
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolypeptide-based multilayer capsules with anti-inflammatory properties: exploring different strategies to incorporate hydrophobic drugs†
Maria Angela Motta ab,
Sergio Martin-Saldañaa,
Ana Beloquiac,
Marcelo Calderón*ac and
Aitor Larrañaga*b
ab,
Sergio Martin-Saldañaa,
Ana Beloquiac,
Marcelo Calderón*ac and
Aitor Larrañaga*b
aPOLYMAT, Applied Chemistry Department, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel de Lardizabal 3, 20018 Donostia-San Sebastián, Spain. E-mail: marcelo.calderonc@ehu.eus
bDepartment of Mining-Metallurgy Engineering and Materials Science, POLYMAT, Bilbao School of Engineering, University of the Basque Country (UPV/EHU), Plaza Torres Quevedo 1, 48013 Bilbao, Spain. E-mail: aitor.larranagae@ehu.eus
cIKERBASQUE, Basque Foundation for Science, Plaza Euskadi 5, 48009 Bilbao, Spain
First published on 28th March 2025
Abstract
More than 90% of drug candidates used in the drug development pipeline and about 40% of drugs on the market are poorly soluble in water based on the definition of the biopharmaceutical classification system. The advent of drug delivery approaches has represented a striking tool to overcome the challenges associated with the use of hydrophobic drugs, such as their low bioavailability and off-target effects. Drug carrier formulations composed of biodegradable and biocompatible polymers, such as polypeptides, have been explored as platforms to host poorly water-soluble drugs to prolong drug circulation, enhance their safety, reduce their immunogenicity, and promote their controlled release. In this work, we evaluated three strategies—co-precipitation, post-encapsulation, and conjugation—to incorporate a hydrophobic model drug, i.e., curcumin (CUR), into biodegradable multilayer capsules fabricated via a layer-by-layer (LbL) approach. Poly(L-lysine) (PLys) and poly(L-glutamic acid) (PGlu) were adopted as building blocks and alternately assembled onto calcium carbonate (CaCO3) microparticles to build a polypeptide-multilayer membrane, which acted as a barrier to control the release of the drug. The application of our three formulations in in vitro inflammatory models of THP-1 derived human macrophages and murine microglia showed a reduction of the inflammation with the suppression of three pivotal pro-inflammatory cytokines (i.e., interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α). Moreover, the intracellular release of CUR detected upon uptake studies on activated microglia suggested that our systems could represent a potential therapeutic approach to reduce acute neuroinflammation and modulate microglia phenotype.
1. Introduction
Drug delivery systems based on biodegradable polymers have found diverse applications in oral, nasal, parenteral, and transdermal administration of small drug molecules, proteins, and nucleic acids. Biodegradable polymers of synthetic (e.g., polyesters, polyanhydrides, and polyamides) or natural origin (e.g., proteins and polysaccharides) have several advantages when used for drug delivery applications due to their biocompatibility and degradation into harmless by-products under specific conditions.1 For instance, poly(lactide-co-glycolide) (PLGA), which is approved by the Food and Drug Administration (FDA), is widely used as a polymeric carrier because it degrades into CO2 and H2O, which are non-toxic for the human body and are removed through the Krebs cycle.2 Synthetic polypeptides are another class of polymers that have been investigated as components of drug delivery systems due to their similarity with proteins, including safety, biocompatibility, biodegradation, and low immunogenicity.3The cornerstone of polymeric carriers is to improve the pharmacological properties of therapeutics, by either encapsulation or conjugation techniques. The encapsulation strategy is very simple and scalable, but it usually suffers from low loading efficiencies and leakage of the payload from the carriers. As an alternative, the covalent conjugation of a drug to a polymer carrier represents an effective strategy to improve the solubility and the bioavailability of the drug, prolong the drug circulation, enhance the safety, reduce the immunogenicity, and promote the controlled release of therapeutics.4 Particularly, synthetic polypeptides, such as poly(L-lysine) (PLys), poly(L-glutamic acid) (PGlu), and poly(L-aspartic acid) (PAsp), which have already reached the pre-clinical or clinical stage, have been widely used as polypeptide–drug conjugates thanks to their conformations (i.e., α-helix, β-sheet, and random coil), ability to self-assemble in versatile architectures, high functionality due to their side chains, and possibility to tailor the responsiveness for biological applications.3 As an illustration, Vivagel®—a PLys-dendrimer used as an antibacterial and an antiviral agent—and Copaxone®—a copolymer of L-glutamic, L-alanine, L-lysine, and L-tyrosine against multiple sclerosis—have already been approved and reached the market. Additionally, PEG-block-copolymers of PLys, PGlu, and PAsp are among the most advanced formulations used in clinical settings.3
Despite the outstanding features of polypeptides, academic research is oriented towards the development of more advanced drug delivery systems to overcome the biological issues associated with several diseases and reduce the off-target effects. With this purpose, the layer-by-layer (LbL) technique has been used to fabricate multifunctional drug delivery systems, which have the unique property to host one or multiple drugs, stimuli-responsive moieties, and additional functionalities for cell targeting in one single entity.5–11 The LbL approach is a strategy that has gained great interest in the field of drug delivery for the fabrication of highly functionalized multilayer capsules by assembling either synthetic or natural polymers as building blocks onto a template. The assembled polymers can act as a barrier membrane, controlling the diffusion of the payload and limiting accordingly the initial burst release that would cause the release of the drug before reaching the target cells.12,13 Polypeptides are attractive LbL building blocks due to their biocompatibility, biodegradability, high functionality, and high tendency to self-assemble into organized multilayer nanostructures.14 One of the most commonly used templates for pharmaceutical applications is calcium carbonate (CaCO3) in both the micro- and nano-meter scales, thanks to its facile synthesis, high porosity, biocompatibility, easy dissolution under mild conditions (i.e., ethylenediaminetetraacetic acid (EDTA) solution, pH < 7.0), and capacity to host molecules of diverse nature, including proteins, growth factors, and small drug molecules.15–17 For instance, lipid-coated CaCO3 nanoparticles, synthesized by a reversed microemulsion method,18 have been reported to efficiently encapsulate a therapeutic peptide for lung cancer treatment.19 CaCO3 vaterite particles represent a promising alternative to hydrogels,20 viral vectors,21 liposomes,22 and micelles.23 Specifically, they can be considered to solve the challenges associated with the delivery of hydrophobic drugs, such as the short residence time in the body due to their low solubility in physiological fluids, and the low bioavailability and rapid clearance that result in reduced efficacy.24 The encapsulation into CaCO3 particles can be performed using three main approaches: (i) adsorption, (ii) infiltration, and (iii) co-precipitation.25,26 The adsorption approach relies on the diffusion of the payload of interest into the pre-synthesized porous particles. Depending on the size of the molecules, this type of encapsulation can result in very low loading capacities and in an inhomogeneous distribution of the cargo molecules due to the steric limitations with respect to the pore size (average of 20–60 nm).27 In the infiltration approach, the loading of the molecules occurs by decreasing their solubility or by solvent evaporation. Nonetheless, the co-precipitation is the most elegant method to load molecules into CaCO3 particles, allowing very high encapsulation efficiencies and very good distribution of the loaded molecules.28,29 This method involves the simultaneous encapsulation of the payload and the assembly of the particles.
In this work, we compared different methodologies to incorporate curcumin (CUR) as a hydrophobic model drug into biodegradable LbL microcapsules. In two systems, we encapsulated CUR into CaCO3 microparticles by co-precipitation or post-encapsulation. In a different approach, we conjugated the drug to one of the layers into the multilayer membrane. The polypeptides, PLys and PGlu, were chosen for their biocompatibility and biodegradability as positively and negatively charged building blocks, respectively, to fabricate a multilayer membrane onto the CaCO3 microparticles via the LbL approach. The LbL systems containing CUR in the core were compared in terms of encapsulation efficiency, drug loading, and drug release under neutral (i.e., pH 7.4) and acidic (i.e., pH 5.0) conditions and in the presence of proteolytic enzymes. However, these systems may cause some concerns about the retention of the drug during the numerous steps of the LbL process and the dissolution of the CaCO3 microparticles. Indeed, She et al. reported that encapsulating a basic fibroblast growth factor into CaCO3 microparticles led to a total loss of 57% of the encapsulated protein after the deposition of six-layers and the core dissolution.17 As an alternative to the encapsulation of CUR, we developed a new LbL system where CUR was included in the multilayer membrane after being conjugated to PGlu (CUR–PGlu). This approach was expected to (i) enhance the solubility of the drug, (ii) control the drug loading quantity into the capsules, and (iii) promote the release of the drug concomitantly with the disassembly of the multilayer nanostructure. Furthermore, taking advantage of the reported pleiotropic properties of CUR (e.g., anti-tumoral, anti-oxidant, anti-bacterial, anti-inflammatory, etc.),30 our LbL-capsules, obtained after core removal, were tested in two in vitro inflammatory models of macrophages derived from human monocytes (THP-1) and murine microglia (BV-2) to highlight their cytocompatibility and the inflammatory response of the cells in the presence of the CUR-loaded microcapsules.
