
Synergistic effects of azobenzene and thiourea backbones in multiresponsive copolymers for sensing and adhesive technologies†
Tse-Yu
Lo
a,
Mei-Li
Li
a,
Chia-Wei
Chang
a,
Tsung-Hung
Tsai
a,
Heng-Hsuan
Su
a,
Chun-Chi
Chang
a,
Yen-Shen
Hsu
a,
Huan-Wei
Lin
a and
Jiun-Tai
Chen
 *ab
*ab
aDepartment of Applied Chemistry, National Yang Ming Chiao Tung University, Hsinchu 300093, Taiwan. E-mail: jtchen@nycu.edu.tw; Tel: +886-3-5731631
bCenter for Emergent Functional Matter Science, National Yang Ming Chiao Tung University, Hsinchu 300093, Taiwan
First published on 4th March 2025
Abstract
Stimuli-responsive polymers have garnered significant attention for their ability to adapt to environmental changes, offering applications in sensing, smart coatings, and adaptive devices. However, challenges remain in developing multifunctional polymers that combine dynamic responsiveness with robust mechanical properties. In this study, we design and synthesize multifunctional azobenzene-based copolymers, poly(thiourea triethylene glycol)-co-azobenzene (PTUEG3-co-Azo) copolymers, through a controlled polycondensation process to address these limitations. The flexible PTUEG3 backbone, with its strong hydrogen-bonding networks, is combined with azobenzene moieties to impart thermal isomerization, acid–base responsiveness, and enhanced adhesion performance. The azobenzene groups exhibited thermally induced cis-to-trans isomerization, leading to structural reorganization, increased molecular packing, and elevated glass transition temperatures (Tg). Additionally, the azobenzene moieties demonstrated reversible acid–base responsiveness, undergoing distinct and repeatable color changes upon protonation and deprotonation. By balancing the flexibility of the PTUEG3 backbone with the rigidity of azobenzene groups, PTUEG3-co-Azo copolymers achieved strong adhesion performance and tunable dynamic properties. The 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 PTUEG3-co-Azo composition demonstrated superior adhesive strength, attributed to the synergistic effects of hydrogen bonding and azobenzene-induced reorganization under thermal activation. These results present PTUEG3-co-Azo as a versatile material, bridging the gap between dynamic responsiveness and mechanical robustness, with potential applications in smart sensing, adhesives, and functional coatings.
:1 PTUEG3-co-Azo composition demonstrated superior adhesive strength, attributed to the synergistic effects of hydrogen bonding and azobenzene-induced reorganization under thermal activation. These results present PTUEG3-co-Azo as a versatile material, bridging the gap between dynamic responsiveness and mechanical robustness, with potential applications in smart sensing, adhesives, and functional coatings.
Introduction
Stimuli-responsive materials have gained significant interest due to their capacity to adapt to environmental changes and display dynamic properties under external stimuli, such as light, heat, mechanical force, and pH, with potential applications in sensors and smart coatings.1–4 Such materials form the basis for a wide range of applications, including sensors, actuators, coatings, and soft robotics, attributed to their reversible and tunable properties.5–8 Among the various stimuli, light is noteworthy for offering a unique combination of precision, non-invasiveness, and versatility in controlling material properties.9,10 Unlike other external triggers, light requires no physical contact or chemical additives, enabling its application in diverse fields such as optical data storage,11 photomechanical devices, and environmental sensors.12–14 Light-responsive materials typically employ photoisomerization, photodimerization, or photoinduced cleavage processes to modulate physical or chemical properties. Among photoswitchable molecules, spiropyran, diarylethene, and azobenzene have been extensively studied for their distinct mechanisms and applications. Spiropyrans exhibit reversible ring-opening and ring-closing reactions under UV and visible light, accompanied by notable color changes and pH sensitivity.15–17 Diarylethenes, known for high fatigue resistance, find applications in optical memory and display systems.18,19 Azobenzene is widely studied for its ability to undergo rapid and reversible cis-to-trans isomerization. This process facilitates molecular reorganization and enables precise modulation of material properties under light exposure, making azobenzene a key component in the design of light-responsive systems.20–23The incorporation of azobenzene into polymer architectures allows tailored responsiveness. Side-chain azobenzene polymers typically exhibit faster isomerization as a result of lower steric hindrance, whereas main-chain azobenzene polymers, despite slower isomerization rates caused by the rigidity of the polymer backbone, provide enhanced mechanical strength and structural stability. However, steric hindrance and restricted mobility in main-chain azobenzene polymers often hinder the efficiency of structural transformations, limiting broader application potential. Incorporating functional groups into the polymer backbone has been shown to improve performance and introduce additional capabilities. For instance, azobenzene-polyurea backbones demonstrate strong hydrogen bonding while maintaining high mechanical strength, enabling broader applications.24 Other studies have demonstrated that main-chain azobenzene polymers exhibit reversible acid–base sensing, utilizing the protonation and deprotonation of azo bonds to produce distinct and rapid color changes.25 These behaviors extend applications in vapochromic sensing for environmental monitoring and detection of volatile organic compounds. By combining photoresponsive behavior with liquid crystalline properties, these materials achieve dynamic structural changes under light stimuli.26,27 The incorporation of reactive groups, such as secondary amines, facilitates hydrogen-bonding interactions among polymer chains, enhancing mechanical strength and making them well-suited for photomechanical actuators and soft robotics.
Despite these studies, it is still challenging to develop multifunctional azobenzene-based polymers. Overcoming these challenges could enable applications in stimuli-responsive adhesives for reconfigurable systems, smart coatings for environmental sensing, and light-controlled actuators for soft robotics. In this work, we present the design and synthesis of multifunctional azobenzene-based copolymers, PTUEG3-co-Azo, through a controlled polycondensation process. Compared to block copolymers, the random copolymer design facilitates a uniform distribution of functional units, enabling synergistic multifunctional properties such as simultaneous acid–base response and light-induced actuation. While block copolymers may offer advantages in microstructural control, their synthesis is more complex. Future studies could explore block copolymer designs to assess their potential benefits for specific applications. The PTUEG3 backbone, derived from thiourea–polyether units, features highly dense yet nonlinear zigzag hydrogen-bonding arrays, which confer flexibility and strong intermolecular interactions. These attributes make PTUEG3 an attractive candidate for applications such as adhesives and self-healing materials.28–30 Despite its advantages, PTUEG3 interacts with moisture, forming hydrogen bonds that weaken intermolecular interactions and compromise mechanical properties, and can be mitigated through copolymerization with hydrophobic groups.31,32 Copolymerization with azobenzene moieties enhances environmental tolerance and imparts dynamic functionalities, including isomerization and acid–base responsiveness. Thermal isomerization of azobenzene groups improves molecular packing, increases the glass transition temperature (Tg), and reduces molecular mobility, which enhances adhesive performance by stabilizing the polymer structure. Copolymerization also balances the flexibility of PTUEG3 with the rigidity of azobenzene units, allowing precise control over composition and mitigating moisture-induced plasticization while maintaining mechanical strength and tunable dynamic properties. By combining the self-healing potential and flexibility of PTUEG3 with the isomerization-driven tunability and acid–base responsiveness of azobenzene, PTUEG3-co-Azo achieves a synergistic combination of robustness, adaptability, and adhesive functionality. These advancements make PTUEG3-co-Azo a material that effectively combines light-responsiveness, mechanical robustness, and adhesive functionality, enabling its use in advanced adhesives, protective coatings, and self-healing systems.
