
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSub-m-benziporphyrin: a subcarbaporphyrinoid and its BIII complex with an unprecedented planar tridentate 14π-aromatic network†
Le
Liu
a,
Shuangji
Song
a,
Jiyeon
Lee
b,
Yutao
Rao
a,
Ling
Xu
a,
Mingbo
Zhou
a,
Bangshao
Yin
a,
Juwon
Oh
c,
Jiwon
Kim
 *b,
Atsuhiro
Osuka
*a and
Jianxin
Song
*a
*b,
Atsuhiro
Osuka
*a and
Jianxin
Song
*a
aKey Laboratory of Chemical Biology and Traditional Chinese Medicine, Ministry of Educational of China, Key Laboratory of the Assembly and Application of Organic Functional Molecules of Hunan Province, College of Chemistry and Chemical Engineering, Hunan Normal University, Changsha 410081, China. E-mail: jxsong@hunnu.edu.cn; atsuhiroosuka@hunnu.edu.cn
bSchool of Integrated Technology, College of Computing, Integrated Science and Engineering Division, Underwood International College, Integrative Biotechnology and Translational Medicine, Graduate School, Yonsei University, Incheon 21983, Korea. E-mail: jiwon.kim@yonsei.ac.kr
cDepartment of Chemistry, Soonchunhyang University, Asan 31538, Korea
First published on 3rd December 2024
Abstract
Sub-m-benziporphyrins were synthesized by Pd-catalyzed cross-coupling of α,α′-diboryl-m-benzitripyrrane with 9,10-bis(1,1-dibromomethylenyl)anthracene. Reaction of sub-m-benziporphyrin with PhBCl2 and triethylamine gave its B-phenyl complex as a tetracoordinate nonaromatic BIII complex. In contrast, the reaction with BBr3 and triethylamine furnished a neutral BIII porphyrinoid with a planar and triangular coordination as the first example, in which the m-phenylene unit was partially reduced, allowing for the global 14π-aromatic circuit. This aromatic BIII complex is stable and inert towards nucleophiles such as pyridine, 4-dimethylaminopyridine, and fluoride anions but undergoes an oxygen-insertion reaction upon refluxing in the air. In addition, this BIII complex displays structured vibronic Q-bands, slow S1-state decay, and fluorescence (ΦF = 0.30 and τF = 9.7 ns), in line with its aromatic nature, while the nonaromatic BIII complexes show ill-defined absorption spectra and very fast S1-state decays.
Introduction
The chemistry of BIII subporphyrins started with our first synthesis in 2006.1 Since then, their appealing attributes have been extensively explored,2–6 which include 14π-aromatic electronic circuit, large substituent effects of meso-aryl substituents,7–9 and relatively strong S2-fluorescence;10 they are used in the synthesis of stable subporphyrin B-hydrides,11 and as effective dyes in dye-sensitized solar cells, achieving over 10% efficiency in the first trial.12 Importantly, BIII porphyrinoids so far explored have all four-coordinated structures, as seen for BIII subporphyrins,1 BIII subphthalocyanines,6 and other BIII porphyrinoids.13,14 The typical structure of BIII subporphyrins is a bowl shape tetracoordinated to three pyrrolic nitrogen atoms and an axial ligand. There has been no example of neutral tridentate BIII complexes of porphyrinoids, while cationic tridentate BIII complexes were reported as borenium cations.15,16On the other hand, sterically non-hindered triarylboranes are usually air and moisture sensitive17 but “structurally constrained” triarylboranes are known as exceptionally stable species,18–22 in which an enforced planar geometry endows a particular stability. This strategy has been extended to directly diphenylborane-fused porphyrins.23 In this paper, we report the first example of a neutral tridentate BIII subporphyrinoid.
