
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity of canonical bacterial cytochrome c peroxidases: insights into the electronic structure of compound I†‡
Patrick Hewitta,
Michael P. Hendrich b and
Sean J. Elliott*a
b and
Sean J. Elliott*a
aDepartment of Chemistry, Boston University, 24 Cummington Mall, Boston, MA 02215, USA. E-mail: elliott@bu.edu
bDepartment of Chemistry, Carnegie Mellon University, Pittsburgh, PA 15213, USA
First published on 26th February 2025
Abstract
Bacterial cytochrome c peroxidase (bCcP) family members include di-heme enzymes that are capable of producing various high oxidation states in their reactions with the substrate H2O2. Canonical family members such as the enzyme from Nitrosomonas europaea (Ne) are responsible for the detoxification of H2O2 in the periplasm of many Gram negative organisms. Elucidation of the electronic structure of the kinetic intermediates for canonical bCcP enzymes has yet to be attained, though the orthologs MauG and others have evoked a Fe(IV)Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species capable of long-range oxidation. Here, we use a combination of optical, electron paramagnetic and Mössbauer spectroscopies to demonstrate that the species produced upon the reaction of the diferric form of Ne bCcP with H2O2 is a ferryl peroxidatic heme that is coupled to a porphyrinyl radical, with an unexpected exchange coupling constant J of −17 cm−1 (Hex = JSFe·Spor).
O species capable of long-range oxidation. Here, we use a combination of optical, electron paramagnetic and Mössbauer spectroscopies to demonstrate that the species produced upon the reaction of the diferric form of Ne bCcP with H2O2 is a ferryl peroxidatic heme that is coupled to a porphyrinyl radical, with an unexpected exchange coupling constant J of −17 cm−1 (Hex = JSFe·Spor).
Introduction
Cytochrome c peroxidases (CcPs) are heme containing enzymes that catalyze the two-electron reduction of hydrogen peroxide to water. Where eukaryotic CcPs contain a single b-type heme, bacterial CcPs (bCcPs) contain 2 c-type hemes, are found in the periplasm of many Gram-negative bacteria, and have been appreciated for their diversity.1,2 The majority of characterized bCcPs are labelled as canonical bCcPs, which are generally classified by efficient peroxidatic activity and as such are purported to function in vivo as H2O2 scavengers in oxidative stress protection roles.3 However, some bCcP family members make use of H2O2 as an oxidant, which we will refer to as MauG-type bCcPs: the mau operon for methylamine utilization in Paracoccus denitrificans contains MauG, which is responsible for the oxidation of the precursor protein of methylamine dehydrogenase;4 methanobactin biosynthesis in Methylosinus trichosporium OB3b requires mbn encoded proteins such as MbnH, another bCcP which is thought to oxidatively install kynurenine on the protein MbnP;5 and BthA is found widely in Burkholderia spp. and possesses a cryptic activity.1 In the case of these enzymes, generally poor dye-linked peroxidase activity is observed, but they are known to produce a remarkable FeIVFeIVO species upon treatment of the diferric enzyme with H2O2. With respect to these enzymes, is the relatively poor understanding of the oxidants that they produce after reacting with H2O2; while the FeIVFeIVO of MauG, MbnH and BthA have been verified,1,4,6 no comparable study has been available for the cognate intermediates of typical bCcP enzymes. Here we begin to fill that knowledge gap.
The canonical bCcPs and MauG-type orthologs share similar overall structural topology, in terms of the core fold, and the presence of two c-type heme cofactors bound by the enzyme in CXXCH motifs (Fig. 1). One heme serves as the peroxidatic active site (the P heme), which is five coordinate in the active state of MauG-like bCcPs, and possess variable coordination in the case of canonical bCcPs; the other heme is six-coordinate (the E heme) and is less solvent accessible. Generally, the E heme is held to act as an electron transfer conduit from partner proteins to the P heme. Both an inter-heme tryptophan residue and a calcium binding site are highly conserved among both MauG-type and canonical bCcPs (Fig. 1). The tryptophan is thought to serve as an electron tunneling conduit for inter-heme electron transfer, and the presence of a bound calcium ion has been shown to be necessary for both dimerization and enzymatic activity in several canonical bCcPs.3 BthA is a notable exception as it does not contain the inter-heme tryptophan or the bound calcium ion.1
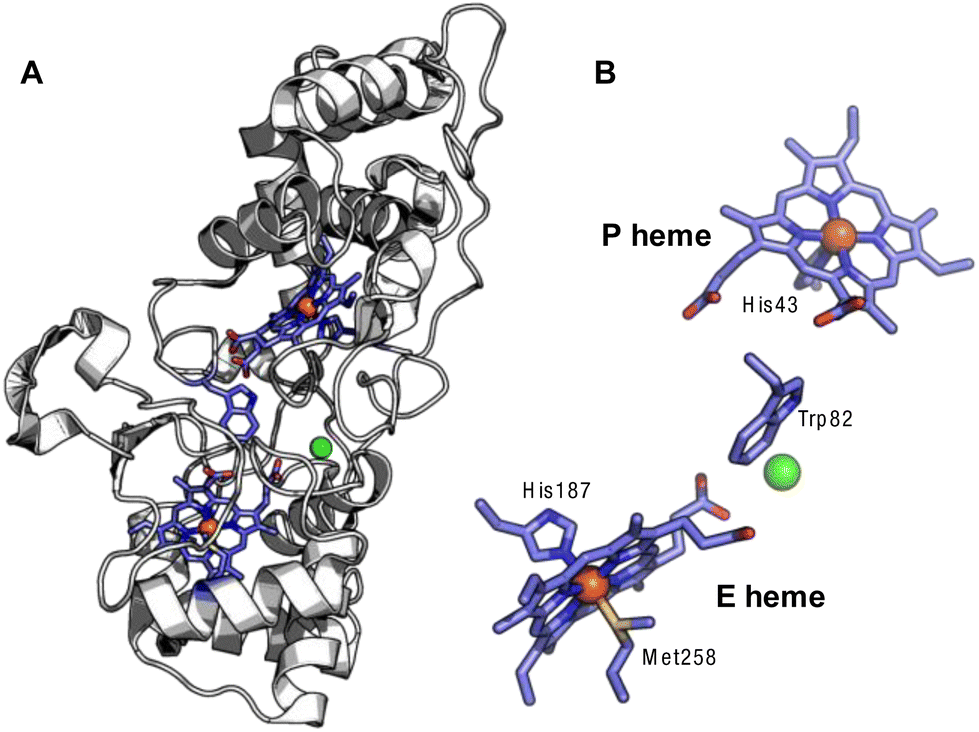 | ||
| Fig. 1 Crystal structure of the Ne bCcP. (A) Full view of the protein backbone with the c-type hemes and intervening tryptophan, with the Ca2+ ion in green (1IQC.pdb).7 (B) View of the E and P hemes with the intervening tryptophan and Ca2+. | ||
Canonical and MauG-type bCcPs are differentiated by the ligands coordinating the E heme. Canonical bCcPs possess His–Met ligation at the E heme, while the same heme of MauG-type bCcPs are His–Tyr ligated. The difference in E-heme coordination is associated with differences in the midpoint potential of the E heme. The E-heme of canonical bCcPs possess a much more positive redox potential (∼+200 to +450 mV vs. SHE, depending on the source) than that of the P-heme (∼−200 mV vs. SHE), while the E heme of MauG-type bCcPs lies closer to the potential of the P heme at ∼−100 to −200 mV vs. SHE. Collectively, this implies that canonical bCcPs can adopt a stable one-electron reduced state, because of the well separated potentials, while the MauG-type enzymes cannot, as their heme redox potentials are very similar.
Canonical bCcPs are subdivided into an activable class and a constitutively active class based on their redox requirements for peroxidatic activity. The activatable enzymes like those from Pseudomonas,8 Paracoccus9 and Shewanella10 require that the E-heme be pre-reduced to the ferrous state in order to trigger a conformational change in which a histidine ligand is displaced from a 6th coordinate position on the P heme, opening up the P heme for peroxide binding and priming one of the two electrons required for peroxide reduction (Scheme 1, top). The constitutively active enzymes (from Nitrosomonas europaea11 and Methylococcus capsulatus12) do not require pre-reduction, and the peroxidatic heme is accessible for peroxide binding when the enzyme is in the fully oxidized state (Scheme 1, middle).