Results and discussion
The synthetic route of the PTUEG3-co-Azo copolymer is illustrated in Fig. 1a. The copolymer is prepared via a straightforward condensation polymerization of 1,2-bis(2-aminoethoxy)ethane (TUDA), 4,4′-azodianiline (Azo-DA), and thiocarbonyldiimidazole (TCDI) in dimethylformamide (DMF) under a nitrogen atmosphere for 48 h. DMF is selected as the reaction medium for its ability to dissolve both monomers and the resulting copolymer, ensuring homogeneous mixing, maintaining an appropriate viscosity, and preventing reductions in the reaction rate and conversion during polymerization. TCDI is widely used as a thiocarbonyl transfer reagent in organic synthesis because its imidazole groups act as excellent leaving groups that can be easily displaced. This property facilitates efficient reactions with amine groups, allowing the reaction to proceed at room temperature without the need for a catalyst, thereby simplifying the experimental procedure and reducing side reactions. The nitrogen atmosphere provides an inert environment, minimizing interference from moisture or oxygen, ensuring the stability of the starting materials throughout the reaction, and contributing to the purity of the resulting copolymer.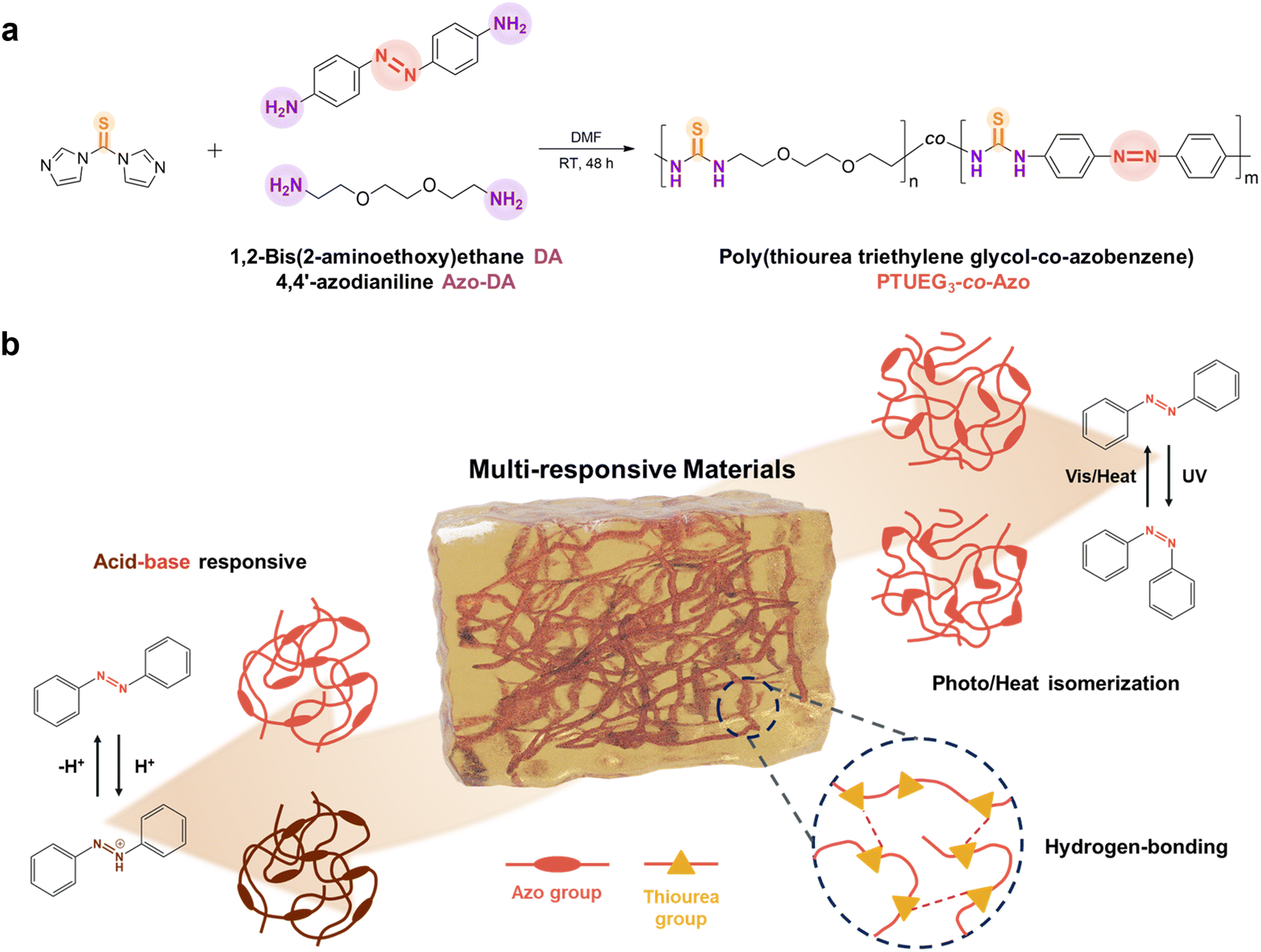 | ||
| Fig. 1 (a) Synthetic scheme of PTUEG3-co-Azo. The polycondensation of 1,2-bis(2-aminoethoxy)ethane (TUEG-DA), 4,4′-azodianiline (Azo-DA) and 1,1′-thiocarbonyldiimidazole (TCDI) can occur under mild conditions. (b) Schematic illustration of PTUEG3-co-Azo as a multiresponsive material, exhibiting acid–base responsiveness via protonation/deprotonation of azobenzene units and thermal isomerization between cis and trans states. | ||
As shown in Fig. 1b, the PTUEG3-co-Azo copolymer demonstrates multiresponsive properties enabled by its molecular structure. The azobenzene groups in PTUEG3-co-Azo provide both acid–base responsiveness and cis/trans isomerization capabilities. Upon exposure to acidic vapors, protonation of the azobenzene groups induces a color change from yellow-orange to deep clay, corresponding to the formation of PTUEG3-co-azonium. This transformation is reversible; exposure to basic vapors deprotonates the azobenzene groups, restoring the original yellow-orange color. This acidochromic behavior demonstrates the potential of PTUEG3-co-Azo films for chemical sensing applications that require visual detection of pH changes in the environment.
In addition, the PTUEG3-co-Azo molecules exhibit cis/trans isomerization. While azobenzene-containing polymers with azobenzene units in the main chain typically show reduced photoresponsiveness because of steric hindrance and restricted segmental motion, the polymers retain thermal responsiveness. Upon heat treatment, the cis form of the azobenzene groups undergoes isomerization to the more stable trans form, leading to changes in molecular conformation and polarity. These structural changes can affect the material's optical and mechanical properties, further enhancing its potential for applications in adaptive systems and thermally controlled devices.
Furthermore, the polyether segments in the PTUEG3 backbone provide flexibility and promote strong intermolecular interactions, which contribute to the adhesive properties on various surfaces. This intrinsic flexibility facilitates chain mobility, enabling effective molecular reorganization under external stimuli such as heat or mechanical force. Moreover, the thiourea groups in PTUEG3 form robust hydrogen bonding networks, where the N–H groups act as hydrogen bond donors, and the sulfur atoms in thiourea and oxygen atoms in polyether segments serve as hydrogen bond acceptors, further strengthening adhesion by enhancing interfacial interactions. While the azobenzene moieties drive the material's responsive behavior, the PTUEG3 backbone ensures reliable adhesive performance and mechanical adaptability. The combination of thermal responsiveness and strong adhesion makes PTUEG3-co-Azo a promising material for applications that require flexibility, durability, and responsiveness to external stimuli.
The synthesis of PTUEG3-co-Azo copolymers introduces azobenzene units into the polymer backbone, as summarized in Table 1. Incorporating Azo-DA as a comonomer presents challenges because its reactivity is lower compared with TUEG-DA. This reduced reactivity is attributed to the conjugation effect of the azobenzene groups, which delocalizes the lone-pair electrons on the amine groups and decreases their nucleophilicity. Additionally, the bulky azobenzene structure introduces steric hindrance, further restricting chain propagation and resulting in reduced molecular weight.
| Entry | TCDI:DA (molar ratio) |
TUEG-DA:Azo-DA (molar ratio) |
T (°C) | Time (h) | Yield of PTU (%) | M w (kg mol−1) | Đ |
|---|---|---|---|---|---|---|---|
| 1 | 1:1 |
1:1 |
RT | 48 | 63.4 | — | — |
| 2 | 1:1 |
2:1 |
RT | 48 | 72 | 3.79 | 1.75 |
| 3 | 1:1 |
3:1 |
RT | 48 | 73.2 | 5.29 | 1.98 |
| 4 | 1:1 |
4:1 |
RT | 48 | 82.8 | 10.2 | 1.97 |
| 5 | 1:1 |
1:0 |
RT | 24 | 88.6 | 27.8 | 2.02 |
The effect of varying TUEG-DA:Azo-DA feed ratios on the polymerization efficiency is systematically investigated. At a 1:1 ratio (entry 1), the yield (63.4%) is significantly reduced, indicating the reactivity limitations of Azo-DA. Increasing the feed ratio to 2:1 (entry 2) improves the yield to 72%, but the molecular weight remains relatively low, likely because of the steric and electronic effects of the azobenzene moieties. Further increasing the TUEG-DA:Azo-DA ratio to 3:1 (entry 3) achieves a balance between yield (73.2%) and molecular weight (5.29 kg mol−1). At a 4:1 ratio (entry 4), the molecular weight increases significantly to 10.2 kg mol−1, suggesting that the higher TUEG-DA content promotes more efficient chain growth by compensating for the lower reactivity of Azo-DA. The results demonstrate that varying the TUEG-DA:Azo-DA feed ratio directly influences the polymer yield and molecular weight. The chemical structure of the resulting PTUEG3-co-Azo copolymers (1:0, 2:1, 3:1, and 4:1) is confirmed by 1H NMR spectroscopy (Fig. S1–S4, ESI†), while the molecular weight distribution is verified through GPC analysis (Fig. S5, ESI†). However, the PTUEG3-co-Azo (1:1) copolymer is excluded from these analyses because it is insoluble in most organic solvents, preventing appropriate characterization. 1H NMR spectra confirm the successful incorporation of both TUEG3-DA and Azo-DA units in the copolymer. Nevertheless, precise integration of proton signals is challenging because of overlapping peaks, residual solvent signals, and hydrogen bonding interactions, particularly from thiourea N–H protons. These findings collectively highlight the role of feed ratio variation in controlling molecular characteristics and optimizing the polymerization efficiency.
