
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeconstructing poplar lignin from ionic liquid pretreatment for biological conversion through sulfonation and Fenton chemistry†
Daniella V. Martinez‡
 a,
Alberto Rodriguez‡ab,
Hemant Choudharyab,
Jay Salinasa,
Estevan J. Martineza,
Oleg Davydovicha,
Gina M. Geiselmanab,
John M. Gladdenab,
Blake A. Simmonsbc and
Michael S. Kent*ab
a,
Alberto Rodriguez‡ab,
Hemant Choudharyab,
Jay Salinasa,
Estevan J. Martineza,
Oleg Davydovicha,
Gina M. Geiselmanab,
John M. Gladdenab,
Blake A. Simmonsbc and
Michael S. Kent*ab
aSandia National Laboratories, Livermore, CA, Albuquerque, NM 87185, USA. E-mail: mmkent88@gmail.com
bJoint BioEnergy Institute, Emeryville, CA 94608, USA
cLawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
First published on 10th February 2025
Abstract
Generating value from lignin through deconstruction and biological conversion is promising but limited by several factors including lack of economically viable deconstruction methods and low bioconversion of the breakdown products. Due to the complex chemical structure of natural lignins, high yield deconstruction requires cleaving both carbon–carbon and ether bonds. The high strength of C–C bonds poses a great challenge for economically viable high conversion of lignin to valuable products or intermediates. Prior work has shown that a Fenton reaction can efficiently cleave C–C bonds in sulfonated polymers at or near room temperature. In the present work, poplar lignin isolated from a cholinium lysinate ionic liquid pretreatment was sulfonated and then treated with a Fenton reaction using conditions that minimized H2O2 and avoided unwanted repolymerization. The deconstruction process was performed at room temperature and ambient pressure. We explored the tradeoff between the extent of deconstruction and the amount of carbon lost as CO2, with total carbon recovered as soluble products ranging up to 40% depending upon conditions. The reaction products were analyzed by size exclusion chromatography, infrared spectroscopy, total dissolved organic carbon and elemental analysis. The results indicated that the products are rich in acid, aldehyde, alcohol, and sulfonate functionalities. A panel of microorganisms were tested for growth using the lignin breakdown products as the sole carbon source and five showed robust growth. A bisabolene-producing strain of Rhodosporidium toruloides was used to demonstrate conversion to product. Several ideas are discussed to improve yields for each step in the process.
Sustainability spotlightLignin is a plentiful feedstock and its conversion to fuels and chemicals is critical for the economic viability of lignocellulosic biofuels. Generating value from lignin through depolymerization and biological conversion holds great promise but is limited by several factors including the lack of cost-effective depolymerization methods and toxicity of the products. Here we employ a method to depolymerize lignin from poplar biomass under mild conditions that produces compounds that are compatible with microbial conversion to bioproducts, which can contribute to the sustainable use of renewable feedstocks. Our work emphasizes the importance of developing technologies to address the UN sustainable development goals on affordable and clean energy (SDG 7). |
1. Introduction
Lignin is an integral component of plant cell walls synthesized from phenylpropanoid units that are linked together through a variety of carbon–carbon (C–C) and carbon–oxygen (C–O) chemical bonds.1 The complex arrangement of the linkages, and specifically the C–C bonds, makes the lignin polymer resistant to chemical and microbial degradation.2 Overcoming this challenge is required for efficient upcycling of this abundant polymer into valuable products such as biofuels, biodegradable polymers, and chemicals, which in turn would promote a sustainable, bio-based economy.3,4Lignin is a renewable source of carbon potentially available in the US at several hundred megatons annually.5 Conversion of this carbon to commodity chemicals could significantly reduce emissions generated in the current production of commodity chemicals from petroleum. For example, plastics are produced from petroleum at 460 MT per year worldwide.6,7 In contrast to sugars from polysaccharides, there is currently little competition for lignin breakdown products. Today most of the lignin available from the pulp and paper industry or from lignocellulosic biorefineries is burned for its energy content. Several reviews of the topic of lignin valorization have been published recently.8–10
A principal goal when deconstructing lignin is to achieve high yields of useful products or intermediates while minimizing the formation of undesirable byproducts, which has proven to be challenging.11 To achieve high conversion of lignins to low molecular weight compounds, it is essential to break C–C bonds.12,13 For example, reductive catalytic fractionation (RCF) largely cleaves ether bonds leaving the C–C bonds intact14 and the yield of aromatic monomers is limited to 15–30%.15,16 Cleaving C–C bonds in lignins can be achieved by catalysis at high temperature and high-pressures but at relatively high cost. This motivates the exploration of alternative approaches.
In prior work we reported an alternative method for breaking C–C bonds in lignin at or near ambient temperature and pressure. This method combines sulfonation with Fenton chemistry for deconstruction. In the Fenton reaction, Fe2+ reacts with hydrogen peroxide to generate Fe3+ and highly potent hydroxyl radical.17–19 Prior work shows that hydroxyl radicals generated by the Fenton reaction efficiently cleave C–C bonds in sulfonated polymers such as lignosulfonate,20,21 sulfonated polyethylene,22 and polystyrene sulfonate.23–25 By adding sulfonate groups to the substrate to chelate iron, the oxidative Fenton reaction is localized to the substrate resulting in efficient breakdown of these polymers to low molecular weight products. The Fenton reaction proceeds at ambient temperature and atmospheric pressure. This is an advantage compared to methods that require energy-intensive processes and a high-pressure reactor. Further, since the Fenton reaction occurs in water with a small amount of biocompatible iron as catalyst, little or no post-processing is needed prior to bioconversion. The extent of deconstruction achieved in the Fenton reaction can be controlled by adjusting the reaction conditions and the amounts of the reagents (iron and H2O2). Extensive deconstruction to low molecular weight products is possible, but a tradeoff exists between the extent of deconstruction and the amount of carbon lost through overoxidation to volatile compounds such as CO2. Overoxidation also results in increased cost through greater consumption of the oxidant H2O2.