2. Materials and methods
2.1 Materials
Calcium chloride anhydrous (CaCl2), ethylenediaminetetraacetic acid disodium salt dehydrate (EDTA), poly(sodium 4-styrene sulfonate) (PSS, molecular weight MW ∼ 70 kDa), sodium carbonate (Na2CO3), sodium chloride (NaCl), potassium chloride (KCl), calcium chloride dihydrate (CaCl2·2H2O), magnesium chloride hexahydrate (MgCl2·6H2O), sodium phosphate monobasic monohydrate (NaH2PO4·H2O), sodium phosphate dibasic heptahydrate (Na2HPO4·7H2O), phosphate buffer saline (PBS), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl), tris(hydroxymethyl)aminomethane hydrochloride (TRIS), phorbol 12-myristate 13-acetate (PMA), lipopolysaccharide (LPS), Hank's Balanced salt solution (HBSS), Triton X-100, Tween 20, bovine serum albumin (BSA), Fluroshield™ with 4′,6-diamidino-2-phenylindole (DAPI), and curcumin (CUR) were purchased from Sigma-Aldrich (Spain). 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid buffer 1 M (HEPES) at pH 7.3 was purchased from Alfa Aesar. 4-Dimethylaminopyridine (DMAP) and dry N,N-dimethylformamide (DMF) were purchased from Acros Organics. N-Butyl-poly(L-lysine hydrobromide) (PLys, MW = 39.9 kDa) and n-butyl-poly(L-glutamate sodium salt) (PGlu, MW = 30.6 kDa) were purchased from Polypeptide Therapeutic Solutions S. L. (Spain). Dichloromethane (DCM), diethyl ether, sodium dodecyl sulfate (SDS), Dulbecco's modified Eagle's medium (DMEM), RPMI 1640 medium, fetal bovine serum (FBS), penicillin–streptomycin solution (P/S), alamarBlue® cell viability reagent, Quant-iT™ PicoGreen™ dsDNA assay kit, tetramethylrhodamine isothiocyanate (TRITC)-phalloidin, 16% formaldehyde solution (w/v), the Griess reagent kit, and Human matrix metalloproteinase (MMP)-2 Recombinant Protein (PeproTech®) were purchased from ThermoFisher Scientific (Spain). DuoSet™ ELISA kits were purchased from R&D Systems (UK). Human monocytes isolated from an acute monocytic leukemia patient were acquired from ATCC, whereas murine microglia (BV-2) from AcceGen Biotech (USA).2.2 Encapsulation of curcumin into the CaCO3 microparticles
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 × g for 2 min) and washed three times with a 5 mM NaCl solution and distilled water. The CUR solution was added either dropwise or directly to the template. In brief, CUR (1 mg) was dissolved in 0.5 mL of ethanol, and the solution was added to the template (30 mg) suspended in 0.5 mL of distilled water. Then, the template was incubated for 30 min sheltered from light. After centrifugation (18800 × g for 2 min), the supernatant was collected to determine the encapsulation efficiency (EE%) (eqn (1)) and the loading capacity (LC%) (eqn (2)) by measuring the absorbance (PerkinElmer PDA Lambda 265 spectrophotometer, λ = 427 nm). Afterwards, the template was washed once with a 5 mM NaCl solution or distilled water and stored for further characterization. All the steps of synthesis were carried out at room temperature.
800 × g for 2 min) and washed three times with a 5 mM NaCl solution and distilled water. The CUR solution was added either dropwise or directly to the template. In brief, CUR (1 mg) was dissolved in 0.5 mL of ethanol, and the solution was added to the template (30 mg) suspended in 0.5 mL of distilled water. Then, the template was incubated for 30 min sheltered from light. After centrifugation (18800 × g for 2 min), the supernatant was collected to determine the encapsulation efficiency (EE%) (eqn (1)) and the loading capacity (LC%) (eqn (2)) by measuring the absorbance (PerkinElmer PDA Lambda 265 spectrophotometer, λ = 427 nm). Afterwards, the template was washed once with a 5 mM NaCl solution or distilled water and stored for further characterization. All the steps of synthesis were carried out at room temperature.
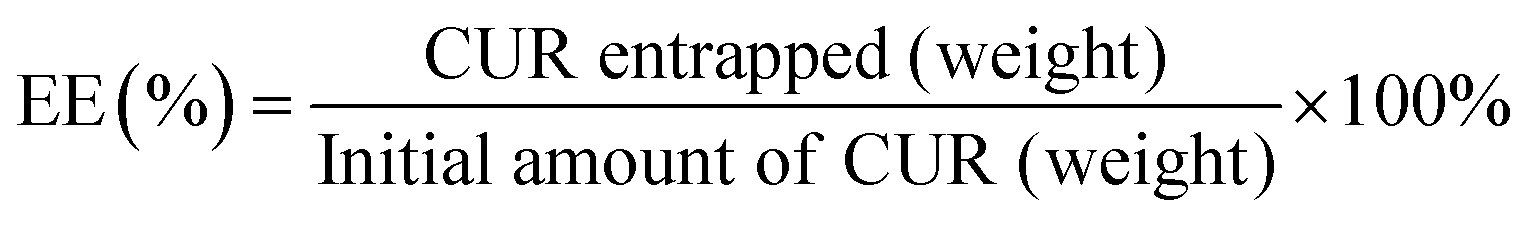 | (1) |
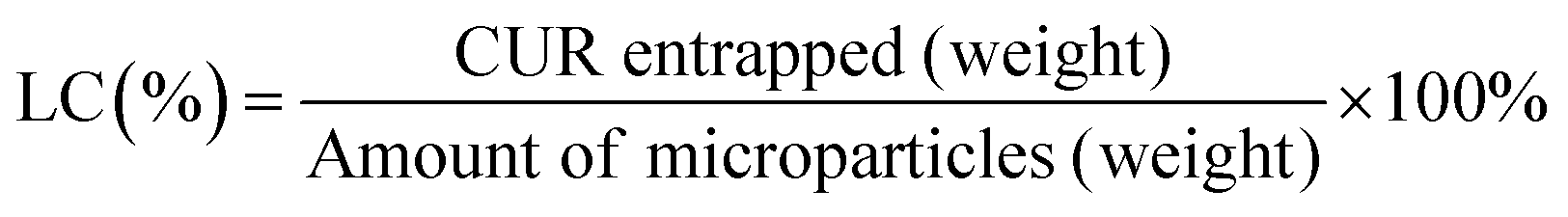 | (2) |
2.3 Conjugation of curcumin to poly(L-glutamic acid)
PGlu (20 mg) was dissolved in 1 mL of dry DMF under a N2 atmosphere. EDC·HCl (5.3 mg, 0.20 eq.) and DMAP (2.4 mg, 0.14 eq.) dissolved in DMF were added, and the mixture was stirred for 20 min to activate the carboxylate groups. Afterwards, CUR (3.5 mg, 0.07 eq., dissolved in dry DMF) was added to the mixture. The mixture was left stirring overnight sheltered from light. Then, 10 mL of distilled water was added to the mixture, and the unreacted reagents were removed by extraction with DCM (×3) and diethyl ether (×2). CUR–PGlu in the aqueous phase was purified using a Sephadex column, lyophilized and stored at −20 °C for further use (yield = 45%).2.4 Fabrication of LbL microcapsule formulations
800 × g for 2 min) and the non-adsorbed polypeptide was removed by washing once with a 5 mM NaCl solution. The ζ-potential was measured to determine the surface charge of the coated microparticles. Following this procedure, the microparticles were coated by three (PLys/PGlu) bilayers. Finally, the core was dissolved by incubating each of the coated microparticles with a 0.1 M EDTA solution for 5 min to obtain unloaded microcapsules (i.e., CPS) and CUR-loaded microcapsules (i.e., COPRE-cps and POST-cps). Then, the capsules were washed once with a 5 mM NaCl solution or distilled water.2.5 Stability studies of LbL microcapsules
The stability of CPS was tested in different media at pH 7.4 and 37 °C at two different time points (i.e., 6 and 24 h). Briefly, microcapsules (about 3 mg) were incubated with 0.2 mL of (i) 0.1 M TRIS, (ii) 0.1 M TRIS + 0.1% or 0.5% SDS, (iii) 0.1 M TRIS + 0.1% or 0.5% SDS + 2 mg mL−1 pronase buffer solutions, and (iv) artificial cerebrospinal fluid (ACSF) in the presence or absence of 0.1% BSA + MMP-2 at different concentrations (i.e., 0, 200, 500, and 1000 ng mL−1). At the selected time points, the capsules were collected by centrifugation (18800 × g for 2 min) and washed once with distilled water before further characterization.