Considering the rich and important chemistry of core-modified porphyrins,24–31 the exploration of core-modified BIII subporphyrins is highly desirable but such compounds have been rare so far. As a core-modified variant, Latos-Grażyński et al. reported sub-m-pyriporphyrin as the first core-modified subporphyrin, which is a stable free base with a non-aromatic character.32
In 2022, we reported the first synthesis of subporphyrin free bases 3 and 4 by the Suzuki–Miyaura cross coupling reaction between α,α′-diborylated tripyrrane 1 and 9,10-bis(1,1-dibromomethylenyl)anthracene 2 (Scheme 1), where the use of 2 was crucial.33 This synthetic success encouraged us to consider that a similar cross coupling reaction of α,α′-diboryl-m-benzitripyrrane 5 with 2 may allow for the synthesis of sub-m-benziporphyrins as an example of subcarbaporphyrinoid. Carbaporphyrins are porphyrin analogues, in which one or more of the pyrrolic nitrogen atoms are replaced by carbon atoms.31,34 These porphyrinoids exhibit a characteristic coordination behavior to stabilize higher oxidation states of transition metals owing to the inner carbon atom.31,34 Carbaporphyrins include N-confused porphyrins (NCP),34,35 benziporphyrins,24–27,30,31 and expanded carbaporphyrins36–38 but subcarbaporphyrinoids have been unprecedented.39
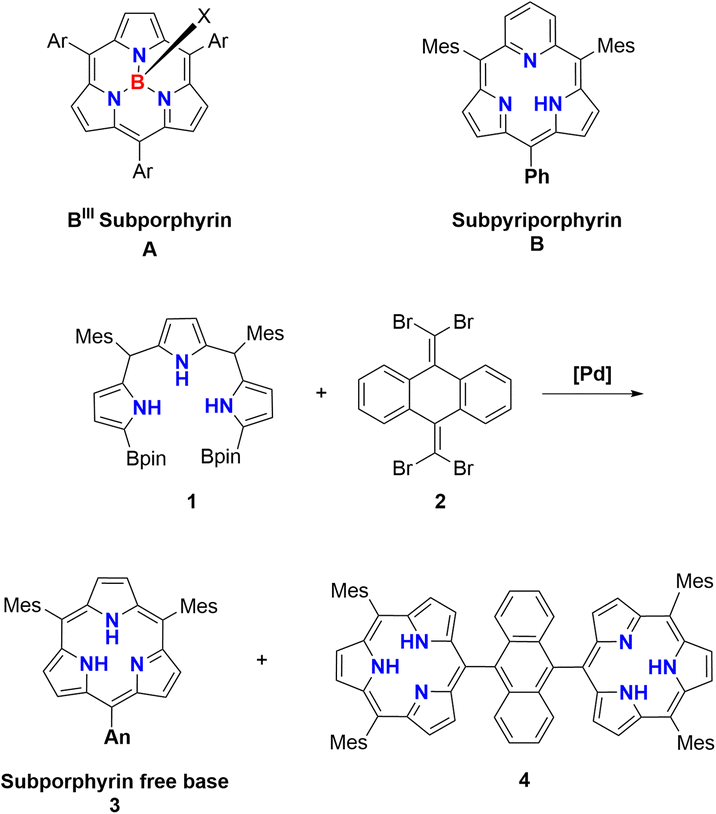 | ||
| Scheme 1 Synthesis of subporphyrin free bases. | ||
Results and discussion
Suzuki–Miyaura cross coupling of 5 with 2 (SPhos Pd G2, K3PO4, toluene/DMF, 115 °C, 48 h) and subsequent oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) gave a complicated mixture, from which sub-m-benziporphyrin 6 (6%) and anthracene-bridged sub-m-benziporphyrin dimer 7 (3%) were isolated (Scheme 2). The structure of 6 has been revealed by single-crystal X-ray diffraction analysis to be nonplanar, being roughly similar to that of the subporphyrin free base33 (Fig. 1a and b). The amino-pyrrole B and the m-phenylene unit C are directed to the opposite side, probably to avoid the steric congestion between the inner NH and CH. The nonplanar structure of 6 is indicated by relatively large dihedral angles: 41.97(6)° between the pyrroles A and B; 40.43(7)° for the pyrrole B and the m-phenylene C; and 39.16(16)° for the m-phenylene and the pyrrole A. The C(1)–C(16) and C(5)–C(6) bond distances are 1.499(3) and 1.497(3) Å, underscoring that the m-phenylene unit is not conjugated with the dipyrrin unit. The 1H NMR spectrum of 6 shows a singlet at 19.54 ppm due to the inner pyrrolic N–proton, a pair of doublets at 6.67 ppm and 6.18 ppm due to the pyrrolic β-protons and a broad signal at 6.65 ppm due to the inner proton of the m-phenylene unit. The NICS value40 in the center of the optimized structure 6 has been calculated to be 1.15 ppm (see ESI Fig. S34†), being consistent with the assignment as a nonaromatic species. High-resolution MALDI-TOF mass spectroscopy detected the parent ion peak of 7 at m/z = 1134.5505 (calcd for (C84H70N4)+ = 1134.5595([M]+). In the 1H NMR spectrum of 7, two singlets are observed for the inner NH protons and the inner CH protons of the m-phenylene unit, reflecting the presence of two tautomers with respect to the positions of the inner NH protons. We tried the coupling reactions with other geminal dibromides such as 1,1-dibromro-2,2-diphenylethene but failed to get sub-m-benziporphyrin. These results underscore the importance of dibromide 2, similar to the synthesis of subporphyrin free bases.33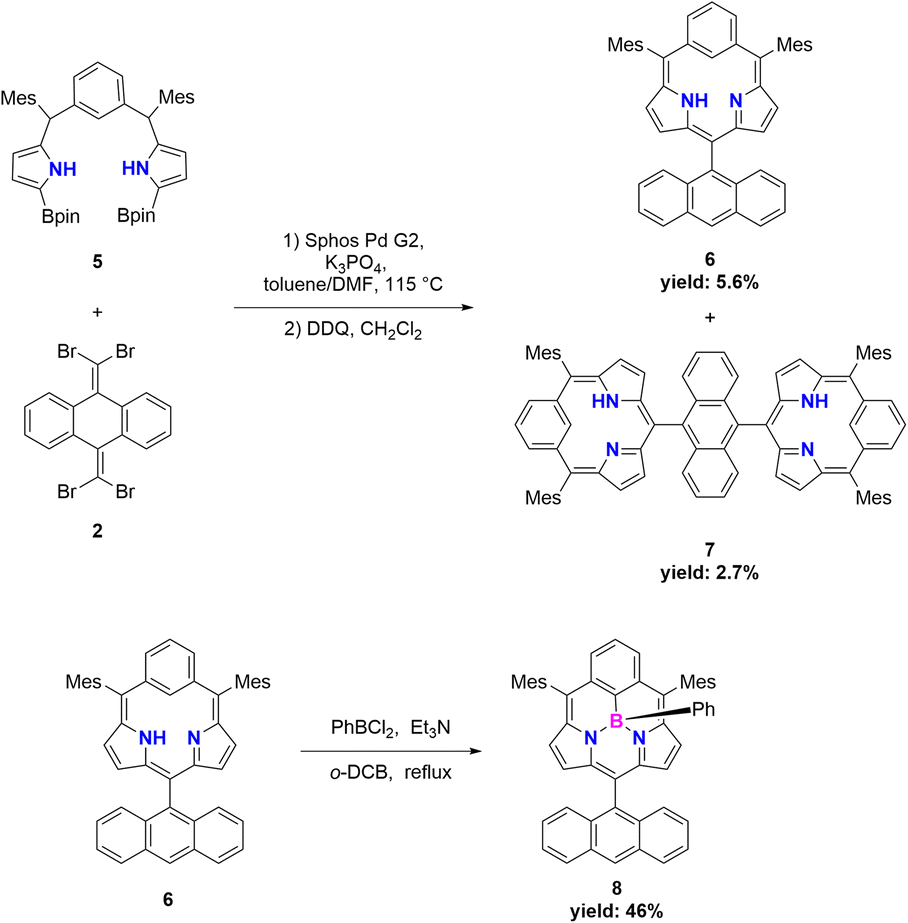 | ||