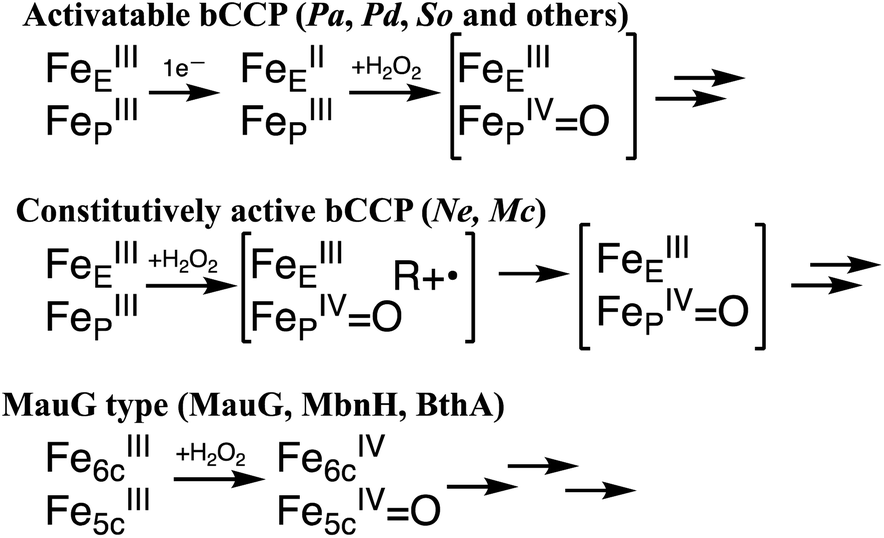 | ||
| Scheme 1 Possible redox states of the bCcP superfamily, where the bracketed intermediates have yet to be concretely identified. | ||
While the proposed high valent species of the constitutively active11 and the activatable13,14 bCcPs are supported by limited spectroscopic evidence, there is a lack of detailed electronic structural characterization of any of the high-valent intermediate species by Mössbauer or EPR spectroscopies. In contrast, the MauG-type of reactivity is well supported,1,4,6 and reaction of the diferric form of the enzyme is known to give rise to a ‘bis FeIV species’ where the six-coordinate (E) heme is formally oxidized to the FeIV oxidation state, and the peroxidatic five-coordinate heme (P) is ferryl (Scheme 1, bottom).
The manner in which the structural and electronic distinctions between MauG-type bCcPs and canonical bCcPs contribute to the observed differences in reactivity is poorly understood. For example, previous attempts by our lab to modulate reactivity of a MauG-type bCcP by mimicking canonical bCcP heme Met–His coordination at the E heme biased the enzyme towards unusual reactivity with molecular oxygen that resulted in the formation of a highly stable FeIVFeIVO species.15 A complementary effort in MauG appeared to eliminate formation of the bis-FeIV state and enable reactivity similar to that of the Ne enzyme.16
Here we utilize optical and magnetic (EPR and Mössbauer) spectroscopies to examine the electronic structure of the high-valent peroxidatic intermediates found in the reaction of the constitutively active Ne bCcP with H2O2. In doing so, we provide evidence for a compound I-like species comprising a porphyrin radical and the ferryl, peroxidatic heme for the first time. In doing so, our work establishes a refined appreciation of electronic structural features of canonical bCcP enzyme intermediates.
Results & discussion
Purification and properties of tag-free WT Ne bCcP
Purified fractions of a tag-free, recombinant WT Ne bCcP were observed to run as a single diffuse band by SDS-PAGE electrophoresis, as long as dithiothreitol (DTT) was used instead of the typical beta-mercapto ethanol (BME), with which samples ran as a closely spaced double band. The recombinant Ne bCcP has been found to be express with a full complement of hemes17 and here the tag-free version showed efficient peroxidase activity with ABTS as an electron donor with a KM of 0.34 μM for H2O2 and a kcat of 14.6 s−1. Ne bCcP purified under ambient conditions exhibited a mixture of di-ferric and semi-reduced states as inferred by inspection of the Soret and Q-bands. Treatment of the as-isolated enzyme with several equivalents of K2Ir(IV)Cl6 was sufficient to prepare the diferric enzyme. Semi-reduced samples (FeIIEFeIIIP) could be prepared by addition of an excess of ascorbate. The semi-reduced form was stable for long periods of time after removal of the ascorbate via a desalting column if the enzyme was kept in an anaerobic environment.Mapping the reactivity of a bacterial cytochrome c peroxidase
Initial probing of the reactivity of the fully oxidized, diferric (FeIIIEFeIIIP) state of Ne bCcP was achieved through optical absorption spectroscopy, and titrations with H2O2 (Fig. 2). Diferric Ne bCcP exhibited a broad Soret band with a peak near 404 nm and broad features in the Q-band region around 525 and 560 nm, with a weak feature at 625 nm attributed to a high-spin population of the P heme. The optical high spin-feature in bCcPs and its contribution to the active state of LP heme are not fully understood. Magnetic circular dichroism (MCD) studies of the Pa bCcP demonstrated that E heme undergoes high to low spin transitions based on both redox state and temperature, while the P heme is low-spin unless the enzyme is in the semi-reduced (activated) at room temperature, in which it is high-spin.18,19 However, resonance raman studies of both the semi-reduced (activated) So bCcP and oxidized Ne bCcP showed that both enzymes possessed predominantly low-spin E heme and P heme environments at room temperature.17 Due to the 625 nm band it is clear that some high-spin heme exists at room temperature, though due to the complexity of the phenomenon we cannot fully describe this behaviour and will attribute the high spin signal to a high-spin population of the P heme.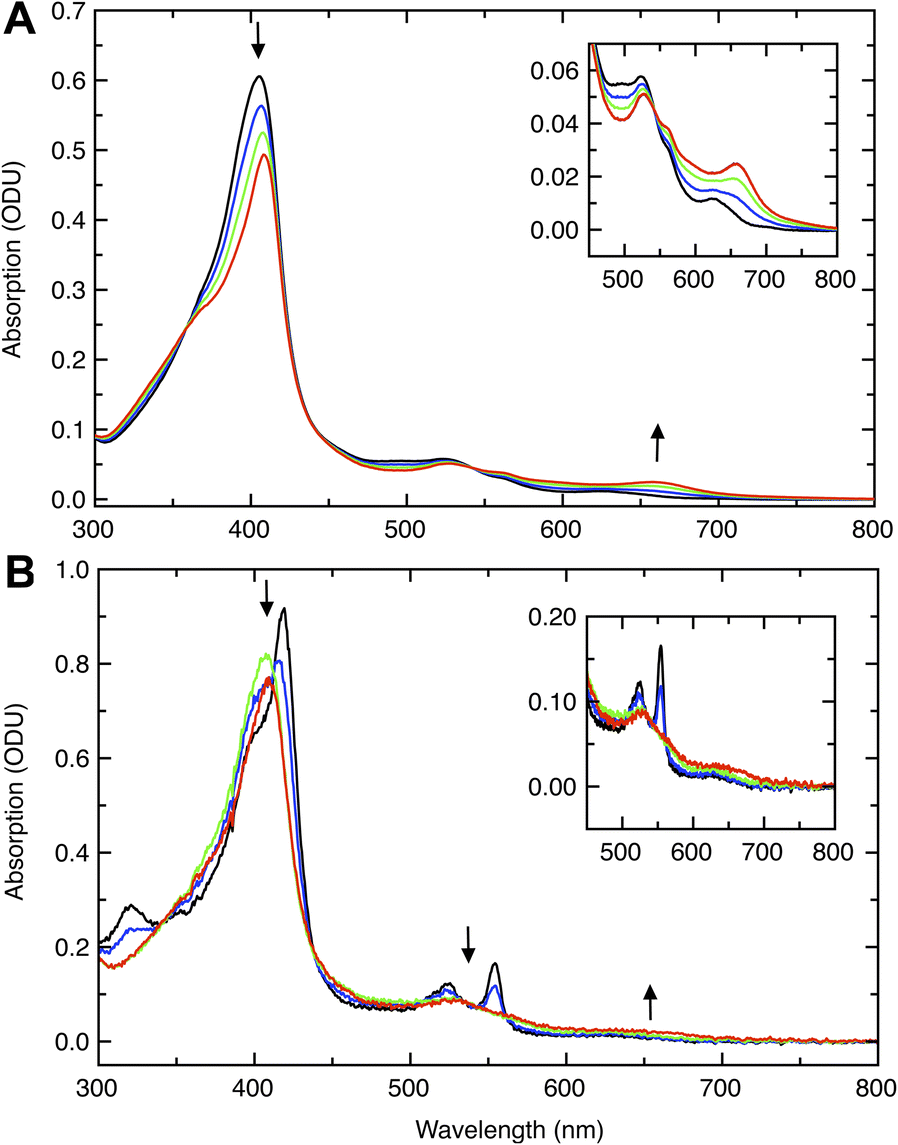 | ||
| Fig. 2 Titrations of Ne CcP with H2O2 in the diferric and half-reduced states. (A) Titration of diferric WT Ne CcP (black) with H2O2. Titration with 0.25 (blue), 0.5 (green) and 1 (red) equivalent H2O2 resulted in pronounced spectral changes at 410 nm and 660 nm (arrows). Inset: Magnified view of the 450–800 nm region. (B) Titration of semi-reduced WT Ne CcP (black) with H2O2. Titration with 0.2 (blue), 0.6 (green), and 1 (red) equivalents H2O2 resulted in pronounced spectral changes at 417 nm, 525 nm, and 553 nm along with a slight increase in intensity near 650 nm (arrows). Inset: Magnified view of the 450–800 nm region. | ||
Upon reduction of the enzyme with ascorbate, features attributed to the reduction of the E heme included a sharp increased intensity feature at 418 nm that was observed to dominate the Soret band, an increase in intensity of Q-band features at 525 nm and 554 nm, and increased intensity near 325 nm presumably from a porphyrin N band.20 The P heme remains oxidized in the presence of ascorbate, and accounts for the Soret band shoulder near 404 nm in the ascorbate reduced enzyme.
