
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePotential theranostics of breast cancer with copper-64/67 sarcophagine-trastuzumab†
Stacey E.
Rudd
 *a,
Jessica
Van Zuylekom
bc,
Carleen
Cullinane‡
bc,
Benjamin J.
Blyth
bc and
Paul S.
Donnelly
*a
*a,
Jessica
Van Zuylekom
bc,
Carleen
Cullinane‡
bc,
Benjamin J.
Blyth
bc and
Paul S.
Donnelly
*a
aSchool of Chemistry and Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Melbourne, Victoria 3010, Australia. E-mail: stacey.rudd@unimelb.edu.au; pauld@unimelb.edu.au
bModels of Cancer Translational Research Centre, Research Division, Peter MacCallum Cancer Centre, 305 Grattan St, Melbourne, Victoria 3000, Australia
cSir Peter MacCallum Department of Oncology, The University of Melbourne, Parkville, Victoria 3010, Australia
First published on 23rd January 2025
Abstract
Over-expression of Human Epidermal Growth Factor Receptor 2 (HER2) is associated with a significant proportion of breast cancers. Targeting HER2 is possible with a monoclonal antibody called trastuzumab but metastatic HER2 positive tumours can develop resistance to this treatment. One approach to develop more potent therapeutic agents which retain the selectivity of trastuzumab is to attach a β− emitting radionuclide, such as copper-67, to the antibody for radioimmunotherapy. It is also possible to attach β+ emitting copper-64 to antibodies for diagnostic imaging with positron emission tomography (PET). In this work, a cage amine sarcophagine (Sar) chelator is conjugated to trastuzumab to give Sar-trastuzumab which can be radiolabelled with either copper-64 or copper-67 at room temperature in minutes to give [64Cu]CuSar-trastuzumab and [67Cu]CuSar-trastuzumab respectively. The diagnostic imaging potential of [64Cu]CuSar-trastuzumab was evaluated in mice bearing HER2+ SKOV-3 tumours showing that the tracer has very high tumour uptake and retention 48 hours after injection. The copper-67 variant, [67Cu]CuSar-trastuzumab, was highly therapeutically efficacious in the same tumour model with no signs of radiotoxicity. The combination of diagnostic PET imaging with [64Cu]CuSar-trastuzumab to guide radionuclide therapy with [67Cu]CuSar-trastzumab has significant potential for theranostic treatment of breast cancer and other HER2+ disease that has become resistant to conventional immunotherapy.
Introduction
Approximately 15–20% of breast cancers over-express the Human Epidermal Growth Factor Receptor 2 (HER2), a receptor tyrosine protein kinase which is involved in activation of cell signalling pathways that promote cell division and growth. Over-expression of HER2 in breast cancer is associated with tumour growth and poor prognosis.1 Targeting HER2 is possible with a humanised monoclonal antibody called trastuzumab (Herceptin).2 The binding of trastuzumab to the extracellular domain of HER2 alters intracellular signalling pathways which can initiate antibody-mediated cytotoxicity. Treatment with trastuzumab, in combination with chemotherapy, has become a common treatment option for patients with HER2 positive (HER2+) metastatic breast cancer leading to significant improvements in survival.3 Unfortunately, some metastatic HER2+ tumours develop resistance to this treatment so there is interest and clinical need in developing more potent therapeutic agents which retain the selectivity of trastuzumab to HER2+ disease.3–7Radioimmunotherapy, where an antibody is used to selectively deliver a cytotoxic radionuclide to tumours, retains the selectivity of antibodies for sites of disease and has the potential to treat tumours that have become resistant to conventional immunotherapy.8–10 Pioneering work on radioimmunotherapy led to the clinical approval of two agents, [90Y]Y-ibritumomab-tiuxetan (Zevalin, approved 2002) and [131I]I-tositumomab (Bexxar, approved 2003), where monoclonal antibodies specific to CD20 were radiolabelled with either β− emitting yttrium-90 or iodine-131.11,12 Both Bexxar and Zevalin performed well in clinical trials but are often considered to be commercially unsuccessful due to a combination of reasons including the logistical challenges associated with radioactive agents, economic concerns and an initial resistance of clinicians to adopt new approaches.9 The relatively recent clinical adoption of peptide targeted radionuclide therapy suggests some of these issues may now represent less of a challenge.10
It is also possible to radiolabel antibodies with either γ (gamma) emitting or β+ (positron) emitting radionuclides to produce tracers that can be used in diagnostic imaging with either Single Photon Emission Computed Tomography (SPECT) or Positron Emission Tomography (PET).10,13,14 The possibility of using the same or similar molecules radiolabelled with two different radionuclides, one for diagnostic imaging and the other for targeted radionuclide therapy, has been termed ‘radiotheranostics’.9,10 An aspiration of radiotheranostics is to use a tracer for diagnostic SPECT or PET imaging to identify patients who could potentially benefit from targeted radionuclide therapy, plan patient-specific dosimetry and monitor the response to treatment. Small molecules radiolabelled with either β+ emitting gallium-68 for diagnostic PET imaging or β− emitting lutetium-177 for targeted radionuclide therapy have gained clinical approvals for the treatment of neuroendocrine tumours and prostate cancer. The clinical and commercial impact of these agents has reinvigorated interest in radiotheranostics.15–19 In contrast to small molecules, radiolabelled full IgG antibodies (MW ∼ 150 kDa) take days to clear the circulation and accumulate in target tissue due to a combination of their relatively high molecular weight and Fc-mediated interactions (Fc = fraction crystallisable). This long circulation time means it is not feasible to radiolabel antibodies with gallium-68 (t1/2 ∼ 68 minutes) due to its relatively short radioactive half-life.