| Scheme 2 Synthesis of 6, 7, and 8. Mes = 2,4,6-trimethylphenyl, Bpin = pinacolatoboryl, DDQ = 2,3-dicyano-5,6-dichlorobenzoquinone. | ||
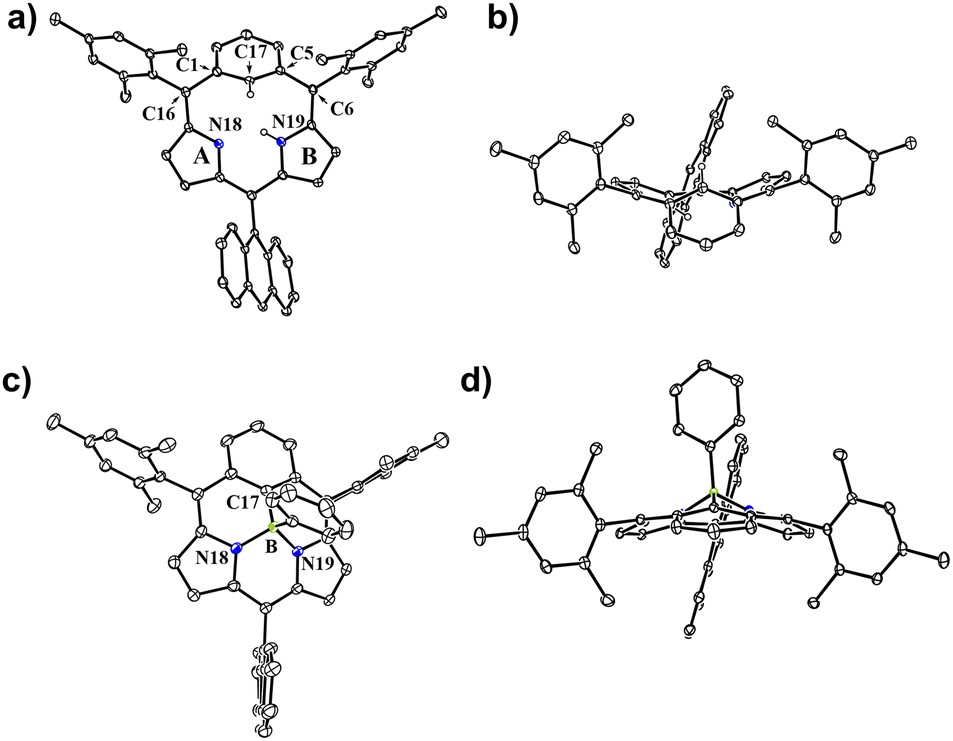 | ||
| Fig. 1 X-ray single-crystal structures of 6 and 8. (a) Top view of 6; (b) side view of 6; (c) top view of 8; (d) side view of 8. The thermal ellipsoids are scaled to the 30% probability level. All hydrogen atoms except those connected to N atoms are omitted for clarity. | ||
The boron-coordinating ability of 6 has been examined by the reaction with freshly distilled PhBCl2 in a boiling mixture of o-dichlorobenzene and triethylamine (TEA), which gave B-phenyl BIII sub-m-benziporphyrin 8 in 46% yield (Scheme 2). The structure of 8 has been confirmed by X-ray analysis to be a tetra-coordinate bowl-shape similar to those of BIII subporphyrins1 (Fig. 1c and d). The bowl-depth, defined by the distance between the boron atom and the mean plane of the four peripheral β-carbons and two carbons at the positions 3 and 5 of the benzene, is 1.321(17) Å, which is distinctly shorter than that (ca. 1.42 Å) of the typical BIII subporphyrins. The bond lengths of the B–N(18), B–N(19), and B–C(17) are 1.515(17) Å, 1.523(17) Å, and 1.575(19) Å, respectively. The B–C(17) bond length is distinctly longer than those (1.500(5) Å) of the B–N in the typical BIII subporphyrins. The 1H NMR spectrum of 8 shows a pair of doublets at 6.64 ppm and 6.10 ppm due to the pyrrolic β-protons, indicating the nonaromatic character of 8.
In the next step, we examined the reaction of 6 with BBr3 in a boiling mixture of o-dichlorobenzene and TEA, which afforded BIII sub-m-benziporphyrin 9 (51%) and B-hydroxy sub-m-benziporphyrin 10 (21%) (Scheme 3). The formation of 9 was unexpected but interesting. The structure of 9 has been unambiguously revealed by X-ray analysis to be a planar triangle coordination structure without an axial group (Fig. 3a and b), entirely different from the bowl-shaped structures of 8 and the usual BIII subporphyrins. Mean-plane deviation (MPD) of 9 is very small (0.15 Å). The bond lengths of the B–N(18), B–N(19), and B–C(17) are 1.396(5) Å, 1.385(5) Å, and 1.474(5) Å, respectively, being noticeably shorter than those of the complex 8. It is worth noting that 9 is the first example of tri-coordinated planar BIIIporphyrinoid. Importantly, the m-phenylene unit is reduced to an exo-methylene cyclohexadiene unit, allowing for the global 14π-aromatic electronic network as indicated in blue (Scheme 3). In line with this 14π-aromatic circuit, the 1H NMR spectrum of 9 displays signals due to the β-protons at 7.58, 7.56, 7.45, and 7.41 ppm apparently in a downfield region, indicating a diatropic ring current, and signals due to the vinylene protons at 6.67 and 6.25 ppm (Fig. 2). The NICS values in the inner region of the optimized structure are calculated to be −9.58 ∼ −11.64 ppm (see ESI Fig. S36†). The structure of 10 has been confirmed by X-ray analysis to be analogous to that of 8 as shown in Fig. 3c and d. We also examined the reaction of 6 with only BBr3, which gave 10 almost quantitatively.