Here we explored the deconstruction of lignin from poplar, a relevant bioenergy feedstock,26 after separation from a sugar-rich stream generated with an ionic liquid-based process.27 We first sulfonated the poplar lignin, following prior work.28 Next, we depolymerized the sulfonated lignin using a Fenton reaction, demonstrating that we can control the extent of deconstruction and repolymerization by varying reagent concentrations. We then explored the biological availability of the breakdown products and demonstrated conversion of the breakdown products to the jet-fuel precursor bisabolene. The goal of this work was to demonstrate proof-of-principle for the entire process including conversion to product. Below we report the results and discuss several ideas to improve the yields for each step in the process.
2. Materials and methods
2.1. Feedstock and chemicals
12 kDa dialysis tubing was purchased from SpectraPor. 98% sulfuric acid, iron(II) sulfate heptahydrate, and hydrogen peroxide were obtained from Fisher Scientific. Lignin was isolated from poplar (sourced from Idaho National Laboratory, Idaho Falls, ID) in a one-pot configuration using cholinium lysinate ([Ch][Lys], Proionic GmbH, Raaba-Grambach, Austria) as described previously.27 Typically, 2 mm poplar samples, [Ch][Lys], and water were mixed in a 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:7.5 ratio (w/w) (15 wt% biomass loading) in a Parr vessel. The slurry was pretreated for 3 h at 140 °C with stirring at 80 rpm powered by process (Parr Instrument Company, model: 4871, Moline, IL) and power controllers (Parr Instrument Company, model: 4875, Moline, IL) using a three-arm, self-centering anchor with PTFE wiper blades. After 3 h, the pretreated slurry was cooled down to room temperature by removing the heating jacket. The pH of the cold pretreated mixture was adjusted to 5 with concentrated hydrochloric acid (J. T. Baker, Inc., Phillipsburg, NJ). Enzymatic saccharification was carried out at 50 °C for 72 h at 80 rpm using enzyme mixtures Cellic® CTec3 and HTec3 (9:1 v/v; Novozymes, North America, Franklinton, NC) at a loading of 10 mg protein per g biomass. After 72 h, the slurry was centrifuged and washed multiple times with DI water to remove any residual sugar (washed until the pH of the washing liquid was neutral). The washed material was freeze dried to obtain [Ch][Lys]-poplar lignin.
:1:7.5 ratio (w/w) (15 wt% biomass loading) in a Parr vessel. The slurry was pretreated for 3 h at 140 °C with stirring at 80 rpm powered by process (Parr Instrument Company, model: 4871, Moline, IL) and power controllers (Parr Instrument Company, model: 4875, Moline, IL) using a three-arm, self-centering anchor with PTFE wiper blades. After 3 h, the pretreated slurry was cooled down to room temperature by removing the heating jacket. The pH of the cold pretreated mixture was adjusted to 5 with concentrated hydrochloric acid (J. T. Baker, Inc., Phillipsburg, NJ). Enzymatic saccharification was carried out at 50 °C for 72 h at 80 rpm using enzyme mixtures Cellic® CTec3 and HTec3 (9:1 v/v; Novozymes, North America, Franklinton, NC) at a loading of 10 mg protein per g biomass. After 72 h, the slurry was centrifuged and washed multiple times with DI water to remove any residual sugar (washed until the pH of the washing liquid was neutral). The washed material was freeze dried to obtain [Ch][Lys]-poplar lignin.
2.2. Sulfonation of lignin
Sulfonation of lignin was performed using pure sulfuric acid following prior work.28 Briefly, lignin was dissolved in concentrated sulfuric acid at a 1:4 ratio. At this ratio the reaction resulted in a paste-like consistency. The temperature of the reaction was maintained below 20 °C. After 10 min, the reaction was quenched with the addition of cold water, and sodium hydroxide was added to adjust the pH to a value between 2.5 and 3.
2.3. Removal of excess sulfuric acid
Dialysis was used to remove excess sulfuric acid from the sulfonated samples. In the first trial (case I), a sulfonated lignin sample was dialyzed at room temperature for 38 h in a 2 L reservoir, with one water change at the halfway point (19 h). Samples were collected at 19 h and 38 h to assess the presence of residual sulfuric acid in the sample using gel permeation chromatography (GPC). At 38 h, residual sulfuric acid was not detected in the sample. The resulting dialyzed, sulfonated lignin product was freeze-dried, and the carbon content determined by elemental analysis. The amount of carbon recovered in the dialysis liquid was determined by analysis of total dissolved organic carbon to close the carbon balance. A significant amount of carbon was present in the dialysis liquid and we subsequently hypothesized that the presence of iron and a lower dialysis temperature would promote aggregation of lignosulfonate and facilitate greater carbon retention during dialysis. Specifically, we hypothesized that iron would be chelated by the functional groups present within the sulfonated lignin resulting in LS–Fe complexes. An additional experiment was conducted to test this hypothesis. In this trial, (case II), iron was directly added to the sulfonated lignin sample to a final concentration of 40 mM and the sample was incubated at room temperature for 1 h. Then the sample was dialyzed for a total of 38 h with the same conditions as for case I except that the dialysis temperature was 4 °C. Upon observing improved carbon recovery for case II, we were encouraged to explore an even higher iron loading concentration prior to dialysis. A third trial (case III) was conducted in which 100 mM Fe was added prior to dialysis. This trial involved a much larger quantity of LS in order to generate sufficient material for microbial studies. The dialysis conditions for this sample differed from those for case I and case II due to the much larger sample size. For case III the sample was split into three dialysis tubes and placed into a single 2 L reservoir. Dialysis was then performed for 48 h at 4 °C with repeated water exchanges until the pH of the dialysis water no longer decreased significantly, indicating that the excess sulfuric acid had been removed from the sample. The removal of excess sulfuric acid from the sample was also confirmed with GPC.2.4. Fenton reaction
