelles in water, dependent on the degree of polymerization (DP) and composition. The copolymers shorter than a threshold DPth exclusively formed multichain micelles and the copolymers longer than the DPth self-folded into unimer micelles. The molecular weight and size of the multichain micelles were determined by the composition, and the aggregation number was controllable by the DP. The critical micelle concentration of the random copolymers was estimated to be approximately 1 × 10−3 mg mL−1, and almost independent of the DP, aggregation number, monomer sequence, and backbone structures. More uniquely, owing to the flexible backbones, the acrylate random copolymer micelles induced the exchange of polymer chains even at a low temperature such as 10 °C (activation energy: Ea = ∼40 kJ mol−1) although their corresponding methacrylate counterparts with relatively rigid backbones required at least 25 °C for polymer chain exchange.
Introduction
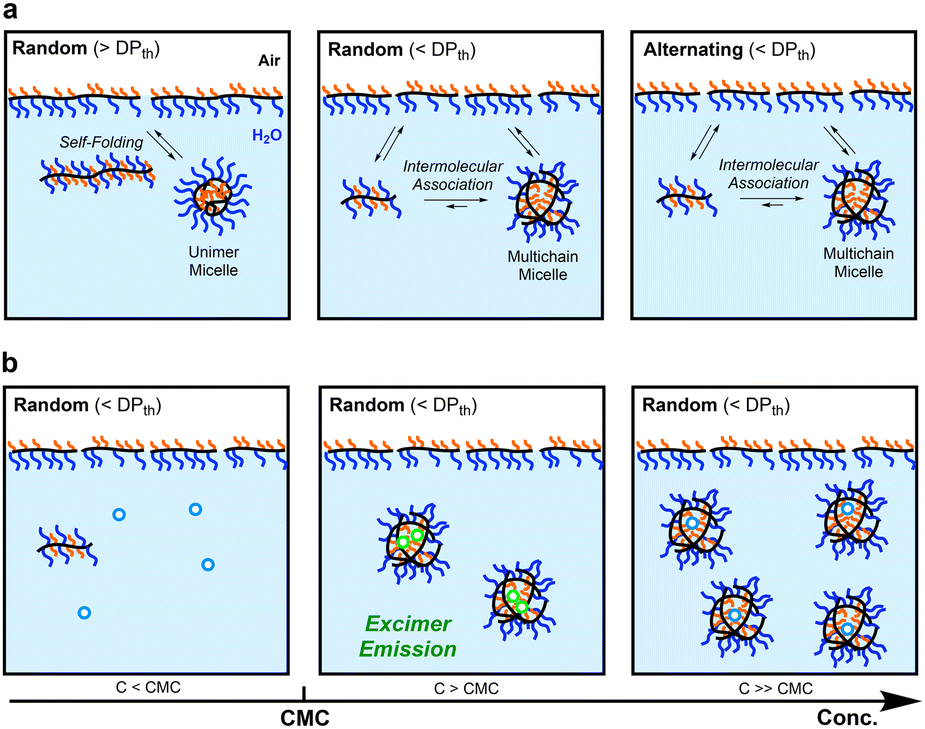
Self-assembly of amphiphilic polymers and molecules in water is a key strategy to produce nanostructured objects such as micelles, vesicles, and single-chain polymer nanoparticles.1–16 These self-assemblies are useful as functional materials including stimuli-responsive materials,17–19 molecular encapsulation/release materials,20,21 and drug-delivery vehicles.22,23 In general, polymer or surfactant micelles and related aggregates are formed in water via hydrophobic effects or physical interactions above the critical micelle or aggregation concentration. These self-assemblies often induce the exchange of the polymer chains or molecules among each other; the dynamic behaviour depends on the molecular structures, solution temperature, solute concentration, and additives in solutions. Controlling not only the size and three-dimensional architectures but also dynamic properties is crucial in designing self-assembled materials with desired properties and functions.To date, various amphiphilic polymers with distinct structures, such as block,1–3,5–8,24–27 random/statictical,28–43 and alternating44–48 copolymers, have been designed for targeted self-assemblies. Among them, random copolymers have attracted attention as scaffolds for small micelles or SCNPs whose size is about 10 nm and close to that of proteins. We have developed self-assembly systems of amphiphilic random copolymers bearing hydrophilic poly(ethylene glycol) (PEG) and hydrophobic alkyl groups [e.g., PEG methyl ether methacrylate (PEGMA)/dodecyl methacrylate (DMA) random copolymers (Fig. 1)].35–43 These copolymers form micelles via chain-folding by the association of hydrophobic side chains in water and show unique self-assembly behaviour in water, depending on the degree of polymerization (DP): the copolymers shorter than a threshold DPth exclusively form multichain micelles via intermolecular self-assembly and those longer than the DPth form unimer micelles via self-folding.35,39,40 The size of the multichain micelles is determined by the copolymer composition and side chain structures, irrespective of the DP. Additionally, the random copolymer micelles show dynamic chain-exchange behaviour, depending on the side chains and temperature.41–43
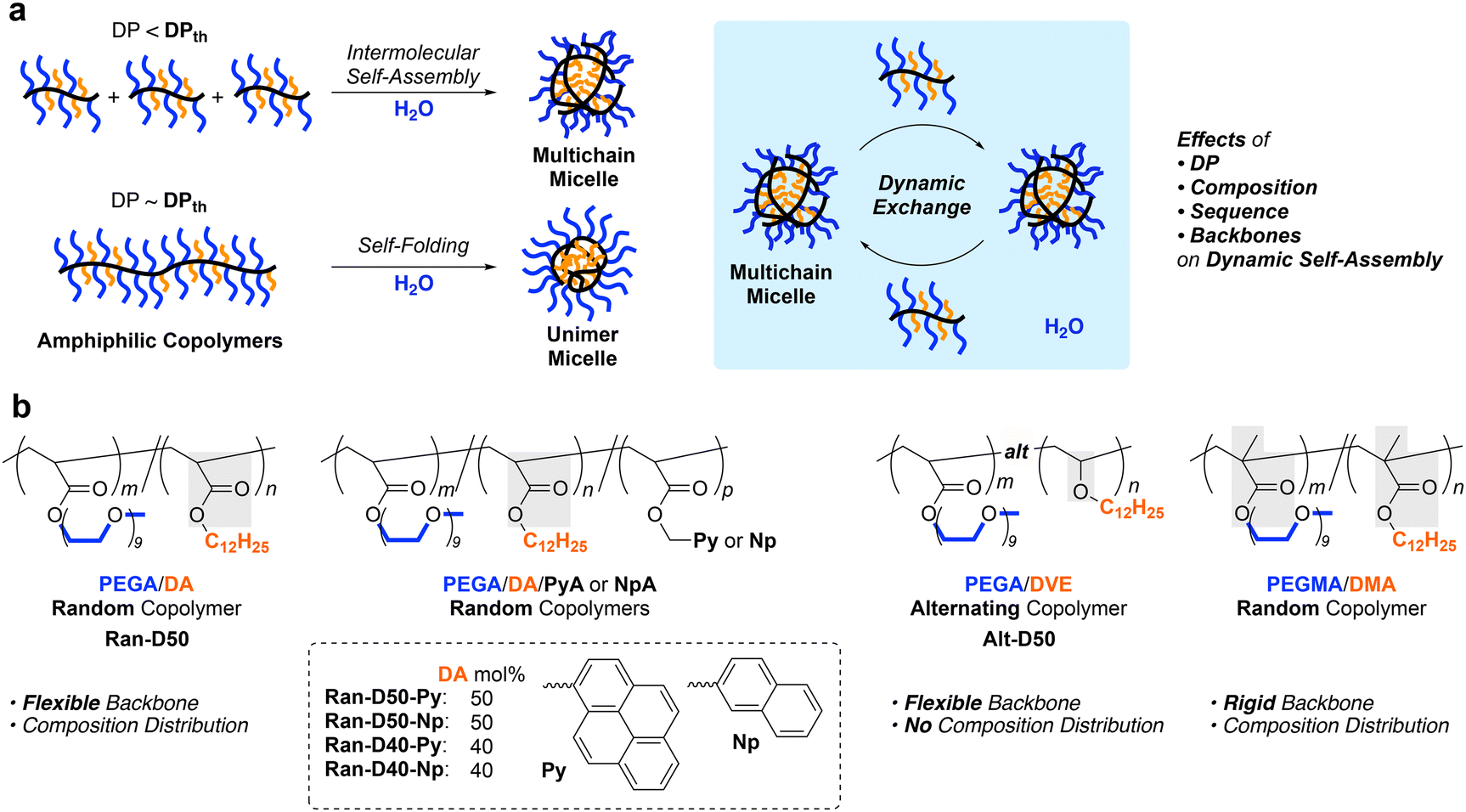 | ||
| Fig. 1 (a) Precision self-assembly of amphiphilic copolymers into multichain or unimer micelles in water, depending on the degree of polymerization (DP), and dynamic exchange of polymer chains between micelles. (b) Design of acrylate random, acrylate/vinyl ether alternating, and methacrylate random copolymers with hydrophilic poly(ethylene)glycol (PEG) chains and hydrophobic dodecyl groups. | ||
Self-assembly of amphiphilic copolymers is also dependent on the main chain structures and monomer sequence. For instance, the alternating copolymer of PEG methyl ether acrylate (PEGA) and dodecyl vinyl ether (DVE)48 forms a multichain micelle with molecular weight lower than the corresponding methacrylate35 or acrylate36 random copolymers with the same composition of PEG and dodecyl side chains. This is probably because the alternating copolymer has a flexible acrylate/vinyl ether backbone and does not contain consecutive dodecyl units. Acrylamide random copolymers consisting of more hydrophilic backbones also formed micelles with molecular weights lower than those of their acrylate counterparts.38 These results suggest that the flexibility and polarity of the polymer backbones affect the dynamic properties of polymer micelles. Owing to the flexible backbones, amphiphilic acrylate random copolymers36,37 are expected to induce precise yet dynamic self-assembly into size-controlled micelles that may promote chain exchange more than methacrylate copolymer micelles.41–43