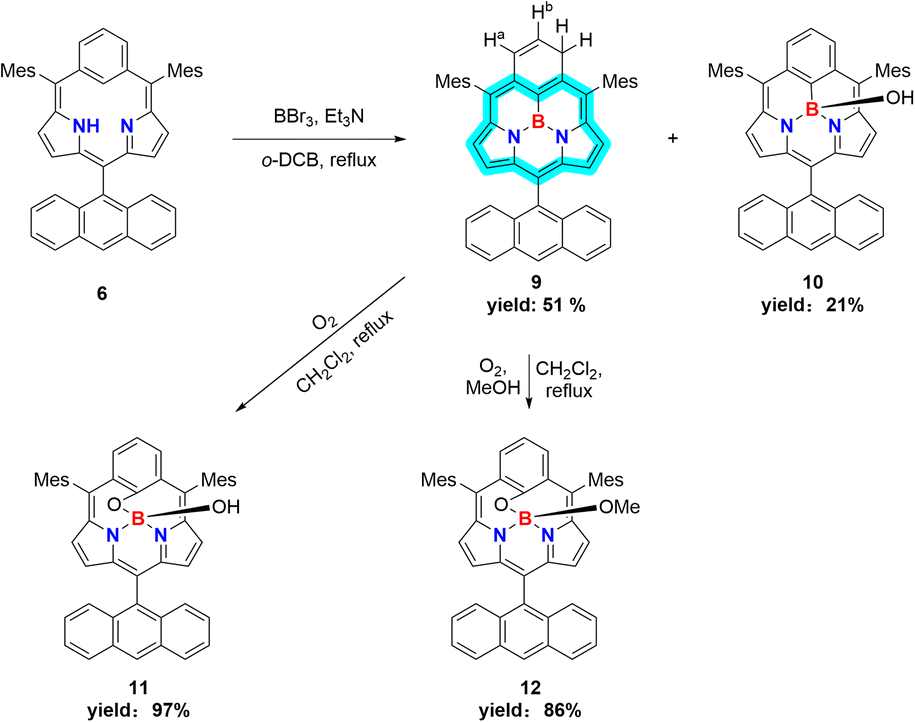 | ||
| Scheme 3 Synthesis of BIII sub-m-benziporphyrins 9, 10, 11, and 12. | ||
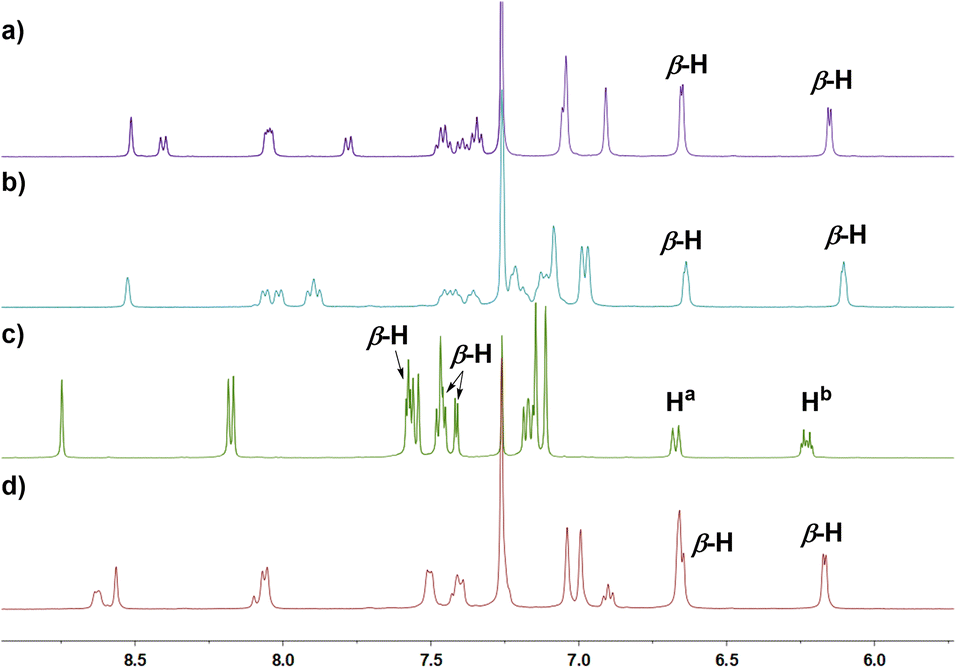 | ||
| Fig. 2 1H NMR spectra of (a) 6 in CDCl3 at 293 K; (b) 8 in CDCl3 at 293 K; (c) 9 in CDCl3 at 293 K; (d) 11 in CDCl3 at 293 K. | ||
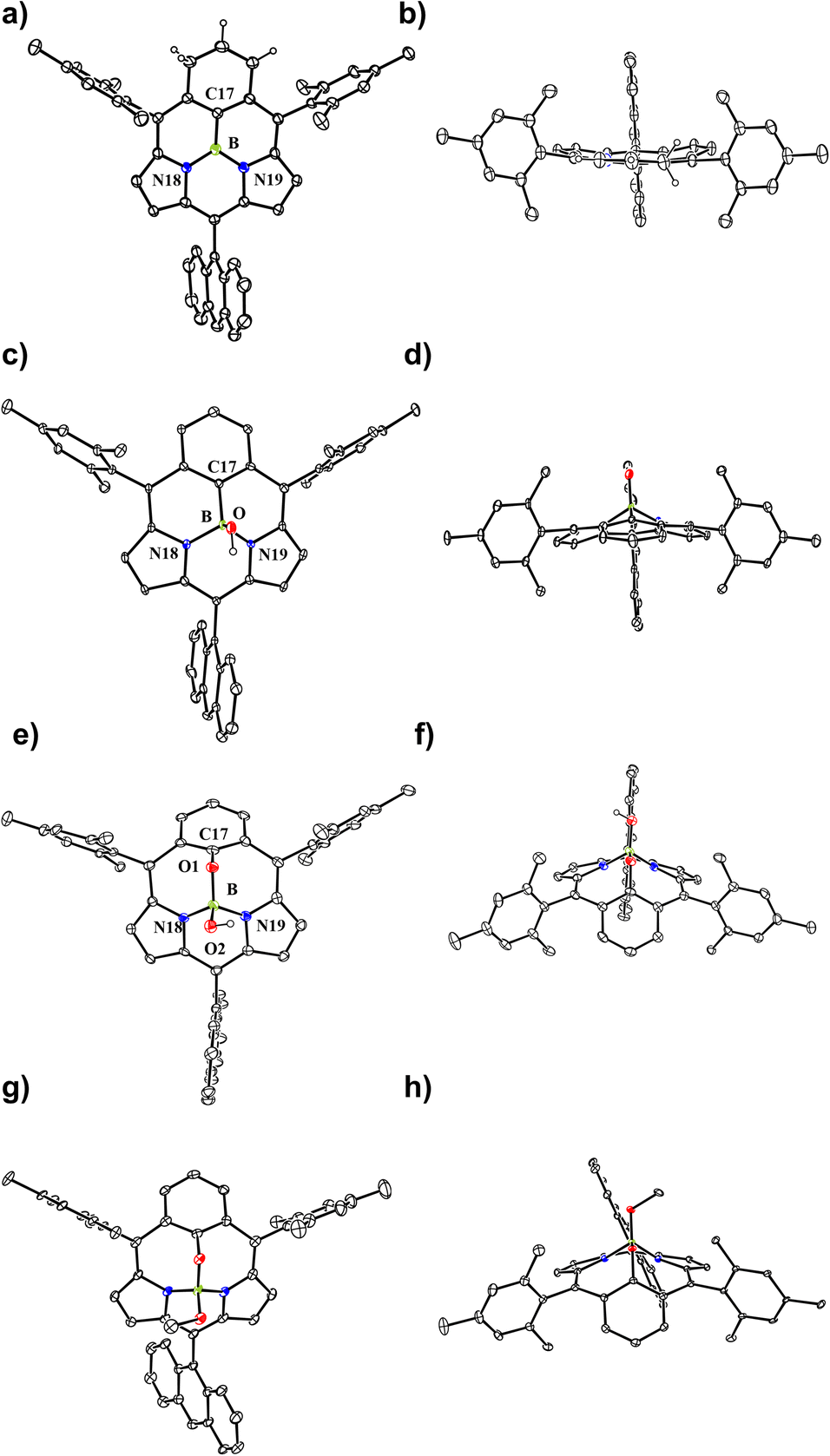 | ||
| Fig. 3 X-ray single-crystal structures of 9, 10, 11, and 12. (a) Top view of 9; (b) side view of 9; (c) top view of 10; (d) side view of 10. (e) Top view of 11; (f) side view of 11; (g) top view of 12; (h) side view of 12. The thermal ellipsoids are scaled to the 30% probability level. All hydrogen atoms except those connected to N and O atoms are omitted for clarity. | ||
In addition, it is worth noting that 9 exhibits high chemical stability under ambient conditions. Interestingly, 9 does not react with nucleophiles such as pyridine, 4-dimethylaminopyridine, or tetrabutylammonium fluoride as a rare case of neutral BIII compounds. On the other hand, refluxing of a solution of 9 in CH2Cl2 in the air for 48 h gave oxygen-inserted product 11 almost quantitatively. The structure of 11 has been revealed by X-ray analysis to have an inserted oxygen atom between the boron and ipso-carbon of the m-phenylene unit and the central BIII has an axial hydroxy group (Fig. 3e and f). The bond lengths are 1.366(4) Å for C(17)–O(1), 1.548(5) Å for B–N(18), 1.551(5) Å for B–N(19), 1.429(5) Å for B–O(1), and 1.416(5) Å for B–O(2). The extremely short C(17)–O(1) bond is arising from the small cavity. The 1H NMR spectrum of 11 shows a pair of doublets at 6.64 ppm and 6.17 ppm due to the pyrrolic