Fig. 2a illustrates the preparation process of PTUEG3-co-Azo films. The PTUEG3-co-Azo copolymer is dissolved in a mixed solvent of DMF and tetrahydrofuran (THF) to form a homogeneous solution. The solution is then drop-casted onto a substrate and dried in a vacuum oven at 85 °C for 5 h, ensuring complete solvent removal and formation of uniform PTUEG3-co-Azo films. The water contact angles (WCA) of PTUEG3 and PTUEG3-co-Azo films with varying TUEG-DA:Azo-DA ratios (2:1, 3:1, and 4:1) are shown in Fig. 2b. Pure Azo-DA does not form a stable and uniform film suitable for direct water contact angle measurements, preventing accurate WCA analysis. The WCA increases progressively with higher azobenzene contents, from 55° for PTUEG3 to 63° (4:1), 76° (3:1), and 92° (2:1), indicating a clear trend of increasing hydrophobicity. This behavior can be attributed to the azobenzene groups, which possess lower surface energy relative to the more hydrophilic PTUEG3 backbone. The rigid and planar structure of azobenzene moieties reduces surface polarity, leading to decreased wettability as the azobenzene content increases. Fig. 2c presents the small-angle X-ray scattering (SAXS) profiles of PTUEG3 and PTUEG3-co-Azo films with varying TUEG-DA:Azo-DA ratios (1:1, 2:1, 3:1, and 4:1). At a ratio of 1:1, a weak scattering peak is observed in the low-q region (q < 0.1 Å−1). This peak suggests the presence of localized aggregation, likely caused by the high azobenzene content at this ratio. The azobenzene groups interact through π–π stacking, forming aggregated domains within the polymer matrix.33 The appearance of a peak in the 1:1 sample may result from enhanced phase separation, which promotes nanoscale aggregation. The absence of higher-order peaks suggests that while aggregation occurs, it does not extend to a well-defined periodic structure, indicating a degree of disorder within the domains. As the Azo-DA ratio decreases, the weak scattering peak disappears, implying a reduced tendency for domain formation and a more homogeneous polymer network. This trend indicates that reduced azobenzene content minimizes π–π interactions and enhances the compatibility between polymer segments, thereby suppressing localized aggregation and promoting a more homogeneous structure. At a higher PTUEG3 content, the flexible polyether chains dominate the polymer matrix, further reducing structural heterogeneities. The weak scattering peak at a ratio of 1:1 thus reflects a transitional state where the high azobenzene content facilitates localized aggregation, while higher PTUEG3 ratios result in a more uniform morphology with minimal phase separation.
 | ||
| Fig. 2 (a) Schematic illustration of the preparation process for PTUEG3-co-Azo films. (b) Water contact angle (WCA) measurements of PTUEG3-co-Azo films. (c) Small-angle X-ray scattering (SAXS) profiles of PTUEG3 and PTUEG3-co-Azo films with different TUEG-DA:Azo-DA ratios (1:1, 2:1, 3:1, and 4:1). (d) Thermogravimetric analysis (TGA) curves of PTUEG3 and PTUEG3-co-Azo films. (e) XPS survey spectra of PTUEG3-co-Azo films. | ||
The thermogravimetric analysis (TGA) results of PTUEG3 and PTUEG3-co-Azo films with varying TUEG-DA:Azo-DA ratios (1:1, 2:1, 3:1, and 4:1) are shown in Fig. 2d. While the thermal degradation temperatures (Td) exhibit no clear trend with an increase in the Azo-DA content, the introduction of azobenzene groups influences the overall thermal behavior, particularly in residue formation. The higher residual content observed with an increase in the azo group content is attributed to the greater thermal stability of azobenzene segments, which decompose at higher temperatures compared to the PTUEG3 backbone. Additionally, the aromatic structure of azobenzene contributes to the formation of thermally stable carbonaceous residues upon degradation. This behavior can be attributed to a combination of factors, including molecular weight variations, the presence of cis–trans isomerization of azobenzene groups, and the random copolymerization of TUEG-DA and Azo-DA. Differences in molecular weight among the copolymers may influence Td, as higher molecular weights typically confer greater thermal stability. Additionally, the cis–trans isomerization of azobenzene moieties introduces structural irregularities, which can disrupt polymer chain packing and reduce thermal resistance. The random distribution of azobenzene units along the PTUEG3 backbone further contributes to heterogeneity in the copolymer structure, resulting in variations in thermal behavior across compositions. The X-ray photoelectron spectroscopy (XPS) analyses of PTUEG3-co-Azo films with varying TUEG-DA:Azo-DA ratios (1:1, 2:1, 3:1, and 4:1) are presented in Fig. 2e. The spectra confirm the presence of key elements, including carbon (C 1s), nitrogen (N 1s), sulfur (S 2p), and oxygen (O 1s), which are consistent with the chemical composition of the copolymer. The N 1s signal originates from the azobenzene and thiourea groups, while the S 2p peak indicates the successful incorporation of thiourea linkages into the polymer backbone. These results verify the successful synthesis of PTUEG3-co-Azo copolymers and confirm the elemental composition, supporting the proposed chemical structure.
The azobenzene groups in the PTUEG3-co-Azo system exhibit a transition between the Azo form and the azonium form under external stimuli, as demonstrated in Fig. 3a. The azobenzene groups undergo protonation upon exposure to acid vapors, converting the Azo form into the azonium form. This transition results in a distinct color change from yellow-orange to deep clay-colored, driven by the protonation process. The protonation-induced color change of PTUEG3-co-Azo occurs upon exposure to strong acids, indicating a relatively low pKa similar to other azobenzene-based systems. Previous studies have shown that azobenzene protonation takes place under acidic conditions within the 0–0.76 M HCl range, which aligns with the acidochromic response observed in this study. The color variation provides a clear indication of the material's response to acidic conditions. Exposing the deep clay-colored films to base vapor, such as ammonium vapor, neutralizes the protonation and restores the original yellow-orange form of the PTUEG3-co-Azo system. The UV-vis absorption spectra in Fig. 3b–d further demonstrate the acid–base responsiveness of PTUEG3-co-Azo films with varying TUEG-DA:Azo-DA ratios (2:1, 3:1, and 4:1). To ensure consistency in optical analysis, the PTUEG3-co-Azo films are prepared with controlled thicknesses: 83 μm (2:1), 80 μm (3:1), and 75 μm (4:1). Upon exposure to HCl vapors, a significant increase in absorbance at ∼540 nm is observed, corresponding to the protonation of azobenzene groups and the formation of the azonium form. This change is particularly prominent for the 2:1 ratio film (Fig. 3b), which contains the highest azobenzene content, providing more active sites for protonation and resulting in a stronger optical response. In comparison, the 3:1 ratio film (Fig. 3c) exhibits a moderate absorbance change, while the 4:1 ratio film (Fig. 3d) shows the weakest response because of the reduced azobenzene content, which limits the protonation capacity.