After dialysis, the three samples (cases I–III) were treated separately with Fenton reactions to deconstruct the sulfonated lignin. The freeze-dried lignin products were dissolved in water to achieve a final concentration of 50 mg mL−1 in separate glass reaction vessels. For case I (0 mM Fe prior to dialysis), a 100 mM stock solution of FeSO4 was added to achieve a final concentration of 15 mM iron in the reaction vessel. For this case Fe was added to the sample after dialysis in order to provide another test condition for the Fenton reaction. In contrast, case II (40 mM Fe prior to dialysis) and III (100 mM Fe prior to dialysis) did not require additional iron because sufficient iron was retained after dialysis to facilitate the Fenton reactions. This provided three different Fenton reaction conditions. For each reaction, the pH was first adjusted to 6 using NaOH. Aliquots of a 35% stock solution of H2O2 were added to all treatments, where each aliquot resulted in 1% H2O2 within the reaction volume. Four H2O2 aliquots were added to each reaction corresponding to a total concentration of 4% H2O2 added. The addition of the first aliquots of H2O2 initiated the reactions, and the mixtures were stirred with magnetic stir bars. Peroxide test strips were used to monitor the progress of the reactions by the decrease in H2O2 concentration. During the reactions the pH decreased and was periodically adjusted back to 6.0 using NaOH. Periodically increasing the pH to 6.0 accelerated H2O2 consumption.2.5. Determination of the fraction of soluble material
For case III, the lignosulfonate and Fenton-treated lignosulfonate samples were freeze-dried to remove all remaining moisture and homogenized using a mortar and pestle. The dried powders were weighed out in separate microcentrifuge tubes and Millipore water added to each tube to achieve concentrations of 50 mg mL−1 and 2 mg mL−1. 50 mg mL−1 corresponds to the concentration used in the Fenton reactions, whereas 2 mg mL−1 corresponded to the concentration used for molecular weight analysis. By preparing the samples at these two concentrations, the concentration dependence of the solubility of the final product was determined over the range of our analyses. The pH was adjusted to 12 using NaOH and the samples were centrifuged for 10 min at room temperature. The supernatant was removed and transferred to a clean tube. The remaining pellets were dried in an oven overnight at 60 °C to remove any remaining liquid. The percent solubilized was calculated using the following formula: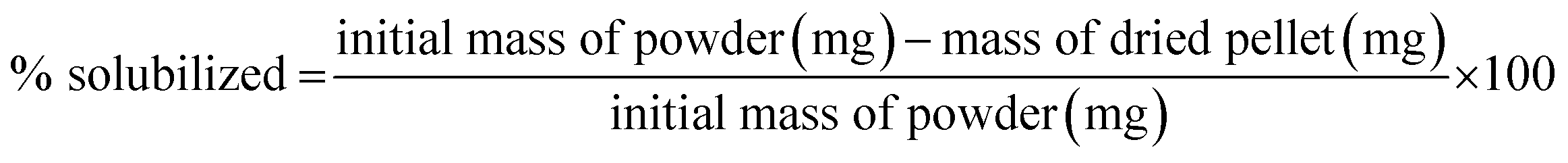
2.6. Size exclusion chromatography (SEC)
Molecular weight distributions were determined using an Agilent 1260 HPLC system with PL Aquagel-OH 30 and PL Aquagel-OH 50 columns in series. The system was equipped with UV detectors at 210 nm and 270 nm, an evaporative light scattering detector (ELSD), and a refractive index detector (RID). Samples were diluted to 2 mg mL−1 using Millipore water. The solutions were centrifuged and filtered using a 0.2-micron filter prior to injecting into the HPLC system. The HPLC system contained an in-line filter in front of the analysis column and guard column. The eluant was 1 mM ammonium bicarbonate buffer at pH 9. Ten polystyrene sulfonate standards with molecular weights ranging from 200–280000 were used for calibration.
2.7. Fourier transform infrared spectroscopy (FTIR)
Samples for FTIR were prepared by pipetting liquid samples onto Teflon pieces to form thin films. The samples were air-dried, and then excess water was removed by vacuum drying for 1 h. FTIR spectra were collected in attenuated total reflectance (ATR) mode using a Bruker LUMOS ATR-FTIR microscope equipped with a germanium probe tip. For each film sample, three spectra were collected and averaged. Each spectrum consisted of 16 averaged scans at a resolution of 4 cm−1. Background spectra were acquired before each sample measurement to ensure accurate baseline correction. Spectral data were processed and peaks corresponding to functional groups of interest were analyzed.2.8. Total dissolved organic carbon analysis
Samples were taken in triplicate (100 μL each) and sent to Hall Environmental Analysis Laboratory (https://www.HallEnvironmental.com) for Total Organic Carbon (TOC) analysis. The samples were diluted with Millipore water as needed (dilution factor ∼ 300) to perform the TOC analysis using Standard Method 5310B: total organic carbon by high-temperature combustion.2.9. Elemental analysis
Elemental analysis (CHNO, Fe, and Ash Content) was performed by ALS Global Company (https://www.alsglobal.com/en/). Samples were oven dried at 60 °C and homogenized using a mortar and pestle prior to sending to ALS for analysis. Solid samples were weighed (2–3 mg) into 6 × 4 mm tin capsules (Alpha Resources; ATD1006) and analyzed with a PerkinElmer Series II 2400 CHNS/O Analyzer on CHN mode. Combustion of the samples occurs at 950 °C and reduction of gases at 640 °C. The instrument uses IR cells to detect the gases. The instrument was calibrated on acetanilide OAS (Elemental Microanalysis; B2000). Before analysis the calibration was verified with acetanilide OAS and EDTA (Alpha Resources; AR2092) and verified again with acetanilide after analysis. Iron content was determined using an Agilent 5110 SVDV ICP-OES Spectrometer. Prior to analysis, samples were prepared using microwave-assisted acid digestion to solubilize the metals, allowing for accurate quantification of Fe.2.10. Microbial conversion
To evaluate microbial conversion of compounds in the depolymerized lignin, microbial cultivations were performed in 48-well plates using 500 μL of culture volume. Yeast nitrogen base (YNB) without amino acids but with 0.1% yeast extract was the medium used in the experiments and was prepared as a 10× stock, pH-adjusted to 6.4 with 10 N NaOH, and filtered (0.22 μm). The 10× stock was mixed with depolymerized lignin (43 g L−1). For positive and negative controls, the 10× stock was combined with a mixture of glucose and fructose, each at 2 g L−1, or pure water, respectively. The resulting solutions were used directly for cultivations.Delftia acidovorans, Exophiala alcalophila, Pseudomonas putida, Cupriavidus necator and Rhodosporidium toruloides were obtained from the strain archive at the Joint BioEnergy Institute https://www.public-registry.jbei.org. The cells were first grown on tryptic soy broth for 48 hours, then centrifuged and washed with sterile water to remove the rich medium. Inoculation was achieved by adding 20 μL of the cell suspension to 480 μL of medium. Cultivations were performed in triplicate and incubated at 30 °C and 200 rpm shaking for 72 h. Cell density (OD 600 nm) was measured directly using a TECAN Spark spectrophotometer (TECAN, Switzerland). The graphs reflect OD 600 values after subtracting readings for media without cells for each condition to correct for coloration effects. Cultivations for bisabolene production were performed and analyzed in quadruplicate using a previously reported strain called GB2 and cultivation protocols29 in 48-well Flower plates with 800 μL culture volume and 200 μL pentadecane overlay.