Titrations of diferric WT Ne bCcP with H2O2 resulted in bleaching and a red-shift of the Soret band to 409 nm, concomitant with the appearance of a feature at 658 nm as was observed with Ne bCcP purified from N. europaea11 suggesting the formation of a porphyrin π-cation radical.21 Titrations of semi-reduced Ne bCcP with peroxide resulted in the bleaching of the characteristic sharp Soret and Q-band features of the semi-reduced enzyme, attributed to the oxidation of the E heme. These changes were accompanied by a shift of the Soret maxima to 409–410 nm. The high-spin feature at 625 nm was replaced by a broad low intensity feature near 650 nm, evocative of compound 1, probed in greater detail below.
Stability of the Ne bCcP compound 1
In the absence of reducing substrates, the Ne bCcP compound 1 decayed slowly enough to permit experiments using hand-mixing methods. Attempts to measure the rate of formation of compound 1 with optically monitored stopped-flow experiments were unsuccessful, as the reaction appeared to be complete within the <2 ms dead time of mixing. The decay of compound 1 was characterized by the Soret band re-gaining intensity at 410 nm and the 658 nm feature losing intensity at approximately the same rate of 0.0443 min−1 (t1/2 ≈ 16 minutes) at pH 7.5 (Fig. S1‡). This suggests that the Ne bCcP compound 1 is more stable than that of mono-heme peroxidases Pisum sativum ascorbate peroxidase,22 Phanerochaete chrysosporium lignin peroxidase,23 both of which fully decay within minutes at pH 7 and pH 6.0 respectively, but less stable than that of manganese peroxidase compound 1 which has a half-life of 89 minutes at pH 4.5.24Addition of the artificial substrate guaiacol to Ne bCcP compound 1 demonstrated that guaiacol could be rapidly oxidized by compound 1. Addition of 2.5 equivalents of guaiacol to compound 1 immediately increased the Soret band intensity at 410 nm and diminished the intensity of the 658 nm band. Incubation of this sample for 7 minutes after guaiacol addition showed a spectrum resembling diferric Ne bCcP, identified by its broad Soret band with an absorbance maxima near 404 nm and the well-defined high-spin band at 625 nm (Fig. S2‡).
The species resulting from both the decay of compound 1 in the absence of reducing equivalents and the reaction of compound 1 with guaiacol was distinct from diferric Ne bCcP, and resembled the species resulting from the addition of H2O2 to the semi-reduced enzyme. This species was distinct from diferric Ne bCcP by nature of its sharper Soret band and absorbance maxima at 410 nm, along with the low intensity 658 nm band and absence of the 625 nm high-spin feature. The 410 nm species was tentatively assigned as the bCcP compound 2 (FeIVO R), as the addition of peroxide to the semi-reduced enzyme supplies the correct amount of reducing and oxidizing equivalents form an FeIIIEFeIVPO intermediate as is proposed in the activatable bCcPs (Scheme 1, top). The decay of compound 1 in the absence of reducing equivalents to compound 2 could also be accomplished by autooxidation of the amino acid side chains of the enzyme, and it has been previously demonstrated that the additions of guaiacol to horseradish peroxidase compound 1 can reduce compound 1 to compound 2, and compound 2 to the ferric enzyme.25
EPR characterization of Ne bCcP treated with H2O2
X-band EPR spectra were acquired for diferric and semi-reduced Ne bCcP, as well as for the enzyme that was treated with a slight excess of H2O2. The spectrum of the diferric enzyme (Fig. 3A) contained 3 major components: a prominent low spin signal at gx,y,z = 1.51, 2.39, and 2.88 assigned to the P heme (blue dashed trace), a HALS low spin signal with the only one observable feature at gz = 3.37 assigned to the E heme (red dashed trace), and an additional minor low spin heme at g = 1.55, 2.39, 2.82 with a concentration 3-fold lower than the gz = 2.88 species (green dashed trace). This gz = 2.82 species was assumed to be minor population of the P heme in a similar state to the major species. A minor high spin feature at g = 6.14 (∼1% of the protein concentration) is assumed to originate from a high-spin population of the P heme, though as noted above, the high-to-low spin equilibria of the Ne bCcP P heme is not fully understood. Ne bCcP purified from the native organism displayed varying amounts of the high-spin species and the minor P heme species between enzyme preparations.11 Overall, our observed values are in good agreement with EPR spectra previously reported for native and recombinant Ne bCcP.11,17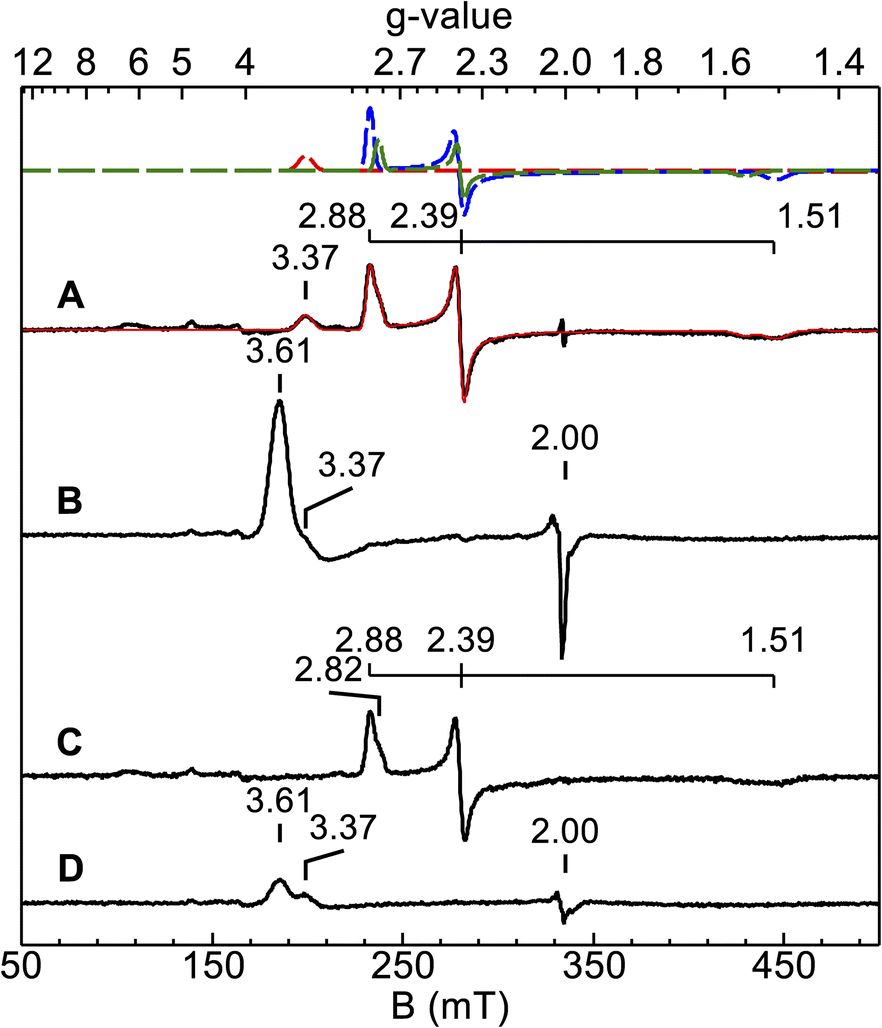 | ||
| Fig. 3 EPR spectra (9.386 GHz) of diferric and semi-reduced Ne bCcP reacted with H2O2 recorded at 12 K. (A) 150 μM diferric Ne bCcP (B) Sample from (A) with 5 equivalents H2O2 added, frozen 2 minutes after peroxide addition. (C) 113 μM semi-reduced Ne bCcP. (D) Sample from (C) with 5 equivalents H2O2 added, frozen 2.5 minutes after peroxide addition. The red trace in (A) is the sum of the simulations of 3 species (dashed traces), see text. | ||
Upon addition of peroxide to the oxidized enzyme (Fig. 3B), the P heme signals were not observed and a new species with features at g = 3.6 and 2 appeared. Ne bCcP reacted with peroxide and frozen at different timepoints showed a decrease in the yield of the g = 3.6, 2 species along with the re-appearance of the P heme signals, and a minor species near g = 2 (Fig. S3‡). As described in greater detail below, the observation of this new EPR-detectable species suggested that Ne bCcP produced a ferryl porphyrin-based radical, similar to compound I (FeIVO R+˙).