There are two copper radionuclides, copper-64 and copper-67, that can be considered an excellent matched pair for radiotheranostics. Copper-64 is a β+ emitting radionuclide (t1/2 = 12.7 h, β+ = 17.4%, Eβ+(mean) = 278 keV) that can be used in diagnostic PET imaging.10 A therapeutic counterpart is copper-67 (t1/2 = 61.9 h, β− = 100%, Eβ− (mean) = 141 keV), a β− emitting radionuclide which has a mean range in tissue of ∼0.2 mm so is well suited to the treatment of small tumours and metastases.10,20 Initial work aimed at taking advantage of the therapeutic potential of copper-67 was hindered by a lack of a reliable supply of high purity copper-67.20 A recent resurgence of interest in the copper-64/67 theranostic pair has stimulated concerted efforts to improve the methods used for the manufacture of copper-67 that have led to the radionuclide now being readily available.10,20
Initial work on copper-64/67 radiopharmaceuticals was also hindered by using chelators that formed copper complexes of insufficient stability in vivo.20–23 Recent human clinical studies have demonstrated that macrobicyclic cage amine ligands, known as sarcophagines (Sar = 3,6,10,13,16,19-hexaazabicyclo[6.6.6]icosane, Fig. 1), form copper-64/67 complexes that are stable in vivo. A bifunctional variant of sarcophagine, (1-CH3-8-NHC(O)(CH2)3CO2H)Sar, abbreviated to ‘MeCOSar’ as an abbreviation of methyl-carboxylate sarcophagine (Fig. 1)24 is used in the peptide conjugates [64/67Cu]CuSarTATE and [64/67Cu]CuSarBombesin. These conjugates have been investigated in human clinical trials and in each case there is no evidence for significant release of the copper radionuclide from the sarcophagine ligand.25,26
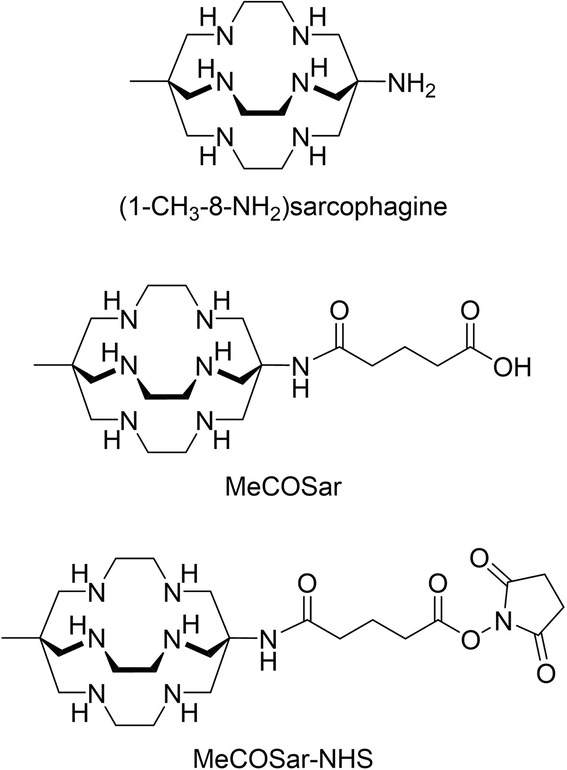 | ||
| Fig. 1 The chemical structures of (1-CH3-8-NH2)sarcophagine, MeCOSar and MeCOSar-NHS. | ||
A particular advantage of sarcophagine ligands for antibody-chelator conjugates is that the ligand can be radiolabelled with either [64Cu]CuII or [67Cu]CuII at room temperature at pH 7.4 to give complexes in high radiochemical yield and purity.27–31 It is important to be able to radiolabel the antibody-chelator conjugate rapidly at room temperature (or <37 °C), as many antibodies are temperature sensitive. The activated N-hydroxysuccinimidyl (NHS) ester of MeCOSar, MeCOSar-NHS (Fig. 1), reacts readily with lysine residues of antibodies to form stable conjugates and was used in a first-in-human PET imaging study with a [64Cu]Cu-labelled HIV-binding antibody, 3BNC117. 32–34
In this work, we use MeCOSar-NHS to form new conjugates with trastuzumab, Sar-trastuzumab, and investigate the potential of the copper-64 and copper-67 complexes as a new theranostic approach for the treatment of HER2+ breast cancer. The tumour uptake and biodistribution of [64Cu]CuSar-trastuzumab was evaluated in HER2+ tumours in two different immune deficient mouse strains. We demonstrate that, as expected, [64Cu]CuSar-trastuzumab and [67Cu]CuSar-trastuzumab have a similar biodistribution in the same tumour model, confirm the tumour uptake of [67Cu]CuSar-trastuzumab with Cherenkov imaging and then evaluate the therapeutic efficacy of [67Cu]CuSar-trastuzumab.
Results
Synthesis and characterisation of [64/67Cu]CuSar-trastuzumab
The reaction of MeCOSar-NHS with trastuzumab led to conjugation of the chelator to the antibody through the reaction of the activated ester with the amine functional groups of the lysine residues. The new bioconjugate was characterised by intact protein electrospray ionisation mass spectrometry (ESI-MS). The ESI mass spectrum of trastuzumab has a signal at m/z ∼148![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 231 Daltons (Da) with different degrees of glycosylation resulting in peaks with a sequential increase in m/z of ∼160 Da (Fig. 2a). Addition of MeCOSar results in sequential additions of ∼410 Da and an average chelator to antibody ratio of 4:1 can be estimated from the ESI-mass spectrum (Fig. 2a). Analysis of Sar-trastuzumab by size exclusion high performance liquid chromatography (SE-HPLC) revealed no changes in the SE-HPLC chromatogram of Sar-trastuzumab when compared to trastuzumab and there is no evidence that attaching MeCOSar at this chelator to antibody ratio causes aggregation of the protein.
231 Daltons (Da) with different degrees of glycosylation resulting in peaks with a sequential increase in m/z of ∼160 Da (Fig. 2a). Addition of MeCOSar results in sequential additions of ∼410 Da and an average chelator to antibody ratio of 4:1 can be estimated from the ESI-mass spectrum (Fig. 2a). Analysis of Sar-trastuzumab by size exclusion high performance liquid chromatography (SE-HPLC) revealed no changes in the SE-HPLC chromatogram of Sar-trastuzumab when compared to trastuzumab and there is no evidence that attaching MeCOSar at this chelator to antibody ratio causes aggregation of the protein.