3. Results
3.1. Lignin deconstruction and characterization
The lignin used in this work was isolated from poplar in a one-pot process using cholinium lysinate as described previously.27 The purity of the lignin generated by this process was found to be 41.8% along with 19.6% glucan and 3.5% xylan. The deconstruction process involved sulfonation of lignin, removal of excess H2SO4, and Fenton reaction. While the degree of sulfonation is likely to affect the extent of deconstruction in the Fenton reaction, the degree of sulfonation was not varied in this initial work. For the results reported below the carbon to sulfur mole ratio was 22.5.Excess sulfuric acid will interact with Fe2+ and negatively impact the Fenton reaction. In this work, dialysis was used to remove excess sulfuric acid after sulfonation. In that regard, the addition of Fe prior to dialysis was explored based on the hypothesis that Fe2+ would bind to LS and cause aggregation, facilitating dialysis and retention of LS. For the first trial (case I) no Fe was added prior to dialysis and the sample was dialyzed at room temperature. For the second trial (case II) 40 mM Fe was added prior to dialysis and the sample was dialyzed at 4 °C. The carbon balance for the dialysis step is shown in Table 1 for case I and case II.
| Case | Fe concentration prior to dialysis (mM) | Initial carbon (mg) | Carbon in sample after dialysis as measured by elemental analysis | Carbon in dialysis liquid as measured by total organic carbon | Carbon in sample + dialysis liquid (%) |
|---|---|---|---|---|---|
| I | 0 | 170 | 65 mg (38%) | 104 mg (61%) | 99.4 |
| II | 40 | 170 | 98 mg (58%) | 72 mg (42%) | 99.9 |
| III | 100 | 2545 | 1694 mg (67%) | 785 mg (31%) | 97.4 |
Iron loading prior to dialysis along with the lower dialysis temperature indeed improved dialysis efficiency and retention of the sulfonated lignin product. Subsequently, a third trial (case III) was performed in which 100 mM Fe was loaded prior to dialysis and the sample was dialyzed at 4 °C. Case III involved a much larger quantity of LS in order to generate sufficient material to use for bioavailability testing. Since the amount of LS used in case III was much greater than for case I and case II, the dialysis conditions differed as described in the Methods section. Analysis of the iron contents in the samples after dialysis (13.5 mM and 7.0 mM for cases II and III, respectively) indicated that the sample for case III was dialyzed more extensively than for case II. As shown in Table 1 the carbon recovery for case III was higher than for case II, confirming the hypothesis that the presence of iron prior to dialysis facilitates carbon recovery upon dialysis.
Following dialysis, Fenton reactions were performed for each case at 50 mg mL−1 LS. For case I, 15 mM Fe was added prior to initiating the Fenton reaction in order to provide a third Fenton reaction sample. For case II and case III, no additional Fe was added prior to the Fenton reactions since these samples retained sufficient Fe for the Fenton reactions to proceed. H2O2 was added up to 4% by weight corresponding to a H2O2-to-lignin monomer mole ratio of 4.7 (assuming 200 g per mol per monomer). The reactions were performed at room temperature. H2O2 was dosed in increments of 1%, and after each increment the reactions were allowed to proceed until all H2O2 was consumed before the next aliquot was added. For each aliquot, after the H2O2 was consumed the molecular weight distribution of the reaction mixtures were determined using SEC and the dissolved carbon contents were determined by elemental analysis. The Fe concentrations of the final solutions were 13.7 mM, 13.5 mM, and 7.0 mM for cases I, II, and III, respectively. 7 mM Fe corresponds to a Fe/S molar ratio of 0.08.
Fig. 1 shows the molecular weight distributions for the three cases after adding 4% H2O2. The data in Fig. 1 show a strong correlation between Fe concentration and extent of deconstruction in the Fenton reaction. Higher Fe concentrations for case I and case II resulted in extensive deconstruction to yield predominantly one main peak at low molecular weight. Lower Fe content for case III, due to removal of most of the Fe during dialysis, resulted in less robust deconstruction and a bimodal molecular weight distribution. We note that the amount of insoluble carbon after the Fenton reaction was negligible for the three cases. The percentages of carbon recovered in the soluble fractions, given in Table 2, indicate that the amount of carbon lost as volatile species occurred in the order case I > case II > case III. These results demonstrate that deconstruction to low molecular weight compounds can be achieved by this approach, but a tradeoff exists between extent of deconstruction and the percentage of carbon lost as CO2 due to overoxidation of low molecular weight compounds.