Herein, we investigated the self-assembly of amphiphilic acrylate random copolymers bearing PEG chains and dodecyl groups into micelles in water, focusing on the effects of the DP, composition, and sequence distribution on the size and aggregation number of the micelles, critical micelle concentration (CMC), and chain exchange properties (Fig. 1). The random copolymers were obtained from free radical copolymerization of hydrophilic PEGA and hydrophobic dodecyl acrylate (DA) in the presence or absence of small amounts of pyrene (Py) or naphthalene (Np)-bearing acrylates (PyA or NpA). The copolymers with broad dispersity (Đ) were fractionated into samples with different molecular weights and narrow Đ by preparative size-exclusion chromatography (SEC). The aqueous solutions of the fractionated copolymers were analyzed by SEC with multiangle laser light scattering (MALLS) to determine the absolute weight-average molecular weight and aggregation number of the micelles. The chain exchange between their micelles was evaluated by fluorescence measurements of the mixtures of Py or Np-labeled copolymers.
The acrylate random copolymers, as well as methacrylate counterparts,35,39,40 induced self-assembly controlled by the DP and composition. The copolymers with a DP smaller than a threshold DPth formed multichain micelles whose size was constant and independent of DP. In contrast, the copolymers with a DP larger than DPth mainly formed unimer micelles. The size of multichain micelles increased with increasing content of dodecyl groups. The CMC of the random copolymers in water was estimated to be approximately 1.0 × 10−3 mg mL−1, independent of the DP (i.e., aggregation number) and monomer sequence distribution (random or alternating). Uniquely, the acrylate random copolymers induced chain exchange even at a low temperature more efficiently than their methacrylate counterparts.
Results and discussion
Synthesis of amphiphilic random copolymers
Amphiphilic random copolyacrylates bearing hydrophilic PEG chains, hydrophobic dodecyl groups, and a small amount (∼1 mol%) of pyrene (Py) or naphthalene (Np) units were synthesized by free radical copolymerization of PEGA, DA, PyA or NpA with 2,2′-azobis(isobutyronitrile) in toluene at 60 °C. The DA content was set to 40 or 50 mol% to examine the effects of composition, main chain structures, and monomer sequence (random vs. alternating) on self-assembly behaviour. The Py or Np fluorophores were used to evaluate the exchange of the polymer chains between their micelles in water by fluorescence resonance energy transfer (FRET). The random copolymers are coded as Ran-D40-Py or Np and Ran-D50-Py or Np, dependent on the DA content and the Py or Np labels. A non-labeled PEGA/DA random copolymer with 50 mol% DA (Ran-D50) was also prepared to investigate the CMC in water.In all the copolymerization, both PEGA and DA were simultaneously consumed at the same rate, regardless of the feed ratio of their monomers. This indicates that the two monomers are randomly distributed in the resulting copolymer chains (Fig. S1†). The random copolymers were analyzed by SEC in N′N′-dimethylformamide (DMF) containing 10 mM LiBr. The copolymers had number-average molecular weight (Mn) values of 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400–34000 g mol−1 and dispersity values (Đ = Mw/Mn: molecular weight distribution) of 2.05–2.43 by poly(methyl methacrylate) (PMMA) standard calibration (Fig. S1†) or Mn values of 10500–15600 g mol−1 and Đ values of 2.36–2.86 by poly(ethylene oxide) (PEO) standard calibration (Fig. 2a, S2 and S3†). To examine the effects of the monomer sequence distribution and backbone structures on the CMC, a PEGA/DVE alternating copolymer (Alt-D50, Mn = 33500 g mol−1, Đ = 1.72 by PMMA calibration or Mn = 15600 g mol−1, Đ = 1.89 by PEO calibration) was also prepared by free radical copolymerization of PEGA in the presence of an excess of DVE according to the literature.46 The alternating sequence of PEGA and DVE was confirmed by 13C NMR spectroscopy (Fig. S4†).
400–34000 g mol−1 and dispersity values (Đ = Mw/Mn: molecular weight distribution) of 2.05–2.43 by poly(methyl methacrylate) (PMMA) standard calibration (Fig. S1†) or Mn values of 10500–15600 g mol−1 and Đ values of 2.36–2.86 by poly(ethylene oxide) (PEO) standard calibration (Fig. 2a, S2 and S3†). To examine the effects of the monomer sequence distribution and backbone structures on the CMC, a PEGA/DVE alternating copolymer (Alt-D50, Mn = 33500 g mol−1, Đ = 1.72 by PMMA calibration or Mn = 15600 g mol−1, Đ = 1.89 by PEO calibration) was also prepared by free radical copolymerization of PEGA in the presence of an excess of DVE according to the literature.46 The alternating sequence of PEGA and DVE was confirmed by 13C NMR spectroscopy (Fig. S4†).
All the copolymers with broad dispersity were fractionated using a preparative SEC into six samples (A–F) with different molecular weights (Fig. 2 and S2, S3†). The chemical structures and compositions of the fractionated copolymers were analyzed by 1H NMR spectroscopy (Fig. S5–S10 and Tables S1–S4†). The absolute weight-average molecular weight (Mw,DMF) and ĐDMF of the fractionated copolymers were determined using an SEC system equipped with a MALLS detector in DMF (10 mM LiBr) as an eluent, where the copolymers are unimolecularly dissolved in DMF. The degree of polymerization (DP) of the fractionated copolymers was calculated from the Mw,DMF and ĐDMF (by SEC-MALLS), the composition (by 1H NMR), and the formula weight of monomers (Tables S1–S4†). Typically, Ran-D50-F–A (Mn = 4500–90700 g mol−1, Đ = 1.16–1.91 by SEC with PEO calibration, Fig. 2c) had a DP of 55–799.