2.6 In vitro release of curcumin from LbL microcapsule formulations
The in vitro release of CUR from each formulation was determined under acidic and neutral environments and in the presence of a mixture of proteolytic enzymes (i.e., pronase) at 37 °C. In brief, the CUR formulations were redispersed in 0.2 mL of (i) 0.1 M acetate buffer containing 0.1% SDS at pH 5.0 or (ii) 0.1 M TRIS containing 0.1% SDS at pH 7.4 or (iii) 0.1 M TRIS containing 0.1% SDS and 2 mg mL−1 pronase at pH 7.4. The release of CUR under the respective conditions was assessed every 15 min for the first 6 h and then every 1 h or 6 h to reach 18 h. At the established time points, the microcapsules were centrifuged (18800 × g for 3 min), and the supernatant was collected to measure the absorbance of the CUR released (λ = 430 nm for COPRE-cps and POST-cps, and λ = 427 nm for CONJ-cps) using a microplate reader (BioTek Synergy H1M). Then, fresh medium was added to the pellet for further incubation times. The release studies were conducted in triplicate for each condition, and a cumulative release of CUR was determined over 18 h (eqn (3)).
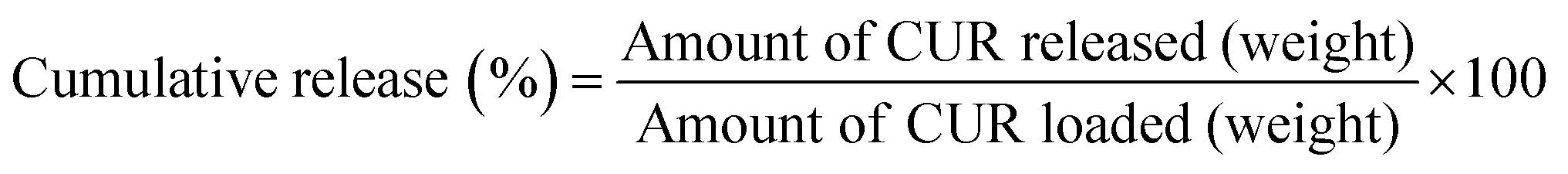 | (3) |
2.7 Physico-chemical and morphological characterization
The size distribution of the unloaded template, COPRE-T, and POST-T was determined using a laser scattering particle size distribution analyzer (HORIBA LA-350) at room temperature.The crystallographic structure of the unloaded template, COPRE-T and POST-T was determined by X-ray diffraction analysis (XRD). The analysis was performed by means of a PHILIPS X’PERT PRO automatic diffractometer operating at 40 kV and 40 mA, in a theta–theta configuration, a secondary monochromator with Cu-Kα (λ = 1.5418 Å) and a PIXcel solid state detector.
The surface charge of the unloaded template, COPRE-T, POST-T, and the corresponding multilayer microparticles after each deposition step was measured using a Malvern Instrument Zetasizer (ZEN 3690) for a minimum of ten runs. The ζ-potential was determined for microparticles dispersed in a 5 mM NaCl solution at room temperature. The mean value and standard deviation (mean ± SD) of the data were calculated from three measurements.
The unloaded template and unloaded LbL microparticles/capsules were characterized by attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR) using a Nicolet AVATAR 370 spectrometer. Spectra were taken with a resolution of 2 cm−1 and averaged over 64 scans.
The morphological analysis of the unloaded template, COPRE-T, POST-T, the corresponding LbL microparticles/capsules, and the CPS monitored during the stability studies was performed by means of scanning electron microscopy (SEM, Hitachi FEG-SEM S-4800) with an acceleration voltage of 5.0 kV. Before taking the images, the samples were coated with a 10 nm layer of gold using an Emitech K550X ion sputter.
Nuclear magnetic resonance (NMR) spectroscopy and high-performance liquid chromatography (HPLC) were used to characterize the conjugated compound CUR–PGlu. In the case of NMR spectroscopy, CUR–PGlu was dissolved in deuterated water (D2O), and 1H-NMR spectra were acquired (128 scans, 64 s of relaxation time) at room temperature using a Bruker Advance DPX 300 spectrometer with 300 MHz of resonance frequency. HLPC measurements were carried out using a HPLC Nexera Lite system (Shimadzu), equipped with a SIL-40C autosampler (Shimadzu), an OHpak SB-803 HQ column (8.0 × 300 mm), and a SPD-M40 photo diode array detector (Shimadzu). The samples analyzed by HPLC were dissolved in PBS (flow rate 0.5 mL min−1, 25 °C, λ = 220 and 427 nm).
2.8 In vitro cellular study
The response of THP-1 differentiated macrophages to two different concentrations of CPS, COPRE-cps, POST-cps, and CONJ-cps (i.e., 100 and 1000 capsules per cell) was measured using an ELISA according to the manufacturer's specification. In brief, three pivotal pro-inflammatory cytokines (i.e., tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6) were quantified in the recovered culture media after the treatment. The release of the cytokines was assessed at least in triplicate (N ≥ 3).
The content of nitrites in the supernatants was measured using a Griess reagent kit, following the protocol provided by the supplier (absorbance 548 nm, room temperature). The TNF-α released by cells was quantified by means of an ELISA, following the protocol provided by the supplier. Both analyses were performed in triplicate.
2.9 Statistical analysis
All quantitative data related to the fabrication and the characterization of LbL microcapsule formulations are represented as the mean ± SD. For the in vitro studies, at least three technical replicates (N ≥ 3) were used, and the results are indicated as the mean ± SD. To process the data of the cell experiments, one-way analysis of variance (ANOVA) followed by the Dunnet post hoc test was used for comparison with the control groups (p < 0.05; p < 0.01, p < 0.00005; p < 0.00001).3. Results and discussion
3.1 Incorporation of curcumin into the calcium carbonate microparticles
CaCO3 vaterite particles represent promising inorganic biomaterials for applications in drug delivery thanks to their biocompatibility, porosity, high drug loading capacity, and preservation of the properties of the payload.26,32,33 Molecules of diverse nature can be incorporated into the particles, whose stability may be enhanced by the presence of additives. For instance, the stability of the CaCO3 vaterite particles in non-polar media has made them good candidates for the inclusion of hydrophobic drugs.15,32,34In this work, CUR, used as a hydrophobic model drug, was incorporated into CaCO3 microparticles through two different approaches: co-precipitation and post-encapsulation. In the co-precipitation strategy, CUR was encapsulated during the formation of the CaCO3 microparticles, whereas in the post-encapsulation approach the loading was carried out after their synthesis by adsorption.