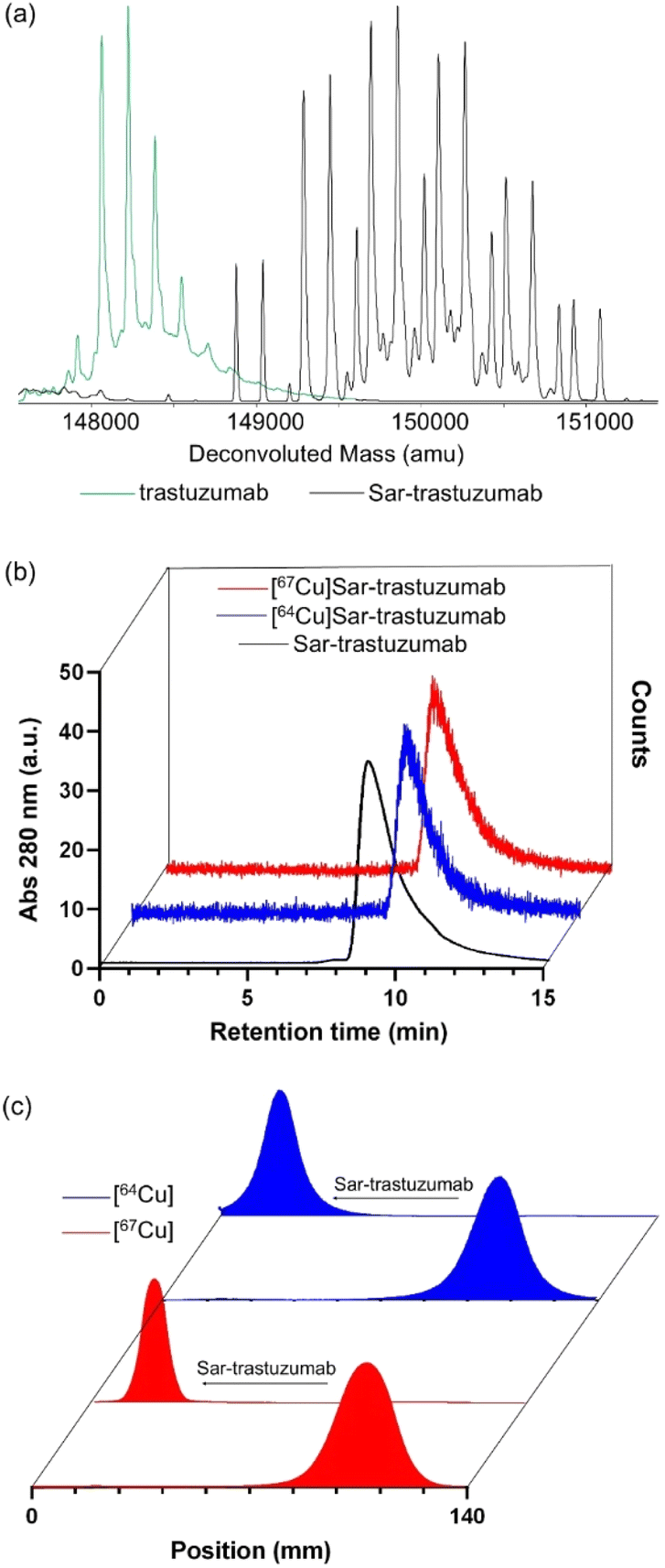 | ||
| Fig. 2 (a) Intact protein ESI-MS of trastuzumab and Sar-trastuzumab, (b) radio-SE-HPLC analysis of Sar-trastuzumab and its [64/67Cu]-labelled conjugates. (c) Radio-TLC of unbound [64/67Cu]CuII and corresponding [64/67Cu]Sar-trastuzumab conjugates. | ||
Following labelling of Sar-trastuzumab with [64Cu]CuII, analysis by radio-SE-HPLC (Fig. 2b) and radio-thin layer chromatography (TLC, Fig. 2c) revealed essentially quantitative radiolabelling within 20 minutes at room temperature to give [64Cu]CuSar-trastuzumab in >99% radiochemical yield and radiochemical purity at a specific activity of 500 MBq mg−1. No evidence for the loss of copper-64 from the conjugate was observed after incubation in human serum for 48 hours. Radiolabelling Sar-trastuzumab with [67Cu]CuII using similar methodology allowed isolation of [67Cu]CuSar-trastuzumab in similar high radiochemical yields and purity at 1000 MBq mg−1 (Fig. 2b and c). Even higher specific activities are likely possible under these conditions, given that they can be achieved even at very low antibody concentrations also at room temperature with longer reaction times (up to 5 GBq mg−1 was achieved with >95% radiochemical yield at an antibody concentration of only 83 μg mL−1). Radiolabelled conjugates were diluted into 0.5% sodium gentisate in phosphate buffered saline (PBS) immediately after labelling to minimise radiolysis.
PET imaging of HER2+ tumours with [64Cu]CuSar-trastuzumab
The tumour uptake and biodistribution of [64Cu]CuSar-trastuzumab was evaluated by PET imaging of HER2+ SKOV-3 xenografts in two different immune compromised mouse strains. Our initial studies used SKOV-3 xenografts in NSG (non-obese diabetic (NOD) scid gamma) mice. Following administration of [64Cu]CuSar-trastuzumab by intravenous tail vein injection PET images were acquired at 24 and 48 hours post injection. Inspection of the images reveals uptake of the tracer in the tumour (mean SUVmax = 8.1 ± 1 at 48 h post injection, where SUV = standard uptake value) and in general low background activity (Fig. 3a). In addition to the high uptake in the tumours, there is also significant uptake of the tracer in the spleen and limb joints/bone. The bone uptake is mainly in the epiphysis of the long bones due to antibody-mediated uptake in the growth plates. The high tumour uptake coupled with significant uptake in the spleen were confirmed by an ex vivo biodistribution study where the amount of radioactivity present in each organ was quantified by gamma-counting (ESI Fig. 4†).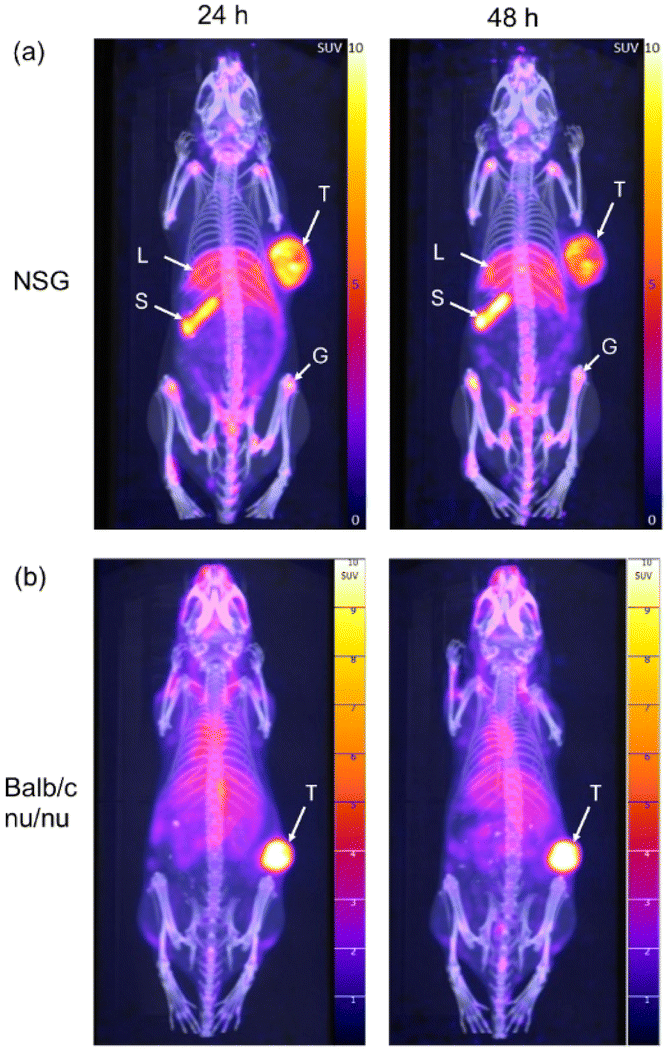 | ||
| Fig. 3 Representative maximum intensity projection PET/CT images of SKOV-3 xenograft tumour-bearing (a) NSG and (b) Balb/c nu/nu mice, 24 and 48 h following administration with [64Cu]CuSar-trastuzumab. Tumour (T), liver (L), spleen (S) and growth plates (G) are indicated. | ||
To investigate the role of the mouse strain and Fc-mediated interactions in the tracer uptake in spleen and joints, the same SKOV-3 tumour model was evaluated in a different immune compromised mouse strain (Balb/c nu/nu). A higher dose (10 MBq) was administered to the Balb/c nu/nu cohort than the NSG mice (3 MBq) to improve the signal to noise ratio at the 48 hour timepoint without increasing scan time for the animals. The PET images following administration of [64Cu]CuSar-trastuzumab in the SKOV-3 Balb/c nu/nu model show very high uptake of the tracer in the tumour (mean SUVmax = 21.0 ± 2.5) and significantly less uptake in both the spleen and joints (Fig. 3b).