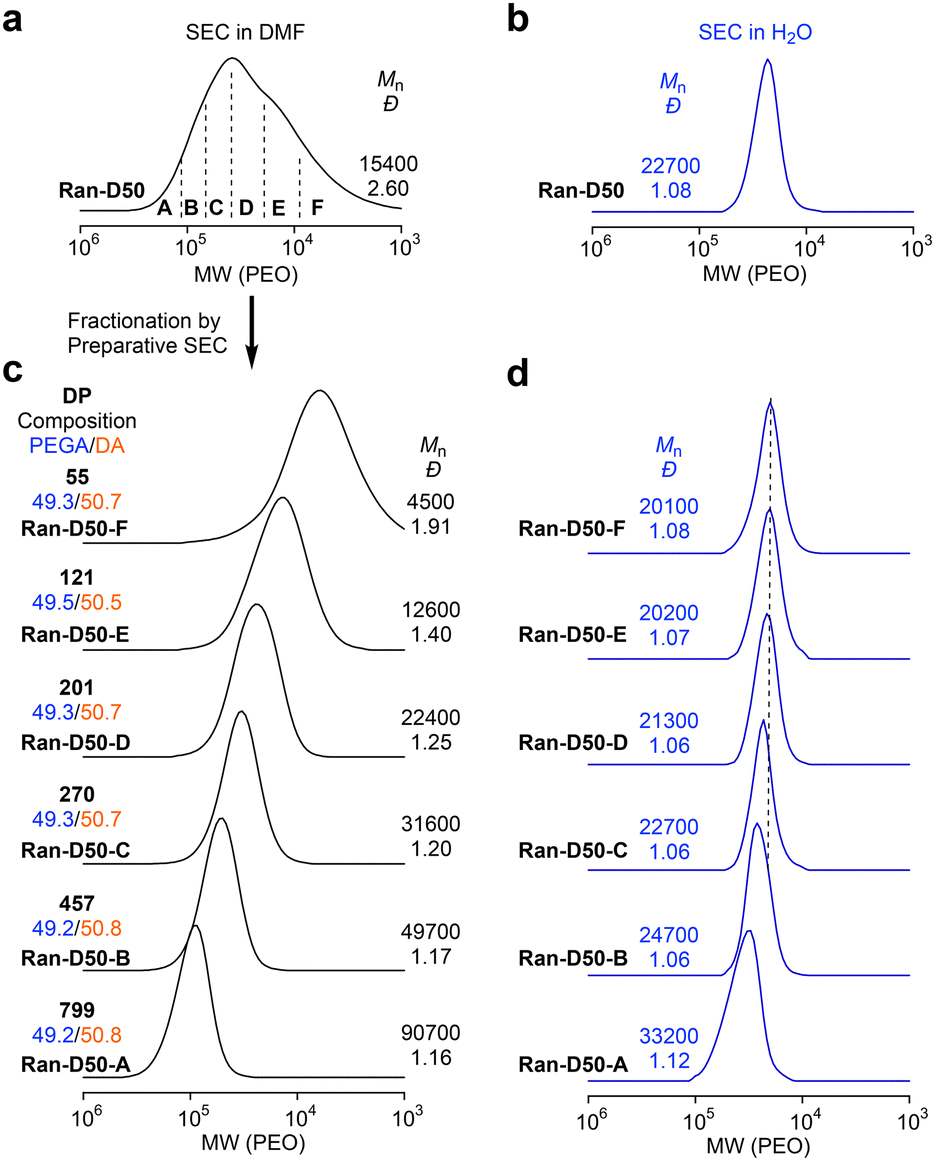 | ||
| Fig. 2 SEC curves of Ran-D50 in (a) DMF (10 mM LiBr) and (b) H2O (100 mM NaCl) with PEO standard calibration. Ran-D50 was fractionated into six samples with different molecular weights (A–F) by preparative SEC. (c and d) SEC curves of fractionated Ran-D50-A–F with different DPs in (c) DMF (10 mM LiBr) and (d) H2O (100 mM NaCl). The Mn and Đ values are determined with PEO standard calibration. | ||
Self-assembly of amphiphilic random copolymers into micelles in water
The self-assembly of amphiphilic random copolymers (Ran-D50, Ran-D50-Py or Np, Ran-D40-Py or Np) into micelles in water was evaluated using SEC-MALLS in H2O containing 100 mM NaCl as an eluent. The aqueous solutions of the copolymers were prepared as follows: in vials, the copolymers were dissolved in water at 25 °C (1 mg mL−1), giving transparent solutions. The solutions were sonicated at 25 °C for 3 min (sonicator: Branson, Bransonic 1510) and filtered through a poly(tetrafluoroethylene) membrane filter (0.45 μm, Merck Millipore) before analysis. The aqueous solutions were injected into the SEC system to determine Mn (apparent size), absolute weight-average molecular weight (Mw,H2O), and Đ of the polymer micelles in water (Tables S1–S4†). The apparent size of the micelles was evaluated with Mn determined by SEC in water with PEO calibration. The Mw,H2O of the micelles was determined by SEC-MALLS in water to estimate the aggregation number (Nagg) of the copolymers [Nagg = Mw,H2O (MALLS)/Mw,DMF (MALLS)]. The Đ of the micelles was determined by PEO calibration or MALLS.100–22700 g mol−1) and narrow dispersity (1.06–1.08) by PEO calibration, whereas Ran-D50-B and A with DP values more than 457 showed a shift of their SEC curves to high molecular weight with increasing DP. This result suggests that Ran-D50 with a DP smaller than 270 forms multi-chain micelles whose size is independent of the DP and Ran-D50 with a DP larger than at least about 450 mostly forms unimer micelles whose size increased with the DP. Fractionated Ran-D50-Py or Np with a DP smaller than approximately 270 also showed SEC curves with almost identical Mn (Fig. 3a and S11†). This supports the fact that a small amount (∼1 mol%) of Py or Np labels hardly affects the self-assembly behaviour of the copolymers and the apparent size of the resulting micelles. In contrast, the copolymer composition affected the self-assembly and apparent size of micelles. Typically, Ran-D40-Py-F–C with a DP smaller than 160 showed SEC curves with almost identical Mn (Fig. 3b and S12), whereas the Mn was smaller than that for constant size micelles of Ran-D50(-Py or Np). In addition, the threshold DP of the constant size micelles for Ran-D40-Py was smaller than that for Ran-D50-Py.