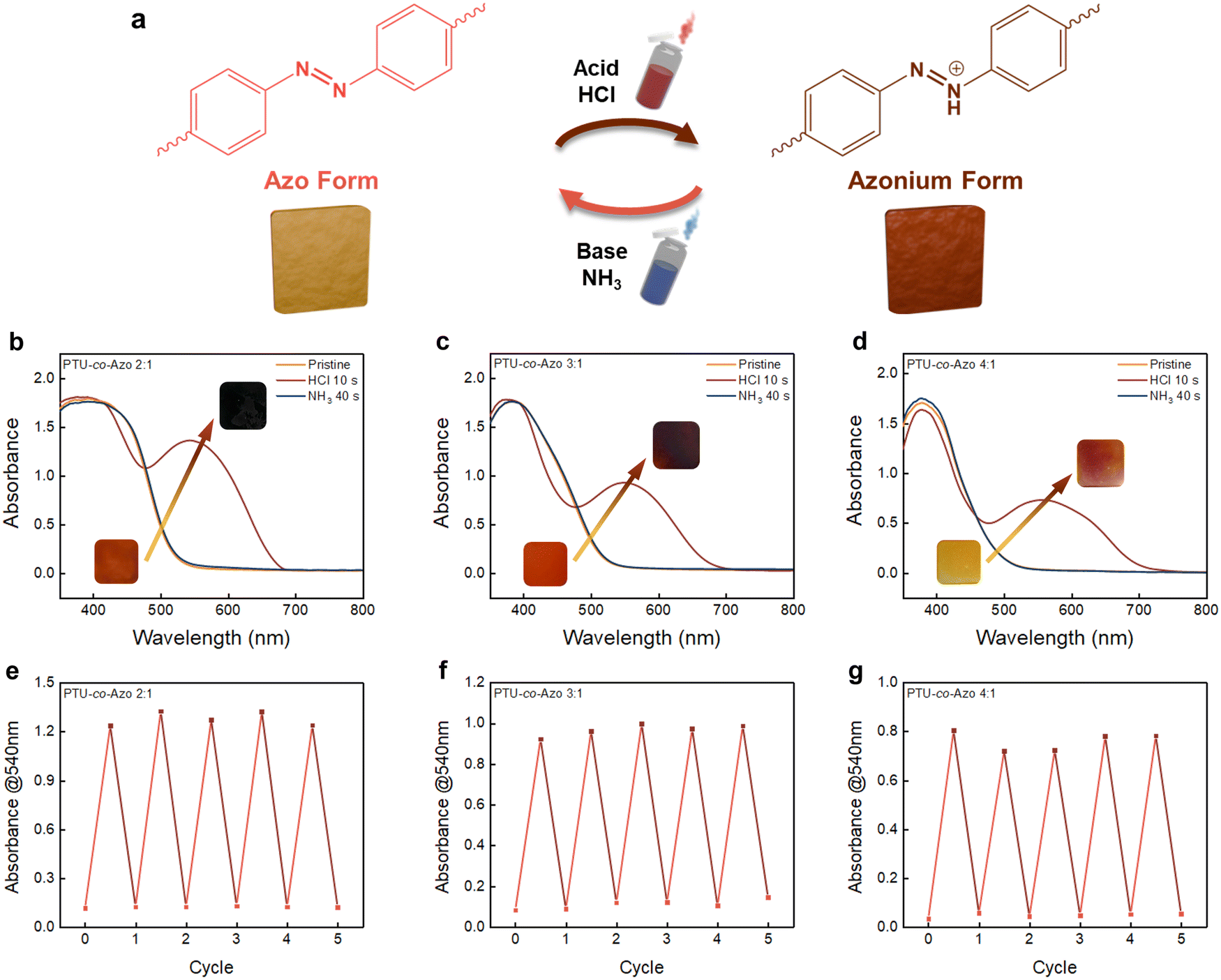 | ||
| Fig. 3 (a) Illustrations and photos of the transformation between the Azo and azonium forms upon exposure to acid and base vapors. (b)–(d) UV-Vis absorption spectra of PTUEG3-co-Azo films with different TUEG-DA:Azo-DA ratios (2:1, 3:1, and 4:1) upon exposure to HCl vapors (10 s) and NH3 vapors (40 s). (e)–(g) Plots of the absorption intensities at 540 nm for PTUEG3-co-Azo films with different TUEG-DA:Azo-DA ratios (2:1, 3:1, and 4:1) during cycles of exposure to HCl vapors (10 s) and NH3 vapors (40 s). | ||
The cycling performance under acid and base vapors (Fig. 3e–g) demonstrates the repeatability and stability of the films’ optical responses. For the 2:1 ratio film (Fig. 3e), consistent and significant absorbance changes at 540 nm over five cycles indicate reliable acid–base cycling stability. The 3:1 ratio film (Fig. 3f) exhibits stable absorbance changes, albeit with slightly lower intensity compared with the 2:1 film, reflecting its lower azobenzene content. In contrast, the 4:1 ratio film (Fig. 3g) shows minimal absorbance changes during cycling, confirming that the reduced azobenzene content weakens the film's acid–base responsiveness. Additional experiments using independently prepared films confirm the reproducibility of the acid–base response. The results show consistent optical transitions upon exposure to acidic and basic vapors, aligning with the trends observed in Fig. 3. The reproduced data is presented in Fig. S6 in the ESI.† To visually demonstrate the acid–base-induced color transitions observed in the UV-vis spectra and cycling tests, a supporting video (Video S1, ESI†) is recorded using trifluoroacetic acid (TFA) and triethylamine (TEA) vapors as acid and base sources, respectively. The use of different acids and bases verifies that the color transition is a general characteristic of the material. Additionally, the stronger basicity of TEA facilitates more rapid deprotonation, ensuring a clearer visualization of the reversible transformation. The video provides a visual representation of the protonation and deprotonation process. Collectively, the results indicate that the acid–base responsiveness of PTUEG3-co-Azo films is strongly dependent on the azobenzene content, with higher azo group concentrations leading to more pronounced color changes and stable cycling performance. These findings highlight the potential of PTUEG3-co-Azo films for pH-responsive color-changing materials in applications such as sensors and smart coatings.
As presented in Fig. 4a, the scheme illustrates the cis–trans isomerization of azobenzene moieties in the PTUEG3-co-Azo films. Upon exposure to external stimuli, such as heat, the azobenzene groups undergo isomerization, transitioning from the cis form to the trans form. The cis form introduces a bent conformation, disrupting the molecular packing and increasing structural irregularity, while the trans form adopts a linear structure that promotes a more ordered arrangement of polymer chains. This structural transformation influences the chain conformation and packing density within the polymer network. However, in the PTUEG3-co-Azo system, the azo groups in the main chain limit their isomerization behavior. The rigidity of the polymer backbone imposes constraints on the large-scale conformational changes required for cis–trans isomerization. Additionally, the azobenzene in the backbone causes steric hindrance and restricts molecular mobility. This lack of flexibility, combined with the reduced free volume in the tightly packed structure, increases the energy barrier for photoinduced isomerization. Typically, azobenzene undergoes UV-induced trans-to-cis isomerization, while heat or visible light facilitates the reverse cis-to-trans transition. However, in this system, the high activation energy required for trans-to-cis conversion limits the efficiency of UV-induced isomerization. As a result, thermal-driven isomerization dominates, making heat the primary stimulus for structural transformation, while UV light has a minimal effect on promoting cis isomerization.
 | ||
| Fig. 4 (a) Cis–trans isomerization of the azobenzene groups in PTUEG3-co-Azo copolymer films under UV and visible light irradiations. (b)–(e) Differential scanning calorimetry (DSC) thermograms of PTUEG3-co-Azo copolymers with TUEG-DA:Azo-DA ratios of (b) 1:1, (c) 2:1, (d) 3:1, and (e) 4:1, showing glass transition temperatures (Tg) during first and second heating cycles. The first heating cycle curves have been vertically offset for clarity. | ||
The differential scanning calorimetry (DSC) thermograms of PTUEG3-co-Azo films with varying TUEG-DA:Azo-DA ratios (1:1, 2:1, 3:1, and 4:1) are presented in Fig. 4b–e. Two Tg values can be observed, which are attributed to the thermally induced isomerization of the azobenzene groups during heating. The Tg variations observed in PTUEG3-co-Azo copolymers correlate with molecular weight differences. Higher molecular weight copolymers tend to exhibit increased Tg values, consistent with the well-established influence of molecular weight on polymer chain mobility. However, additional factors such as hydrogen bonding interactions, chain rigidity, and azobenzene cis–trans isomerization also play a role in modifying the thermal properties, leading to a complex Tg response. In the first heating cycle, the azobenzene groups are predominantly in the cis form, exhibiting poor packing efficiency due to their bent conformation. This structural irregularity disrupts chain packing and reduces intermolecular interactions, leading to weaker restriction of chain segmental motion and a lower Tg. Upon heating, thermal energy induces the cis-to-trans isomerization of azobenzene groups. The trans form adopts a more linear and thermodynamically stable structure, which facilitates improved chain packing and enhances intermolecular interactions, including hydrogen bonding among thiourea groups and π–π stacking between azobenzene moieties. Consequently, the Tg values observed in the second heating cycle are larger, attributed to the formation of trans azobenzene moieties, which enhance chain alignment and intermolecular interactions, thereby reducing chain mobility and increasing structural order. At the condition of 1:1 ratio, the two Tg values show the largest difference between heating cycles, with 45 °C in the first cycle and a significant increase to 113 °C in the second cycle. As the azobenzene content decreases to the 2:1 ratio, the more flexible PTUEG3 backbone becomes dominant, lowering the Tg in the first heating cycle to 26.7 °C. However, the Tg increases to 44.9 °C during the second cycle, indicating that the remaining azobenzene moieties still undergo cis-to-trans isomerization, contributing to improved chain packing. Compared with the condition of 1:1 ratio, the reduced azobenzene content in the 2:1 ratio limits the extent of this transition-induced enhancement. With a further reduction in azobenzene content at the condition of 3:1 ratio, the Tg drops slightly to 23.8 °C in the first heating cycle, reflecting the increasing influence of PTUEG3 segments and their greater chain flexibility. Upon heating, the Tg rises to 50.4 °C as the trans form of the azobenzene groups facilitates better chain packing. While this increase is still noticeable, it is less pronounced than at higher azobenzene ratios due to the lower content of azobenzene moieties. At the condition of 4:1 ratio, where azobenzene content is minimal, the Tg reaches its lowest value of 10.3 °C in the first heating cycle, highlighting the dominant role of the PTUEG3 backbone in promoting chain mobility. Despite this condition, the Tg rises to 58.2 °C in the second heating cycle, showing that even a small fraction of azobenzene groups can contribute to improved chain packing through thermally induced cis-to-trans isomerization.