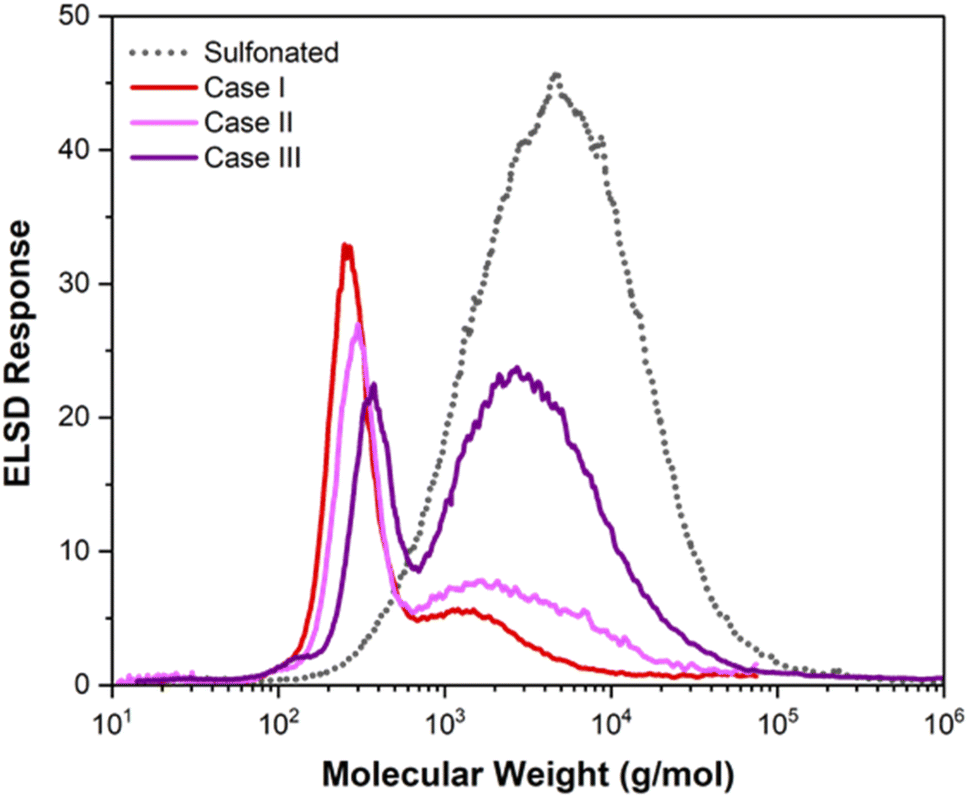 | ||
| Fig. 1 Molecular weight distribution of sulfonated lignin after Fenton reaction for the three dialyzed samples (4% H2O2). | ||
| Case | Concentration for Fenton (mg mL−1) | Carbon before Fenton (mg) | Soluble carbon after Fenton (mg) | Soluble carbon recovered after Fenton (%) | Overall soluble carbon recovered (%) |
|---|---|---|---|---|---|
| I | 50 | 65 | 18 | 28 | 11 |
| II | 50 | 98 | 47 | 48 | 28 |
| III | 50 | 1694 | 1030 | 61 | 40 |
Finally, we note that the molecular weight distributions in Fig. 1 and the percents of carbon lost as volatile species inferred from the data in Table 2, indicate that the Fenton reaction for case I was more robust than that for case II, despite comparable iron concentrations. This suggests that the iron added prior to the Fenton reaction for case I was more available for the reaction than the iron of case II that was retained in the sample after dialysis of the sulfuric acid. Some of the iron retained in the sample of case II after dialysis appears to have been in a form that was less productive for the Fenton reaction.
For case III, Fig. 2 shows the molecular weight distributions and the fractions of carbon recovered as soluble species by TOC analysis as a function of H2O2 applied during the Fenton reaction. Analysis of soluble carbon was not performed for H2O2 < 2% as those samples were not fully soluble. The value at 0% H2O2 in Fig. 2a reflects the fact that 33% of the original carbon was lost during the dialysis step (see Table 1). Fig. 2b shows that as the amount of H2O2 increased from 0–4%, the high molecular weight peak shifted to lower molecular weight and a separate low molecular weight peak was generated in increasing amounts. At 4% H2O2 loading, 40% of the original carbon was recovered as soluble species with a substantial fraction of products with molecular weight less than 500 g mol−1. For case III, the soluble fractions for the original poplar lignin, after sulfonation, and after Fenton reaction with 4% H2O2 are given in Fig. S1.† Tables of Mw, Mn and PDI for all the samples are given in the ESI.†
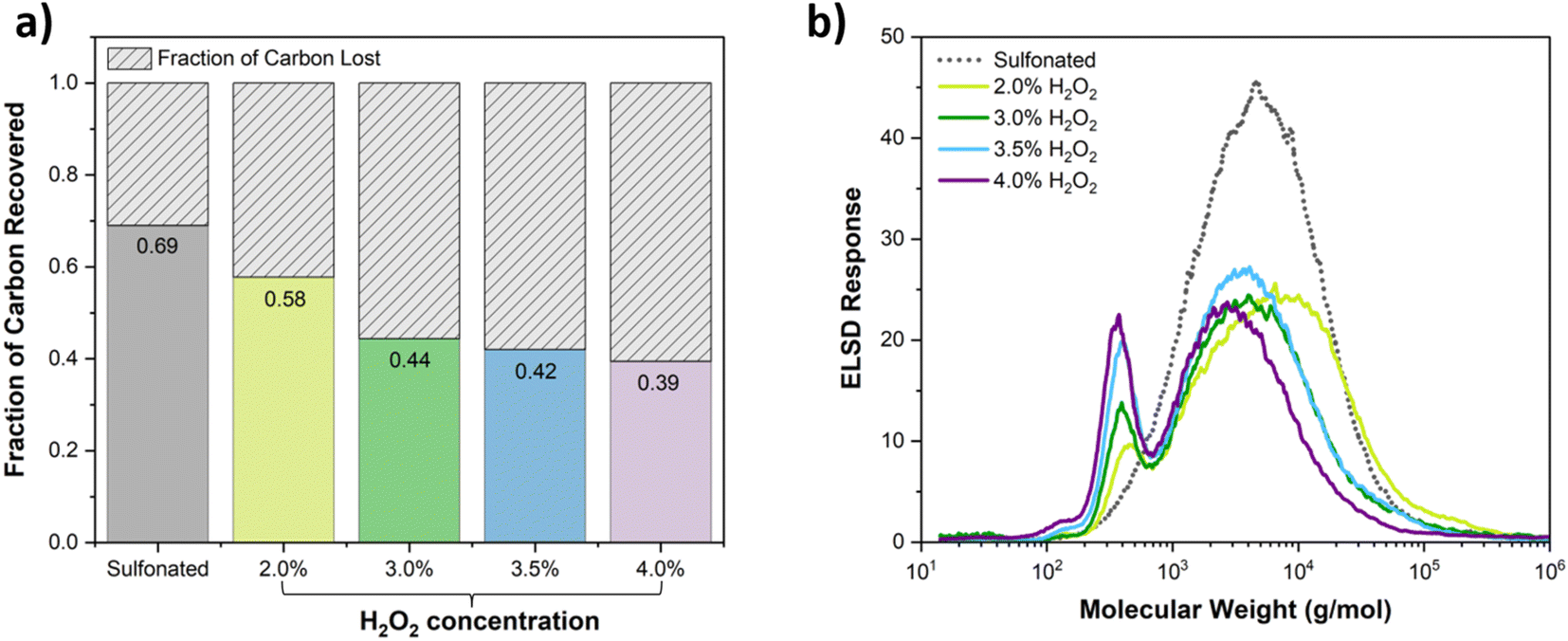 | ||
| Fig. 2 (a) Fraction of carbon recovered as dissolved carbon by TOC relative to the total H2O2 concentration added in the Fenton reaction. (b) Molecular weight distributions after Fenton reaction as a function of H2O2 added starting from 2% at which point nearly all the material was soluble. | ||
The soluble material was analyzed by FTIR-ATR after adjusting portions to pH 2 and pH 12 and then drying the solutions to form films. The FTIR data (Fig. S2a†) indicate that a large amount of carboxylic acid groups were present in the reaction products reflected by the shift in the carbonyl band from carboxylate at 1600 cm−1 at pH 12 to carboxylic acid at 1700 cm−1 at pH 2. The spectra also indicate that the reaction products contain a large amount of secondary or tertiary C–OH groups shown by the C–O stretch band at 1150 cm−1 and the broad OH band at 3600–3400 cm−1 for the sample adjusted to pH 12 where no carboxylic acids are present (Fig. S2b†).