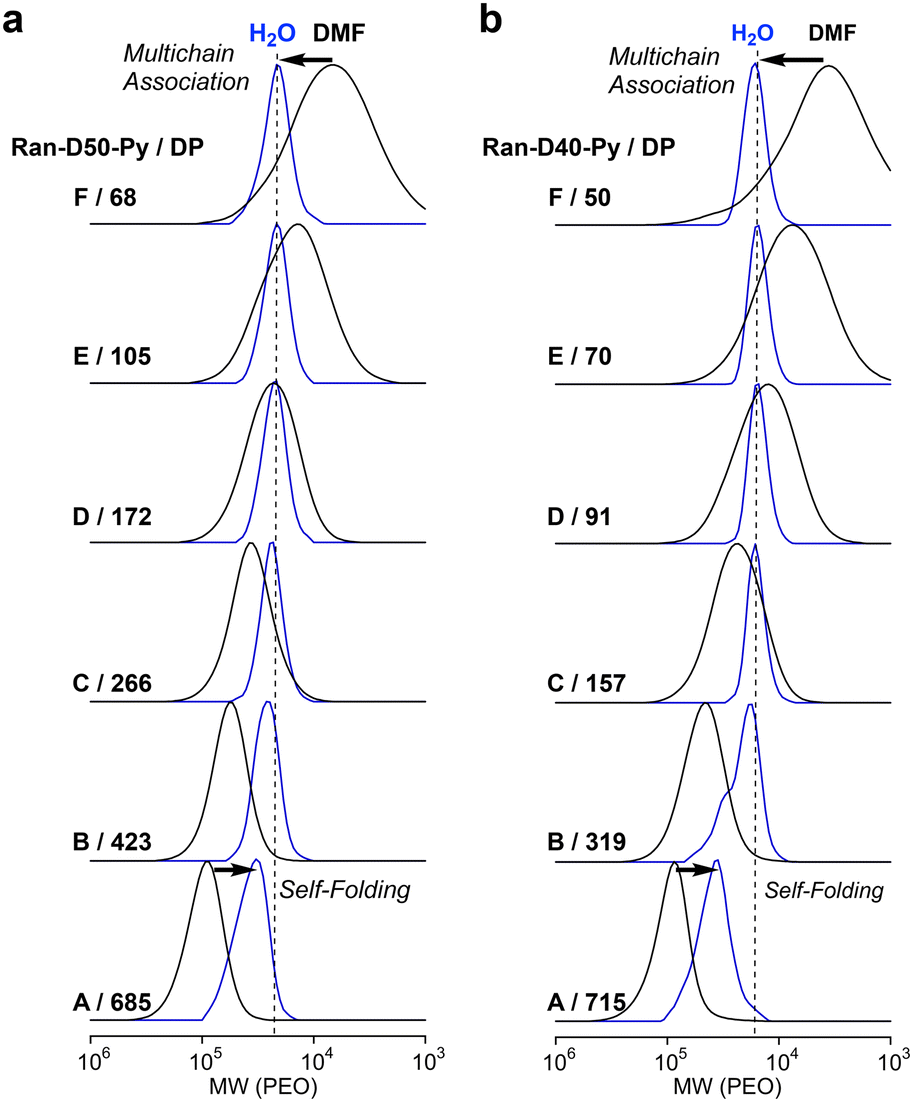 | ||
| Fig. 3 SEC curves of (a) Ran-D50-Py-A–F and (b) Ran-D40-Py-A–F with different molecular weights in DMF (black lines) and H2O (blue lines). | ||
000 g mol−1 and their Nagg values decreased from 6.6 to 1.9 with increasing DP (Fig. 4a). In contrast, the Mw,H2O for Ran-D50-B and A with a DP more than 457 increased with increasing DP and their Nagg values were close to 1. As confirmed by dynamic light scattering in water, the hydrodynamic radius (Rh) of Ran-D50-F–C micelles was also almost constant in the range of 5.7–6.1 nm, independent of the DP of the copolymers, whereas the Rh for Ran-D50-B and A micelles increased from 6.2 nm to 7.7 nm with increasing DP (Fig. S13 and Table S1†). These results demonstrate that, as implied by Mn (apparent size by PEO calibration), Ran-D50 copolymers have a threshold DP (DPth) of approximately 400 between multichain micelles and unimer micelles: (1) the copolymers with a DP smaller than that of DPth exclusively induce the intermolecular association of the polymer chains to form multichain micelles (Nagg > 2) with constant Mw,H2O and Rh, where the Nagg of the micelles decreases with increasing DP. (2) The copolymers with a DP larger than the DPth mostly induce self-folding into unimer micelles whose Mw,H2O and Rh increase with increasing DP. Py or Np-labeled Ran-D50-F–C with a DP smaller than DPth also formed multichain micelles with Mw,H2O (∼200000 g mol−1) that is close to those of non-labelled Ran-D50-F–C (Fig. 4b and c).
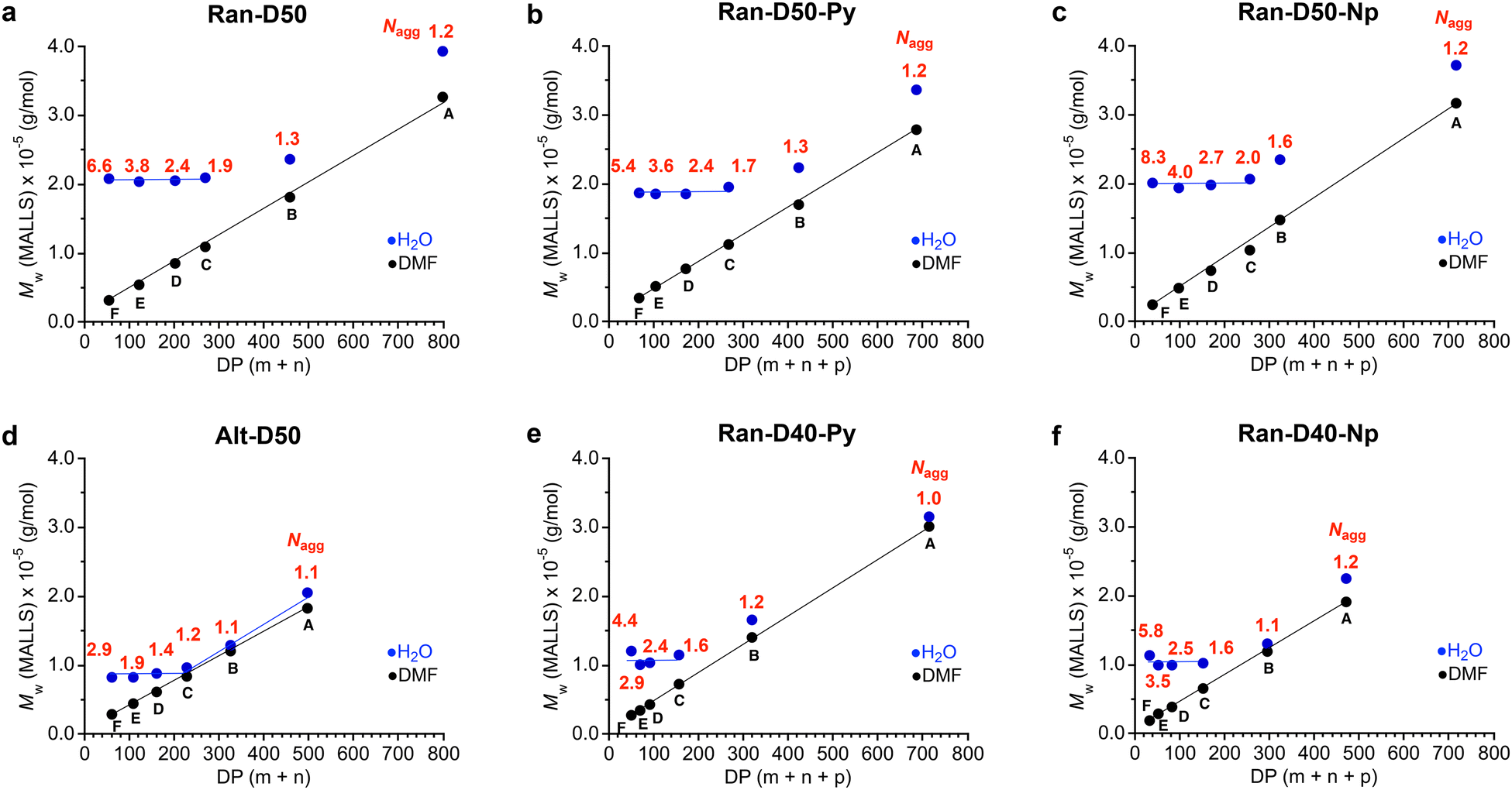 | ||
| Fig. 4 Absolute weight-average molecular weight (Mw, determined by SEC-MALLS) of (a) Ran-D50-A–F, (b) Ran-D50-Py-A–F, (c) Ran-D50-Np-A–F, (d) Alt-D50-A–F, (e) Ran-D40-Py-A–F, and (f) Ran-D40-Np-A–F in water (100 mM NaCl, blue) or DMF (10 mM LiBr, black) as a function of the DP. Aggregation number: Nagg = Mw,H2O/Mw,DMF. | ||
In contrast, Ran-D40-Py (or Np)-F–C with a DP smaller than about 160 formed multichain micelles with a constant Mw,H2O of approximately 100000 g mol−1 and Ran-D40-Py (or Np)-B and A with a DP larger than about 300 formed unimer micelles whose Mw,H2O increased with increasing DP (Fig. 4e and f). The constant Mw,H2O and DPth of their Ran-D40 multichain micelles were smaller than those of their Ran-D50 counterparts, indicating that more hydrophilic polymers form smaller micelles. This trend is consistent with the self-assembly of methacrylate-based amphiphilic random copolymers into micelles.35,39 Furthermore, the constant Mw,H2O of Alt-D50 multichain micelles (∼90000 g mol−1, Fig. 4d) was smaller than that of their Ran-D50 counterparts (∼200000 g mol−1). This is probably because the alternating copolymers have flexible vinyl ether backbones and have no consecutive sequence of dodecyl side chains to afford the efficient folding of polymer chains into more compact micelles.48
Critical micelle concentration of amphiphilic random or alternating copolymers