Overall, the Tg behavior across all ratios reflects a balance between the flexible PTUEG3 backbone and the rigid azobenzene moieties. The 1:1 ratio shows the largest Tg increase, highlighting the strong influence of azobenzene content and its structural transformation. As the azobenzene content decreases (2:1, 3:1, and 4:1), the Tg in the first heating cycle decreases because of the greater flexibility of the copolymer backbone. Nevertheless, the Tg increase in the second heating cycle consistently demonstrates the role of cis-to-trans isomerization in improving chain packing and structural order, even at lower azobenzene levels. The observed Tg values exhibit a generally consistent trend across multiple scans. Minor variations likely result from the inherent randomness of the copolymerization process and differences in sample preparation. Despite these slight deviations, the overall thermal behavior remains reproducible.
The preparation process of the adhesion sample is demonstrated in Fig. 5a, where adhesive polymer layers are applied between two glass plates and cured under controlled conditions. The lap-shear configuration ensures uniform stress distribution across the adhesive layer during mechanical testing, enabling a reliable assessment of adhesive performance. Fig. 5b displays photographs of the lap shear test, demonstrating that the polymer layers are applied between two 1 × 1 cm2 glass plates, which are separately fixed onto substrates. This configuration ensures uniform stress distribution across the adhesive layer during mechanical testing. The image shows the stretching process during the lap shear test, where tensile forces are applied to the bonded interface to measure shear strength and evaluate maximum strength. The strong chain entanglement of PTUEG3 segments results in a highly cohesive polymer network that remains structurally robust below Tg. By increasing the curing temperature, molecular rearrangement is facilitated, allowing the polymer chains to adopt more favorable configurations. This structural reorganization elevates the Tg, enhancing adhesion performance without the need for chemical crosslinking. Combining hydrogen bonding and π–π stacking interactions contributes to the improved shear strength observed at elevated curing temperatures. The lap shear test involves PTUEG3 and the PTUEG3-co-Azo (4:1) copolymer. The selection of the 4:1 ratio is considered by the balance of functionality and synthetic accessibility, providing an integration of hydrogen bonding interactions and azo group characteristics. Fig. 5c compares the shear strength of PTUEG3 and PTUEG3-co-Azo (4:1) at various curing temperatures. The results show that both materials exhibit significant improvements in shear strength with increasing curing temperatures, attributed to enhanced chain mobility and the cis-to-trans transition of azobenzene moieties, which reduce steric hindrance and bring functional groups on different polymer chains closer together. This structural reorganization strengthens hydrogen bonding among thiourea groups and π–π stacking between azobenzene moieties, collectively improving adhesion performance. At 100 °C, the PTUEG3-co-Azo (4:1) copolymer demonstrates a maximum shear strength exceeding that of PTUEG3, attributed to the combined effects of chain reorganization via azo group isomerization and strengthened hydrogen bonding interactions. This enhanced performance highlights the adhesive capabilities of the PTUEG3-co-Azo copolymer, making it particularly suitable for applications requiring robust adhesion.
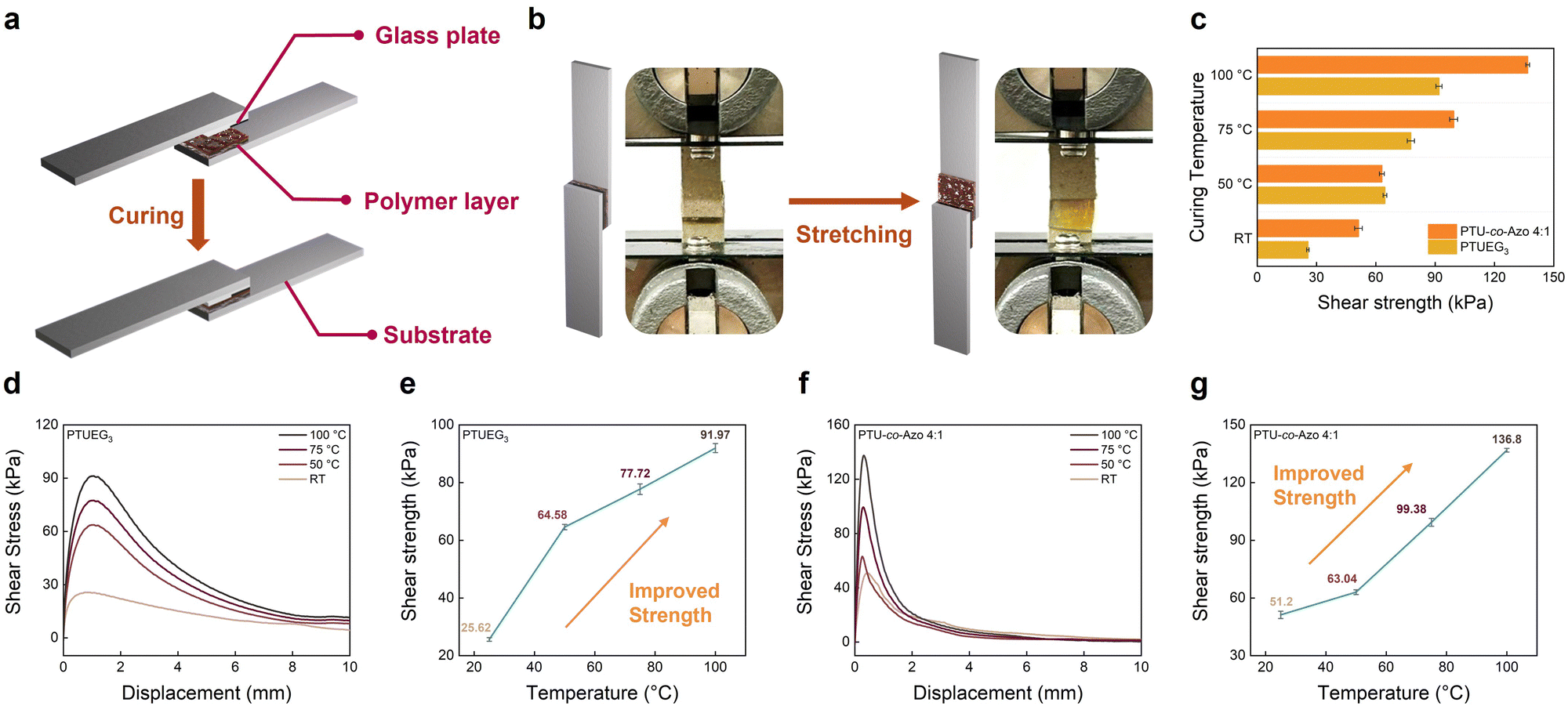 | ||
| Fig. 5 (a) Schematic of the preparation of the adhesion samples, where PTUEG3 and PTUEG3-co-Azo layers are applied between two glass plates and cured under controlled conditions. (b) Photographs of the stretching process during lap-shear testing. (c) Comparison of shear strength for PTUEG3 and PTUEG3-co-Azo (4:1) at different curing temperatures. (d) and (f) Shear stress–displacement curves of PTUEG3 and PTUEG3-co-Azo (4:1) at different curing temperatures. (e) and (g) Plot of the shear strength for different curing temperatures corresponding to (d) and (f). | ||
To evaluate the adhesion ability, shear stress–displacement curves of PTUEG3 and PTUEG3-co-Azo (4:1) at different curing temperatures are presented in Fig. 5d and f, illustrating the temperature-dependent variation in adhesive shear strength. For PTUEG3, the curves in Fig. 5d show a gradual increase in peak shear stress and displacement as the curing temperature rises. At room temperature (25 °C), PTUEG3 demonstrates a relatively low maximum shear stress of 25.62 kPa, which increases to 64.58 kPa at 50 °C, 77.72 kPa at 75 °C and further to 91.97 kPa at 100 °C. The observed trend demonstrates the effect of thermal activation on improving adhesive performance. Increased curing temperatures promote greater chain mobility and hydrogen bonding interactions within the adhesive layer, resulting in stronger interfacial adhesion and improved mechanical performance. PTUEG3-co-Azo (4:1) exhibits superior adhesive performance compared with PTUEG3, as shown in the shear stress–displacement curves in Fig. 5f. At room temperature (25 °C), the copolymer achieves a maximum shear stress of 51.20 kPa, which increases to 63.04 kPa at 50 °C, 99.38 kPa at 75 °C and further to 136.8 kPa at 100 °C. The results mark improvement because of the synergistic effect of azobenzene moieties and the flexible PTUEG3 backbone. The rigid azobenzene groups enhance interfacial adhesion by increasing mechanical strength and enabling thermally induced chain reorganization, which collectively contribute to improved cohesive strength within the adhesive layer. The PTUEG3 backbone complements these effects by providing structural flexibility and chain mobility during the reorganization process, facilitating efficient stress distribution and ensuring enhanced mechanical stability.