As CUR is poorly soluble in water and the synthesis of the microparticles is performed in aqueous solution, an optimization of the encapsulation procedure was carried out to achieve a high loading efficiency and a control on the morphology of the microparticles. In the case of the co-precipitation approach, two different parameters, i.e., the volume of ethanol used as co-solvent to dissolve CUR and the speed of its addition to the CaCl2 solution, were evaluated for the synthesis of the microparticles. Specifically, when 1 mL of CUR in ethanol was mixed with 0.5 mL of 0.2 M CaCl2 via direct or dropwise addition, the final CaCO3 microparticles were characterized by more or less defined flower-like shapes, respectively (Fig. 1(A) ad (B)). Furthermore, no significant changes were observed in the encapsulation efficiency that was about 70% in both cases. Contrarily, by decreasing the volume of ethanol in the mixture of CUR/CaCl2, the SEM images showed particles with spherical morphology (Fig. 1(C) and (D)). Specifically, when CUR was dissolved in 0.5 mL of ethanol and directly added to 1 mL of 0.2 M CaCl2, final CaCO3 microparticles with a spongy surface were observed in SEM (Fig. 1(C)). When CUR in 0.3 mL of ethanol was directly added to 1.5 mL of 0.2 M CaCl2, the final CaCO3 microparticles appeared porous but smoother than the previous ones (Fig. 1(D)). Despite the different morphologies of the synthesized particles, the XRD peaks revealed the formation of pure vaterite particles (Fig. S1, ESI†). Therefore, we hypothesized that the volume of ethanol could alter the morphology of the microparticles without affecting their crystallinity state. Traditionally, equimolar concentrations of CaCl2 and Na2CO3 in equal volumes lead to the formation of isotropic CaCO3 particles. The synthesis is driven by nucleation, growth, and aggregation of calcium carbonate grains at the microscale.35 In a reported study, it was corroborated that the initial salt concentration had a strong impact on the nucleation process of the CaCO3 particles, allowing the generation of spherical particles when an equilibrium between ions was reached.36 However, the addition of different additives, such as macromolecules, divalent cations, or organic solvents, affects the morphology of the CaCO3 microparticles.33 In particular, in a reported study, it was stated that ethanol, isopropanol, and diethylene glycol could affect the crystal growth rate and stabilize the vaterite crystals preventing the transformation into calcite crystals.37 The different morphologies (non-spherical) of the particles are reported to be caused by the inhomogeneity of the solvent alcohol–water mixture and the preferential solvation of the ions.38 In fact, as CO32− ions are preferentially solvated by water and Ca2+ ions both by water and ethanol, a compartmentalized water-rich microphase is formed influencing the shape and size of the primary CaCO3 particles in the first phase of the reaction.38 In our work, we could observe that the reduction of the volume of ethanol to the minimum amount to dissolve the hydrophobic molecules of CUR (i.e., 0.3 mL) allowed to obtain spherical-like and stable CaCO3 microparticles with high encapsulation efficiency (Table 1). The speed of addition of CUR did not have a relevant impact on the morphology of the particles and on the encapsulation efficiency. However, it is not excluded that the crystals of CUR formed during the synthesis could influence the process of the formation of the particles.
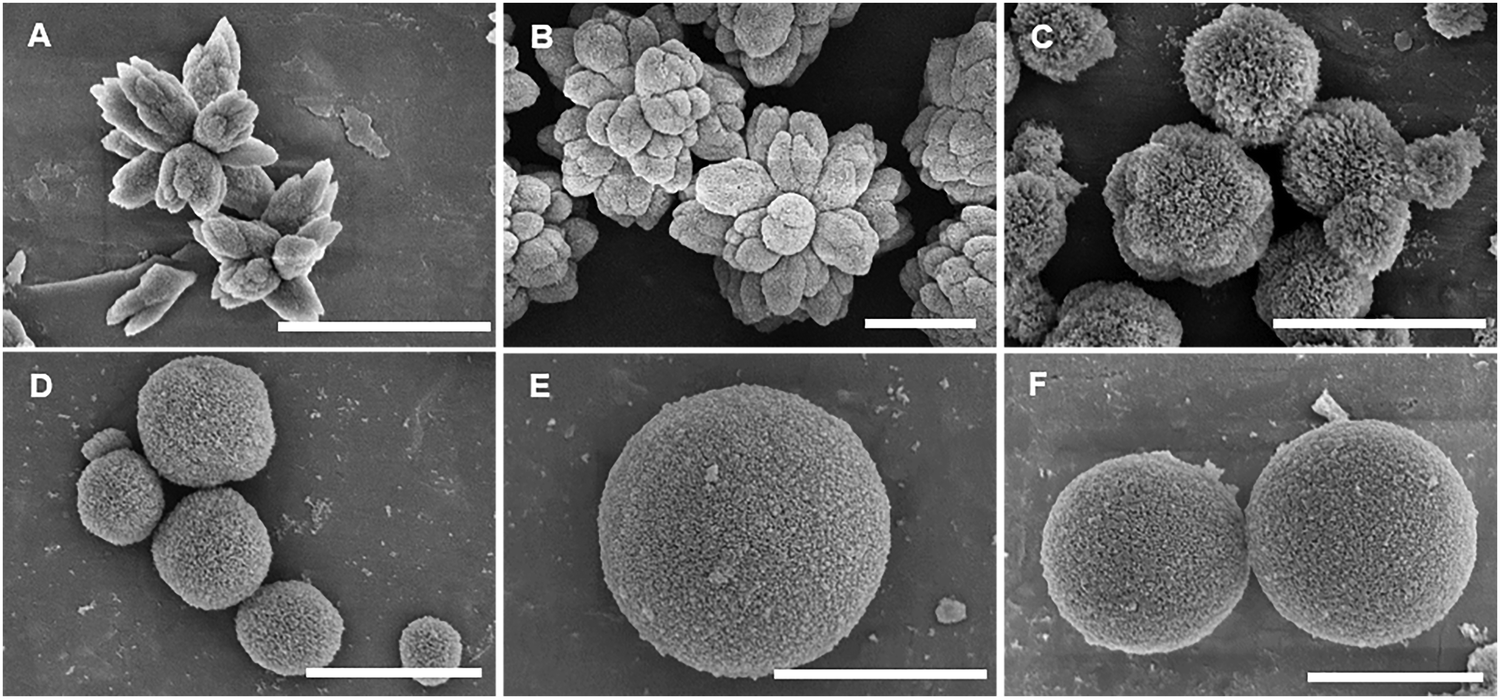 | ||
| Fig. 1 SEM images showing the morphology of the curcumin (CUR)-loaded calcium carbonate (CaCO3) microparticles synthesized during the optimization of the procedure. (A) and (B) Flower-like CaCO3 microparticles obtained by the co-precipitation approach after directly (A) or dropwise (B) adding CUR (1 mg) dissolved in the mixture ethanol/0.2 M calcium chloride (CaCl2) (1 mL/0.5 mL) to 1.5 mL of a 0.2 M sodium carbonate (Na2CO3) solution. (C) and (D) Spherical-like CaCO3 microparticles obtained by pouring the mixture composed of CUR (1 mg) in 0.5 mL (C) or in 0.3 mL (D) of ethanol and 1 mL or 1.5 mL of a 0.2 M CaCl2 solution, respectively, to 1.5 mL of a 0.2 M Na2CO3 solution. (E) and (F) Spherical-like CaCO3 microparticles synthesized by the post-encapsulation approach, resuspending the pre-synthesized CaCO3 microparticles (30 mg) in distilled water and adding directly (E) or dropwise (F) CUR (1 mg) dissolved in 0.5 mL ethanol. The scale bar is 3 μm. | ||
| Method of encapsulation | Type of addition | Shape | Encapsulation efficiency (EE%) | Loading capacity (LC%) |
|---|---|---|---|---|
| a The co-precipitation approach was performed at room temperature by directly adding curcumin (CUR, 1 mg) dissolved in 0.3 mL of ethanol to 1.5 mL of a 0.2 M calcium chloride (CaCl2) solution. The mixture CUR/CaCl2 was then added to 1.5 mL of a 0.2 M sodium carbonate (Na2CO3) solution containing 4 mg mL−1 poly(sodium 4-styrene sulfonate) (PSS) at high stirring speed for 1 min. The particles were allowed to precipitate for 30 min.b The post-encapsulation approach was performed at room temperature by directly or dropwise adding CUR (1 mg) dissolved in 0.5 mL of ethanol to the pre-synthesized CaCO3 microparticles (30 mg) re-dispersed in 0.5 mL of distilled water. The CaCO3 microparticles were incubated for 30 min.c Direct addition of CUR.d Dropwise addition of CUR.e The pellet of the synthesized CUR-loaded CaCO3 microparticles was collected to measure the absorbance of the supernatant (λ = 427 nm) and calculate the encapsulation efficiency (EE%) and the loading capacity (LC%). | ||||
| Co-precipitationa | Direct | Spherical | 87.3 ± 1.0e | 2.9 ± 0.0e |
| Post-encapsulationb | Directc | Spherical | 51.8 ± 1.4e | 1.7 ± 0.1e |
| Dropwised | Spherical | 71.7 ± 4.7e | 2.4 ± 0.2e | |
In the case of the post-encapsulation approach, the pre-synthesized microparticles were resuspended in distilled water and incubated with CUR dissolved in ethanol in a final v/v ratio of 1:1. The dropwise or direct addition of CUR to the microparticles led to a difference of 20% in the encapsulation efficiency, resulting in a higher value in the case of the dropwise addition (Table 1). The preservation of the spherical-like shape of the original microparticles after the incubation with CUR in ethanol was corroborated by the SEM images (Fig. 1(E) and (F)), and the stability of the vaterite crystals was further confirmed by the XRD peaks (Fig. S1, ESI†).
