
Towards degradable and functionalizable polymers: alternating ring-opening metathesis copolymerization of oxanorbornadiene dicarboxylate and 2,3-dihydrofuran†
Tarek
Ibrahim
 ,
Kaia
Kendzulak
,
Syrena
Carver
,
Tamara
Perez
and
Hao
Sun
*
,
Kaia
Kendzulak
,
Syrena
Carver
,
Tamara
Perez
and
Hao
Sun
*
Department of Chemistry and Chemical & Biomedical Engineering, Tagliatela College of Engineering, University of New Haven, West Haven, CT 06516, USA. E-mail: hasun@newhaven.edu
First published on 25th March 2025
Abstract
Here we report the design and synthesis of acid-degradable and functionalizable polymers via alternating ring-opening metathesis copolymerization of oxanorbornadiene dicarboxylate and 2,3-dihydrofuran. The resulting polymers can undergo post-polymerization modification through aza-Michael and thia-Michael additions, leading to a diverse set of degradable polymer structures with various functional groups and tunable properties.
Olefin metathesis polymerization techniques have recently emerged as a powerful synthetic approach for creating degradable polymer materials with enhanced sustainability and eco-friendliness.1,2 Moreover, the degradability of these novel polyolefin materials makes them highly promising for biomedical applications, such as stimuli-responsive drug delivery systems.3–5 To date, a library of cyclic olefin monomers containing degradable groups (termed degradable monomers) have been designed to incorporate degradable moieties into the polymer backbones, giving rise to polyesters,6,7 polyacetals,8–11 poly(silyl ether)s,3,12 poly(diazanorbornene)s,13 polyoxazinones,4,14 polyphosphoramidates,5,15 poly (phosphonate)s,16 polydisulfides,17,18 and poly(enol ether)s.19–26 The properties of these degradable polymers are tunable by either modifying the functional groups on the degradable monomers,3 or through a copolymerization strategy that combines degradable monomers with non-degradable monomers bearing different functional groups.27 Nevertheless, gaining access to a series of degradable polymers with different functional groups typically requires the synthesis and purification of multiple monomers, a process that can be cumbersome and inefficient.
Compared to the monomer modification approach (vide supra), post-polymerization modification (PPM) enables the functionalization of a pre-synthesized polymer, thereby providing access to a diverse set of polymer structures without the need to synthesize various monomers.28 Recent studies by Kiessling and Gutekunst have demonstrated degradable and functionalizable polyolefins through the design of a single monomer that contains both a degradable group and a functional handle for post-polymerization modification (Fig. 1).4,9 While click-type PPM has proven highly efficient in these pioneering works, multi-step monomer synthesis is still required due to the complexity of the monomer structures (Fig. 1A and B).