Tumour uptake, biodistribution and therapeutic efficacy of [67Cu]CuSar-trastuzumab in HER2+ mouse model
The SKOV-3 Balb/c nu/nu model was selected to evaluate the therapeutic efficacy of [67Cu]CuSar-trastuzumab. The tumour uptake and biodistribution of [67Cu]CuSar-trastuzumab was compared with [64Cu]CuSar-trastuzumab by an ex vivo biodistribution study where radioactivity in each organ was quantified by gamma counting and expressed as a percentage of injected activity per gram of tissue (% IA g−1). As expected, conjugates prepared with the two different radionuclides displayed similar tumour uptake and biodistribution (Fig. 4a). The tumour uptake of [67Cu]CuSar-trastuzumab at 48 hours post injection was 59 ± 7% IA g−1 and the tumour uptake of the [64Cu]CuSar-trastuzumab was 61 ± 6% IA g−1.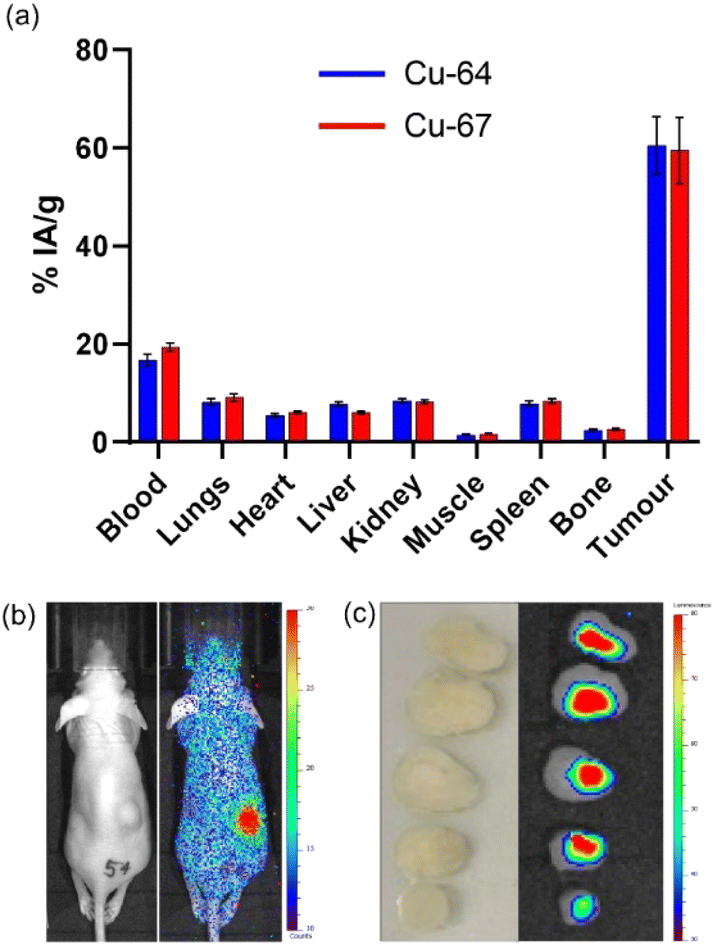 | ||
| Fig. 4 (a) Ex vivo biodistribution of [64Cu]CuSar-trastuzumab and [67Cu]CuSar-trastuzumab 48 hours after administration into SKOV-3 tumour-bearing Balb/c nu/nu mice. (b) 2D planar Cherenkov image of a mouse 24 hours following administration with [67Cu]CuSar-trastuzumab. The Cherenkov light image (colour scale) is overlaid on an optical image (left). (c) 2D planar Cherenkov image of tumour slices resected 48 hours following administration of [67Cu]CuSar-trastuzumab. The Cherenkov luminescence image (colour scale) is overlaid on an optical image. | ||
A qualitative indication of the tumour uptake of [67Cu]CuSar-trastuzumab was obtained by Cherenkov imaging. Following injection with [67Cu]CuSar-trastuzumab, mice were imaged using a bioluminescence imager that can detect the Cherenkov radiation emitted by the beta-particle emitting copper-67 (Fig. 4b). The uptake and retention of copper-67 in the tumours was further demonstrated by Cherenkov imaging of tumour slices that had been resected 48 hours after administration and fixed in formalin (Fig. 4c).
The therapeutic efficacy of a single administration of three different radioactive doses of [67Cu]CuSar-trastuzumab (4.5 MBq, 6.7 MBq and 9.0 MBq, all with a total of 20 μg Sar-trastuzumab) were compared to the same amount of non-labelled Sar-trastuzumab (20 μg) as well as a saline (vehicle) control. Mice were inoculated with SKOV-3 cells, and once the tumours reached a mean volume of approximately 250 mm3, the mice were randomized to receive either saline, non-labelled Sar-trastuzumab or one of three doses of [67Cu]CuSar-trastuzumab via intravenous injection (n = 9–10 per treatment group). All treatments were well tolerated causing only a transient reduction in animal body weight that did not exceed 7% of that at baseline, and which did not differ significantly from the vehicle control group (p = 0.6889, Fig. 5a). No overt signs of toxicity were observed in any of the treatment groups. Four mice (1 in each treatment group) were euthanized prior to tumour endpoint on welfare grounds due to skin or eye irritations that are more common in Balb/c nu/nu mice. The mice administered saline were culled at 18–39 days as the tumours had reached tumour endpoint due to disease progression (Fig. 5). All doses of [67Cu]CuSar-trastuzumab resulted in significant partial tumour growth inhibition or complete tumour regression (P < 0.0001, two-way ANOVA with Dunnett test). Percentage tumour growth inhibition was calculated for each dose at day 18 (corresponding to the first control group endpoint) using the formula (1 − ΔT/ΔC) × 100), where ΔT = difference between day 0 and day 18 mean tumour volumes of the test group, and ΔC = difference between day 0 and day 18 mean tumour volumes of the control group. Administration of a single dose of [67Cu]CuSar-trastuzumab (4.5 MBq) inhibited tumour growth by 88% at 18 days post treatment and median survival was extended from 32 days (saline) to 46 days. Two mice from this treatment group exhibited complete regression for the duration of the study. A clear dose response was evident with the mid-range dose of [67Cu]CuSar-trastuzumab (6.7 MBq) leading to a 120% tumour growth inhibition by day 18 (>100% growth inhibition indicates a decrease in tumour size compared to day 0), and median survival extension out to 81 days. Administration of the highest dose (9.0 MBq) also led to a 119% tumour growth inhibition by day 18 and extension of median survival to 102 days. Two mice in this treatment group also exhibited complete regression. Administration of an equal mass dose of unlabelled Sar-trastuzumab did not result in a therapeutic effect with median survival of this cohort matching that of the saline control (32 days).