We examined the effects of the monomer sequence and DP (related to Nagg) on the CMC of amphiphilic random or alternating copolymers. Ran-D50 and Alt-D50, both of which have no fluorescent labels, were used. The CMC of the copolymers in water was determined by fluorescence spectroscopy with pyrene as follows:41,49,50 aqueous mixtures of the copolymers with different concentrations (1.0 × 10−4–1.0 × 10−1 mg mL−1) and a small amount of pyrene (5 × 10−7 M) were prepared, and the fluorescence spectra of the mixtures were measured at 25 °C (Fig. 5 and S15†). The emission intensity ratio of I1 (at 373 nm) and I3 (at 384 nm) [I1/I3] was plotted as a function of the polymer concentration. CMC was defined as a concentration at which I1/I3 starts to decrease with increasing polymer concentration (Fig. 5).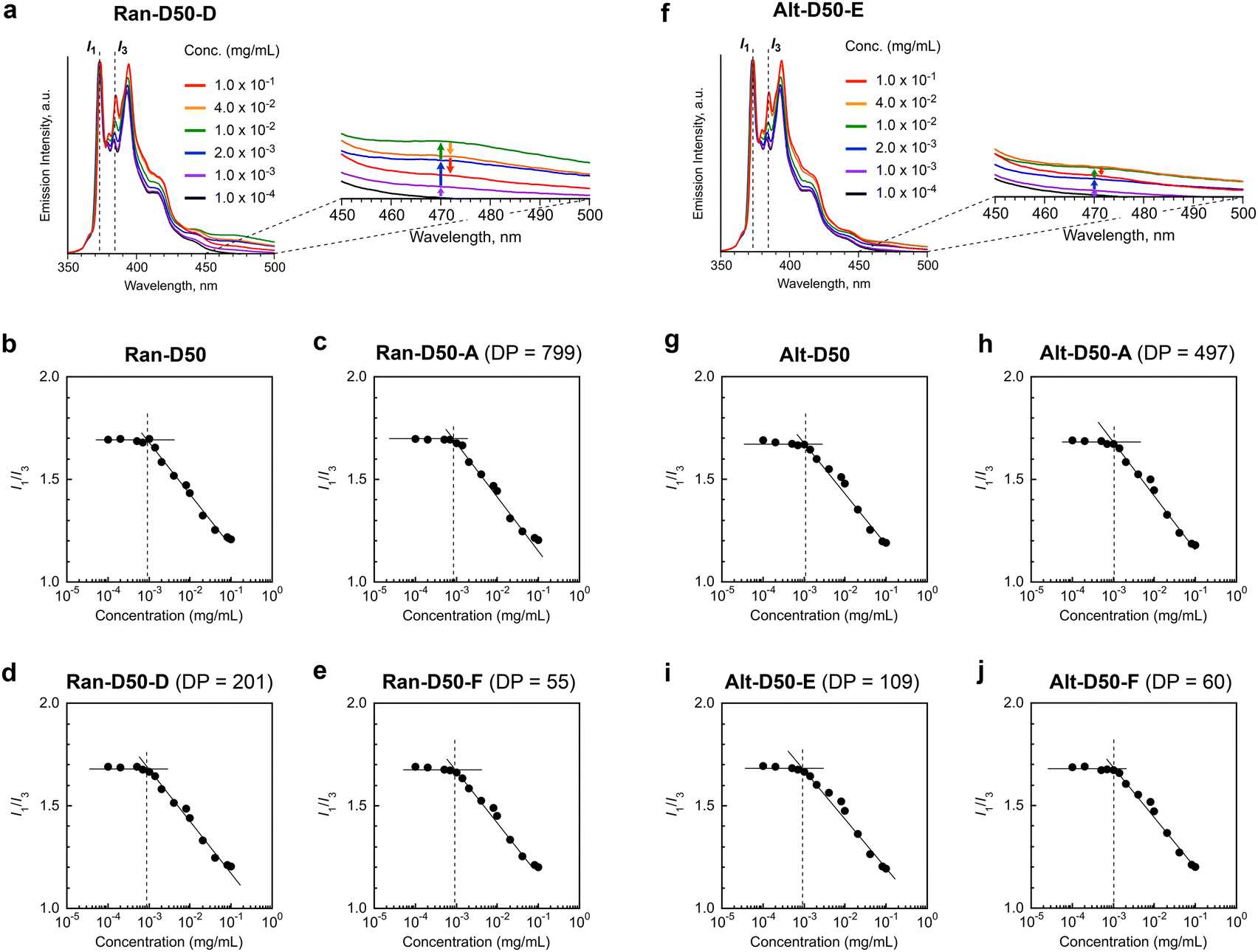 | ||
| Fig. 5 CMC of amphiphilic random or alternating copolymers in water. (a and f) Emission spectra of pyrene (5 × 10−7 M: normalized by I1) containing (a) Ran-D50-D and (f) Alt-D50-E in water. (b–e and g–j) I1/I3 plots as a function of the polymer concentration (1.0 × 10−4–1.0 × 10−1 mg mL−1) of (b) Ran-D50, (c) Ran-D50-A, (d) Ran-D50-D, (e) Ran-D50-F, (g) Alt-D50, (h) Alt-D50-A, (i) Alt-D50-E, and (j) Alt-D50-F. | ||
As shown in Fig. 5a, the I3 intensity (normalized by I1) for the aqueous solutions of Ran-D50-D with DP 201 was almost constant up to approximately 1 × 10−3 mg mL−1 and increased with increasing polymer concentration above 1 × 10−3 mg mL−1. From the intersection concentration of the constant or decreasing I1/I3 values, the CMC was determined to be approximately 1 × 10−3 mg mL−1 (Fig. 5d). Similarly, the CMC of Ran-D50 with broad dispersity, Ran-D50-A with a DP of 799, and Ran-D50-F with a DP of 55 was approximately 1 × 10−3 mg mL−1 (Fig. 5b, c and e). Though Ran-D50-A, D, and F form micelles with Nagg values of 1.2, 2.4, and 6.6 in water, respectively, the CMC was independent of their Nagg values. This is consistent with the fact that a Ran-D50 micelle with broad Nagg distribution due to broad dispersity (Đ = 2.60) also has a close CMC. The CMC of amphiphilic alternating copolymers (Alt-D50 with broad dispersity, Alt-D50-A with a DP of 497, Alt-D50-E with a DP of 109, and Alt-D50-F with a DP of 60) was also estimated to be 1 × 10−3 mg mL−1, independent of Nagg (Fig. 5f–j). These CMC values were close to those of poly(styrene-ethylene oxide) block copolymer micelles.50
In general, amphiphilic copolymers bearing hydrophilic and hydrophobic segments in water are placed at the air/water interface and dispersed as unimer chains or self-assemblies of multiple polymer chains in water.51,52 The three modes are dynamically exchanged in the equilibrium state, depending on the polymer concentration (Fig. 6a). At very low concentration, amphiphilic polymer chains are primarily located at the air/water interface, where the hydrophobic alkyl groups with low surface free energy are directed to the air. As the concentration increases, the amount of polymer chains at the interface increases. Once the interface is saturated, the polymer chains are not only dispersed as unimer chains in water but also form self-assemblies such as micelles via the association of hydrophobic groups above a concentration called CMC.