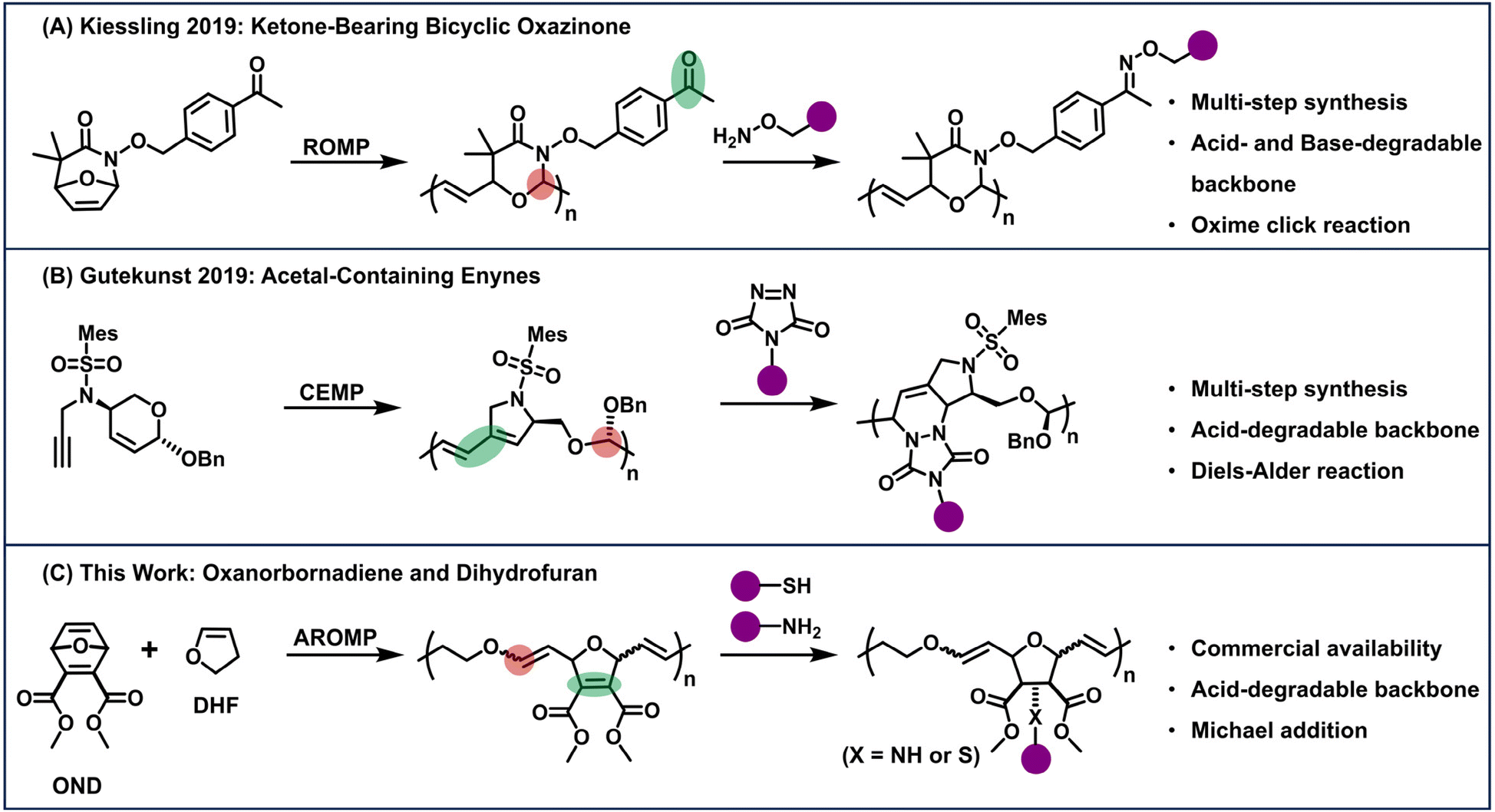 | ||
| Fig. 1 Degradable and functionalizable polymers via olefin metathesis polymerization strategy. (A) ROMP of ketone-bearing bicyclic oxazinone followed by an oxime click reaction. (B) Cascade enyne metathesis polymerization (CEMP) of acetal-containing enyne monomers, followed by Diels–Alder reaction. (C) This work: alternating ring-opening metathesis copolymerization (AROMP) of oxanorbornadiene dicarboxylate and 2,3-dihydrofuran. The resulting polymers are capable of post-polymerization modification via Michael addition. | ||
With the goal of reducing the synthetic effort in achieving degradable and functionalizable polymers, we reasoned that alternating ring-opening metathesis copolymerization (AROMP) of a degradable monomer and a functional comonomer would lead to a target polymer structure, where each repeating unit contains both a degradable linkage and a functional handle (Fig. 1C). 2,3-Dihydrofuran (DHF) was chosen as the degradable monomer due to its commercial availability and unique reactivity, which allows it to form alternating copolymers with various comonomers.20–24 Moreover, we selected oxanorbornadiene dicarboxylate (OND) as the functional comonomer to pair with DHF for AROMP. Notably, OND is also commercially available and contains an activated alkene group that can serve as a Michael acceptor.29
The ROMP reactivity of OND was first investigated in the presence of second-generation Grubbs catalyst (G2), with an initial OND concentration of 0.15 M (section 3.2 in ESI†). After 24 hours, only 14% of OND was converted into polymer, suggesting its low activity in homopolymerization. Next, we performed ring-opening metathesis copolymerization of OND and DHF, using the same initial OND concentration (0.15 M) as in its homopolymerization. The DHF-to-OND ratio was set at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to further suppress the homoaddition of OND. Based on the copolymerization results (Table S1†), both G2 and third-generation Grubbs catalyst (G3) were effective in catalyzing the copolymerization of OND and DHF, showing OND conversions over 75%. In particular, quantitative monomer conversions of OND were achieved when G2 was used in all cases (entries 4–6 in Table S1†). Given that the homoaddition of OND was sluggish, the high conversions of OND observed in the copolymerization study can be attributed to its heteroaddition with DHF.