 | ||
| Fig. 5 (a) Change in body weight, (b) mean tumour volume to first endpoint and (c) survival probability for the range of doses of [67Cu]CuSar-trastuzumab as well as the saline and unlabelled Sar-trastuzumab controls. (a and b) are given as mean ± SEM, n = ≥9 at day 0. | ||
Discussion
PET imaging with [64Cu]CuSar-trastuzumab in HER2+ SKOV-3 xenografts in NSG mice showed high tumour uptake but also significant uptake in the spleen and joints. PET imaging with radiolabelled humanised IgG antibodies in immune compromised mice can give anomalous biodistribution due to Fc-mediated interactions leading to accumulation in the spleen and bone marrow.35 This uptake in non-target organs could be due to the lack of endogenous IgG in mice bearing the scid mutation.35 PET imaging following administration of [64Cu]CuSar-trastuzumab using the same tumour model, SKOV-3, but in a different mouse strain (Balb/c nu/nu) also revealed very high uptake of the tracer in the tumour but significantly less uptake in both the spleen and joints. The lower spleen and off-target uptake in the Balb/c nu/nu model led to this model being selected to study the therapeutic efficacy of [67Cu]CuSar-trastuzumab.Aa expected, the tumour uptake of [64Cu]CuSar-trastuzumab and [67Cu]CuSar-trastuzumab are similar (Fig. 4a). Confirmation of the uptake and retention of radioactivity in the tumour following administration of [67Cu]CuSar-trastuzumab was obtained by Cherenkov luminescence imaging (Fig. 4b and c). Cherenkov radiation is emitted when a charged particle, such as a β− particle emitted from copper-67, travels through a dielectric medium faster than the speed of light in that medium.36 It is possible to detect the Cherenkov radiation emitted from [67Cu]CuSar-trastuzumab using a luminescence imaging scanner.
[67Cu]CuSar-trastuzumab displays very high therapeutic efficacy against HER2+ SKOV-3 tumours. All three doses of [67Cu]CuSar-trastuzumab were well tolerated by the mice. There were no significant changes in weight when comparing the treated cohorts to the control group (saline) consistent with no systemic radiation toxicity. Future translation of this work to clinical studies would require assessment of stability with respect to radiolysis when formulated with higher radiation doses and quantification of the immunoreactive fraction.
In our previous work, we used a bifunctional sarcophagine variant with an isothiocyanate functional group to radiolabel trastuzumab but were unable to evaluate the therapeutic efficacy of the copper-67 conjugate as at that time we were unable to access the radionuclide.37
The therapeutic potential of copper-67 has been known for several decades but initial studies were hampered by limited supply and low specific activity of the radionuclide. Pioneering studies also used derivatives of the tetraazamacrocycle cyclam as a chelator and the copper(II) complexes formed were of insufficient stability in vivo so dissociation of the copper-67 from the chelator led to accumulation of the radionuclide in the liver and non-target tissue.10,38 Recent breakthroughs in the production of copper-67 means the radionuclide can now be produced with high specific activity (>5.6 MBq mg−1) and >99% radionuclide purity.20 The shorter radioactive half-life of copper-67 (t1/2 = 2.58 days) also has potential advantages with respect to patient management when compared to lutetium-177 (t1/2 = 6.7 days). The increased availability of copper-67 and the use of macrobicyclic cage amine sarcophagine chelators, such as MeCOSar (Fig. 1), to form copper-64/67 complexes that are stable in vivo has led to several human clinical studies that have been characterised by high quality images.25,26,34,39
The potential for diagnostic imaging and radionuclide therapy with a combination of copper-64 and copper-67 has also been demonstrated using a tumour pre-targeting approach with a derivative of MeCOSar bearing a tetrazine functional group and a huA33 antibody that was modified to incorporate a trans-cyclooctene functional group.40
The work presented here adds to mounting evidence supporting the use of copper-67 labelled antibodies for radioimmunotherapy. During the course of this work a separate study documenting the therapeutic efficacy of copper-67 labelled trastuzumab was published where the NOTA (1,4,7-triazacyclononane-1,4,7-triacetic acid) chelator was conjugated to trastuzumab ([67Cu]CuNOTA-trastuzumab). Administration of [67Cu]CuNOTA-trastuzumab was therapeutically efficacious in two different HER2+ models (BT-474 and JIMT-1) to what we used in our work but a significantly higher radioactive dose was administered (∼16.8 MBq).41 In an additional separate study, where NOTA was conjugated to pertuzumab, [67Cu]CuNOTA-pertuzumab was found to have some therapeutic effect at a low dose of 3.7 MBq in a HER2+ HCC1954 mouse xenograft model but higher doses (7.4 MBq and 14.8 MBq) resulted in systemic radiation induced toxicity.42
Conclusions
This pre-clinical data suggests that diagnostic PET imaging with [64Cu]CuSar-trastuzumab has the potential to be of use in selecting patients that may be suitable for radionuclide therapy with β− emitting [67Cu]CuSar-trastuzumab and for monitoring treatment. [67Cu]CuSar-trastuzumab is highly therapeutically efficacious against HER2+ SKOV-3 tumours in mice. Administration of a single relatively low dose of [67Cu]CuSar-trastuzumab (4.5 MBq) inhibited tumour growth by 88% at 18 days post treatment and two mice in this cohort demonstrated complete tumour regression. Administration of a higher dose of [67Cu]CuSar-trastuzumab (9.0 MBq) led to a 119% tumour growth inhibition and two mice exhibited complete tumour regression with no signs of radiation toxicity.The use of a theranostic pair, where the same antibody conjugate labelled with two different isotopes of the same element with the same biodistribution and pharmacokinetics, has potential advantages over the use of chemically distinct antibody conjugates that are radiolabelled with different elements. The combination of diagnostic PET imaging with [64Cu]CuSar-trastuzumab to guide radionuclide therapy with [67Cu]CuSar-trastuzumab has significant potential to be translated to the clinic for genuine theranostic treatment of breast cancer and other HER2+ disease that has become resistant to conventional immunotherapy.