 | ||
| Fig. 6 (a) Equilibrium of amphiphilic random or alternating copolymers in water among three states: polymer chains placed at the air/water interface and unimer chains/micelles or multichain micelles dispersed in water. (b) Effects of concentration on the encapsulation of pyrene molecules into micelles in water during CMC measurements. | ||
The CMC of both the acrylate-random and acrylate/vinyl ether alternating copolymers (∼1.0 × 10−3 mg mL−1) was close to that of a methacrylate-random copolymer bearing PEG chains and dodecyl groups.41 This result importantly demonstrates that the CMC of their amphiphilic copolymers is mainly determined by the structure and composition of the side chains, and is independent of the backbone structures and monomer sequence. This is because the mass of copolymer chains potentially filling at the air/water interface and averaged hydrophobic/hydrophilic balance of the copolymer chains are independent of the DP and monomer sequence as far as the composition (the molar ratio of PEG and dodecyl groups) of the copolymers is identical. Another interesting finding is that unimer micelles of Ran-D50-A or Alt-D50-A also have a CMC of ∼1.0 × 10−3 mg mL−1 as determined by the fluorescence measurements using pyrene. This implies that the amphiphilic copolymers are primarily placed at the air/water interface below the CMC (<∼1.0 × 10−3 mg mL−1) and immediately form unimer micelles in water above the concentration.
The aqueous solutions of Ran-D50 and Alt-D50 further showed fluorescence intensity stemming from the excimer emission of pyrene at around 470 nm (I470) above their CMC (∼1.0 × 10−3 mg mL−1). However, the emission intensity changed, in response to the polymer concentration (Fig. 5a and f). For example, the I470 of Ran-D50-D increased with increasing concentration up to 1.0 × 10−2 mg mL−1 and again decreased with increasing concentration. This suggests that the number of pyrene molecules enclosed within each micelle changes as the number of micelles increases (Fig. 6b). When the polymer concentration is below the CMC, pyrene is molecularly dispersed in the water phase. This is also confirmed by the fact that the I1/I3 ratio of the aqueous solutions of pyrene containing the copolymers (below the CMC) is identical to that of the aqueous solution of pyrene alone (5 × 10−7 M, Fig. S15†). At a concentration slightly above the CMC, a few micelles formed in water enclose multiple pyrene molecules within the hydrophobic cores, leading to excimer emission. At a concentration much higher than the CMC, the pyrene molecules were fully dispersed within the multiple micelle cores, decreasing the excimer emission. These results indicate that amphiphilic random or alternating copolymer micelles can encapsulate hydrophobic molecules within their cores and are potentially applicable as nanocapsules.
Dynamic exchange of polymer chains between micelles
The dynamic properties of Ran-D50 or Ran-D40 micelles in water were investigated, focused on the effects of copolymer composition (dodecyl groups: 50 or 40 mol%), DP (related to Nagg), and temperature on the exchange of the acrylate-copolymer chains. The chain exchange was analyzed by FRET in the mixtures of a Py-labeled micelle and a Np-labeled micelle.41,42 The aqueous solution of a Py-labeled micelle was mixed with that of a Np-labeled micelle ([polymer] = 1.0 mg mL−1, Fig. 7a). Then, fluorescence measurement of the resulting mixture was immediately performed at various temperatures (10–35 °C), where the excitation wavelength for the Np units was set at 290 nm. The fluorescence intensity was monitored at 336 nm for the Np units (INp) and at 396 nm for the Py units (IPy). The Förster radius of Np and Py is 2.86 nm.30 Since the labeled Np or Py units are located within the hydrophobic cores of small micelles (Rh = ∼6 nm), the coexistence of Py-labeled copolymers and Np-labeled copolymers within single micelles can be evaluated by FRET.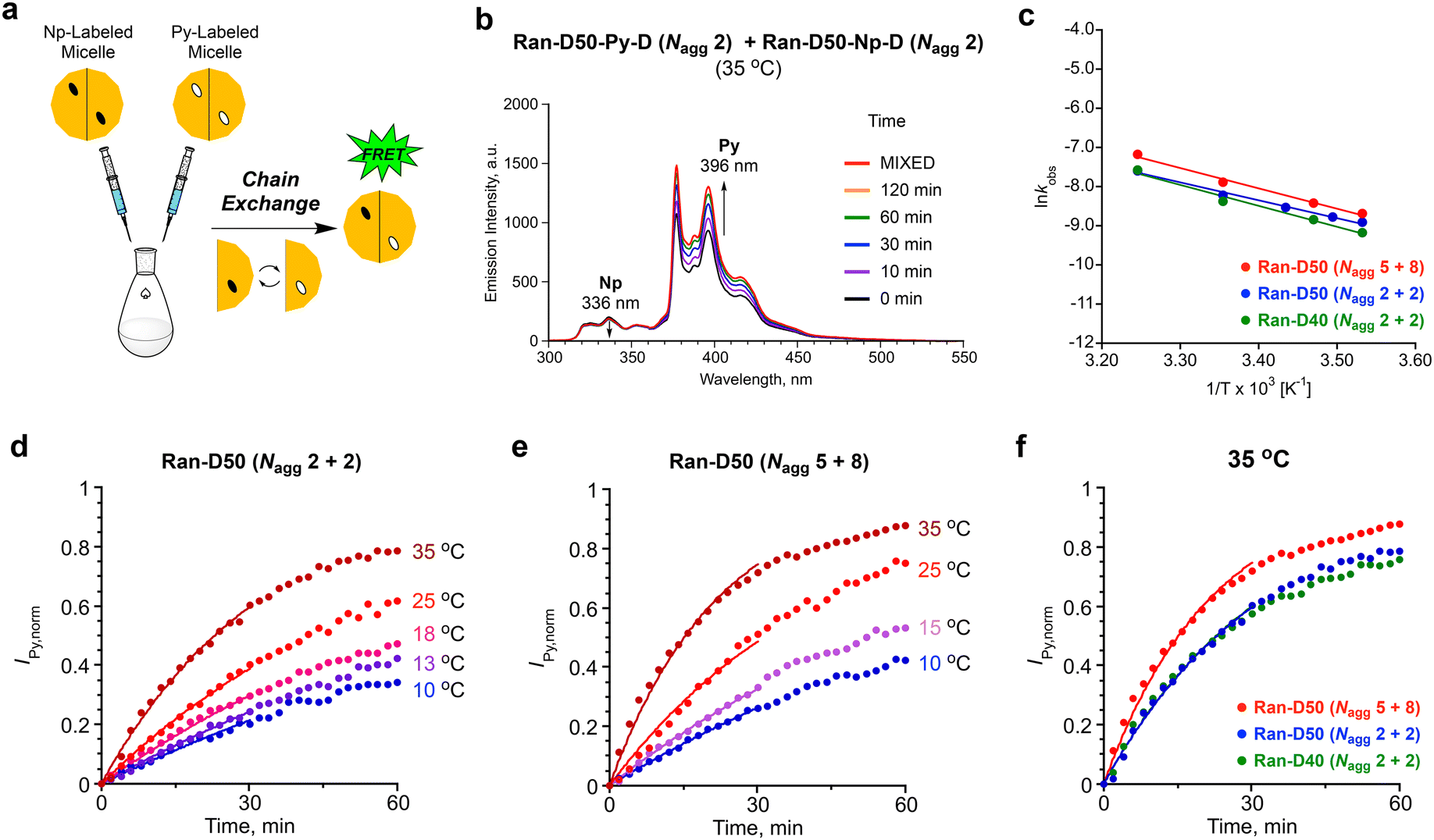 | ||