:1 to further suppress the homoaddition of OND. Based on the copolymerization results (Table S1†), both G2 and third-generation Grubbs catalyst (G3) were effective in catalyzing the copolymerization of OND and DHF, showing OND conversions over 75%. In particular, quantitative monomer conversions of OND were achieved when G2 was used in all cases (entries 4–6 in Table S1†). Given that the homoaddition of OND was sluggish, the high conversions of OND observed in the copolymerization study can be attributed to its heteroaddition with DHF.
To evaluate the degree of alternation, we analyzed the copolymer structure using NMR spectroscopy (Fig. 2A and Fig. S1–3†). As shown in 1H NMR analysis (Fig. 2A), the characteristic olefinic proton signals of polyDHF (highlighted in blue) are absent in the 1H NMR spectrum of copolymer poly(OND-alt-DHF), indicating that no homoaddition of DHF took place during the polymerization. In addition, the olefinic proton signal from polyOND (highlighted in green) is notably reduced in the copolymer, indicating suppression of the homoaddition of OND throughout the AROMP process. The 13C NMR data further corroborate these findings by showing the absence of alkene carbon signals of polyDHF in the copolymer's 13C NMR spectrum (Fig. S2†). Critically, the integration of proton signals from the DHF and OND repeating units reveals high degrees of alternation (>98% alternating dyads) in all the poly(OND-alt-DHF) copolymers, demonstrating an exceptional sequence control in the copolymer structure (Fig. S3 and Table S1†).
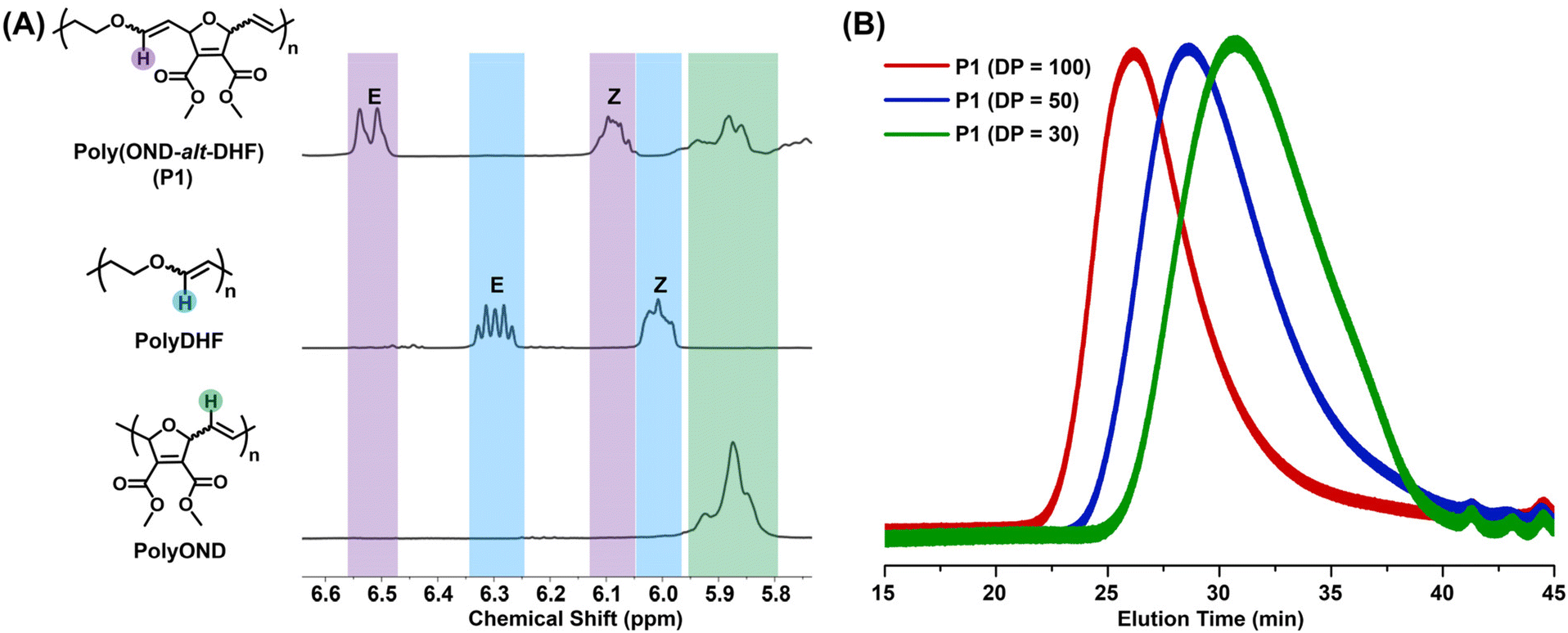 | ||
| Fig. 2 Characterization of the alternating copolymers. (A) Partial 1H NMR spectra of poly(OND-alt-DHF)100, polyDHF, and polyOND. (B) Size exclusion chromatography traces of poly(OND-alt-DHF) with varying degrees of polymerization (DP). These copolymers were prepared using G2. | ||