Overall, by comparing the co-precipitation and post-encapsulation strategies, the differences in the encapsulation efficiencies shown in Table 1 may be due to the different mechanisms of inclusion of CUR into the CaCO3 microparticles. Indeed, the entrapment of CUR during the formation of the microparticles in the case of the co-precipitation approach determined a higher encapsulation efficiency compared to the post-encapsulation approach, where the CUR molecules might simply diffuse in and out through the pores of the particles. Therefore, this aspect might influence the successive release studies of CUR.
3.2 Conjugation of curcumin to poly(L-glutamic acid)
The poor water-solubility of CUR is one of the drawbacks that limit its use as a drug, despite its excellent properties, including anti-cancer, anti-oxidant, and anti-inflammatory effects.39 The conjugation to polymers to form polymer–drug conjugates offers the advantage to enhance the solubility, bioavailability, and plasma half-life of hydrophobic drugs. Prior works have reported the conjugation of CUR to PGlu or amino acids.40–43 For instance, star-shaped polyglutamate conjugates of bisdemethoxycurcumin have shown extended blood circulation times, exhibiting a potential nephroprotection against acute kidney injury42 and neuroprotection against Alzheimer's diseases.43 In this work, we conjugated CUR to PGlu to enhance its solubility, included it in a drug delivery formulation, and explore its potential anti-inflammatory properties. Specifically, the conjugation of CUR to PGlu was carried out via an ester bond by means of an esterification reaction to obtain CUR–PGlu with an overall yield after purification of 45% (Fig. 2).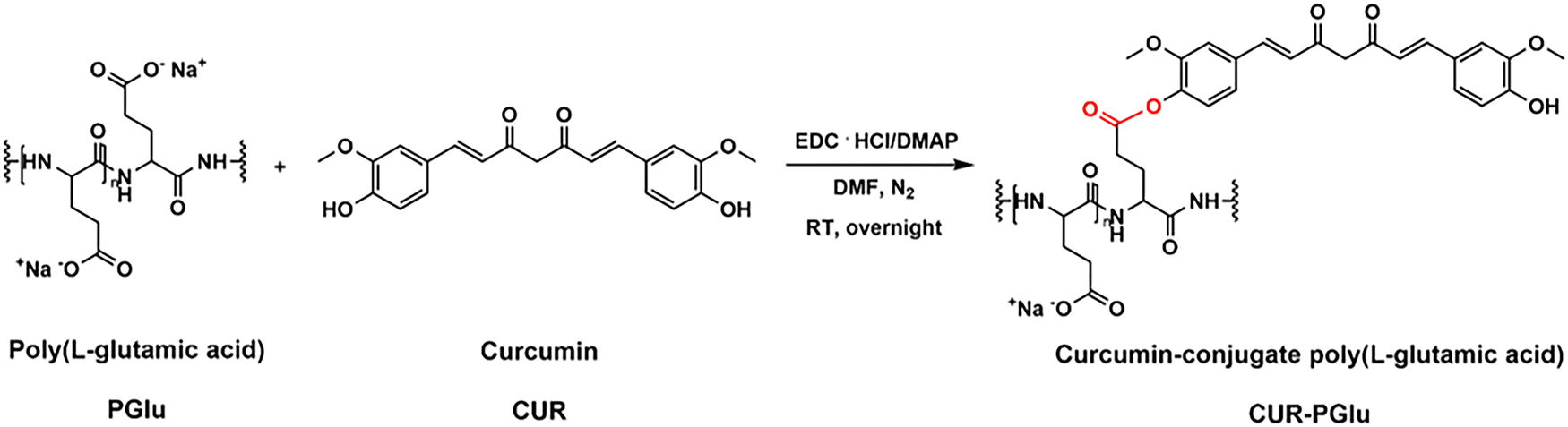 | ||
| Fig. 2 Scheme of the synthesis of curcumin–conjugate poly(L-glutamic acid) (CUR–PGlu) via an esterification reaction between curcumin (CUR) and poly(L-glutamic acid) (PGlu). The reaction was performed overnight at room temperature (RT) after activating the side chains of PGlu with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl) and dimethylaminopyridine (DMAP). | ||
After the purification step, CUR–PGlu was characterized by 1H-NMR spectroscopy, where, in addition to the typical peaks of PGlu taken as a reference, the peaks in the range of 2.8–3.3 ppm and 6.8–7.9 ppm were attributed to CUR (Fig. S2A, ESI†). The UV-Vis spectra of CUR–PGlu were characterized by a slight shift of the absorption band (420 nm) with respect to the absorbance of CUR (λ = 427 nm) probably due to the conjugation to PGlu (Fig. S2B, ESI†). Additionally, in the chromatogram obtained by HPLC analysis, a main peak appeared in the case of the conjugated compound at a retention time of 6.7 min (λ = 220 nm and λ = 427 nm) with respect to PGlu, which was characterized by a retention time of 14.1 min (λ = 220 nm) (Fig. S2C, ESI†). The shorter retention time of CUR–PGlu with respect to the one of PGlu highlighted the successful reaction. However, a small and broad peak at around 15 min may be attributed to a variety of combinations of CUR–PGlu (Fig. S2C, ESI†). Indeed, due to the lack of selectivity of the reaction, all the carboxylic groups could be activated during the first step of the reaction; therefore a different number of CUR molecules could be conjugated at the end of the process. Moreover, it is not excluded that, being CUR a symmetric molecule bearing two OH groups, it can react together with two activated carboxylic groups in the same polypeptide chain or between two polypeptide chains, giving different conjugated compounds.
The ζ-potential of the final compound was −11.0 ± 2.2 mV against −20.3 ± 1.7 mV for PGlu. This highlighted that most of the carboxylic groups of the PGlu chains were free from the conjugation. Hence, due to the negative charge, CUR–PGlu could be further used as one of the building blocks in the multilayer network of the capsules.
3.3 Fabrication of the curcumin-loaded LbL microcapsule formulations
The LbL technique was subsequently used to fabricate multilayer biodegradable microcapsules containing CUR. Three different systems were synthesized: COPRE-cps, POST-cps, and CONJ-cps (Fig. 3). | ||
| Fig. 3 (A) and (B) Schematic illustration of the layer-by-layer (LbL) process to synthesize curcumin (CUR)-loaded particles (i.e., COPRE-particles, POST-particles, and CONJ-particles) via co-precipitation (i), post-encapsulation (ii), and conjugation (iii) approaches. COPRE-T was synthesized by directly adding 0.3 mL of CUR in ethanol to 1.5 mL of 0.2 M CaCl2 solution. POST-T was synthesized by dropwise addition of CUR in ethanol to pre-synthesized CaCO3-T. (C) Core removal to obtain the corresponding capsules: COPRE-cps (i), POST-cps (ii), and CONJ-cps (iii). Poly(L-lysine) (PLys) was used as the positively charged building block. Poly(L-glutamic acid) (PGlu) and CUR–PGlu were used as the negatively charged building blocks. | ||
To fabricate the biodegradable microcapsules, two polypeptides were used as building blocks, i.e., PLys and PGlu. Generally, due to their side chains and pKa values, polypeptides are characterized by a pH-dependent charge, which can be exploited to use them as polyelectrolytes in the LbL methodology. Specifically, under our working pH conditions (i.e., pH 6.5), PLys (pKa = 9.0) and PGlu (pKa = 4.8) bear positive and negative charges, respectively. Therefore, by alternating the positively charged PLys and the negatively charged PGlu onto the template, we could build a multilayer membrane made of three (PLys/PGlu) bilayers mainly interacting each other by electrostatic interactions (Fig. 3(B)-i–iii).