β-protons, indicating the nonaromatic character. The oxygenation of 9 was conducted in the presence of methanol to give 12, which carries an axial methoxy group as evinced by X-ray analysis. The bond length of C(17)–O(1) is also short (1.358(4) Å).
The UV-vis absorption spectra of 6, 7, 8, 9, 11, and 12 are shown in Fig. 4. Monomer 6 shows broad bands at 384 and 673 nm and dimer 7 shows nearly an identical spectrum with twice the extinction coefficients, indicating negligible electronic interaction between the two sub-benziporphyrin units. Compared to 6, 8 exhibits a similar band at 388 nm and a red-shifted and broad band at 803 nm. In contrast, the absorption spectrum of 9 is quite different, displaying sharp bands at 335 and 356 nm and sharp vibronic-structured bands at 511 and 546 nm. Moreover, 9 emits fluorescence at 559 nm with ΦF = 0.30 and τF = 9.7 ns (see ESI Fig. S38 and S46†). These contrasting optical properties of 9 can be attributed to its distinct aromatic nature. The electron density delocalized bond (EDDB) analysis visualizes nonaromatic cyclic π-conjugation for 6 and 8 (see ESI Fig. S40 and S41†).41,42 On the basis of these electronic structures, the broad lowest absorption bands of 6 and 8 are ascribed primarily to the HOMO–LUMO transitions (see ESI Fig. S43 and S44†). On the other hand, the cyclic delocalization feature is shown for 9 by EDDB (see ESI Fig. S42†), indicating that its vibronic-structured absorption bands and intense fluorescence come from the aromatic nature.43–45 In this regard, the similar absorption spectral features of 11 and 12 to 6 are consistent with their nonaromatic nature.