Size exclusion chromatography (SEC) analysis further shows that the molecular weights of alternating copolymers can be tuned by adjusting the ratio of monomer to catalyst (Fig. 2B, Fig. S4 and Table S1†). While the molecular weights of copolymers made with G2 or G3 are similar at the same monomer-to-catalyst ratio, the copolymers made with G2 exhibit relatively narrower molecular weight distributions compared to those made with G3, suggesting its better control over the AROMP of OND and DHF (Table S1†).
Since each OND repeating unit contains two electron-withdrawing ester groups attached to the alkene, we hypothesized that these activated alkenes could act as functional handles for post-polymerization modifications via Michael addition reactions. Moreover, poly(enol ether)s are known to be stable under basic conditions.27 Therefore, the backbone enol ether groups would remain unreactive towards amines. To verify this hypothesis, we evaluated the Michael addition efficiency of poly(OND-alt-DHF)50 (denoted as P1, entry 5 in Table S1†) using a wide range of nucleophiles, including primary amines and thiols (Fig. 3). Through aza-Michael addition, primary amines with various functional groups were attached to the polymer side chains, resulting in P2, P3, and P4. 1H NMR analysis shows that the aza-Michael addition efficiency of ethanolamine (50%) was markedly higher than that of n-butyl amine (22%) and benzylamine (16%) (Fig. S5–7†). This difference is likely due to the lower steric hindrance encountered by ethanolamine during the Michael addition process, compared to n-butylamine and benzylamine. Furthermore, thia-Michael addition was employed to broaden the scope of Michael donors for the post-polymerization modification. The addition of 1-thioglycerol and benzyl mercaptan to P1 led to P5 (54%) and P6 (33%), respectively (Fig. S8 and S9†).