The temperature-dependent shear strength, as summarized in Fig. 5e and g, further underscores the advantages of PTUEG3-co-Azo (4:1) over PTUEG3. This variation in adhesive strength across curing temperatures can be attributed to the interplay of molecular structure and thermal effects in the materials. At room temperature, PTUEG3-co-Azo (4:1) exhibits higher shear strength compared to PTUEG3 because of the presence of azobenzene moieties, which contribute to a more robust network, providing an advantage at lower temperatures where chain mobility is limited. At 50 °C, the adhesive strengths of both materials become comparable, as the increased thermal energy facilitates greater chain mobility in PTUEG3, reducing the initial gap in performance. The thermal activation allows for more effective molecular alignment and bonding at the interface, diminishing the advantage provided by the azobenzene groups under these conditions. At 100 °C, PTUEG3-co-Azo (4:1) outperforms PTUEG3, achieving higher shear strength. This outcome can be attributed to the thermally induced molecular reorganization of azobenzene moieties, which strengthens the adhesive layer by enhancing interfacial interactions and cohesive strength. Additionally, the higher curing temperature promotes optimal chain alignment and bonding in the copolymer, leveraging the combined effects of hydrogen bonding and azo group reorganization. These results underscore the role of thermal activation in modulating the adhesive properties of these materials, particularly the unique advantages conferred by the copolymer structure. The adhesive performance of PTUEG3-co-Azo (4:1) demonstrates a clear advantage over PTUEG3, particularly under elevated curing temperatures. The synergistic effects of hydrogen bonding interactions, structural flexibility, and azobenzene-induced reorganization contribute to its superior performance across a range of conditions. These findings highlight the potential of PTUEG3-co-Azo (4:1) for advanced adhesive applications, particularly in environments where thermal activation can be leveraged to optimize adhesion strength and durability.
Conclusions
In this study, a multifunctional PTUEG3-co-Azo copolymer is successfully synthesized via a controlled polycondensation process, incorporating azobenzene moieties into the polymer backbone. The azobenzene groups undergo thermally induced cis-to-trans isomerization, which promotes structural reorganization, enhances molecular packing, and significantly increases the Tg values. This transformation reduces segmental mobility, improving the mechanical properties under thermal treatment. Meanwhile, the flexible PTUEG3 backbone facilitates molecular rearrangement, while the thiourea groups form robust hydrogen bonding networks, contributing to enhanced interfacial adhesion. These combined features make PTUEG3-co-Azo a promising material for durable and flexible adhesive applications, where heat-induced structural changes improve the bonding performance. Additionally, the azobenzene groups exhibit reversible protonation and deprotonation, enabling distinct color changes with durability and repeatable acid–base responsiveness over multiple cycles. This behavior is influenced by the azobenzene content, where higher concentrations enable more pronounced responses and stable performance under repeated acid–base exposure. With heat-induced structural reorganization enhancing thermal properties and interfacial adhesion, PTUEG3-co-Azo copolymers demonstrate versatility for functional applications. These properties make them suitable for adhesive applications and pH-responsive systems, supporting the development of advanced functional materials.Experimental
Materials
1,2-Bis(2-aminoethoxy)ethane and 4,4′-azodianiline were bought from Tokyo Chemical Industry (TCI). 1,1′-Thiocarbonyldiimidazole (TCDI, 95%) was bought from Nova Materials. Dimethylformamide (DMF, 99.8%) was procured from Thermo Scientific. Methanol (MeOH, 99.5%) was procured from Echo Chemical Co. Ltd. Diethyl ether was obtained from DUKSAN Pure Chemicals Co. Ltd. Tetrahydrofuran (THF) was obtained from Tedia.Synthesis of poly(thiourea triethylene glycol) (PTUEG3)
1,2-Bis(2-aminoethoxy)ethane (1.468 mL, 10 mmol) was dissolved in DMF (3 mL) in a double-necked flask. Separately, 1,1′-thiocarbonyldiimidazole (TCDI, 1.782 g, 10 mmol) was dissolved in DMF (2 mL) in a glass sample bottle. Once both solutions were fully dissolved, the TCDI solution was transferred into the double-necked flask. The reaction mixture was stirred under a nitrogen atmosphere at room temperature for 24 h. After completion, the reaction mixture was poured into stirred diethyl ether (200 mL) to induce precipitation. The precipitate was further reprecipitated in a DMF/MeOH (50 mL) solution to obtain the polymer product. The final product was dried in a vacuum oven at 140 °C for 24 h to remove the residual solvent, yielding a yellow to orange glass-like material.Synthesis of the random copolymer poly(thiourea triethylene glycol)-co-Azo (PTUEG3-co-Azo)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1).
A mixture of 1,2-bis(2-aminoethoxy)ethane (0.293 mL, 2 mmol) and 4,4′-azodianiline (0.425 g, 2 mmol) was dissolved in DMF (1 mL) in a double-necked flask. Separately, 1,1′-thiocarbonyldiimidazole (TCDI, 0.713 g, 4 mmol) was dissolved in DMF (1.5 mL) in a glass sample bottle. Once both solutions were fully dissolved, the TCDI solution was transferred into the double-necked flask. The reaction mixture was stirred under a nitrogen atmosphere at room temperature for 48 h. After completion, the reaction mixture was poured into stirred diethyl ether (100 mL) to induce precipitation. The precipitate was further reprecipitated in a DMF/MeOH (50 mL) solution to obtain the polymer product. The final product was dried in a vacuum oven at 140 °C for 24 h to remove the residual solvent, yielding a yellow to orange glass-like material.
:1).
A mixture of 1,2-bis(2-aminoethoxy)ethane (0.293 mL, 2 mmol) and 4,4′-azodianiline (0.212 g, 1 mmol) was dissolved in DMF (0.5 mL) in a double-necked flask. Separately, 1,1′-thiocarbonyldiimidazole (TCDI, 0.535 g, 3 mmol) was dissolved in DMF (1.5 mL) in a glass sample bottle. Once both solutions were fully dissolved, the TCDI solution was transferred into the double-necked flask. The reaction mixture was stirred under a nitrogen atmosphere at room temperature for 48 h. After completion, the reaction mixture was poured into stirred diethyl ether (100 mL) to induce precipitation. The precipitate was further reprecipitated in a DMF/MeOH (50 mL) solution to obtain the polymer product. The final product was dried in a vacuum oven at 140 °C for 24 h to remove the residual solvent, yielding a yellow to orange glass-like material.
:1).
A mixture of 1,2-bis(2-aminoethoxy)ethane (0.44 mL, 3 mmol) and 4,4′-azodianiline (0.212 g, 1 mmol) was dissolved in DMF (1 mL) in a double-necked flask. Separately, 1,1′-thiocarbonyldiimidazole (TCDI, 0.713 g, 4 mmol) was dissolved in DMF (1.5 mL) in a glass sample bottle. Once both solutions were fully dissolved, the TCDI solution was transferred into the double-necked flask. The reaction mixture was stirred under a nitrogen atmosphere at room temperature for 48 h. After completion, the reaction mixture was poured into stirred diethyl ether (100 mL) to induce precipitation. The precipitate was further reprecipitated in a DMF/MeOH (50 mL) solution to obtain the polymer product. The final product was dried in a vacuum oven at 140 °C for 24 h to remove residual solvent, yielding a yellow to orange glass-like material.
:1).
A mixture of 1,2-bis(2-aminoethoxy)ethane (0.587 mL, 4 mmol) and 4,4′-azodianiline (0.212 g, 1 mmol) was dissolved in DMF (1 mL) in a double-necked flask. Separately, 1,1′-thiocarbonyldiimidazole (TCDI, 0.891 g, 5 mmol) was dissolved in DMF (2 mL) in a glass sample bottle. Once both solutions were fully dissolved, the TCDI solution was transferred into the double-necked flask. The reaction mixture was stirred under a nitrogen atmosphere at room temperature for 48 h. After completion, the reaction mixture was poured into stirred diethyl ether (100 mL) to induce precipitation. The precipitate was further reprecipitated in a DMF/MeOH (50 mL) solution to obtain the polymer product. The final product was dried in a vacuum oven at 140 °C for 24 h to remove the residual solvent, yielding a yellow to orange glass-like material.
:1).