The optimization of the methodology to encapsulate CUR into CaCO3 microparticles by the co-precipitation approach described in Section 3.1 allowed exploration of the optimal conditions to obtain spherical-like CaCO3 microparticles. Importantly, the morphology of the template affects the shape of the particles after applying the LbL approach. We could confirm this phenomenon through the SEM micrographs after the deposition of the multilayer membrane onto the spherical-like and flower-like CaCO3 templates. The obtained LbL-particles kept the morphology of the corresponding uncoated microparticles, either flower-like (Fig. S3A and B, ESI†) or spherical-like (Fig. 4(B)) shape. Moreover, after the dissolution of the template, the LbL-capsules looked collapsed, but no relevant changes were observed for both morphologies (Fig. S3C and D, ESI,† and Fig. 4(B), respectively). As the morphology of the LbL systems may also determine the kinetics of the release of the payload, and/or the interactions with cells (e.g., uptake), in this work, we performed a comparative study between the three LbL systems with the spherical-like shape (Fig. 3(A)-i–iii).
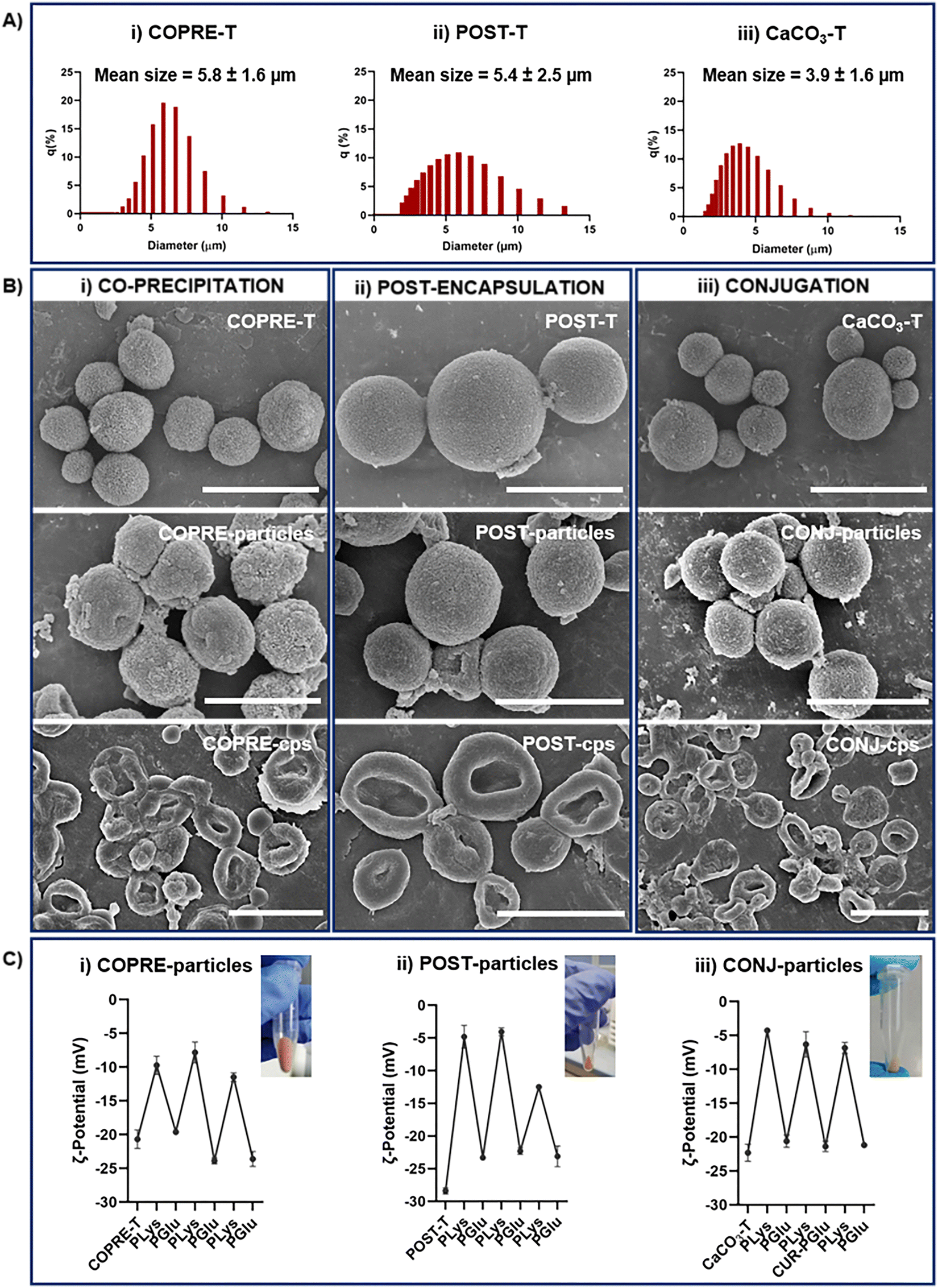 | ||
| Fig. 4 Characterization of curcumin (CUR)-loaded templates (i.e., COPRE-T and POST-T), calcium carbonate template (CaCO3-T), and the corresponding layer-by-layer (LbL)-particles and capsules synthesized according to the co-precipitation, post-encapsulation, and conjugation approaches. (A) Size distribution of COPRE-T (i), POST-T (ii), and CaCO3-T (iii), respectively. (B) Micrographs of the templates, LbL-particles, and LbL-capsules (from top to bottom) obtained for each formulation. The scale bar is 5 μm. (C) ζ-potential values measured during the LbL process after the deposition of each polypeptide layer in COPRE-particles (i), POST-particles (ii), and CONJ-particles (iii). Insets show photographs of the LbL-particles, highlighting the yellowish color associated with the presence of CUR. The values are reported as mean ± standard deviation (SD); N = 3. | ||
To proceed with the fabrication of the CUR-loaded LbL systems, we selected the CUR-loaded CaCO3 microparticles that showed the highest encapsulation efficiency in the case of co-precipitation and post-encapsulation (Table 1).
The mean sizes of 5.8 ± 1.6 μm, 5.4 ± 2.5 μm, and 3.9 ± 1.6 μm for the COPRE-T, POST-T, and the CaCO3-T, respectively, were observed by measuring the particle size distribution (Fig. 4(A)) and further confirmed by the SEM micrographs (Fig. 4(B)). Once synthesized the CaCO3 microparticles for each formulation, the LbL approach was followed by depositing three (PLys/PGlu) bilayers on their surface to fabricate a polypeptide-based multilayer membrane. In the case of COPRE-T and POST-T, during each incubation step with the polypeptide solution, no significant release of CUR was detected, which can also be ascribed to its poor water solubility. Therefore, we assumed that most of the encapsulated CUR was retained in the systems during the LbL process. The successful deposition of each polypeptide layer onto the particle surface was monitored by measuring the values of the surface charge of the particles. Due to the negative surface charge of the COPRE-T, POST-T, and CaCO3-T, the positively charged PLys was deposited as the first layer allowing a change in the ζ-potential value that confirmed the successful coating (Fig. 4(C)). Furthermore, the adsorption of the subsequent layers by alternating PGlu and PLys up to six-layers was highlighted by a zig-zag trend of the ζ-potential values for all the formulations under study. In the particular case of the CONJ-particles, the fourth layer was substituted by CUR–PGlu to accommodate CUR in the polypeptide-multilayer membrane. Being CUR–PGlu soluble in water, 25 mM HEPES/20 mM NaCl buffer solution at pH 6.5 was used to dissolve it, as in the case of PLys and PGlu. The negative ζ-potential value achieved after the deposition of the fourth layer (i.e., −21.4 ± 0.8 mV) corroborated the successful adsorption of the conjugated polypeptide onto the particles. Moreover, the SEM micrographs of the three synthesized LbL-particles (i.e., COPRE-particles, POST-particles, and CONJ-particles) (Fig. 4(B)) showed that after the LbL process the particles retained the size and shape of the corresponding template (i.e., COPRE-T, POST-T, and CaCO3-T). The core removal was finally performed in the presence of 0.1 M EDTA solution to obtain the corresponding capsules (i.e., COPRE-cps, POST-cps, and CONJ-cps), and no relevant release of CUR was detected after collecting the supernatants. Additionally, the SEM images clearly demonstrated the dissolution of the CaCO3 core from all the formulations with the formation of collapsed membrane structures (Fig. 4(B)). A complete characterization of the LbL-capsules was carried out in the absence of CUR (Fig. S4, ESI†), where the core removal was further investigated by ATR-FTIR spectroscopy. Indeed, by comparing the spectra of the template, LbL-particles, and LbL-capsules, the disappearance of the two strong absorbance bands at 1390 cm−1 of νas (CO32−) and 877 cm−1 of δout-of-plane (CO32−) (Fig. S4E, ESI†) in the case of the LbL-capsules with respect to the corresponding template and LbL-particles confirmed the complete removal of the core.