The EPR spectrum of the ascorbate-reduced Ne bCcP appeared similar to that of the diferric enzyme, with the absence of the E heme signal at g = 3.37 as the E heme is now in the EPR silent FeII state. Taking the semi-reduced enzyme (Fig. 3C), exposing it to 5 equivalents of H2O2 and freezing within 2.5 minutes yielded a lower intensity g = 3.6, 2 species, as well as the feature attributed to the E heme at g = 3.37 (Fig. 3D). Here, we presume that the P heme is in the FeIV state and EPR silent. A similar sample frozen 5 minutes after peroxide addition led to a further decrease in the intensity of the g = 3.6, 2 signal, and reappearance of low intensity signals attributed to the P heme (Fig. S4‡).
Mössbauer spectroscopy of the Ne bCcP enzyme
Mössbauer spectra of 57Fe-enriched oxidized Ne bCcP, pH 7.5, were recorded at 4 K and various magnetic fields (Fig. 4). The spectra are composed of 3 species which are the simulated filled areas of Fig. 4, with the resultant sums being the black lines overlaid on the data. The simulations are global least-squares fits of the data. The E heme (axial Met/His ligation) was partially reduced and thus shows spectra from both S = 0 (violet, E FeII) and S = 1/2 (red, E FeIII) species with the parameters and percentages of total iron given in Table 1. The E FeII species shows a well resolved quadrupole doublet in low magnetic field from 26% of the iron in the sample. The P heme (blue, P FeIII) shows a S = 1/2 spectrum with significantly less overall splitting relative to the E FeIII heme owing to a smaller hyperfine tensor (A). All 3 species have parameters typical of 6-coordinate FeII (S = 0) or FeIII (S = 1/2) heme centers.26 The E FeIII heme is a highly anisotropic low-spin (HALS) center with one g-value near 4. The larger A-tensor for the E FeIII heme has been observed for other HALS hemes.26 The ratio of total E to P hemes is 62/38 indicating that the protein sites were less than fully occupied with both hemes. The presence of some reduced E heme indicates that the oxidant treatment did not fully oxidize the E heme.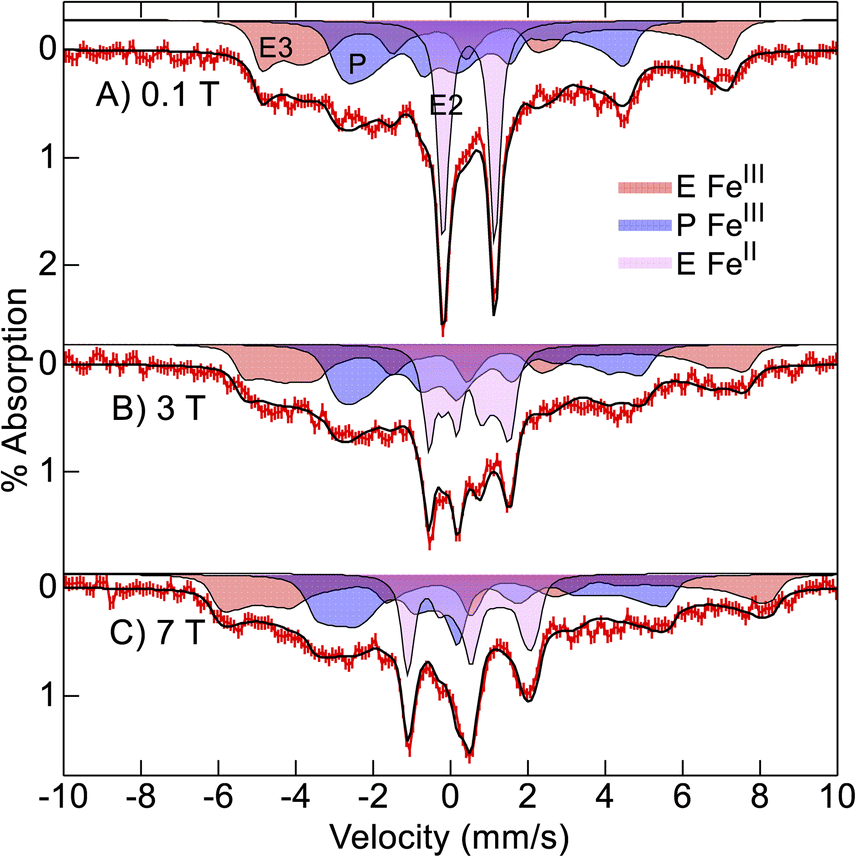 | ||
| Fig. 4 Mössbauer spectra ((A–C) red vertical bars) of Ne bCcP at pH 7.5 recorded at 4 K in the magnetic fields listed. The black traces and filled areas are simulations (see text) for the parameters of Table 1. | ||
| Species | Spin | % | δ, mm s−1 | ΔEQ, mm s−1 | η | A, T |
|---|---|---|---|---|---|---|
| E FeII | 0 | 26 | 0.49 | 1.33 | ||
| E FeIII | 1/2 | 36 | 0.25 | 2.00 | −0.8 | −38, 35, 76 |
| P FeIII | 1/2 | 38 | 0.29 | 2.07 | −1.4 | −28, 15, 43 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| +H2O2 | ||||||
| E FeIII | 1/2 | 57 | 0.28 | 2.22 | −1.1 | −35, 29, 77 |
| Cmpd 2 | 1 | 18 | 0.05 | +1.67 | 0.1 | −23, −23, −3 |
| Cmpd 1 | 1 × 1/2 | 25 | 0.09 | +1.21 | 0 | −23, −23, −5 |
Mössbauer spectra of Ne bCcP, pH 7.5, after addition of H2O2 showed a mixture of S = 1/2 FeIII E and P hemes, compound 2 and compound 1 in amounts relative to total iron of 56%, 16%, 17%, 11%, respectively (Fig. S5‡). The amount of compound 1 in the sample was low and difficult to identify. The significant amount of compound 2 was possibly owing to the presence of the reduced E heme in the sample prior to H2O2 addition. The reduced heme could supply a reducing equivalent to generate compound 2 from compound 1.