| Fig. 7 (a) Evaluation of polymer chain exchange between Py-labeled micelles and Np-labeled micelles by FRET. (b) Fluorescence spectra obtained from the aqueous mixture of a Ran-D50-Py-D micelle and a Ran-D50-Np-D micelle by excitation at 290 nm at 35 °C: [polymer] = 1.0 mg mL−1. The fluorescence intensity was monitored at 396 nm (IPy) and 336 nm (INp). (c) Arrhenius plots of kobs for the chain exchange of the mixtures of (blue) Ran-D50-Py-D (Nagg = 2) and Ran-D50-Np-D (Nagg = 2), (green) Ran-D40-Py-D (Nagg = 2) and Ran-D40-Np-D (Nagg = 2), and (red) Ran-D50-Py-F (Nagg = 5) and Ran-D50-Np-F (Nagg = 8). (d and e) Effect of temperature on the chain exchange: normalized IPy (IPy,norm) and fitting curves for the chain exchange of the binary mixtures of (d) a Ran-D50-Py-D micelle (Nagg = 2) and a Ran-D50-Np-D micelle (Nagg = 2) or (e) a Ran-D50-Py-F micelle (Nagg = 5) and a Ran-D50-Np-F micelle (Nagg = 8) in water at 10–35 °C. (f) Effect of Nagg and composition on the chain exchange at 35 °C: the mixtures of (blue) Ran-D50-Py-D (Nagg = 2) and Ran-D50-Np-D (Nagg = 2), (green) Ran-D40-Py-D (Nagg = 2) and Ran-D40-Np-D (Nagg = 2), and (red) Ran-D50-Py-F (Nagg = 5) and Ran-D50-Np-F (Nagg = 8). | ||
For example, in the mixture of a Ran-D50-Py-D (DP = 172, Nagg = 2) micelle and a Ran-D50-Np-D (DP = 170, Nagg = 2) micelle at 35 °C, IPy increased and INp slightly decreased with increasing measurement time (Fig. 7b). This indicates that FRET from the Np units to the Py units occurs with time progress: namely, the Np-labeled polymer chains are gradually mixed with their Py-labeled counterparts to form Np- and Py-mixed micelles in water. Additionally, the mixture of Ran-D40-Py-D (DP = 91, Nagg = 2) micelle and Ran-D40-Np-D (DP = 82, Nagg = 2) or that of Ran-D50-Py-F (DP = 68, Nagg = 5) and Ran-D50-Np-F (DP = 38, Nagg = 8) also showed similar increase of IPy and decrease of INp with time progress (Fig. S16–S18†). The chain exchange kinetics were thus evaluated from the IPy values of the mixtures normalized over time {IPy(t)} using eqn (1):24
 | (1) |
In all the cases, IPy,norm increased with time, and the increase of IPy,norm turned fast on increasing temperature (Fig. 7). This indicates that the exchange of the polymer chains is promoted upon heating the solutions. The initial IPy,norm values were fitted by eqn (1) to determine the apparent rate constant (kobs) for the exchange of polymer chains between their micelles. The kobs for the mixture of a Ran-D50-Py-D (DP = 172, Nagg = 2) micelle and a Ran-D50-Np-D (DP = 170, Nagg = 2) micelle at 35 °C (3.0 × 10−2 min−1) was almost the same as that for a Ran-D40-Py-D (DP = 91, Nagg = 2) micelle and a Ran-D40-Np-D (DP = 82, Nagg = 2) micelle (3.0 × 10−2 min−1), indicating that the 10 mol% difference in copolymer composition had little effect on the apparent exchange rate of polymer chains. In contrast, the kobs for the mixture of a Ran-D50-Py-F (DP = 68, Nagg = 5) micelle and a Ran-D50-Np-F (DP = 38, Nagg = 8) micelle at 35 °C (4.6 × 10−2 min−1) was larger than that for the mixtures of micelles with Nagg = 2. This means that the exchange rate of polymer chains increased with increasing Nagg;42 namely, small DP polymers are more easily exchanged between micelles. In general, exchange of polymer chains between micelles occurs via unimer release/insertion, fragmentation of micelles, and micelle/micelle collision processes.24,27,43 Since the CMC of the random copolymers was independent of the DP, faster chain exchange of micelles with a large Nagg may be promoted by fragmentation and/or collision processes of their micelles.
The kobs values were applied to Arrhenius plots (Fig. 7c). The activation energy (Ea) for the chain exchange processes of Ran-D50-Py-D (Nagg = 2)/Ran-D50-Np-D (Nagg = 2) micelles, Ran-D40-Py-D (Nagg = 2)/Ran-D40-Np-D (Nagg = 2) micelles, and Ran-D50-Py-F (Nagg = 5)/Ran-D50-Np-F (Nagg = 8) micelles was estimated to be 35, 47, and 43 kJ mol−1, respectively. The Ea values were almost independent of the composition and DP of their copolymers but significantly lower than the Ea for the chain exchange of methacrylate-based PEGMA/DMA (50/50 mol%) random copolymer micelles carrying identical PEG chains and dodecyl groups (141 kJ mol−1, Fig. 1b).53 This notably demonstrates that the flexible polyacrylate backbones easily induce the exchange of polymer chains between micelles with less temperature effects although rigid polymethacrylate backbones have a higher energy barrier for the exchange of polymer chains. In fact, the acrylate-random copolymer micelles induced chain exchange even at 10 °C (Fig. 7), whereas their methacrylate-random copolymer counterparts required a temperature at least above 25 °C for efficient chain exchange.53 We thus revealed that the flexible acrylate random copolymers were effective for the design of size- and aggregation number-controlled micelles with low CMC yet dynamic chain exchange properties even at low temperature.