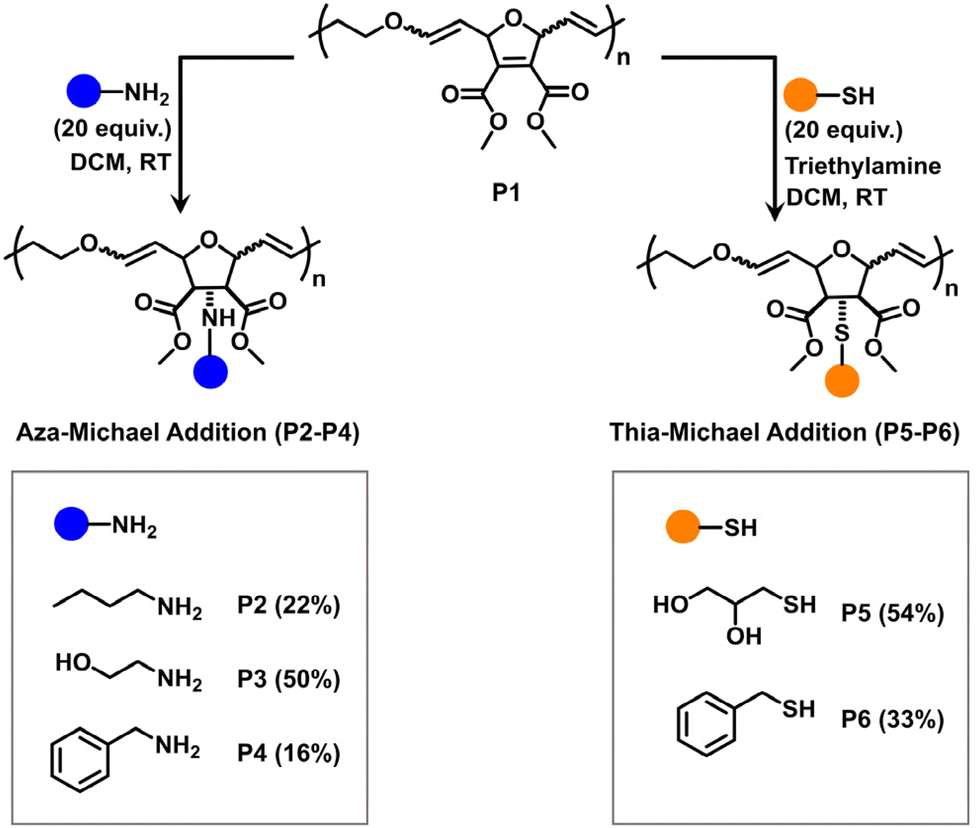 | ||
| Fig. 3 Post-polymerization modification of poly(OND-alt-DHF) (P1). Both aza-Michael addition and thia-Michael addition were employed to functionalize P1 with various nucleophiles. | ||
To shed light on the chain integrity during the PPM process, SEC was used to monitor changes in the polymer chain size (Fig. S10†). While no aldehyde signals were detectable in the 1H NMR analysis of P2–P6, a modest shift to longer elution times (indicating lower molecular weights) was observed in the SEC traces of these functional polymers. This slight degradation is presumably due to the acidity of adventitious ammonium formed during the Michael addition, as observed by the Xia group in their norbornene-DHF copolymer system.27
To elucidate the impact of functional groups on the thermal properties of polymers, we performed thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) on P1–P6. As shown in the TGA thermograms, all the polymers demonstrated excellent thermal stability, with decomposition temperatures (Td,5%) ranging from 228 to 280 °C (Fig. S11†). Because of the thermally reversible nature of aza-Michael and thia-Michael adducts,30,31P2–P6 exhibited slightly lower Td,5% values compared to P1.
DSC further revealed the glass transition temperatures (Tg) of the alternating copolymers (Fig. 4 and Fig. S12–14†). The Tg values of P2, P4, and P6 are similar to that of P1, due to their modest degrees of functionalization (16–33%) (Fig. S12–14†). Interestingly, P3 and P5, which contain abundant side-chain hydroxyl groups, displayed notably higher Tg values than P1 (Fig. 4). The increased glass transition temperatures observed in P3 and P5 can be ascribed to their ability to form hydrogen bonds, which enhance the intermolecular interactions and thus reduce the molecular mobility of the polymer chains.32,33
 | ||
| Fig. 4 Differential scanning calorimetry thermograms of P1, P3, and P5. Glass transition temperatures (Tg) were determined by identifying the midpoint of the sigmoidal change in heat capacity. | ||
To evaluate the mechanical properties of the investigated polymers for potential industrial applications, we conducted tensile tests on the original polymer (P1, DP = 100) and the modified polymer (P5). As shown in Fig. 5, P1 shows a low Young's modulus (6.6 MPa), an ultimate tensile strength (UTS) of 4.6 MPa, and an elongation at break exceeding 100%. By contrast, the hydroxyl-functionalized P5 exhibits limited stretchability (elongation at break of 15%), but significantly enhanced mechanical properties, including a Young's modulus of 163 MPa and a UTS of 35.2 MPa. These properties are of the same magnitude as those of commercial polystyrene, which has a Young's modulus of 693 MPa and a UTS of 41.2 MPa (Fig. 5).