A mixture of 1,2-bis(2-aminoethoxy)ethane (0.293 mL, 2 mmol) and 4,4′-azodianiline (0.425 g, 2 mmol) was dissolved in DMF (1 mL) in a double-necked flask. Separately, 1,1′-thiocarbonyldiimidazole (TCDI, 0.713 g, 4 mmol) was dissolved in DMF (1.5 mL) in a glass sample bottle. Once both solutions were fully dissolved, the TCDI solution was transferred into the double-necked flask. The reaction mixture was stirred under a nitrogen atmosphere at room temperature for 48 h. After completion, the reaction mixture was poured into stirred diethyl ether (100 mL) to induce precipitation. The precipitate was further reprecipitated in a DMF/MeOH (50 mL) solution to obtain the polymer product. The final product was dried in a vacuum oven at 140 °C for 24 h to remove the residual solvent, yielding a yellow to orange glass-like material.
:1).
A mixture of 1,2-bis(2-aminoethoxy)ethane (0.293 mL, 2 mmol) and 4,4′-azodianiline (0.212 g, 1 mmol) was dissolved in DMF (0.5 mL) in a double-necked flask. Separately, 1,1′-thiocarbonyldiimidazole (TCDI, 0.535 g, 3 mmol) was dissolved in DMF (1.5 mL) in a glass sample bottle. Once both solutions were fully dissolved, the TCDI solution was transferred into the double-necked flask. The reaction mixture was stirred under a nitrogen atmosphere at room temperature for 48 h. After completion, the reaction mixture was poured into stirred diethyl ether (100 mL) to induce precipitation. The precipitate was further reprecipitated in a DMF/MeOH (50 mL) solution to obtain the polymer product. The final product was dried in a vacuum oven at 140 °C for 24 h to remove the residual solvent, yielding a yellow to orange glass-like material.
:1).
A mixture of 1,2-bis(2-aminoethoxy)ethane (0.44 mL, 3 mmol) and 4,4′-azodianiline (0.212 g, 1 mmol) was dissolved in DMF (1 mL) in a double-necked flask. Separately, 1,1′-thiocarbonyldiimidazole (TCDI, 0.713 g, 4 mmol) was dissolved in DMF (1.5 mL) in a glass sample bottle. Once both solutions were fully dissolved, the TCDI solution was transferred into the double-necked flask. The reaction mixture was stirred under a nitrogen atmosphere at room temperature for 48 h. After completion, the reaction mixture was poured into stirred diethyl ether (100 mL) to induce precipitation. The precipitate was further reprecipitated in a DMF/MeOH (50 mL) solution to obtain the polymer product. The final product was dried in a vacuum oven at 140 °C for 24 h to remove residual solvent, yielding a yellow to orange glass-like material.
:1).
A mixture of 1,2-bis(2-aminoethoxy)ethane (0.587 mL, 4 mmol) and 4,4′-azodianiline (0.212 g, 1 mmol) was dissolved in DMF (1 mL) in a double-necked flask. Separately, 1,1′-thiocarbonyldiimidazole (TCDI, 0.891 g, 5 mmol) was dissolved in DMF (2 mL) in a glass sample bottle. Once both solutions were fully dissolved, the TCDI solution was transferred into the double-necked flask. The reaction mixture was stirred under a nitrogen atmosphere at room temperature for 48 h. After completion, the reaction mixture was poured into stirred diethyl ether (100 mL) to induce precipitation. The precipitate was further reprecipitated in a DMF/MeOH (50 mL) solution to obtain the polymer product. The final product was dried in a vacuum oven at 140 °C for 24 h to remove the residual solvent, yielding a yellow to orange glass-like material.
Fabrication process of the PTUEG3 and PTUEG3-co-Azo films
Initially, 1 g of polymers (PTUEG3 and PTUEG3-co-Azo with ratios of 1:1 to 4:1), 2.8 mL of DMF, and 1.2 mL of THF were added to a parafilm-sealed glass bottle. The mixture was stirred for 2–3 h at room temperature to obtain a 20 wt% homogeneous polymer solution. Subsequently, the polymer solutions were drop-cast onto 2.5 × 2.5 cm2 glass substrates. Once the substrates were completely covered with the solutions, the substrates were placed in a vacuum oven at 85 °C for 5 h to remove the residual solvents.
Acidochromic experiments of the PTUEG3-co-Azo films
The yellow-orange PTUEG3-co-Azo films were exposed to hydrochloric acid vapors for 10 s, transforming into deep clay-colored PTUEG3-co-azonium films. These deep clay PTUEG3-co-azonium films could be reverted to the original yellow-orange PTUEG3-co-Azo form after being placed in ammonium vapors for a minimum of 40 s.Adhesion test of the PTUEG3-co-Azo copolymers
Two 1 × 1 cm2 glass plates were used for adhesion testing. The copolymer film was prepared by casting and curing directly on one glass plate under controlled conditions to ensure uniform thickness. The second glass plate was placed on top of the cured film, and the plates were adhered together by curing at room temperature, 50, and 100 °C for 2 h. To prevent direct clamping of the glass plates during testing, the plates were fixed onto supporting substrates, and the tensile testing machine's grips were attached to the substrates. Adhesion strength was evaluated by pulling the plates apart at a constant rate of 2 mm min−1 until failure occurred. The maximum force recorded during the test was used to calculate the shear adhesive strength. Tests were performed under room temperature conditions to assess the adhesion performance.Structure analysis and characterization
The chemical structures of PTUEG3 and PTUEG3-co-Azo were analyzed using a 400 MHz 1H NMR spectrometer (Varian). Size exclusion chromatography (SEC) was performed with a JASCO RI-4030 refractive index detector and an AS-4050 autosampler to evaluate the apparent weight-average molecular weight (Mw). Narrowly distributed polystyrene (PS) standards were used for calibration, and dimethylformamide (DMF) was employed as the mobile phase at a flow rate of 0.1 mL min−1. The water contact angles of the samples were evaluated using a first ten angstroms (FTA125) contact angle meter, which was equipped with a CCD camera. Attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy was performed with a PerkinElmer Spectrum One FTIR spectrometer, operating at a resolution of 4 cm−1. The absorption spectra of PTUEG3-co-Azo films with varying Azo ratios were recorded in the range of 350 to 800 nm using an ultraviolet-visible (UV-vis) spectrometer (Hitachi U4100). X-Ray scattering measurements were performed at the TPS 25A1 coherent X-ray scattering beamline of the National Synchrotron Radiation Research Center (NSRRC), Hsinchu, Taiwan. The incident photon energy, corresponding to a wavelength of 0.8269 Å, was tuned using a double crystal monochromator (DCM). Film samples were analyzed using small-angle X-ray scattering (SAXS), with scattering patterns captured using an Eiger X 1 M photon-counting detector. A hexapod stage was utilized to ensure precise sample alignment. The thermal properties of PTUEG3 and PTUEG3-co-Azo were evaluated using a thermogravimetric analyzer (TGA, TA 55, TA Instruments). The samples were subjected to heating under a nitrogen atmosphere at a rate of 20 °C min−1, reaching a maximum temperature of 700 °C. The glass transition temperatures of PTUEG3 and PTUEG3-co-Azo were determined using a differential scanning calorimeter (DSC, TA Q200, TA Instruments) equipped with a refrigerated cooling system (RCS90). The analyses were carried out under a nitrogen atmosphere over a temperature range of −20 to 200 °C with both heating and cooling rates set at 5 °C min−1. The adhesion properties of PTUEG3 and PTUEG3-co-Azo copolymer films were evaluated using uniaxial elongation tests performed with a tensile testing machine (Shimadzu EZ Test).Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the 2030 Cross-Generation Young Scholars Program of the National Science and Technology Council, Taiwan (NSTC), under Grant No. NSTC 113-2628-E-A49-006, the Center for Emergent Functional Matter Science of National Yang Ming Chiao Tung University from the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan. The authors thank Dr Li-Ching Shen (Center for Advanced Instrumentation at NYCU) for assistance with NMR experiments, and Swee-Lan Cheah of the NTHU Instrumentation Center for assistance with the XPS measurements.References
- C.-T. Chang, K.-C. Kuo, J.-H. Ho, L.-R. Lee, B. Gautam, J.-H. Ciou, Y.-H. Tseng, C.-W. Chang, C.-T. Wu, C.-T. Lin and J.-T. Chen, Reversible Sensing Technologies Using Upcycled TPEE: Crafting pH and Light Responsive Materials towards Sustainable Monitoring, Small, 2024, 20(28), 2400491 CrossRef CAS PubMed
.