Conclusions
In summary, we investigated the self-assembly of acrylate-based amphiphilic random copolymers into micelles in water, focusing on the micelle size and aggregation number, CMC, and dynamic exchange behaviour of polymer chains. Multichain micelles with almost constant size (Mw and Rh) were formed below a threshold DPth and unimer micelles were mainly formed above the DPth. The constant Mw and DPth for the multichain micelles increased with the content of hydrophobic dodecyl groups. The CMC of the random copolymers in water was approximately 1 × 10−3 mg mL−1 regardless of the DP and monomer sequence, indicative of the stable formation of micelles at low concentration. Uniquely, the flexible acrylate random copolymer micelles induced the exchange of polymer chains in water even at a low temperature such as 10 °C, though relatively rigid methacrylate random copolymers required at least 25 °C for efficient chain exchange. The chain exchange was promoted by increasing temperature and Nagg. Thus, the self-assembly of flexible acrylate-based random copolymers in water is suitable for the design of size-controlled micelles with dynamic chain exchange properties even at low temperature; such dynamic yet precise self-assemblies would be useful as encapsulation/release materials or delivery vessels for various applications.Data availability
The data supporting this study have been included as part of the ESI.† Experimental details of the synthesis and characterization of polymers and SEC, NMR, DLS, and fluorescence spectroscopy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Japan Society for the Promotion of Science KAKENHI grants (JP19K22218, JP20H02787, JP20H05219, JP22H04539, JP23H02008, and JP23K26701), JST PRESTO grant number JPMJPR24M6, Ogasawara Foundation for the Promotion of Science & Engineering, the Noguchi Institute, the Institute for Chemical Fibers, Japan, Iketani Science and Technology Foundation, Kurita Water and Environment Foundation.References
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS PubMed.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- Z. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98–101 CrossRef CAS PubMed.
- R. K. O'reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2012, 45, 2–19 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- Y. Lu, J. Lin, L. Wang, L. Zhang and C. Cai, Chem. Rev., 2020, 120, 4111–4140 CrossRef CAS PubMed.
- T. Terashima, Polym. J., 2014, 46, 664–673 CrossRef CAS.
- S. Mavila, I. Rozenberg and N. G. Lemcoff, Chem. Sci., 2014, 5, 4196–4203 RSC.
- M. Gonzalez-Burgos, A. Latorre-Sanchez and J. A. Pomposo, Chem. Soc. Rev., 2015, 44, 6122–6142 RSC.
- P. W. Roesky and C. Barner-kowollik, Polym. Chem., 2015, 6, 4358–4365 RSC.
- C. Song, L. Li, L. Dai and S. Thayumanavan, Polym. Chem., 2015, 6, 4828–4834 RSC.
- A. M. Hanlon, C. K. Lyon and E. B. Berda, Macromolecules, 2016, 49, 2–14 CrossRef CAS.
- S. Mavila, O. Eivgi, I. Berkovich and N. G. Lemcoff, Chem. Rev., 2016, 116, 878–961 CrossRef CAS PubMed.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2016, 37, 29–46 CrossRef CAS PubMed.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- M. I. Gibson and R. K. O'reilly, Chem. Soc. Rev., 2013, 42, 7204–7213 RSC.
- A. P. Esser-Kahn, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Macromolecules, 2011, 44, 5539–5553 CrossRef CAS.
- Z. Jiang, H. Liu, H. He, A. E. Ribbe and S. Thayumanavan, Macromolecules, 2020, 53, 2713–2723 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64, 37–48 CrossRef.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
- Y. Wang, C. M. Kausch, M. Chun, R. P. Quirk and W. L. Mattice, Macromolecules, 1995, 28, 904–911 CrossRef CAS.
- P. Bhargava, J. X. Zheng, P. Li, R. P. Quirk, F. W. Harris and S. Z. D. Cheng, Macromolecules, 2006, 39, 4880–4888 CrossRef CAS.
- R. Lund, L. Willner, J. Stellbrink, P. Lindner and D. Richter, Phys. Rev. Lett., 2006, 96, 1–4 CrossRef PubMed.
- T. P. Lodge, C. L. Seitzinger, S. C. Seeger, S. Yang, S. Gupta and K. D. Dorfman, ACS Polym. Au, 2022, 2, 397–416 CrossRef CAS PubMed.
- H. Yamamoto and Y. Morishima, Macromolecules, 1999, 32, 7469–7475 CrossRef CAS.
- H. Yamamoto, I. Tomatsu, A. Hashidzume and Y. Morishima, Macromolecules, 2000, 33, 7852–7861 CrossRef CAS.
- S. Yusa, A. Sakakibara, T. Yamamoto and Y. Morishima, Macromolecules, 2002, 35, 10182–10188 CrossRef CAS.
- T. Kawata, A. Hashidzume and T. Sato, Macromolecules, 2007, 40, 1174–1180 CrossRef CAS.
- K. Dan, N. Bose and S. Ghosh, Chem. Commun., 2011, 47, 12491–12493 RSC.
- L. Li, K. Raghupathi, C. Song, P. Prasad and S. Thayumanavan, Chem. Commun., 2014, 50, 13417–13432 RSC.
- R. Nakahata and S. I. Yusa, Langmuir, 2019, 35, 1690–1698 CrossRef CAS PubMed.
- Y. Hirai, T. Terashima, M. Takenaka and M. Sawamoto, Macromolecules, 2016, 49, 5084–5091 CrossRef CAS.
- G. Hattori, Y. Hirai, M. Sawamoto and T. Terashima, Polym. Chem., 2017, 8, 7248–7259 RSC.
- G. Hattori, M. Takenaka, M. Sawamoto and T. Terashima, J. Am. Chem. Soc., 2018, 140, 8376–8379 CrossRef CAS PubMed.
- Y. Kimura, T. Terashima and M. Sawamoto, Macromol. Chem. Phys., 2017, 218, 1700230 CrossRef.
- S. Imai, Y. Hirai, C. Nagao, M. Sawamoto and T. Terashima, Macromolecules, 2018, 51, 398–409 CrossRef CAS.
- M. Shibata, M. Matsumoto, Y. Hirai, M. Takenaka, M. Sawamoto and T. Terashima, Macromolecules, 2018, 51, 3738–3745 CrossRef CAS.
- S. Imai, M. Takenaka, M. Sawamoto and T. Terashima, J. Am. Chem. Soc., 2019, 141, 511–519 CrossRef CAS PubMed.
- M. Hibino, K. Tanaka, M. Ouchi and T. Terashima, Macromolecules, 2022, 55, 178–189 CrossRef CAS.
- M. Hibino, S. I. Takata, K. Hiroi, H. Aoki and T. Terashima, Macromolecules, 2023, 56, 2955–2964 CrossRef CAS.
- D. Taura, A. Hashidzume, Y. Okumura and A. Harada, Macromolecules, 2008, 41, 3640–3645 CrossRef CAS.
- D. Taura, Y. Taniguchi, A. Hashidzume and A. Harada, Macromol. Rapid Commun., 2009, 30, 1741–1744 CrossRef CAS PubMed.
- M. Ueda, A. Hashidzume and T. Sato, Macromolecules, 2011, 44, 2970–2977 CrossRef CAS.
- K. Uramoto, R. Takahashi, K. Terao and T. Sato, Polym. J., 2016, 48, 863–867 CrossRef CAS.
- H. Kono, M. Hibino, D. Ida, M. Ouchi and T. Terashima, Macromolecules, 2023, 56, 6086–6098 CrossRef CAS.
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS.
- M. Wilhelm, C. Le Zhao, Y. Wang, R. Xu, M. A. Winnik, J. L. Mura, G. Riess and M. D. Croucher, Macromolecules, 1991, 24, 1033–1040 CrossRef CAS.
- P. Raffa, D. A. Z. Wever, F. Picchioni and A. A. Broekhuis, Chem. Rev., 2015, 115, 8504–8563 CrossRef CAS PubMed.
- A. Goswami, G. Verma, P. A. Hassan and S. S. Bhagwat, J. Dispersion Sci. Technol., 2015, 36, 885–891 CrossRef CAS.
- R. Kanno, H. Kono, A. Ryoki, M. Ouchi and T. Terashima, J. Am. Chem. Soc., 2024, 146, 30848–30859 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of the synthesis and characterization of polymers, and SEC, NMR, DLS, and fluorescence spectroscopy. See DOI: https://doi.org/10.1039/d4py01272k |
| This journal is © The Royal Society of Chemistry 2025 |