3.2. Microbial growth and substrate conversion
The solution of breakdown products from case III was used to screen a panel of microorganisms for growth on this substrate as sole carbon source. This is an important step to validate the applicability of the generated streams as microbial substrates due to the expected heterogeneity and potential toxicity of compounds that can be produced from lignin.30 Microbes that have previously shown promise as bioconversion hosts for depolymerized lignin streams21 were selected for this assay. The post-reaction solution was used directly in the screening without any post-processing other than combining it with growth medium salts containing ammonium sulfate and 0.1 g L−1 of yeast extract.Fig. 3 shows robust growth from all organisms on the reaction product as main carbon source, reaching OD values from 0.9 to 1.9 after 3 days of cultivation. This demonstrates that the material is not highly toxic to these organisms and specifically that the leftover iron and sulfonated compounds that are generated do not strongly inhibit growth. Media containing 2 g L−1 of glucose and 2 g L−1 of fructose instead of depolymerized lignin was used as positive control, and the resulting OD values were comparable to those obtained from the lignin breakdown stream for three of the five organisms. It is important to highlight that the employed bacteria and yeasts are known to have different growth requirements and therefore the media and conditions used here may not be optimal for all of them and were used for preliminary bioavailability testing purposes only. A negative control using the same medium without any additional carbon supplementation confirmed that the observed cell growth in presence of supplemented substrates is due to assimilation of sugars or lignin-derived compounds.
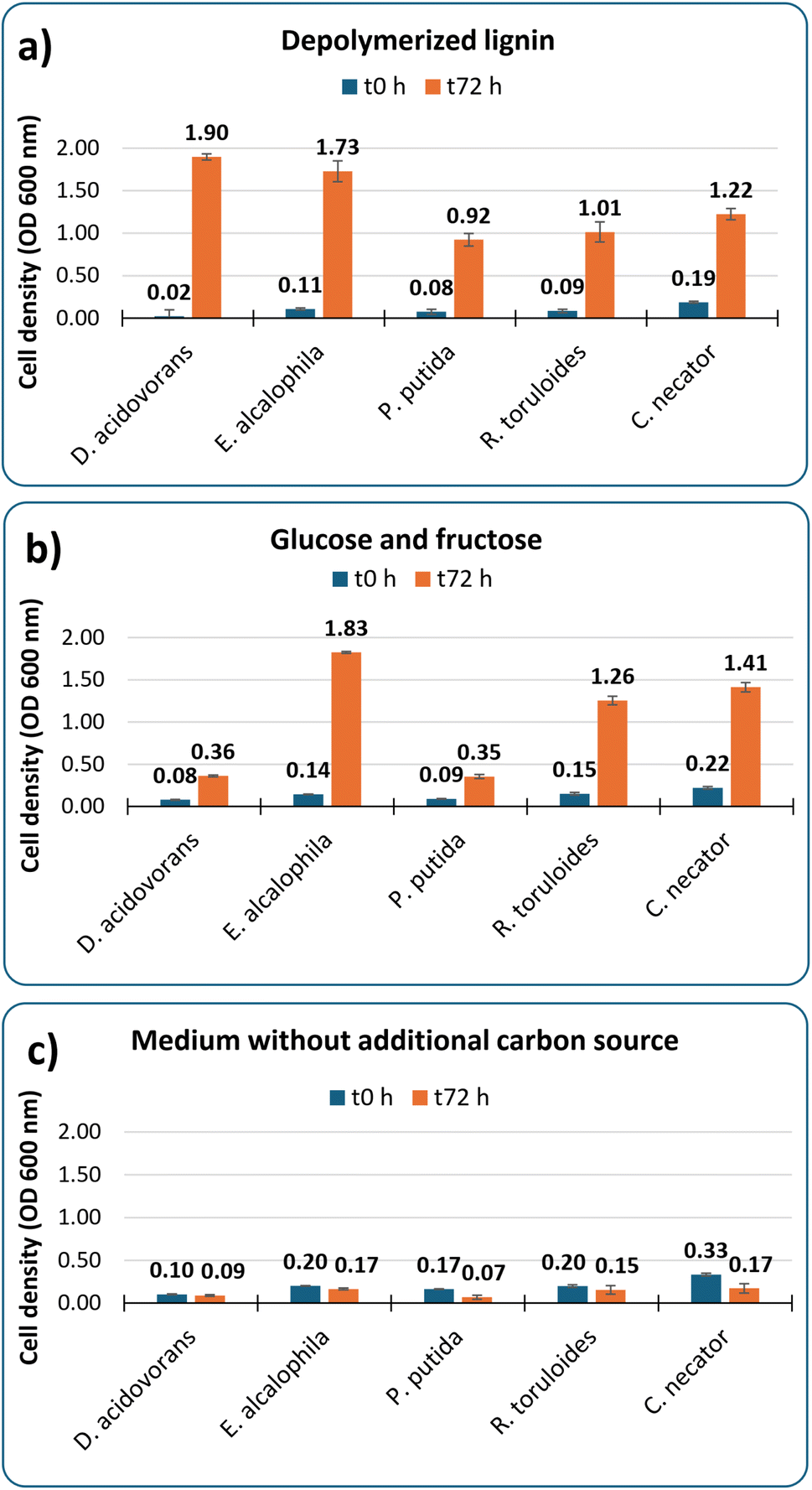 | ||
| Fig. 3 Growth of five microorganisms on medium containing depolymerized lignin, sugars, or no additional substrate. (a) lignin from case III at a concentration of 43 g L−1; (b) a mixture of glucose and fructose (2 g L−1 each); (c) no additional carbon source. | ||
Following this survey of bioavailability, a bisabolene-producing strain of R. toruloides called GB2 (ref. 29) was used to demonstrate conversion of the lignin breakdown products to bisabolene. After a 5 day cultivation, bisabolene was detected at 22 mg L−1 and a final OD of 0.6 was observed. SEC analysis before and after incubation shown in Fig. S3† indicates that both peaks of the molecular weight distribution decreased slightly during incubation, consistent with the observed cell growth and bisabolene production.