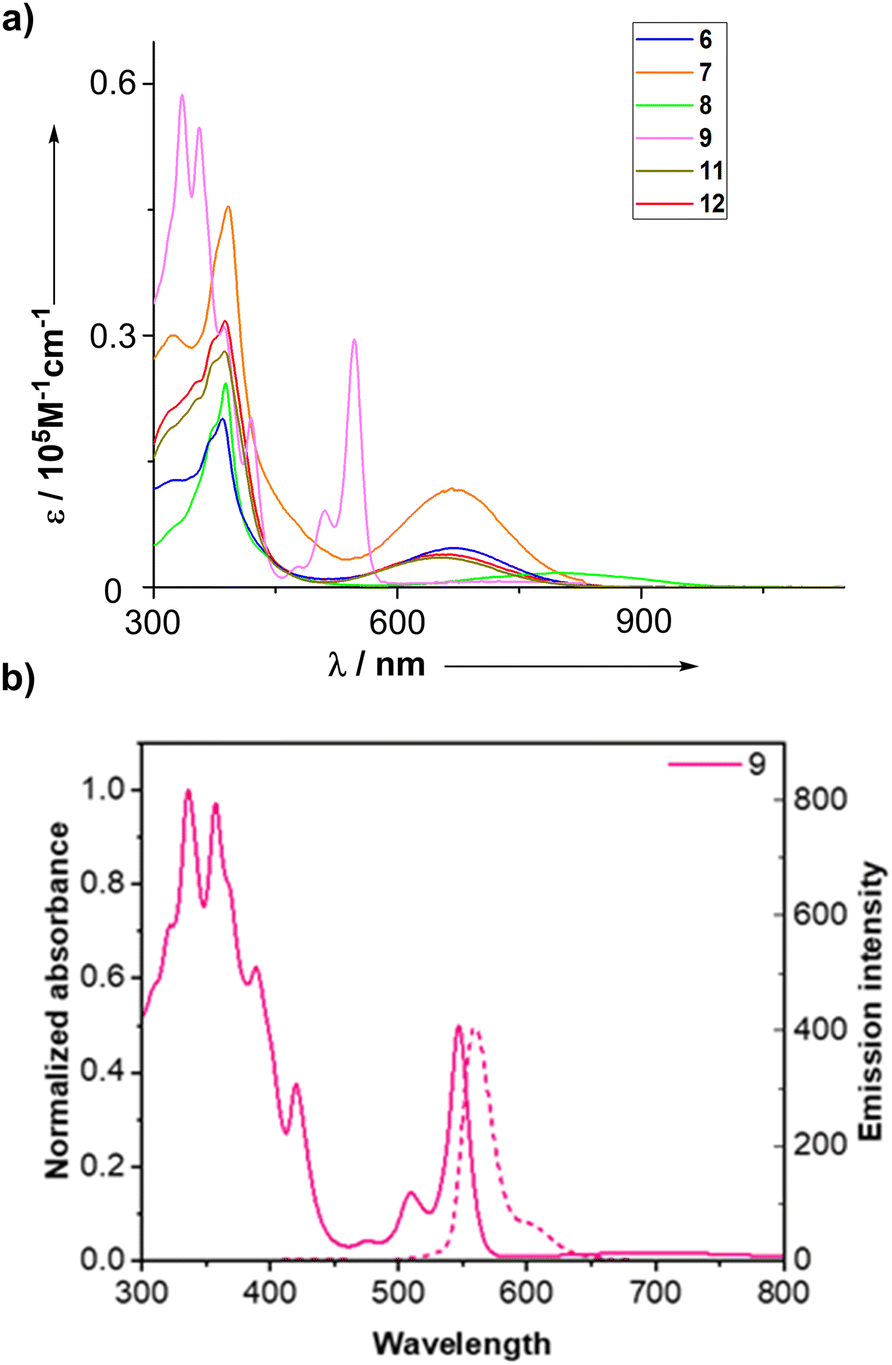 | ||
| Fig. 4 (a) UV absorption spectra of 6, 7, 8, 9, 11, and 12. (b) UV-vis and fluorescence spectra of 9 with solid and dotted lines, respectively. | ||
We further measured transient absorption (TA) spectra of 6, 8, and 9 to investigate their excited-state dynamics (Fig. 5). The TA spectra of nonaromatic 6 and 8 rapidly decayed within tens of picoseconds. This is consistent with their nonaromatic nature, since such species show fast non-radiative decays. In sharp contrast, substantially long-live TA spectra were observed for 9, which decayed double exponentially with time constants of 10.7 ns and longer than 5 μs. Here, the faster decay is well matched with its fluorescence lifetime, being assigned as the S1 state decay. The long-lived TA signal can be assigned as the T1 state, since it is considered that the long-lived S1-state may have an ample time to undergo intersystem crossing to give the T1 state.