3.4 Multilayer capsule stability
Stability studies of the carrier, i.e., CPS, were carried out to assess the integrity of the systems at pH 7.4 in TRIS buffer and ACSF. In both cases, the SEM was used to monitor any change in the shape and morphology at two different time points (i.e., 6 and 24 h). The experiments in 0.1 M TRIS buffer were carried out with and without the addition of SDS and/or pronase. The SDS was included in the media thinking on the subsequent release studies of CUR, as it acts as a surfactant capable of enhancing the solubility of the poorly water soluble CUR. For this reason, we first evaluated the preservation of the integrity of the capsules in the presence of SDS. In the case of (PSS/poly(allylamine hydrochloride)(PAH))3 microcapsules, it has been recently observed that the disassembly of the microcapsules was dependent on the concentration of SDS. Specifically, with a concentration of 3 mg mL−1 SDS, the microcapsules started to disassemble after 3 h, while with a concentration of 0.1 mg mL−1 SDS, this process was prolonged in time.44 In our work, two different percentages of SDS (i.e., 0.1% and 0.5%, corresponding to 1 mg mL−1 and 5 mg mL−1, respectively) were used to evaluate whether the presence of the surfactant could affect the stability of the polypeptide-multilayer membrane, and, therefore, have a role in the subsequent controlled release of the payload. In contrast to the case of (PSS/PAH)3 microcapsules previously reported, our (PLys/PGlu)3 microcapsules did not show any significant disassembly after 6 and 24 h of incubation, regardless of the concentration of SDS used (Fig. S5, ESI†). Moreover, since PLys and PGlu used as building blocks in the LbL process are biodegradable, an enzymatic degradation of CPS was followed by taking SEM images after a further incubation of the systems with 2 mg mL−1 pronase for 6 and 24 h (Fig. S6, ESI†). Although specific enzymes could be used to degrade specific biopolymers, such as polypeptides, proteins, or polysaccharides, in this work we used pronase as non-specific proteolytic enzymes to explore the biodegradability of the systems under study. Pronase is a mixture of enzymes containing numerous proteases and peptidases capable of hydrolyzing most of the proteins into individual amino acids. In our study, it was observed that after the incubation with pronase, CPS mostly lose their shape and morphology giving a film of aggregated capsules (Fig. S6, ESI†). Therefore, we could assume that a degradation of our capsules was occurring. However, we could not exclude that the presence of SDS on the one hand and the formation of aggregates on the other hand could affect the subsequent release profile of CUR.In the presence of specific proteolytic enzymes, such as MMP-2 that is overexpressed in neuroinflammation, the degradation profile of CPS may be different due to the complex structure of the multilayer membrane and the selectivity of the enzyme for the two polypeptides. In particular, by incubating our microcapsules in ACSF supplemented with 0.1% BSA and with different concentrations of MMP-2 (i.e., 0, 200, 500, and 1000 ng mL−1) for 6 and 24 h, the SEM images showed a partial degradation of the systems (Fig. S8, ESI†). While the BSA was not affecting the structural integrity of the systems (Fig. S8, ESI†) compared to the control without BSA (Fig. S7, ESI†), the increased concentration of MMP-2 and incubation time gradually intensified their degradation profile. Therefore, we could conclude that under these conditions the disruption of the multilayer membrane might be slower than in the case of the incubation with pronase. As a consequence, this might affect the release profile of the payload incorporated into the microcapsules. Therefore, these studies allowed us to highlight that our multilayer systems may also undergo degradation under specific pathological conditions, such as neuroinflammation.
3.5 In vitro release of curcumin from the LbL microcapsule formulations
The presence of PLys and PGlu in the multilayer membrane is a key factor to explore how the biodegradable capsules may affect the release of the payload from the microcarriers under healthy and pathological conditions. Specifically, three different conditions were investigated to assess the release of CUR from the three formulations of LbL-capsules under study: neutral (i.e., pH 7.4) and acidic (i.e., pH 5.0) conditions, and in the presence of 2 mg mL−1 pronase at pH 7.4. As pronase is stable in the pH range of 6.0 to 9.0, as specified by the supplier, the effect of pronase on the release was studied at pH 7.4 to ensure the activity of the enzymes during the incubation time.By comparing the release of CUR from POST-cps and COPRE-cps, where the payload was included into the core before performing the LbL process, a difference in the released amount of CUR was observed throughout the entire study (i.e., 18 h) (Fig. 5(A) and (B)). In the case of the POST-cps, CUR was almost completely released under all the conditions after 18 h (i.e., 87.5% at pH 7.4, 98.7% at pH 5.0, and 85.2% in the presence of proteolytic enzymes). Under neutral and acidic conditions, the release of CUR was very low during the first hour of incubation (i.e., 0.5% and 1.8%, respectively), probably due to the presence of the multilayer membrane that prevented the initial burst release of the CUR molecules. Contrarily, the presence of enzymes facilitated the release of 13.8% of CUR in 1 h (Fig. 5(A)). This phenomenon might be ascribed to the initial degradation and/or disassembly of the polypeptides, which promoted an accelerated diffusion of CUR through the multilayer membrane. In fact, during the first 5 h, the release of CUR in the presence of proteolytic enzymes resulted faster than the one at pH 7.4 and 5.0. In the case of the COPRE-cps, CUR was not completely released in the time period studied. Particularly, COPRE-cps showed a higher cumulative release under neutral pH conditions with respect to the release under acidic conditions or in the presence of enzymes. Furthermore, the release of CUR at pH 7.4 and pH 5.0 was slower during the first time points with respect to the POST-cps. Specifically, the first 2.5% of CUR was released only after 75 min of incubation at pH 7.4, while the first 0.2% of release was observed after 45 min at pH 5.0. In contrast, in the presence of enzymes, a burst release of the CUR molecules (i.e., 5.8%) was observed after 15 min of incubation. Therefore, similar to the POST-cps formulation, the presence of the enzymes promoted an initial fast release that can be attributed to the degradation of the multilayer membrane.