 | ||
| Fig. 5 Tensile properties of polymers. Representative stress–strain curves of P1, P5, and commercial polystyrene at room temperature. | ||
Finally, the degradability of the alternating copolymers was examined. Poly(enol ether)s are well known for their acid-sensitivity.19,34 Since the polymers developed in this study contain an enol ether structure in every repeating unit, they were expected to be fully degradable under acidic conditions. In a proof-of-concept degradation study, P1, P2, and P5 were treated with hydrochloric acid in a THF solution (section 3.6 in ESI†). In all cases, polymers decomposed into small fragments (<500 g mol−1) upon the acid treatment (Fig. 6 and Fig. S15†). This result is in good agreement with previous degradation studies of DHF-based copolymers in the literature.22,23,27
 | ||
| Fig. 6 Degradation study of alternating copolymers. (A) Schematic illustration of the acid-promoted degradation of P1. (B) SEC traces of P1 as a function of the degradation time. (C) SEC traces of P5 as a function of degradation time. The concentrations of HCl and water in THF are 0.02 and 1.1 M, respectively. | ||
Conclusions
In summary, we present a novel synthetic approach to degradable and functionalizable polymer materials via AROMP of two commercially available monomers: OND and DHF. High degrees of alternating dyads (>98%) were achieved in the copolymers of OND and DHF, incorporating both a degradable group and a functional handle in each repeating unit. The resulting copolymers can be further modified through Michael additions of amines and thiols, yielding degradable polymers with diverse functional groups and tunable properties. Given the widespread applications of degradable polymers in fields such as lithography and biomedicine, we envision that the polymers developed in this work will give rise to a new class of degradable and functional materials. Importantly, the ease of polymer synthesis and the wide availability of the investigated monomers will facilitate their scalability and applications.Author contributions
Tarek Ibrahim: data curation, formal analysis, investigation, writing – original draft preparation. Kaia Kendzulak: data curation, investigation. Syrena Carver: data curation, investigation. Tamara Perez: data curation, investigation. Hao Sun: conceptualization, supervision, funding acquisition, methodology, project administration, resources, visualization, writing – original draft preparation, writing – review and editing.Data availability
All the data relevant to the current submission has been collated in the ESI.† Raw data, such as NMR, SEC, TGA, and DSC data can be obtained upon request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by a LEAPS-MPS award from the National Science Foundation under Grant No. [CHE-2316842].References
- J. Xu and N. Hadjichristidis, Prog. Polym. Sci., 2023, 139, 101656 CrossRef CAS.
- H. Sun, Y. Liang, M. P. Thompson and N. C. Gianneschi, Prog. Polym. Sci., 2021, 120, 101427 CAS.
- P. Shieh, H. V. Nguyen and J. A. Johnson, Nat. Chem., 2019, 11, 1124–1132 CrossRef CAS PubMed.
- J. M. Fishman, D. B. Zwick, A. G. Kruger and L. L. Kiessling, Biomacromolecules, 2019, 20, 1018–1027 CrossRef CAS PubMed.
- Y. Liang, H. L. Sullivan, K. Carrow, J. M. Mesfin, J. Korpanty, K. Worthington, C. Luo, K. L. Christman and N. C. Gianneschi, J. Am. Chem. Soc., 2023, 145, 11185–11194 CrossRef CAS PubMed.
- W. R. Gutekunst and C. J. Hawker, J. Am. Chem. Soc., 2015, 137, 8038–8041 CrossRef CAS PubMed.
- T. G. Hsu, J. Zhou, H. W. Su, B. R. Schrage, C. J. Ziegler and J. Wang, J. Am. Chem. Soc., 2020, 142, 2100–2104 CrossRef CAS PubMed.
- T. Debsharma, F. N. Behrendt, A. Laschewsky and H. Schlaad, Angew. Chem., Int. Ed., 2019, 58, 6718–6721 CrossRef CAS PubMed.
- L. B. Fu, X. L. Sui, A. E. Crolais and W. R. Gutekunst, Angew. Chem., Int. Ed., 2019, 58, 15726–15730 CrossRef CAS PubMed.