Experimental
General experimental
Trastuzumab was purchased as a lyophilised powder and reconstituted in MilliQ H2O prior to use. MeCOSar-NHS was synthesised according to reported procedures.32 Copper-64 was supplied as no carrier added [64Cu]CuCl2 in H2O by Austin Health Molecular Imaging & Therapy, Victoria, Australia. Copper-67 was supplied as no carrier added [67Cu]CuCl2 in 0.01 M HCl by Idaho Accelerator Centre, Idaho, USA. ESI-MS protein samples were analysed using an Agilent 6220 ESI-TOF LC/MS mass spectrometer coupled to an Agilent 1200 LC system (Agilent, Palo Alto, CA). All data were acquired and reference mass corrected using a dual-spray electrospray ionisation (ESI) source. Acquisition was performed using the Agilent Mass Hunter Acquisition software version B.02.01 (B2116.30). Protein desalting and chromatographic separation was performed using a Phenomenex Jupiter C5 5 5 μm 300 Å 2.1 × 50 mm column using 5% (v/v) acetonitrile ported to waste (0–5 min). Upon desalting of sample the flow was ported back into the ESI source for subsequent gradient elution with (5% (v/v) to 100% (v/v)) acetonitrile/0.1% formic acid over 8 min at 0.25 mL min−1. Data analysis was performed using Mass Hunter version B.06.00 with BioConfirm software. Protein concentrations were determined using a Thermo Fisher Scientific NanoDrop instrument (model NanoDrop One). All protein yields were calculated using the volume of solution obtained and the protein concentration of these solutions. For analysis of radioactive samples, size exclusion HPLC was performed on an AKTA Purifier P-900 system with an in-line Berthold Technologies Flowstar LB513 Radiodetector followed by an Agilent 1100 Series G1315B Diode Array Detector. Samples were analysed using a Phenomenex Yarra SEC-3000 3 μm 4.6 × 300 mm column (0.35 mL min−1 flow rate, 0.1 M phosphate buffered saline pH 6.8 with 5% isopropanol as mobile phase). Radio-iTLC were performed using an Elysia Raytest MiniGita TLC scanner system with MiniGita OFA Probe using silica gel impregnated glass fibre instant TLC (iTLC) plates eluted with 10 mM ethylenediaminetetraacetic acid in Dulbecco's PBS.Preparation of Sar-trastuzumab
Trastuzumab (∼6.1 mg) was ‘buffer exchanged’ into sodium bicarbonate (0.1 M, pH 8.2) using an Amicon Ultra-0.5 centrifuge filter with a 50 kDa molecular weight cutoff (MWCO), then adjusted to a concentration of 10 mg mL−1. An aliquot of MeCOSar-NHS in dimethyl sulfoxide (11.13 μL, 55.6 mM, 15 molar eq.) was added and the mixture heated at 37 °C for 3 h. The mixture was then purified by filtration through a 10 kDa MWCO centrifuge filter, washed twice with 4% dimethyl sulfoxide in ammonium acetate buffer (10 mM, pH 5.6), then twice with ammonium acetate buffer (10 mM pH 5.6). The concentrate was further diluted with buffer and analysed by NanoDrop, ESI-MS and SE-HPLC to give Sar-trastuzumab (35 mg mL−1, 3.2 mg).Synthesis of [64Cu]CuSar-trastuzumab
Copper-64 was supplied as copper chloride solution (4.35 GBq mL−1 in H2O). An aliquot (235 MBq, 54 μL) was removed and buffered to pH 5.6 by addition of ammonium acetate buffer (50 μL, 1 M). Sar-trastuzumab (13.4 μL, 35 mg mL−1, 470 μg) was added and the reaction mixture allowed to stand at ambient temperature. Radio-iTLC after 6 minutes indicated quantitative radiochemical yield/purity without any purification steps, and this was confirmed with radio-SEHPLC after 19 minutes. The reaction mixture was then diluted with 0.5% (w/v) sodium gentisate in Dulbecco's PBS (286 μL) to give [64Cu]CuSar-trastuzumab (500 MBq mg−1). Serum stability testing was performed by mixing [64Cu]CuSar-trastuzumab (25 MBq) in human serum (250 μL) and incubating at 37 °C. Aliquots of the sample were analysed by radio-iTLC after 0, 1, 18, 24 and 48 hours.Synthesis of [67Cu]CuSar-trastuzumab
Copper-67 was supplied as copper chloride solution (18.8 GBq mL−1 in 0.01 M HCl). An aliquot (301 MBq, 16 μL) was removed and buffered to pH 5.6 by addition of ammonium acetate buffer (16 μL, 1 M). Sar-trastuzumab (8.6 μL, 35 mg mL−1, 301 μg) was added and the reaction mixture allowed to stand at ambient temperature. Radio-iTLC after 6 minutes indicated ∼99% radiochemical yield/purity without any purification steps, and this was confirmed with Radio-SEHPLC after 17 minutes. The reaction mixture was then diluted with 0.5% (w/v) sodium gentisate in Dulbecco's PBS (100 μL) to give [67Cu]CuSar-trastuzumab. The radiolabelled product was adjusted to the required specific activity by the addition of Sar-trastuzumab to ensure each mouse received a 20 μg protein dose (i.e. a specific activity of 225 MBq mg−1 for the 4.5 MBq therapy cohort, 335 MBq mg−1 for the 6.7 MBq cohort, and 450 MBq mg−1 for the 9.0 MBq cohort).Mouse imaging (PET/CT & Cherenkov) and biodistribution
All animal experiments were performed with the approval of the Peter MacCallum Cancer Centre Animal Ethics Committee and in accordance with the Australian code for the care and use of animals for scientific purposes, 8th Edition, 2013. Balb/c nu/nu and NSG mice were sourced from Animal BioResources, (Moss Vale, New South Wales). For each mouse 3 × 106 SKOV-3 cells (American Type Culture Collection) were implanted subcutaneously onto the right flank in a 1:1 mix of PBS and Matrigel basement membrane matrix (Corning) and once tumours were well established, the animals were used for imaging and biodistribution studies. Doses were drawn up into 0.3 mL insulin syringes and administered via tail vein injection (MBq doses for each study are given in ESI Table 2†). For the biodistribution study with [67Cu]CuSar-trastuzumab, all mice received approximately 4.5 MBq (20 μg) doses. For PET-CT imaging and biodistribution with [64Cu]CuSar-trastuzumab, NSG mice received 3 MBq (6 μg) and Balb/c nu/nu mice received 10 MBq (20 μg) doses. Animals were anaesthetised with 1.5% isoflurane and placed in the imaging chamber of a PerkinElmer/Sofie Biosciences small animal G8 PET/CT. A 10 min PET acquisition was performed followed by a CT scan. Images were acquired using the G8 acquisition engine software and reconstructed using a 3D MLEM algorithm. Quantification was performed using VivoQuant software, Version 3.0 (inviCRO Imaging Services and Software). After imaging the final time point mice were euthanized and tissues excised, weighed and counted using a Capintec (Captus 4000e) gamma counter. For Cherenkov imaging animals were anaesthetised with 2.5% isoflurane and placed in the imaging chamber of a PerkinElmer IVIS Lumina III pre-clinical imager. Photon emissions were acquired over 10 min in Cherenkov mode with open filter position, and counts overlaid on a photographic reference image.