To increase the yield of compound 1 for Mössbauer studies, we attempted to optimize sample conditions to both increase the initial yield of compound 1 and slow the decay of compound 1 to compound 2. We first investigated the effects of pH on the conversion of compound 1 to compound 2 as this has been demonstrated to be proton dependent in HRP.27 Initial experiments on the Ne bCcP compound 1 stability suggested that an increase in the pH from 7.5 to 8.6 slowed compound 1 decay, so it was decided to use a sample pH of 8.6 to obtain a higher yield of compound 1. Importantly, diferric Ne bCcP showed similar UV-vis and EPR spectra at pH 7.5 and pH 8.6 (Fig. S6‡). However, it was later found that the apparent compound 1 decay rates were essentially identical at both pH 7.5 and pH 8.6 (Fig. S7‡).
Consequently, we buffer exchanged the partially reduced 57Fe-enriched Ne bCcP sample of Fig. 4 into pH 8.6 buffer and treated the sample with additional oxidant to fully oxidize the E heme before peroxide addition with the intent of maximizing compound 1 yield. Mössbauer spectra of the oxidant treated, pH 8.6, Ne bCcP sample after addition of H2O2 were recorded at 4 K and various magnetic fields (Fig. 5). The spectra are composed of 3 species which are the simulated filled areas of Fig. 5, with the resultant sums being the black lines overlaid on the data. The simulations are global least-squares fits of the data giving the parameters of Table 1. The S = 1/2 FeIII E heme is present in the same amount (red, 57%) as the lower pH sample, but the S = 1/2 FeIII P heme is now absent.
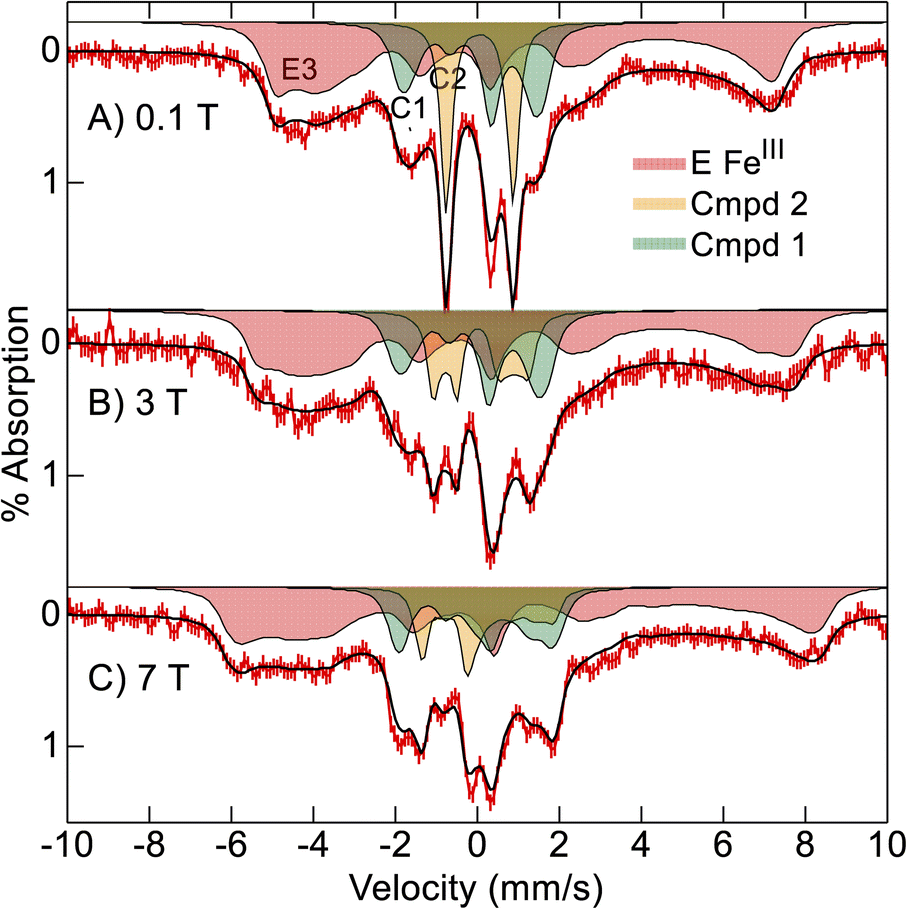 | ||
| Fig. 5 Mössbauer spectra ((A–C) red vertical bars) of oxidized Ne bCcP at pH 8.6 reacted with 15 equivalents H2O2 recorded at 4 K in the magnetic fields listed. The black traces and filled areas are simulations (see text) for the parameters of Table 1. | ||
The orange area (C2, 18%) has parameters indicative of compound 2 (Table 1) in the same amount as the lower pH sample. The green area (C1, 25%) has parameters indicative of compound 1 and indeed the yield of compound 1 has increased relative to the lower pH sample. In particular, the low field spectrum of Fig. 5A accentuates the different spectra of compounds 1 and 2. Compound 2 is an intermediate spin (S = 1) FeIVO (ferryl) species with a large positive D-value (28 cm−1) and an Ms = 0 level low in energy. Thus, compound 2 displays a quadrupole doublet at low magnetic field (orange, Fig. 5A). Compound 1 also has an S = 1 FeIV O heme with a large positive D-value, and in addition contains a porphyrin π-cation radical (S = 1/2). The iron and porphyrin spins exchange couple to give a system spin which is half integer. Half-integer spin states are magnetic at low field and thus show magnetic splitting (green, Fig. 5A).
The variable magnetic field data allowed measurement of the nuclear and electronic parameters of the compound 1 and 2 species. The values of δ and ΔEQ for both species are close to those of horse radish peroxidase (HRP). The increase in δ and decrease in ΔEQ of compound 1 relative to compound 2 is also observed for HRP (Table 2). Ne bCcP, like HRP, has an His coordinated axial to the heme and the value of ΔEQ < 2.0 mm s−1 for compound 2 is consistent with an FeIVO that is not protonated.
| Species | SFe:Spor |
δ, mm s−1 | ΔEQ, mm s−1 | J, cm−1 | D, cm−1 | A, T | Ref. |
|---|---|---|---|---|---|---|---|
| a t.w. refers to this work. | |||||||
| Ne bCcP 1 | 1:1/2 |
0.09 | + 1.21 | −17 | 28 | −23, −23, −5 | t.w. |
| Ne bCcP 2 | 1:0 |
0.05 | +1.67 | 28 | −23, −23, −3 | t.w. | |
| HRP 1 | 1:1/2 |
0.08 | 1.25 | <±4 | 26 | −19, −19, −6 | 28 |
| HRP 2 | 1:0 |
0.03 | 1.61 | 22 | −19, −19, −3 | 29 | |
| CPO 1 | 1:1/2 |
0.14 | 1.02 | 37 | 36 | −20, −20, −1 | 30 |
| P450 1 | 1:1/2 |
0.11 | 0.90 | 47 | 36 | −20, −23, −3 | 31 |
| Catalase 1 | 1:1/2 |
0.12 | 1.09 | −5 | 17 | −19, −19, −6 | 32 |
Further work is needed to determine whether raising pH or additional oxidant treatment were more significant in the improved compound 1 yield. Nevertheless, the higher yield allowed determination of the exchange coupling constant, J = −17 cm−1 (Hex = JSFe·Spor). The ferromagnetic interaction places the approximate S = 3/2 multiplet low in energy, but owing to the large D value (28 cm−1), the approximate Ms states are widely split in zero magnetic field. The energies of the doublets are: 0 (S = 3/2, Ms = ±1/2), 23 cm−1 (3/2, ±3/2), 44 cm−1 (1/2, ±1/2).
Further corroboration of these values comes from simulation of the EPR spectrum. Fig. 6A shows the EPR signal of the sample after H2O2 addition (blue trace) and a simulation (black trace) using the Mössbauer parameters. The compound 1 (green dash) and HALS (red dash) components of the simulation are also shown. The simulation matches the data and the intensity predicts an amount of compound 1 that is 25% of the enzyme concentration, which is similar to that of the Mössbauer samples. Also shown is a simulation calculated for large negative J (|J| ≫ |D|, Fig. 6B) with all other parameters the same. This is the signal predicted from an isolated S = 3/2 multiplet. The significant shift of the g⊥-value from 4.3 to 3.5 and g‖-value from 2.025 to 2.007 is owing to the ratio J/D = −0.61 determined from the Mössbauer simulations. The Ms = ±3/2 doublet is predicted to have negligible intensity in EPR spectroscopy, but both Ms = ±1/2 doublets have high EPR transition probabilities. We attempted to locate the EPR signal from the doublet at 44 cm−1. However, at temperatures well below that needed to populate the excited states, the observed EPR signal from the ground doublet was greatly broadened, implying that an excited state signal would also be very broad, and thus no excited state signals were detectable. The broadening is likely from enhanced relaxation owing to Orbach relaxation with the nearby excited states.