- B. R. Elling, J. K. Su and Y. Xia, ACS Macro Lett., 2020, 9, 180–184 CrossRef CAS PubMed.
- F. O. Boadi, J. Zhang, X. Yu, S. Bhatia and N. S. Sampson, Macromolecules, 2020, 53, 5857–5868 CrossRef CAS PubMed.
- P. Shieh, W. Zhang, K. E. L. Husted, S. L. Kristufek, B. Xiong, D. J. Lundberg, J. Lem, D. Veysset, Y. Sun, K. A. Nelson, D. L. Plata and J. A. Johnson, Nature, 2020, 583, 542–547 CrossRef CAS PubMed.
- X. Wang, Y. Wen, Y. Wang, W. Li, X. Lu and W. You, CCS Chem., 2024, 6, 2305–2317 CrossRef CAS.
- J. M. Fishman and L. L. Kiessling, Angew. Chem., Int. Ed., 2013, 52, 5061–5064 CrossRef CAS PubMed.
- Y. Liang, H. Sun, W. Cao, M. P. Thompson and N. C. Gianneschi, ACS Macro Lett., 2020, 9, 1417–1422 CrossRef CAS PubMed.
- T. Steinbach, E. M. Alexandrino, C. Wahlen, K. Landfester and F. R. Wurm, Macromolecules, 2014, 47, 4884–4893 CrossRef CAS.
- H. G. Seong, T. P. Russell and T. Emrick, Chem. Sci., 2024, 15, 17084–17091 Search PubMed.
- C.-C. Chang and T. Emrick, Macromolecules, 2014, 47, 1344–1350 CAS.
- J. D. Feist and Y. Xia, J. Am. Chem. Soc., 2020, 142, 1186–1189 CrossRef CAS PubMed.
- X. Sui, T. Zhang, A. B. Pabarue, L. Fu and W. R. Gutekunst, J. Am. Chem. Soc., 2020, 142, 12942–12947 Search PubMed.
- X. Sui and W. R. Gutekunst, ACS Macro Lett., 2022, 11, 630–635 CrossRef CAS PubMed.
- K. Tashiro, M. Akiyama, K. Kashiwagi and T. Okazoe, J. Am. Chem. Soc., 2023, 145, 2941–2950 CrossRef CAS PubMed.
- H. Sun, T. Ibrahim, A. Ritacco and K. Durkee, ACS Macro Lett., 2023, 12, 1642–1647 CrossRef CAS PubMed.
- T. An, H. Ryu and T. L. Choi, Angew. Chem., Int. Ed., 2023, 62, e202309632 CrossRef CAS PubMed.
- A. Mandal and A. F. M. Kilbinger, ACS Macro Lett., 2023, 12, 1372–1378 CrossRef CAS PubMed.
- T. Ibrahim and H. Sun, ACS Appl. Polym. Mater., 2024, 6, 14076–14083 CrossRef CAS.
- J. D. Feist, D. C. Lee and Y. Xia, Nat. Chem., 2022, 14, 53–58 CrossRef CAS PubMed.
- M. A. Gauthier, M. I. Gibson and H. A. Klok, Angew. Chem., Int. Ed., 2008, 48, 48–58 CrossRef PubMed.
- A. A. Kislukhin, C. J. Higginson, V. P. Hong and M. G. Finn, J. Am. Chem. Soc., 2012, 134, 6491–6497 CrossRef CAS PubMed.
- N. Kuhl, R. Geitner, J. Vitz, S. Bode, M. Schmitt, J. Popp, U. S. Schubert and M. D. Hager, J. Appl. Polym. Sci., 2017, 134, 44805 CrossRef.
- C. Taplan, M. Guerre and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 9140–9150 CrossRef CAS PubMed.
- K. Nakamura, T. Hatakeyama and H. Hatakeyama, Polym. J., 1983, 15, 361–366 CrossRef CAS.
- P. Siachouli, K. S. Karadima, V. G. Mavrantzas and S. N. Pandis, Soft Matter, 2024, 20, 4783–4794 RSC.
- H. Shimomoto, T. Mori, T. Itoh and E. Ihara, Macromolecules, 2019, 52, 5761–5768 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00176e |
| This journal is © The Royal Society of Chemistry 2025 |