Therapeutic efficacy study
SKOV-3 tumours were implanted into Balb/c nu/nu mice as per the imaging and biodistribution studies, and tumour-bearing mice were randomised into groups with matched tumour volumes. Doses were drawn up into 0.3 mL insulin syringes and administered via tail vein injection. Mice received either saline, cold Sar-trastuzumab (20 μg), or radiolabelled [67Cu]CuSar-trastuzumab at doses of 4.5 MBq (20 μg), 6.7 MBq (20 μg), or 9.0 MBq (20 μg). Tumour volumes (from electronic calliper measurements of length and width) and body weights were measured at least twice per week until mice reached tumour volume endpoint (planned as 1200 mm3). As tumours grew rapidly and began to ulcerate once above 800 mm3, the tumour volume endpoint was brought forward on ethical grounds to severe ulceration or 1000 mm3. Survival to tumour endpoint is the day tumours reached the revised endpoint criteria for all mice.Data availability
All experimental data is provided within the manuscript or the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The University of Melbourne has received research funding from Clarity Pharmaceuticals and this company has potential interests in this work. P. S. D. has a financial interest in Clarity Pharmaceuticals.Acknowledgements
Greg Santamaria and Cyclotek Pty Ltd (Australia) are thanked for the provision of radiochemistry laboratories that supported this research. The copper-67 used in this study was kindly provided by Clarity Pharmaceuticals. This research was funded in part by the Australian Research Council and the National Health and Medical Research Council. We acknowledge the Bio21 Mass Spectrometry and Proteomics Facility for instrumentation and technical support. Financial support from the Australian Cancer Research Foundation supported the purchase of some of the equipment used in this work. We thank Kai Jhuang, Stephanie Jansen, Danielle Norman, Kerry Warren and Rachael Walker of the Models of Cancer Translational Research Centre (Peter MacCallum Cancer Centre) for their technical contributions to this work.References
- S. M. Swain, M. Shastry and E. Hamilton, Nat. Rev. Drug Discovery, 2023, 22, 101–126 Search PubMed.
- H. M. Shepard, Nat. Rev. Cancer, 2024, 24, 287–288 Search PubMed.
- J. T. Jørgensen, Ann. Transl. Med., 2023, 12, 53 Search PubMed.
- A. Ahad, H. K. Saeed, V. del Solar, J. E. Lopez-Hernandez, A. Michel, J. Mathew, J. S. Lewis and M. Contel, ACS Pharmacol. Transl. Sci., 2023, 6, 1972–1986 Search PubMed.
- L. Qian, X. Lin, X. Gao, R. U. Khan, J.-Y. Liao, S. Du, J. Ge, S. Zeng and S. Q. Yao, Chem. Rev., 2023, 123, 7782–7853 Search PubMed.
- C. Dumontet, J. M. Reichert, P. D. Senter, J. M. Lambert and A. Beck, Nat. Rev. Drug Discovery, 2023, 22, 641–661 Search PubMed.
- K. Tsuchikama, Y. Anami, S. Y. Y. Ha and C. M. Yamazaki, Nat. Rev. Clin. Oncol., 2024, 21, 203–223 Search PubMed.
- J. P. Pouget, C. Lozza, E. Deshayes, V. Boudousq and I. Navarro-Teulon, Front. Med., 2015, 2, 12 Search PubMed.
- L. Bodei, K. Herrmann, H. Schoder, A. M. Scott and J. S. Lewis, Nat. Rev. Clin. Oncol., 2022, 19, 534–550 Search PubMed.
- K. A. Morgan, S. E. Rudd, A. Noor and P. S. Donnelly, Chem. Rev., 2023, 123, 12004–12035 Search PubMed.
- H. A. Jacene, R. Filice, W. Kasecamp and R. L. Wahl, J. Nucl. Med., 2007, 48, 1767 Search PubMed.
- D. Rizzieri, Crit. Rev. Oncol. Hematol., 2016, 105, 5–17 Search PubMed.
- W. Wei, Z. T. Rosenkrans, J. Liu, G. Huang, Q.-Y. Luo and W. Cai, Chem. Rev., 2020, 120, 3787–3851 Search PubMed.
- S. E. Rudd, A. Noor, K. A. Morgan and P. S. Donnelly, Acc. Chem. Res., 2024, 57, 1421–1433 Search PubMed.
- H. R. Maecke and J. C. Reubi, J. Nucl. Med., 2011, 52, 841–844 Search PubMed.
- U. Hennrich and K. Kopka, Pharmaceuticals, 2019, 12, 114 Search PubMed.
- O. Sartor, J. de Bono, K. N. Chi, K. Fizazi, K. Herrmann, K. Rahbar, S. T. Tagawa, L. T. Nordquist, N. Vaishampayan, G. El-Haddad, C. H. Park, T. M. Beer, A. Armour, W. J. Perez-Contreras, M. DeSilvio, E. Kpamegan, G. Gericke, R. A. Messmann, M. J. Morris and B. J. Krause, N. Engl. J. Med., 2021, 385, 1091–1103 Search PubMed.
- U. Hennrich and M. Eder, Pharmaceuticals, 2022, 15, 1292 Search PubMed.
- L. Emmett, S. Subramaniam, M. Crumbaker, A. Nguyen, A. M. Joshua, A. Weickhardt, S.-T. Lee, S. Ng, R. J. Francis, J. C. Goh, D. A. Pattison, T. H. Tan, I. D. Kirkwood, C. Gedye, N. K. Rutherford, S. Sandhu, A. R. Kumar, D. Pook, S. Ramdave, D. P. Nadebaum, M. Voskoboynik, A. D. Redfern, W. Macdonald, L. Krieger, G. Schembri, W. Chua, P. Lin, L. Horvath, P. Bastick, P. Butler, A. Y. Zhang, S. Yip, H. Thomas, A. Langford, M. S. Hofman, M. McJannett, A. J. Martin, M. R. Stockler and I. D. Davis, Lancet Oncol., 2024, 25, 563–571 Search PubMed.
- L. Mou, P. Martini, G. Pupillo, I. Cieszykowska, C. S. Cutler and R. Mikołajczak, Molecules, 2022, 27, 1501 Search PubMed.