- W. Qu, Z. Bi, C. Zou and C. Chen, Light, Heat, and Force-Responsive Polyolefins, Adv. Sci., 2024, 11(11), 2307568 CrossRef CAS PubMed
- X. Li, Y. L. Zhan, W. Li, Z. H. Huang and A. Amirfazli, Light- and Heat-Responsive Superhydrophobic Surfaces with Shape Memory Capacity Prepared by 4D Printing, Adv. Eng. Mater., 2024, 26(22), 2401415 CrossRef CAS
- L. Li, J. M. Scheiger and P. A. Levkin, Design and Applications of Photoresponsive Hydrogels, Adv. Mater., 2019, 31(26), 1807333 CrossRef PubMed
- X. Li, J. Liu, D. Li, S. Huang, K. Huang and X. Zhang, Bioinspired Multi-Stimuli Responsive Actuators with Synergistic Color- and Morphing-Change Abilities, Adv. Sci., 2021, 8(16), 2101295 CrossRef PubMed
- P. She, Y. Qin, X. Wang and Q. Zhang, Recent Progress in External-Stimulus-Responsive 2D Covalent Organic Frameworks, Adv. Mater., 2022, 34(22), 2101175 CrossRef CAS PubMed
- S. Kim, S.-N. Lee, A. A. Melvin and J.-W. Choi, Stimuli-Responsive Polymer Actuator for Soft Robotics, Polymers, 2024, 16(18), 2660 CrossRef CAS PubMed
- Z. Shen, F. Chen, X. Zhu, K.-T. Yong and G. Gu, Stimuli-responsive functional materials for soft robotics, J. Mater. Chem. B, 2020, 8(39), 8972–8991 RSC
- S. Chatani, C. J. Kloxin and C. N. Bowman, The power of light in polymer science: photochemical processes to manipulate polymer formation, structure, and properties, Polym. Chem., 2014, 5(7), 2187–2201 RSC
- Z.-H. Liao and F. Wang, Light-controlled smart materials: Supramolecular regulation and applications, Smart Mol., 2024, 2(4), e20240036 CrossRef CAS
- D. Dai, Y. Zhang, S. Yang, W. Kong, J. Yang and J. Zhang, Recent Advances in Functional Materials for Optical Data Storage, Molecules, 2024, 29(1), 254 CrossRef CAS PubMed
- F. Caroleo, G. Magna, M. L. Naitana, L. Di Zazzo, R. Martini, F. Pizzoli, M. Muduganti, L. Lvova, F. Mandoj, S. Nardis, M. Stefanelli, C. Di Natale and R. Paolesse, Advances in Optical Sensors for Persistent Organic Pollutant Environmental Monitoring, Sensors, 2022, 22(7), 2649 CrossRef CAS PubMed
- Y. Wu, Q. Zhang, Y. Zhao, K. Zhuang, Y. Fan, S. Zhang, X. Zhang, K. Huang and Z. Yao, A Sensitive and Selective Fluorescent Sensor for Berberine Chloride Based on the Supramolecular Self-Assembly of Perylene Diimide in Aqueous Solution, ACS Sustainable Chem. Eng., 2020, 8(16), 6517–6523 CrossRef CAS
- J. Ledesma, P. L. Pisano, D. M. Martino, C. E. Boschetti and S. A. Bortolato, Thymine based copolymers: feasible sensors for the detection of persistent organic pollutants in water, RSC Adv., 2017, 7(77), 49066–49073 RSC
- R. Klajn, Spiropyran-based dynamic materials, Chem. Soc. Rev., 2014, 43(1), 148–184 RSC
- C. Li, A. Iscen, L. C. Palmer, G. C. Schatz and S. I. Stupp, Light-Driven Expansion of Spiropyran Hydrogels, J. Am. Chem. Soc., 2020, 142(18), 8447–8453 CrossRef CAS PubMed
- L. Kortekaas and W. R. Browne, The evolution of spiropyran: fundamentals and progress of an extraordinarily versatile photochrome, Chem. Soc. Rev., 2019, 48(12), 3406–3424 RSC
- D. Bléger and S. Hecht, Visible-Light-Activated Molecular Switches, Angew. Chem., Int. Ed., 2015, 54(39), 11338–11349 CrossRef PubMed
- C. C. Jia, A. Migliore, N. Xin, S. Y. Huang, J. Y. Wang, Q. Yang, S. P. Wang, H. L. Chen, D. M. Wang, B. Y. Feng, Z. R. Liu, G. Y. Zhang, D. H. Qu, H. Tian, M. A. Ratner, H. Q. Xu, A. Nitzan and X. F. Guo, Covalently bonded single-molecule junctions with stable and reversible photoswitched conductivity, Science, 2016, 352(6292), 1443–1445 CrossRef CAS PubMed
- A.-L. Yan, C.-W. Chang, T.-H. Tsai, M.-R. Huang, K.-T. Lin, V. K. Karapala, J.-H. Ho, W.-Y. Dai and J.-T. Chen, Fabrication of patterned and anti-patterned azopolymer nanoarrays via the light-induced nanowetting method, J. Polym. Sci., 2024, 62(16), 3708 CrossRef CAS
- H. Zhou, C. Xue, P. Weis, Y. Suzuki, S. Huang, K. Koynov, G. K. Auernhammer, R. Berger, H.-J. Butt and S. Wu, Photoswitching of glass transition temperatures of azobenzene-containing polymers induces reversible solid-to-liquid transitions, Nat. Chem., 2017, 9(2), 145–151 CrossRef CAS PubMed
- W. C. Xu, S. D. Sun and S. Wu, Photoinduced Reversible Solid-to-Liquid Transitions for Photoswitchable Materials, Angew. Chem., Int. Ed., 2019, 58(29), 9712–9740 CrossRef CAS PubMed
- S. F. Liang, C. R. Yuan, C. Nie, Y. Z. Liu, D. C. Zhang, W. C. Xu, C. W. Liu, G. F. Xu and S. Wu, Photocontrolled Reversible Solid-Fluid Transitions of Azopolymer Nanocomposites for Intelligent Nanomaterials, Adv. Mater., 2024, 36(38), 2408159 CrossRef CAS PubMed
- S. Erekath, K. Chordiya, K. V. Vidhya, M. U. Kahaly and S. K. Kalpathy, Self-aggregation, H-bonding, and photoresponse in film and solution states of azobenzene containing polyurea, Phys. Chem. Chem. Phys., 2022, 24(38), 23447–23459 RSC
- M. Younis, J. Long, S.-Q. Peng, X.-S. Wang, C. Chai, N. Bogliotti and M.-H. Huang, Reversible Transformation between Azo and Azonium Bond Other than Photoisomerization of Azo Bond in Main-Chain Polyazobenzenes, J. Phys. Chem. Lett., 2021, 12(14), 3655–3661 CrossRef CAS PubMed
- L. Fang, H. Zhang, Z. Li, Y. Zhang, Y. Zhang and H. Zhang, Synthesis of Reactive Azobenzene Main-Chain Liquid Crystalline Polymers via Michael Addition Polymerization and Photomechanical Effects of Their Supramolecular Hydrogen-Bonded Fibers, Macromolecules, 2013, 46(19), 7650–7660 CrossRef CAS
- X. Pang, J.-A. Lv, C. Zhu, L. Qin and Y. Yu, Photodeformable Azobenzene-Containing Liquid Crystal Polymers and Soft Actuators, Adv. Mater., 2019, 31(52), 1904224 CrossRef CAS PubMed
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Mechanically robust, readily repairable polymers via tailored noncovalent cross-linking, Science, 2018, 359(6371), 72–76 CrossRef CAS PubMed
- K. Kikkawa, Y. Sumiya, K. Okazawa, K. Yoshizawa, Y. Itoh and T. Aida, Thiourea as a “Polar Hydrophobic” Hydrogen-Bonding Motif: Application to Highly Durable All-Underwater Adhesion, J. Am. Chem. Soc., 2024, 146(30), 21168–21175 CrossRef CAS PubMed
- T.-Y. Lo, H.-H. Su, J.-H. Ho, C.-W. Chang, H.-R. Chen, H.-H. Hsu, K.-J. Chang and J.-T. Chen, Intrinsic Repeated Self-Healing Textiles: Developing Electrospun Fabrics for Enhanced Durability and Stretchability, ACS Omega, 2024, 9(52), 51623 CrossRef CAS PubMed
- Y. Fujisawa, A. Asano, Y. Itoh and T. Aida, Mechanically Robust, Self-Healable Polymers Usable under High Humidity: Humidity-Tolerant Noncovalent Cross-Linking Strategy, J. Am. Chem. Soc., 2021, 143(37), 15279–15285 CrossRef CAS PubMed
- Y. Fujisawa, Y. Nan, A. Asano, Y. Yanagisawa, K. Yano, Y. Itoh and T. Aida, Blending to Make Nonhealable Polymers Healable: Nanophase Separation Observed by CP/MAS 13C NMR Analysis, Angew. Chem., Int. Ed., 2023, 62, e202214444 CrossRef CAS PubMed
- S. Wu and C. Bubeck, Macro- and Microphase Separation in Block Copolymer Supramolecular Assemblies Induced by Solvent Annealing, Macromolecules, 2013, 46(9), 3512–3518 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01536c |
| This journal is © The Royal Society of Chemistry 2025 |