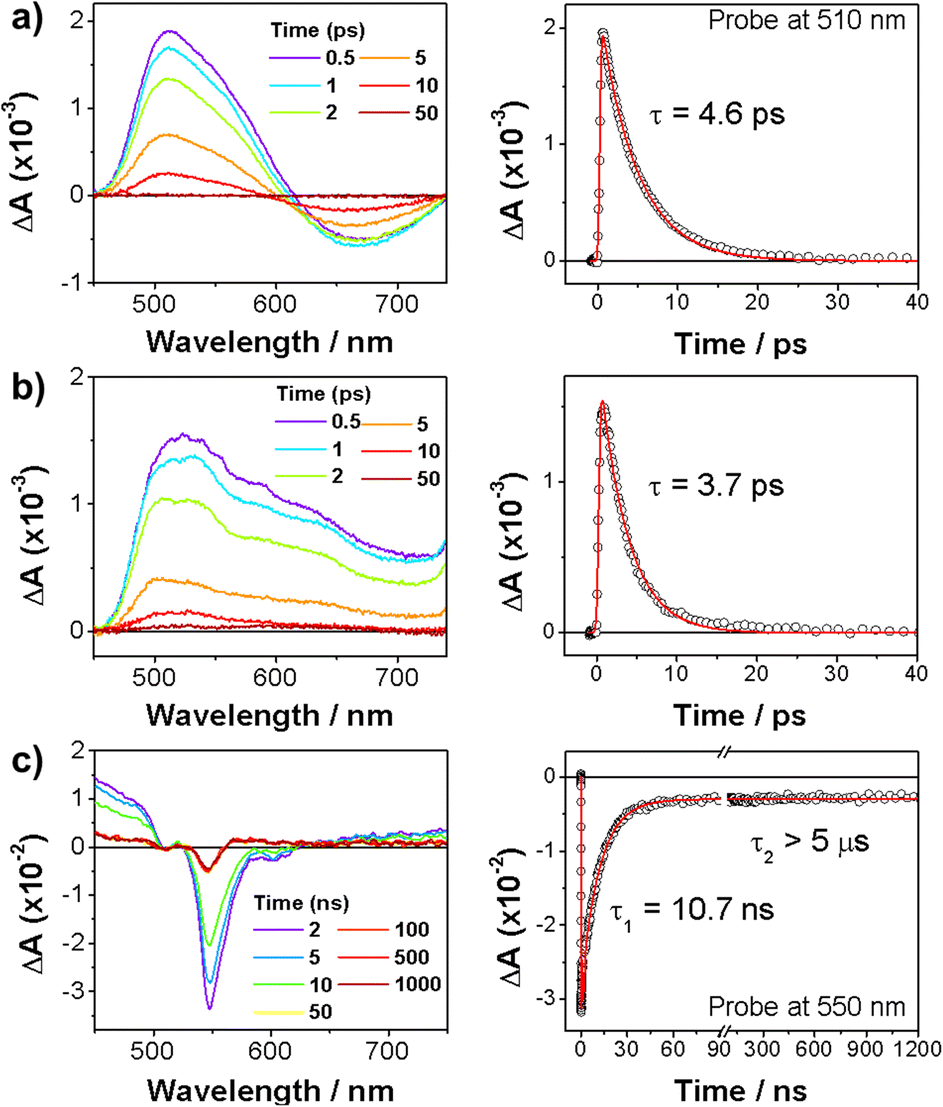 | ||
| Fig. 5 TA spectra (left) and decay profiles (right) of (a) 6, (b) 8, and (c) 9 in Ar-bubbled toluene with photoexcitation at 400 nm. | ||
Conclusions
In summary, sub-m-benziporphyrins were synthesized by the cross-coupling of α,α′-diboryl-m-benzitripyrrane with 9,10-bis(1,1-dibromomethylenyl)anthracene as an example of subcarbaporphyrinoid. The sub-m-benziporphyrin was converted to a B-phenyl BIII complex upon treatment with PhBCl2 and TEA. These sub-m-benziporphyrins and their BIII complexes are nonaromatic owing to the cross-conjugated networks as evinced by their 1H NMR spectra and absorption spectra. In contrast, the reaction of sub-m-benziporphyrin with BBr3 and TEA gave a BIII complex that showed a planar and tridentate coordination of BIII as the first example of neutral BIII porphyrinoid. In addition, the m-phenylene unit is concurrently reduced to an exo-methylene cyclohexadiene unit, which allows the global 14π-aromatic circuit. The aromatic BIII complex is stable towards nucleophiles but undergoes oxygen-insertion reaction upon treatment with the atmospheric oxygen at reflux in CH2Cl2. While the nonaromatic BIII complexes 6 and 8 show ill-defined absorption spectra and very fast S1-state decays, the aromatic BIII complex 9 displays structured Q-bands, slow S1-state decay, and fluorescence. The exploration of novel ring-contracted carbaporphyrins with novel reactivities and properties is ongoing in our laboratory.Data availability
The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2379624 (for 6), 2379625 (for 8), 2379626 (for 9), 2379627 (for 10), 2379628 (for 11), and 2379629 (for 12). All experimental data and detailed experimental procedures are available in the ESI.†Author contributions
J. Song designed and conducted the project. L. Liu, S. Song, J. Lee, J. Oh, and Y. Rao performed the synthesis and characterization, and measured the optical and electrochemical properties. L. Xu, M. Zhou, and B. Yin performed X-ray diffraction analysis and DFT calculations. J. Kim, A. Osuka, and J. Song prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work is supported by the National Natural Science Foundation of China (grant no. 22471070, 22471071, 22071052, 22201072, and 22271091), Science and Technology Planning Project of Hunan Province (2018TP1017), and Science and Technology Innovation Program of Hunan Province (2021RC4059). The work is also supported by the NRF funded by the Korea Government (MSIT) (no. RS-2023-00240624, RS-2024-00343229, and 2021R1A6A1A03039503).Notes and references
- Y. Inokuma, J.-H. Kwon, T. K. Ahn, M.-C. Yoo, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2006, 45, 961 CrossRef CAS PubMed.
- Y. Inokuma and A. Osuka, Dalton Trans., 2008, 2517 RSC.
- A. Osuka, E. Tsurumaki and T. Tanaka, Bull. Chem. Soc. Jpn., 2011, 84, 679 CrossRef CAS.
- C. G. Claessens, D. González Rodríguez, M. S. Rodríguez-Morgade, A. Medina and T. Torres, Chem. Rev., 2014, 114, 2192 CrossRef CAS.
- S. Shimizu, Chem. Rev., 2017, 117, 2730 CrossRef CAS PubMed.
- G. Lavarda, J. Labella, M. V. Martínez-Díaz, M. S. Rodríguez-Morgade, A. Osuka and T. Torres, Chem. Soc. Rev., 2022, 51, 9482 RSC.
- Y. Inokuma, Z. S. Yoon, D. Kim and A. Osuka, J. Am. Chem. Soc., 2007, 129, 4747 CrossRef CAS.
- Y. Inokuma, S. Easwaramoorthi, Z. S. Yoon, D. Kim and A. Osuka, J. Am. Chem. Soc., 2008, 130, 12234 CrossRef CAS.