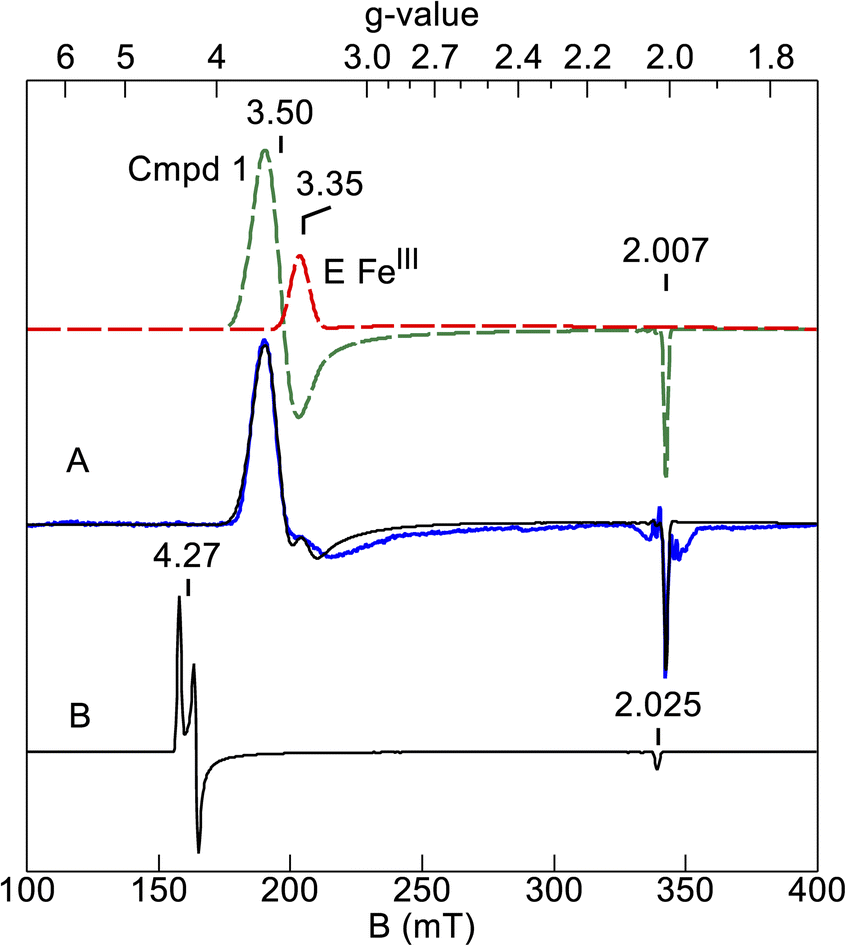 | ||
| Fig. 6 (A) Simulation (black trace) of the EPR spectrum of Ne bCcP compound 1 (blue trace) calculated from the Mössbauer parameters. Spectral conditions: microwaves 2 mW at 9.621 GHz; temperature 5 K. The dashed lines are the species which sum to give the black trace (see text). (B) Simulation for the Mössbauer parameters, but with large negative J. | ||
The exchange coupling constants (J) for the compound 1 state of several other enzymes are listed in Table 2. The variation in J has long been thought to correlate with the axial protein residue coordinated to the heme. These axial residues are: Tyr (J = −5 cm−1, catalase), His (J ≈ 0, HRP), and Cys (J ≈ +40 cm−1, CPO, cyt P450). However, the value J = −17 cm−1 for Ne bCcP is significantly more negative than these enzymes and does not follow the correlation for an axial His residue. Whether this value is a strong function of some other heme property, such as the thioether linkages, is at present unknown.
The analysis of the EPR signal with the Mössbauer parameters provides an interesting retrospective view of early mechanistic investigations of the activatable Pseudomonas aeruginosa bCcP with H2O2. In these previous studies, an EPR signal with g-values near 3.4 and 2 was observed to form in the seconds after the decay of a primary ferryl species, and was presumed to be a signal arising from a magnetic interaction between two low spin ferric hemes.13,14 The similarity in lineshape and g-values of the published signals to the one described in this work suggest that this species may have also been a ferryl-porphyrin radical.
Physiological relevance of the species
The data presented here also inform our understanding of traditional bCcP enzymes that detoxify H2O2 versus those enzymes that seem to use H2O2 as an oxidant to achieve even more elaborate redox states (i.e., MauG, BthA, MbnH, Scheme 1). As additional bCcP family members are discovered, their reactivities and ability to produce compound 1 or “bis-Fe(IV)” intermediates will be a critical distinction in their characterization. Here, considering just the traditional bCcPs, the propensity for the Ne bCcP to form a compound 1 intermediate in the oxidized state provides an interesting point of comparison between the constitutively active enzymes and activatable class of bCcPs. As highly oxidizing intermediates like compound 1 may be oxidatively damaging to the enzyme, the two sub-classes of traditional bCcPs may achieve oxidative protection through two distinct methods: E heme linked reductive activation, or a high potential at the E heme. The activatable enzymes likely do not form a significant amount of compound 1 during turnover, as peroxide binding is only possible when the E heme is reduced. The Ne bCcPs E heme potential is so high, it is likely reduced whenever sufficient electrons are available from redox protein donors. In either scenario the resulting compound 1 generated here would be consumed under native turnover conditions.Again in the context of the traditional bCcP enzymes, assigning a specific physiological significance of the functional differences is difficult as relatively few studies have been done to interrogate bCcP physiological functions. Speaking broadly, the activatable enzymes are sourced from organisms with flexible metabolisms capable of sustained growth under anoxic and microoxic conditions utilizing a variety of terminal electron acceptors. Among the activatable enzymes, the Paracoccus denitrificans bCcP is upregulated under conditions of low oxygen tension,33 and the Shewanella oneidensis bCcP has been shown to confer a selective advantage to cells under micro-aerobic dissimilatory iron reducing conditions,34 under which it is highly expressed.35 While not an activatable enzyme, the triheme bCcP YhjA from Escherichia coli is upregulated under low oxygen tension and downregulated under aerobic conditions,36 and enables E. coli to utilize H2O2 as a terminal electron acceptor during anaerobic growth.37 In contrast, the two constitutively active canonical bCcPs are sourced from N. europaea and M. capsulatus bath, which utilize aerobic ammonia and aerobic methane oxidation respectively as their primary pathway for producing energy. Recent transcriptomic studies show that Ne bCcP is upregulated 2-fold in the transition from oxygen limited to ammonia limited growth in pure cultures of N. europaea,38 while mixed partial nitration anammox communities containing Nitrosomonas sp. also show upregulation of a gene homologous to Ne bCcP in response to the transition from anoxic to low-oxygen conditions.39 In both cases the pattern of Ne bCcP upregulation appears to coincide with an increase in the O2 dependent oxidation of ammonia, suggesting that Ne bCcP fulfills an oxidative stress protection role during aerobic metabolism. In this context, the ability to react with peroxide in the diferric state may impart the Ne bCcP with a more promiscuous peroxidatic activity than its activatable orthologues, as it can likely function as a small molecule oxidizing peroxidase even when redox carrier proteins capable of reducing the E heme are not present. Given these correlations, we suspect that when subjected to the same methods used here, the activatable bCcP will reveal little to no compound 1, whereas the enzyme from M. capsulatus will display the same reactivity as the enzyme studied here.