4. Discussion
The lignin deconstruction approach described here based on sulfonation and Fenton reaction has several attractive features. The deconstruction reaction is performed at or near room temperature without the need for a pressure reactor, and the reaction products can be used in a bioreactor directly without any post-processing. While the Fenton reaction is likely to be too slow at room temperature for industrial application, the reaction rate can be accelerated greatly by heating slightly or by exposure to UV light.31–38 As shown in this work, the presence of sulfonated compounds in the breakdown stream does not appear to hinder growth for some potential conversion hosts.The present work has also revealed several challenges. The overall yield of carbon to product is very low in this initial proof-of-principle work. To address this issue, improvements in yield must be made at each step in the process. Several changes to the process are likely to improve the overall efficiency and yield. First, it is important to achieve extensive deconstruction to low molecular weight species while minimizing the H2O2 requirement, as H2O2 is the major reagent cost, and while also avoiding the loss of carbon as volatile species due to overoxidation. Both of these goals can be achieved if low molecular weight products are continuously removed during the Fenton reaction. This would allow extensive deconstruction to low molecular weight compounds and would improve the efficiency of H2O2 usage by avoiding unproductive consumption of H2O2 in overoxidating low molecular weight compounds. Removing low molecular weight compounds could conceivable be achieved within the design of a coupled reactor system, taking advantage of the fact that the Fenton reaction product is directly biocompatible. Coupling reactors for deconstruction and bioconversion would allow for semi batch processing and would likely lead to further process cost reduction. Second, a more efficient process is needed to remove and recycle excess H2SO4. In the present work, 31% of the carbon was lost during dialysis (case III). It is likely that some of the carbon lost at this stage is due to hydrolysis of polysaccharides, and that more extensive purification of the lignin stream could lead to higher carbon recovery upon dialysis. Nevertheless, dialysis is unlikely to be a scalable separation technology so a more efficient and cost-effective approach to recover and recycle H2SO4 will be required that minimizes carbon losses.
This work showed substantial deconstruction for a low extent of sulfonation (C/S mole ratio of 22.5). More efficient deconstruction is likely to be achieved with greater extent of sulfonation. A higher Fe/S ratio and a higher lignosulfonate loading during the Fenton reaction may also improve deconstruction efficiency. While the results for growth of monocultures indicate that all tested microorganisms grow well on the deconstructed lignin, bisabolene production by the engineered R. toruloides strain was relatively low compared to reports that used concentrated sugars as substrates.29,39 This suggests that further strain engineering and adaptation may improve conversion of the compounds in the depolymerized lignin material to bisabolene. D. acidovorans and E. alcalophila showed high OD values in the depolymerized lignin and deserve to be explored further since they are promising yet underdeveloped bioconversion hosts, known for their resilience to environmental hazards.40,41 Deconstructing the substrate to yield a larger amount of lower molecular weight species, as discussed above, may be the most critical factor for improved utilization and conversion to product. Finally, industrial application of this process may also require removing and recycling iron from the waste stream after the microbial conversion step to avoid adverse environmental impacts.
Data availability
All data for this article has been included in the manuscript and the ESI.†Author contributions
D. M. V.: investigation, validation, visualization, writing – original draft; A. R.: investigation, validation, visualization, writing – review & editing; H. C.: investigation, methodology, validation; J. S.: investigation, validation; E. J. M.: investigation, validation; O. D.: methodology, project administration; G. M. G.: investigation, validation; J. M. G.: resources, supervision; B. A. S.: funding acquisition, resources; M. S. K.: concept generation, methodology, investigation, funding acquisition, resources, supervision, writing – original draft, writing – review & editing.Conflicts of interest
B. A. S. has a financial interest in Caribou Biofuels, Illium Technologies, and Erg Bio.Acknowledgements
This material is based upon work supported by the Joint BioEnergy Institute, U.S. Department of Energy, Office of Science, Biological and Environmental Research Program under Award Number DE-AC02-05CH11231 with Lawrence Berkeley National Laboratory. This work was also partially supported by the Laboratory Directed Research and Development program at Sandia National Laboratories. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan. Sandia National Laboratories is a multimission laboratory managed and operated by National Technology & Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International Inc., for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-NA0003525. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.References
- X. Liu, F. P. Bouxin, J. Fan, V. L. Budarin, C. Hu and J. H. Clark, ChemSusChem, 2020, 13, 4296–4317 CrossRef CAS PubMed.
- M. Li, Y. Pu and A. J. Ragauskas, Front. Chem., 2016, 4, 45 Search PubMed.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- S. Shrestha, S. Goswami, D. Banerjee, V. Garcia, E. Zhou, C. N. Olmsted, E. L.-W. Majumder, D. Kumar, D. Awasthi, A. Mukhopadhyay, S. W. Singer, J. M. Gladden, B. A. Simmons and H. Choudhary, ChemSusChem, 2024, 17, e202301460 CrossRef CAS.
- M. Langholtz, Oak Ridge National Laboratory (ORNL), Oak Ridge, TN (United States), Bioenergy Knowledge Discovery Framework (BEKDF), DOI: DOI:10.23720/bt2023/2316165.
- OECD, Global Plastics Outlook: Economic Drivers, Environmental Impacts and Policy Options, OECD Publishing, Paris, 2022 Search PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. da Costa Lopes, R. M. Łukasik and P. T. Anastas, Green Chem., 2017, 19, 4200–4233 RSC.
- L. Cao, I. K. M. Yu, Y. Liu, X. Ruan, D. C. W. Tsang, A. J. Hunt, Y. S. Ok, H. Song and S. Zhang, Bioresour. Technol., 2018, 269, 465–475 Search PubMed.