- Y. Inokuma, S. Easwaramoorthi, S. Y. Jang, K. S. Kim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 4840 CrossRef CAS.
- J. Sung, P. Kim, S. Saga, S. Hayashi, A. Osuka and D. Kim, Angew. Chem., Int. Ed., 2013, 52, 12632 CrossRef CAS.
- E. Trurumaki, J. Sung, D. Kim and A. Osuka, Subporphyrinato boron(III) hydrides, J. Am. Chem. Soc., 2015, 137, 1056 CrossRef.
- G. Copley, D. Hwang, K. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 10287 CrossRef CAS.
- W. J. Belcher, P. D. W. Boyd, P. J. Brothers, M. J. Liddle and C. E. F. Rickard, J. Am. Chem. Soc., 1994, 116, 8416 CrossRef CAS.
- P. J. Brothers, Chem. Commun., 2008, 2090 RSC.
- E. Tsurumaki, S. Hayashi, F. S. Tham, C. A. Reed and A. Osuka, J. Am. Chem. Soc., 2011, 133, 11956 CrossRef CAS PubMed.
- T. Kato, F. S. Tham, P. D. Boyd and C. A. Reed, Heteroat. Chem., 2006, 17, 209 CrossRef CAS.
- F. Zettler, H. D. Hausen and H. Hess, Organomet. Chem., 1974, 72, 157 CrossRef CAS.
- Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529 CrossRef CAS.
- S. Saito, K. Matsuo and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 9130 CrossRef CAS PubMed.
- C. Dou, S. Saito, K. Matsuo, I. Hisaki and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 12206 CrossRef CAS PubMed.
- T. Kushida, C. Camacho, A. Shuto, S. Irle, M. Muramatsu, T. Katayama, S. Ito, Y. Nagasawa, H. Miyasaka, E. Sakuda, N. Kitamura, Z. Zhou, A. Wakamiya and S. Yamaguchi, Chem. Sci., 2014, 5, 1296 RSC.
- K. Matsuo, S. Saito and S. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 12580 CrossRef CAS PubMed.
- K. Fuimoto, J. Oh, H. Yorimitsu, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 3196 CrossRef.
- K. Berlin and E. Breitmaier, Angew. Chem., Int. Ed., 1994, 33, 1246 CrossRef.
- M. Stȩpień and L. Latos-Grażyński, Acc. Chem. Res., 2005, 38, 88 CrossRef.
- M. Stȩpień and L. Latos-Grażyński, Chem.–Eur. J., 2001, 7, 5113 CrossRef.
- M. Stȩpień and L. Latos-Grażyński, J. Am. Chem. Soc., 2002, 124, 3838 CrossRef PubMed.
- B. Szyszko, M. J. Bialek, E. Pacholska-Dudziak and L. Latos-Grażyński, Chem. Rev., 2017, 117, 2839 CrossRef CAS PubMed.
- T. D. Lash, S. T. Chaney and D. T. Richter, J. Org. Chem., 1998, 63, 9076 CrossRef CAS.
- T. D. Lash, Org. Biomol. Chem., 2015, 13, 7846 RSC.
- T. D. Lash, Chem. Rev., 2017, 117, 2313 CrossRef CAS PubMed.
- R. Mysliborski, L. Latos-Grażyński, L. Szterenberg and T. Lis, Angew. Chem., Int. Ed., 2006, 45, 3670 CrossRef CAS PubMed.
- L. Liu, J. Kim, L. Xu, Y. Rao, M. Zhou, B. Yin, J. Oh, D. Kim, A. Osuka and J. Song, Angew. Chem., Int. Ed., 2022, 61, e202214342 CrossRef CAS.
- N. J. Bialek, K. Hurej, H. Furuta and L. Latos-Grażyński, Chem. Soc. Rev., 2023, 52, 2082 RSC.
- M. Toganoh and H. Furuta, Chem. Commun., 2012, 48, 937 RSC.
- Y. Hisamune, K. Nishimura, K. Isakari, M. Ishida, S. Mori, S. Karsawa, T. Kato, S. Lee, D. Kim and H. Furuta, Angew. Chem., Int. Ed., 2015, 54, 7323 CrossRef CAS PubMed.
- T. Tanaka and A. Osuka, Chem. Rev., 2017, 117, 2584 CrossRef CAS.
- F. Luo, L. Liu, H. Wu, L. Xu, Y. Rao, M. Zhou, A. Osuka and J. Song, Nat. Commun., 2023, 14, 5028 CrossRef CAS PubMed.
- T. D. Lash, L. M. Stateman and D. I. AbuSalim, J. Org. Chem., 2019, 84, 14733 CrossRef CAS.
- Z. Cen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef.
- D. W. Szczepanik, M. Andrzejak, K. Dyduch, E. Żak, M. Makowski, G. Mazur and J. Mrozek, Phys. Chem. Chem. Phys., 2014, 16, 20514 RSC.
- D. W. Szczepanik, M. Andrzejak, J. Dominikowska, B. Pawełek, T. M. Krygowski, H. Szatylowicz and M. Solá, Phys. Chem. Chem. Phys., 2017, 19, 28970 RSC.
- J. Y. Shin, K. S. Kim, M. C. Yoon, J. M. Lim, Z. S. Yoon and D. Kim, Chem. Soc. Rev., 2010, 39, 2751 RSC.
- J. Oh, Y. M. Sung, Y. Hong and D. Kim, Acc. Chem. Res., 2018, 51, 1349 CrossRef CAS PubMed.
- J. Kim, J. Oh, A. Osuka and D. Kim, Chem. Soc. Rev., 2022, 51, 268 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2379624 (for 6), 2379625 (for 8), 2379626 (for 9), 2379627 (for 10), 2379628 (for 11), and 2379629 (for 12). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07199a |
| This journal is © The Royal Society of Chemistry 2025 |