Conclusions
In summary, we have investigated the peroxide reactivity of the Ne bCcP and in doing so have presented the first characterization of the electronic structure of high-valent iron species from a canonical bCcP. Members of the bCcP superfamily continue to be discovered, revealing unforeseen reactivity (i.e. BthA, MbnH). These data allow for a comparison of the best characterized bCcP family member MauG, and will enable the comparison between canonical bCcPs from across the super-family, as we and others try to understand their reactivity with H2O2 and varied substrates.Experimental methods
Cloning, protein expression and purification
A plasmid encoding a tag-free wild type (WT) Ne bCcP construct (pETSN-Ne bCcP) was used as in Wolf et al.17 for the expression of WT Ne bCcP. Purification of untagged WT Ne bCcP was conducted as previously described17 with the exception that 1 mM EDTA was included in the lysis buffer, AEBSF was used in place of PMSF and that bound Ne bCcP on the MonoQ column was washed with one column volume of 0.5 mM EDTA prior to elution. Ne bCcP was found to elute in a broad band from 40–60 mM NaCl. For the size exclusion step, a buffer of 20 mM HEPES 100 mM NaCl 10% w/v. Glycerol pH 7.5 was used on a Cytiva Sephacryl S-100 column. Purity of SEC fractions was assessed through SDS-PAGE.Preparation of Ne bCcP in oxidized and semi-reduced states for UV-vis H2O2 titrations
The diferric state of Ne bCcP was prepared by titrating the enzyme with an stoichiometric equivalents of K2(IrIV)Cl6 and monitoring by UV-vis spectroscopy until the enzyme appeared oxidized and desalting on a PD-10 column, and subsequently concentrating diluted protein in a 30 kDa MWCO centrifugal concentrator. Alternatively, the diferric state could be prepared by diluting the as-isolated enzyme in a 100-fold excess of the assay buffer and stirring vigorously under a stream of air for 1 hour, and subsequently concentrating to the required volume.The semi-reduced Ne bCcP was prepared under anaerobic conditions by incubating the enzyme with excess (1–5 mM) ascorbate for 30 minutes and desalting on a PD-10 column to remove ascorbate. When prepared using oxygen-free buffer under anaerobic conditions, the semi-reduced state was stable for hours. When the semi-reduced state was prepared on the benchtop and exposed to oxygen it would slowly oxidize over the course of hours.
ABTS assays
ABTS peroxidase assays were performed on the benchtop at 25 °C in a buffer consisting of 50 mM sodium phosphate at pH 6.5. Reactions contained 3 mM ABTS and varying concentrations of H2O2. The background oxidation of ABTS by H2O2 was allowed to proceed for several seconds to obtain a baseline rate of ABTS oxidation and then the enzyme was added to the cuvette and mixed. The formation of the oxidized ABTS was monitored at 420 nm and quantified using the extinction coefficient for ABTS at 420 nm of 36000 M−1 cm−1.40 Assays were monitored with monitored with a refurbished Hewlett-Packard 8453 spectrophotometer (On-Line Instrument Systems).
Optical titrations with H2O2
Ne bCcP was quantified spectrophotometrically by determining an approximate extinction coefficient at 280 nm using the Bradford assay as a reference. Reactions of the diferric Ne bCcP with H2O2 were conducted on the benchtop at 25 °C and monitored with a refurbished Hewlett-Packard 8453 spectrophotometer (On-Line Instrument Systems). Reactions of the semi-reduced Ne bCcP with H2O2 were conducted in an anaerobic coy tent and monitored using an Avantes Avaspec spectrometer at room temperature, with buffer that had been made anaerobic by several cycles of sonication under vacuum and purging with N2. All UV-vis experiments were conducted in a buffer consisting of 20 mM MOPS, 100 mM NaCl, 10% w/v glycerol, and 0.1 mM CaCl2 unless otherwise noted. Rates of 658 nm absorbance decay were fit using the Curve Fitter application in MATLAB. Hydrogen peroxide solutions for substrate titrations were quantified by UV-vis using the extinction coefficient at 240 nm of 43.6 M−1 cm−1, and guaiacol was quantified using the extinction coefficient at 274 nm of 2550 M−1 cm−1.EPR spectroscopy
EPR samples were prepared in 20 mM MOPS pH 7.5, 100 mM NaCl, 10% w/v glycerol 0.1 mM CaCl2 unless specified otherwise. Ne bCcP samples were prepared in the diferric state and semi-reduced states as described above, though it should be noted that the chemical reduction and oxidations of the samples were done with dilute (<10 μM) enzyme to ensure efficient mixing of the enzyme and oxidant. For peroxide-reacted samples, a pre-peroxide addition spectrum was recorded, then the EPR sample of the enzyme without substrate was thawed at room temperature, and sample was then transferred from the EPR tube to an Eppendorf tube using PTFE tubing. The specified equivalent of H2O2 was then added to the sample and mixed extensively with a pipette. The sample was then transferred back to the EPR tube and frozen in LN2.EPR spectra were recorded on a Bruker Elexsys E-500 spectrometer with an Oxford ESR 910 cryostat and a Bruker bimodal cavity using a modulation amplitude of 10 G, 100 kHz modulation frequency, and receiver gain of 60 dB. All signals were recorded at a temperature of 12 K and a power of 0.2 mW unless otherwise specified.
Mössbauer spectroscopy
57Fe WT Ne bCcP was expressed in a defined media containing 1× M9 salts: (33.7 mM Na2HPO4, 22 mM KH2PO4, 8.55 mM NaCl, 9.35 mM NH4Cl), 0.4% w/v glucose, 0.2% w/v casmino acids (Difco), trace elements: 1 mM MgSO4, 0.3 mM CaCl2, 6.2 μM ZnCl2, 0.76 μM CuSO4, 0.42 μM CoCl2, 1.6 μM H3BO3, 0.81 μM MnCl2, and vitamins: 1 g per L pyridoxine HCl, 0.5 g per L thiamine HCl, 0.5 g per L riboflavin, 0.5 g per L nicotinic acid, 0.5 g per L calcium D-pantothenate, p-aminobenzoic acid, 0.2 g per L biotin, 0.2 g per L folic acid, 0.01 g per L vitamin B12, along with 35 μg per mL chloramphenicol and 100 μg per mL ampicillin. For each liter of culture, 1 mL of 2 mg per mL 57Fe in 4 M HCl with 5 mg mL−1 ascorbate was added to cultures and mixed prior to the addition of 5 mL of overnight seed culture. The remainder of the expression and purification procedure was carried out as stated with WT Ne bCcP grown in 2× YT. Mössbauer samples were prepared by buffer exchanging purified protein into 20 mM MOPS pH 7.5 for the as-isolated sample and as-isolated + H2O2 sample or 20 mM TAPS pH 8.6 in the case of the high yield compound 1 sample. Preparation of diferric Ne bCcP for Mössbauer experiments was done as described above for EPR experiments.Mössbauer spectra were recorded on a variable field spectrometer operating in a constant acceleration mode in a transmission geometry using Janis Research Inc. cryostat. The Dewar housed a superconducting magnet that allowed for the application of magnetic fields up to 7 T parallel to the γ-radiation. Isomer shifts are reported relative to Fe metal at 298 K.
The processing of Mössbauer and EPR data and calculation of simulations use SpinCount software written by one of the authors. The software diagonalizes the electronic terms of the spin Hamiltonian and performs least-squares fitting of simulations to the spectra. The quantitative simulations are generated with consideration of all intensity factors, which allows computation of simulated spectra for a specified sample concentration (EPR). The simulations use the following spin Hamiltonian:
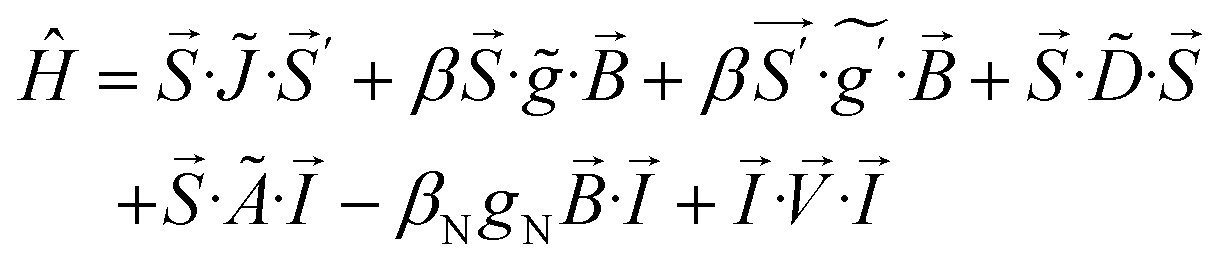
Data availability
The data supporting this article, are included in the main text, and additional data have been included as part of the ESI.‡Author contributions
PH was responsible for conception, methodology, investigation, analysis and writing; MH was responsible for investigation, analysis and writing; and SJE was involved with conception, funding, writing, and project supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of General Medical Sciences, National Institutes of Health through grants GM141948 (MPH) and GM136294 (SJE).Notes and references
- K. Rizzolo, S. E. Cohen, A. C. Weitz, M. M. López Muñoz, M. P. Hendrich, C. L. Drennan and S. J. Elliott, Nat. Commun., 2019, 10(1), 1–10, DOI:10.1038/s41467-019-09020-4.