- P. M. Smith-Jones, R. Fridrich, T. A. Kaden, I. Novak-Hofer, K. Siebold, D. Tschudin and H. R. Maecke, Bioconjugate Chem., 1991, 2, 415–421 Search PubMed.
- T. M. Jones-Wilson, K. A. Deal, C. J. Anderson, D. W. McCarthy, Z. Kovacs, R. J. Motekaitis, A. D. Sherry, A. E. Martell and M. J. Welch, Nucl. Med. Biol., 1998, 25, 523–530 Search PubMed.
- A. B. Delaloye, B. Delaloye, F. Buchegger, C. A. Vogel, M. Gillet, J. P. Mach, A. Smith and P. A. Schubiger, J. Nucl. Med., 1997, 38, 847–853 Search PubMed.
- B. M. Paterson, P. Roselt, D. Denoyer, C. Cullinane, D. Binns, W. Noonan, C. M. Jeffery, R. I. Price, J. M. White, R. J. Hicks and P. S. Donnelly, Dalton Trans., 2014, 43, 1386–1396 Search PubMed.
- R. J. Hicks, P. Jackson, G. Kong, R. E. Ware, M. S. Hofman, D. A. Pattison, T. A. Akhurst, E. Drummond, P. Roselt, J. Callahan, R. Price, C. M. Jeffery, E. Hong, W. Noonan, A. Herschtal, L. J. Hicks, A. Hedt, M. Harris, B. M. Paterson and P. S. Donnelly, J. Nucl. Med., 2019, 60, 777–785 Search PubMed.
- K. Wong, G. Sheehan-Dare, A. Nguyen, B. Ho, V. Liu, J. Lee, L. Brown, R. Dear, L. Chan, S. Sharma, A. Malaroda, I. Smith, E. Lim and L. Emmett, Pharmaceuticals, 2022, 15, 772 Search PubMed.
- N. M. Di Bartolo, A. M. Sargeson, T. M. Donlevy and S. V. Smith, J. Chem. Soc., Dalton Trans., 2001, 2303–2309 Search PubMed.
- N. Di Bartolo, A. M. Sargeson and S. V. Smith, Org. Biomol. Chem., 2006, 4, 3350–3357 Search PubMed.
- S. D. Voss, S. V. Smith, N. DiBartolo, L. J. McIntosh, E. M. Cyr, A. A. Bonab, J. L. J. Dearling, E. A. Carter, A. J. Fischman, S. T. Treves, S. D. Gillies, A. M. Sargeson, J. S. Huston and A. B. Packard, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17489–17493 Search PubMed.
- M. T. Ma, J. A. Karas, J. M. White, D. Scanlon and P. S. Donnelly, Chem. Commun., 2009, 3237–3239 Search PubMed.
- M. T. Ma, M. S. Cooper, R. L. Paul, K. P. Shaw, J. A. Karas, D. Scanlon, J. M. White, P. J. Blower and P. S. Donnelly, Inorg. Chem., 2011, 50, 6701–6710 Search PubMed.
- K. Alt, B. M. Paterson, K. Ardipradja, C. Schieber, G. Buncic, B. Lim, S. S. Poniger, B. Jakoby, X. Wang, G. J. O'Keefe, H. J. Tochon-Danguy, A. M. Scott, U. Ackermann, K. Peter, P. S. Donnelly and C. E. Hagemeyer, Mol. Pharmaceutics, 2014, 11, 2855–2863 Search PubMed.
- S. E. Rudd, J. K. Van Zuylekom, A. Raicevic, L. A. Pearce, C. Cullinane, C. C. Williams, T. E. Adams, R. J. Hicks and P. S. Donnelly, Chem. Sci., 2021, 12, 9004–9016 Search PubMed.
- J. H. McMahon, J. M. Zerbato, J. S. Y. Lau, J. L. Lange, M. Roche, C. Tumpach, A. Dantanarayana, A. Rhodes, J. Chang, T. A. Rasmussen, C. A. Mackenzie, K. Alt, M. Hagenauer, J. Roney, J. O'Bryan, A. Carey, R. McIntyre, P. Beech, G. J. O'Keefe, C. W. Wichmann, F. E. Scott, N. Guo, S.-T. Lee, Z. Liu, M. Caskey, M. C. Nussenzweig, P. S. Donnelly, G. Egan, C. E. Hagemeyer, A. M. Scott and S. R. Lewin, EBioMedicine, 2021, 65, 103252 Search PubMed.
- S. K. Sharma, A. Chow, S. Monette, D. Vivier, J. Pourat, K. J. Edwards, T. R. Dilling, D. Abdel-Atti, B. M. Zeglis, J. T. Poirier and J. S. Lewis, Cancer Res., 2018, 78, 1820–1832 Search PubMed.
- D. Thorek, R. Robertson, W. A. Bacchus, J. Hahn, J. Rothberg, B. J. Beattie and J. Grimm, Am. J. Nucl. Med. Mol. Imaging, 2012, 2, 163–173 Search PubMed.
- B. M. Paterson, G. Buncic, L. E. McInnes, P. Roselt, C. Cullinane, D. S. Binns, C. M. Jeffery, R. I. Price, R. J. Hicks and P. S. Donnelly, Dalton Trans., 2015, 44, 4901–4909 Search PubMed.
- P. M. Smith-Jones, R. Fridrich, T. A. Kaden, I. Novak-Hofer, K. Siebold, D. Tschudin and H. R. Maecke, Bioconjugate Chem., 1991, 2, 415–421 Search PubMed.
- D. L. Bailey, K. P. Willowson, M. Harris, C. Biggin, A. Aslani, N. A. Lengkeek, J. Stoner, M. E. Eslick, H. Marquis, M. Parker, P. J. Roach and G. P. Schembri, J. Nucl. Med., 2023, 64, 704 Search PubMed.
- O. Keinanen, K. Fung, J. M. Brennan, N. Zia, M. Harris, E. van Dam, C. Biggin, A. Hedt, J. Stoner, P. S. Donnelly, J. S. Lewis and B. M. Zeglis, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 28316–28327 Search PubMed.
- J. Pougoue Ketchemen, F. N. Njotu, H. Babeker, S. Ahenkorah, A. F. Tikum, E. Nwangele, N. Henning, F. Cleeren and H. Fonge, Eur. J. Nucl. Med. Mol. Imaging, 2024, 51, 2070–2084 Search PubMed.
- G. Hao, T. Mastren, W. Silvers, G. Hassan, O. K. Öz and X. Sun, Sci. Rep., 2021, 11, 3622 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Mass spectra, mouse imaging and biodistribution data. See DOI: https://doi.org/10.1039/d4sc06969b |
| ‡ Deceased. |
| This journal is © The Royal Society of Chemistry 2025 |