- R.-C. Sun, J. S. M. Samec and A. J. Ragauskas, ChemSusChem, 2020, 13, 4175–4180 CrossRef CAS.
- R. Davis and A. Bartling, Biochemical Conversion of Lignocellulosic Biomass to Hydrocarbon Fuels and Products: 2022 State of Technology and Future Research, National Renewable Energy Laboratory (NREL), Golden, CO (United States), 2023 Search PubMed.
- T. Phongpreecha, N. C. Hool, R. J. Stoklosa, A. S. Klett, C. E. Foster, A. Bhalla, D. Holmes, M. C. Thies and D. B. Hodge, Green Chem., 2017, 19, 5131–5143 RSC.
- C. T. Palumbo, N. X. Gu, A. C. Bleem, K. P. Sullivan, R. Katahira, L. M. Stanley, J. K. Kenny, M. A. Ingraham, K. J. Ramirez, S. J. Haugen, C. R. Amendola, S. S. Stahl and G. T. Beckham, Nat. Commun., 2024, 15, 862 CrossRef CAS.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- G. G. Facas, D. G. Brandner, J. R. Bussard, Y. Román-Leshkov and G. T. Beckham, ACS Sustain. Chem. Eng., 2023, 11, 4517–4522 CrossRef CAS.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- C. Walling, Acc. Chem. Res., 1975, 8, 125–131 CrossRef CAS.
- R. Chen and J. J. Pignatello, Environ. Sci. Technol., 1997, 31, 2399–2406 CrossRef CAS.
- N. Kang, D. S. Lee and J. Yoon, Chemosphere, 2002, 47, 915–924 CrossRef CAS PubMed.
- D. Areskogh and G. Henriksson, Nord. Pulp Pap. Res. J., 2011, 26, 90–98 CrossRef CAS.
- D. V. Martinez, A. Rodriguez, M. A. Juarros, E. J. Martinez, T. M. Alam, B. A. Simmons, K. L. Sale, S. W. Singer and M. S. Kent, Green Chem., 2022, 24, 1627–1643 RSC.
- C.-F. Chow, W.-L. Wong, K. Y.-F. Ho, C.-S. Chan and C.-B. Gong, Chem.–Eur. J, 2016, 22, 9513–9518 CrossRef CAS.
- H.-M. Feng, J.-C. Zheng, N.-Y. Lei, L. Yu, K. H.-K. Kong, H.-Q. Yu, T.-C. Lau and M. H. W. Lam, Environ. Sci. Technol., 2011, 45, 744–750 CrossRef CAS PubMed.
- M. C. Krueger, U. Hofmann, M. Moeder and D. Schlosser, PLoS One, 2015, 10, e0131773 CrossRef PubMed.
- A. Landera, D. V. Martinez, J. Salinas, A. Rodriguez, E. J. Martinez, O. Davydovich and M. S. Kent, Polym. Degrad. Stab., 2023, 215, 110451 CrossRef CAS.
- A. N. Chowyuk, H. El-Husseini, R. R. Gustafson, N. Parker, R. Bura and H. L. Gough, Biomass Bioenergy, 2021, 153, 106213 CrossRef.
- H. Choudhary, L. Das, J. G. Pelton, L. Sheps, B. A. Simmons, J. M. Gladden and S. Singh, Chem.–Eur. J, 2023, 29, e202300330 CrossRef CAS PubMed.
- P. Dilling, Sulfonation of Lignins, US Pat., 5049661A, Westvaco Corporation, 1991 Search PubMed.
- J. Kirby, G. M. Geiselman, J. Yaegashi, J. Kim, X. Zhuang, M. B. Tran-Gyamfi, J.-P. Prahl, E. R. Sundstrom, Y. Gao, N. Munoz, K. E. Burnum-Johnson, V. T. Benites, E. E. K. Baidoo, A. Fuhrmann, K. Seibel, B.-J. M. Webb-Robertson, J. Zucker, C. D. Nicora, D. Tanjore, J. K. Magnuson, J. M. Skerker and J. M. Gladden, Biotechnol. Biofuels, 2021, 14, 101 CrossRef CAS PubMed.
- A. K. Verma, D. Chettri, A. K. Verma, M. Selvaraj and M. A. Assiri, Biomass Bioenergy, 2024, 181, 107052 CrossRef CAS.
- E. Neyens and J. Baeyens, J. Hazard. Mater., 2003, 98, 33–50 CrossRef CAS PubMed.
- D. Ortiz, M. Munoz, J. Garcia, S. Cirés, Z. M. de Pedro, A. Quesada and J. A. Casas, Environ. Sci. Pollut. Res. Int., 2023, 30, 21598–21607 CrossRef CAS PubMed.
- N. López-Vinent, A. Cruz-Alcalde, J. Giménez, S. Esplugas and C. Sans, Appl. Catal., B, 2021, 290, 120066 Search PubMed.
- G. Subramanian and G. Madras, Water Res., 2016, 104, 168–177 CrossRef CAS.
- A. De Luca, R. F. Dantas and S. Esplugas, Water Res., 2014, 61, 232–242 CrossRef CAS.
- W. Huang, M. Brigante, F. Wu, K. Hanna and G. Mailhot, J. Photochem. Photobiol., A, 2012, 239, 17–23 CrossRef CAS.
- B. Faust and R. Zepp, Environ. Sci. Technol., 1993, 27, 2517–2522 CrossRef CAS.
- P. Carneiro, R. Nogueira and M. Zanoni, Dyes Pigm., 2007, 74, 127–132 CrossRef CAS.
- H. D. Magurudeniya, N. R. Baral, A. Rodriguez, C. D. Scown, J. Dahlberg, D. Putnam, A. George, B. A. Simmons and J. M. Gladden, Green Chem., 2021, 23, 3127–3140 RSC.
- S. V. Bhat, H. Maughan, A. D. S. Cameron and C. K. Yost, Microb. Genomics, 2022, 8, 000864 CAS.
- A. M. Borman, M. Fraser, A. Szekely, D. E. Larcombe and E. M. Johnson, J. Clin. Microbiol., 2017, 55, 1162–1176 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5su00039d |
| ‡ Equal-contributing first authors. |
| This journal is © The Royal Society of Chemistry 2025 |