- D. S. Barreiro, R. N. S. Oliveira and S. R. Pauleta, Coord. Chem. Rev., 2023, 485, 215114 CrossRef CAS.
- G. W. Pettigrew, A. Echalier and S. R. Pauleta, J. Inorg. Biochem., 2006, 100(4), 551–567, DOI:10.1016/j.jinorgbio.2005.12.008.
- X. Li, R. Fu, S. Lee, C. Krebs, V. L. Davidson and A. Liu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(25), 8597–8600, DOI:10.1073/pnas.0801643105.
- G. E. Kenney, L. M. K. Dassama, A. C. Manesis, M. O. Ross, S. Chen, B. M. Hoffman and A. C. Rosenzweig, J. Biol. Chem., 2019, 294(44), 16141–16151, DOI:10.1074/jbc.RA119.010202.
- A. C. Manesis, J. W. Slater, K. Cantave, J. M. Bollinger, C. Krebs and A. C. Rosenzweig, Biochemistry, 2023, 62(5), 1082–1092 Search PubMed.
- H. Shimizu, D. J. Schuller, W. N. Lanzilotta, M. Sundaramooorthy, D. M. Arciero, A. B. Hooper and T. L. Poulos, Biochemistry, 2001, 40, 13483–13490 Search PubMed.
- M. Rönnberg and N. Ellfolk, Biochim. Biophys. Acta, Bioenerg., 1978, 504(1), 60–66, DOI:10.1016/0005-2728(78)90006-3.
- R. Gilmour, C. F. Goodhew, G. W. Pettigrew, S. Prazeres, I. Moura and J. J. Moura, Biochem. J., 1993, 294(Pt 3), 745–752, DOI:10.1042/bj2940745.
- G. S. Pulcu, K. E. Frato, R. Gupta, H. R. Hsu, G. A. Levine, M. P. Hendrich and S. J. Elliott, Biochemistry, 2012, 51(5), 974–985, DOI:10.1021/bi201135s.
- D. M. Arciero and A. B. Hooper, J. Biol. Chem., 1994, 269(16), 11878–11886, DOI:10.1016/s0021-9258(17)32655-8.
- J. A. Zahn, D. M. Arciero, A. B. Hooper, J. R. Coats and A. A. DiSpirito, Arch. Microbiol., 1997, 168(5), 362–372, DOI:10.1007/s002030050510.
- N. Ellfolk, M. Rönnberg, R. Aasa, L. E. Andréasson and T. Vänngård, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1983, 743(1), 23–30, DOI:10.1016/0167-4838(83)90413-2.
- C. Greenwood, N. Foote, P. M. A. Gadsby and A. J. Thomson, Chem. Scr., 1988, 28, 79–84 Search PubMed.
- K. Rizzolo, A. C. Weitz, S. E. Cohen, C. L. Drennan, M. P. Hendrich and S. J. Elliott, J. Am. Chem. Soc., 2020, 142(28), 11978–11982, DOI:10.1021/jacs.0c04023.
- N. Abu Tarboush, L. M. R. Jensen, M. Feng, H. Tachikawa, C. M. Wilmot and V. L. Davidson, Biochemistry, 2010, 49(45), 9783–9791, DOI:10.1021/bi101254p.
- M. W. Wolf, K. Rizzolo, S. J. Elliott and N. Lehnert, Biochemistry, 2018, 57(45), 6416–6433, DOI:10.1021/acs.biochem.8b00732.
- N. Foote, J. Peterson, P. M. Gadsby, C. Greenwood and A. J. Thomson, Biochem. J., 1984, 223(2), 369–378 CrossRef CAS PubMed.
- N. Foote, J. Peterson, P. M. A. Gadsby, C. Greenwood and A. J. Thomson, Biochem. J., 1985, 230(1), 227–237 CrossRef CAS PubMed.
- W. S. Caughey, R. M. Deal, C. Weiss and M. Gouterman, J. Mol. Spectrosc., 1965, 16, 451–463 CrossRef CAS.
- D. Dolphin, A. Forman, D. C. Borg, J. Fajer and R. H. Felton, Proc. Natl. Acad. Sci. U. S. A., 1971, 68(3), 614–618 CrossRef CAS PubMed.
- W. R. Patterson, T. L. Poulos and D. B. Goodin, Biochemistry, 1995, 34(13), 4342–4345, DOI:10.1021/bi00013a024.
- V. Renganathan and M. H. Gold, Biochemistry, 1986, 25(7), 1626–1631, DOI:10.1021/bi00355a027.
- K. Kishi, D. P. Hildebrand, M. K. V. Someren, J. Gettemy, A. G. Mauk and M. H. Gold, Biochemistry, 1997, 36(14), 4268–4277, DOI:10.1021/bi962627t.
- M. Santimone, Can. J. Biochem., 1975, 53, 649–657 CrossRef CAS PubMed.
- P. G. Debrunner, in Iron Porphyrins, ed. A. B. P. Lever and H. B. Gray, VCH Publishers, New York, Mössbauer Spectroscopy of Iron Porphyrins, 1989, vol. 3, pp. 137–234 Search PubMed.
- H. Yamada and I. Yamazaki, Arch. Biochem. Biophys., 1974, 165(2), 728–738, DOI:10.1016/0003-9861(74)90301-4.
- C. E. Schulz, P. W. Devaney, H. Winker, P. G. Debrunner, N. Doan, R. Chiang, R. Rutter and L. P. Hager, FEBS Lett., 1979, 103(1), 102–105 CrossRef CAS PubMed.
- C. Schulz, R. Chiang and P. G. Debrunner, International Conference on the Applications of the Mössbauer Effect, ed. L. Ulis, EDP Sciences, 1979, vol. 40, pp. C2-534-C2-536 Search PubMed.
- R. Rutter, L. P. Hager, H. Dhonau, M. Hendrich, M. Valentine and P. Debrunner, Biochemistry, 1984, 23(26), 6809–6816 CrossRef CAS PubMed.
- J. Rittle and M. T. Green, Science, 2010, 330(6006), 933–937 CrossRef CAS PubMed.
- O. Horner, J. M. Mouesca, P. L. Solari, M. Orio, J. L. Oddou, P. Bonville and H. M. Jouve, J. Biol. Inorg. Chem., 2007, 12(4), 509–525 CrossRef CAS PubMed.
- R. J. M. Van Spanning, A. P. N. De Boer, W. N. M. Reijnders, H. V. Westerhoff, A. H. Stouthamer and J. Van Der Oost, Mol. Microbiol., 1997, 23(5), 893–907, DOI:10.1046/j.1365-2958.1997.2801638.x.
- B. Schütz, J. Seidel, G. Sturm, O. Einsle and J. Gescher, Appl. Environ. Microbiol., 2011, 77(17), 6172–6180, DOI:10.1128/AEM.00606-11.
- B. Schuetz, M. Schicklberger, J. Kuermann, A. M. Spormann and J. Gescher, Appl. Environ. Microbiol., 2009, 75(24), 7789–7796, DOI:10.1128/AEM.01834-09.
- J. D. Partridge, R. K. Poole and J. Green, Microbiology, 2007, 153(5), 1499–1507, DOI:10.1099/mic.0.2006/004838-0.
- M. Khademian and J. A. Imlay, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(33), E6922–E6931, DOI:10.1073/pnas.1701587114.
- C. Sedlacek, D. Woebken, L. A. Sayavedra-soto, P. J. Bottomley, H. Daims and M. Wagner, mSystems, 2020, 5(5), e00562 Search PubMed.
- N. K. Beach, K. S. Myers, B. R. Owen, M. Seib, T. J. Donohue and D. R. Noguera, mSystems, 2021, 6(5), e00906 Search PubMed.
- T. Kenzom, P. Srivastava and S. Mishra, Appl. Environ. Microbiol., 2014, 80(24), 7484–7495 Search PubMed.
Footnotes |
| † We dedicate this manuscript to the memory of Prof. Alan Hooper, a great champion of those who worked with enzymes and proteins from organisms such as Nitrosomonas europaea. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07339h |
| This journal is © The Royal Society of Chemistry 2025 |