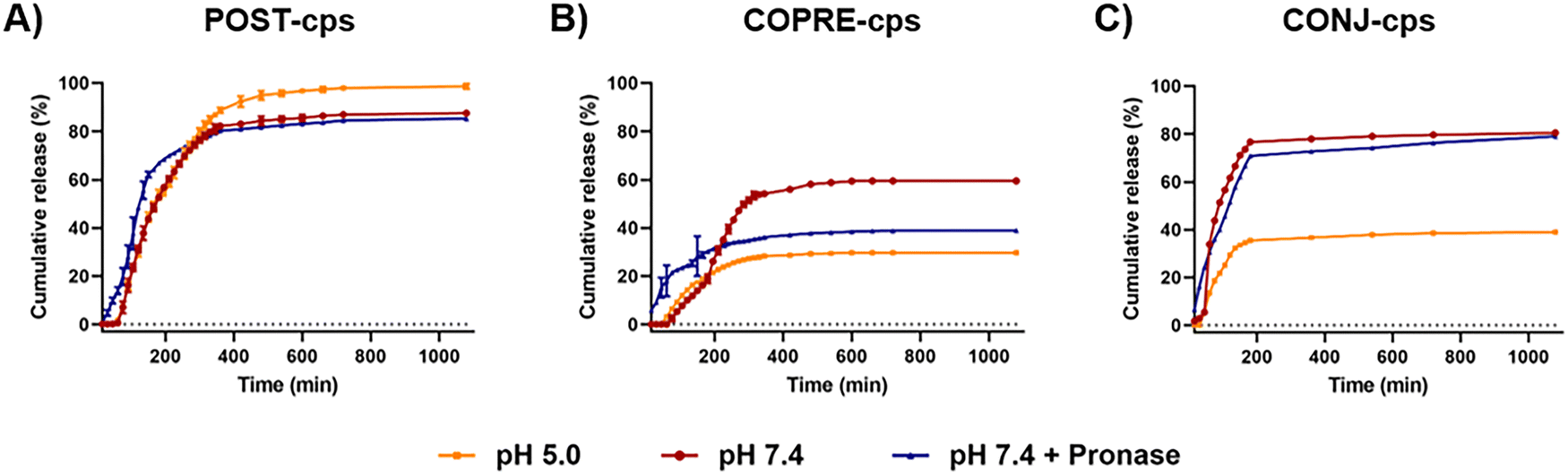 | ||
| Fig. 5 Release studies of curcumin (CUR) from CUR-loaded capsules by post-encapsulation (POST-cps) (A), co-precipitation (COPRE-cps) (B), and conjugation (CONJ-cps) (C) methods under acidic (pH 5.0, orange line) and physiological (pH 7.4, red line) conditions and in the presence of 2 mg mL−1 pronase (blue line). The studies were performed in triplicate (N = 3). Each time point is expressed as mean ± standard deviation (SD). | ||
The difference in the amount of CUR released by POST-cps and COPRE-cps may be due to the approach used to encapsulate the payload into the template. During the loading by the post-encapsulation approach, CUR molecules could be less anchored to the CaCO3 microparticles compared to the co-precipitation approach, where they are directly involved in the generation of the crystals of the template. In the case of the POST-cps, this phenomenon may lead to an easier diffusion of the CUR molecules through the first layers during the LbL procedure that may facilitate the release of the drug. It has been reported, in fact, that the imbalance of the CUR molecules between the interior of the capsules and the bulk solution may promote a diffusion mechanism.45 In contrast, in the case of the COPRE-cps, CUR crystals remain predominantly entrapped into the core after the coating with the first layer of PLys and the subsequent layers. Therefore, the time that the CUR molecules may take to cross the multilayer barrier during the release could be longer. This phenomenon might explain the absence of the released drug from COPRE-cps after 1 h at pH 7.4. However, due to the complexity of the systems, the drug release profile might be affected by other parameters, such as the colloidal stability, the solubility of the drug, the ionic strength, the presence of surfactants (i.e., SDS), and the characteristics of the LbL assembly. For example, the capsules may suffer aggregation during the incubation time, which may bring to the formation of packed structures that prevent the complete release of the drug. Moreover, the assembled polypeptides in the multilayer membrane may affect the release of the payload due to their pH-responsiveness.46 The polypeptides might undergo a rearrangement due to the pH change with respect to the pH conditions where the synthesis occurred (i.e., pH 6.5), determining the release profile of the drug.
In the case of CONJ-cps, the CUR–PGlu conjugate was loaded as a building block in the polypeptide membrane. Compared to the POST-cps and COPRE-cps, the loading of the drug in CONJ-cps can be precisely controlled in the multilayer membrane. In our systems, we were able to load 1.2 μg of CUR per mg of the template. Therefore, this suggested that the amount of the loaded CUR could be tuned by the deposition of the CUR–PGlu in different locations of the multilayer structure. Moreover, by performing the release studies over 18 h, it was observed that CUR was not completely released in any of the explored conditions (i.e., 80.4% at pH 7.4, 38.9% at pH 5.0, and 79.0% in the presence of pronase). Unexpectedly, a high release of the drug was detected under neutral conditions (Fig. 5(C)). Hence, we hypothesize that as the multilayer membrane is dominated by electrostatic forces, there might be a rearrangement of the polypeptide chains of the last layers of the membrane at pH 7.4, triggering a slight change in the charge density of the chains. Therefore, this phenomenon might promote the disassembly of CUR–PGlu and its release over time. In the presence of proteolytic enzymes, the cumulative release is similar, but a burst release (i.e., 6.6%) is detected in the first 15 min of incubation, which might be due to the degradation of the external layers. On the other hand, the amount of CUR released at pH 5.0 was about half in the first 2 h (i.e., 29.3%) compared to the other two tested conditions, reaching afterward a sustained release over the explored time. This phenomenon might be due to the stronger interactions between the layers that prevented the disassembly of the external layers.
Therefore, the three explored formulations represent a very complex scenario, where several parameters could affect the release profile of CUR, which may also degrade during the in vitro studies.45
3.6 Study of the inflammatory response of THP-1 in the presence of multilayer capsules
Inflammation plays a pivotal role in the resolution of any disease in the body, responding to harmful stimuli and recognizing pathogens and toxins.47,48 When the innate immune response fails in containing the initial acute inflammation, a chronic pathological response can lead to tissue damage in many disorders, such as acute lung and liver injury, rheumatoid arthritis, neurodegenerative diseases, and so on.48 Drug delivery systems, including synthetic polymer-based drug carriers,49 may provide a valid solution to treat the inflammation thanks to the controlled release of anti-inflammatory therapeutics. In this work, we leveraged the anti-inflammatory properties of CUR to investigate the potential of our LbL formulations in downregulating pro-inflammatory cytokines secreted by inflamed THP-1 derived macrophages. THP-1 is a human leukemia monocytic cell line that has been extensively used in in vitro models of monocytes and macrophages to study inflammatory diseases.50 THP-1 cells are commonly differentiated into macrophages-like cells by incubation with PMA.51 Stimulation with LPS, a potent activator of the immune system derived from the outer membrane of Gram-negative bacteria, once combined with the toll-like receptors TLR4 expressed by cells, promotes the production of the pro-inflammatory cytokines IL-1β, IL-6, and TNF-α.52,53 Specifically, it has been reported that, when exposed to LPS, THP-1 cells, both in the monocyte phenotype and as derived macrophages, become reactive through the activation of the nuclear factor-κB (NF-κB) pathway, triggering a robust and reproducible inflammatory response.54In our study, we first determined the metabolic activity of both resting and LPS-activated THP-1 derived macrophages over 24 h in the presence of our LbL-capsules by means of an alamarBlue® assay. Cells were treated with three different concentrations of LbL-capsules (i.e., 10, 100, and 1000 capsules per cell) and any detrimental effect induced by the carrier and CUR was evaluated (Fig. 6(A) and (B)). In the presence of the unloaded capsules (i.e., CPS), used as the control, the normalized metabolic activity to the dsDNA content of both resting and LPS-activated THP-1 macrophages was higher than 70% with respect to the healthy and LPS controls, respectively. No statistically significant differences were observed in terms of dsDNA content (Fig. S9A and B, ESI†). Hence, these results confirmed that CPS did not exhibit any detrimental effect over THP-1 cells at any of the tested concentrations. Furthermore, when resting macrophages were exposed to CUR-loaded capsules at the concentrations under study, they were able to maintain a metabolic activity higher than 70% with respect to the healthy control (Fig. 6(A)). In contrast, the metabolic activity slightly decreased to approximately 70% with significant differences compared to the LPS control when LPS-treated macrophages were exposed to CUR-loaded capsules (Fig. 6(B)). Despite the statistically significant difference in terms of dsDNA content measured in the case of COPRE-cps at 10 and 1000 capsules per cell with respect to the LPS control (Fig. S9B, ESI†), in general, we could state that the cells maintained a good metabolic activity (i.e., ≥70%) in the presence of our capsules at any of the tested concentrations and a similar dsDNA content with respect to the healthy control. Therefore, we could corroborate that our CUR-loaded formulations did not produce any significant detrimental effect on these cells.
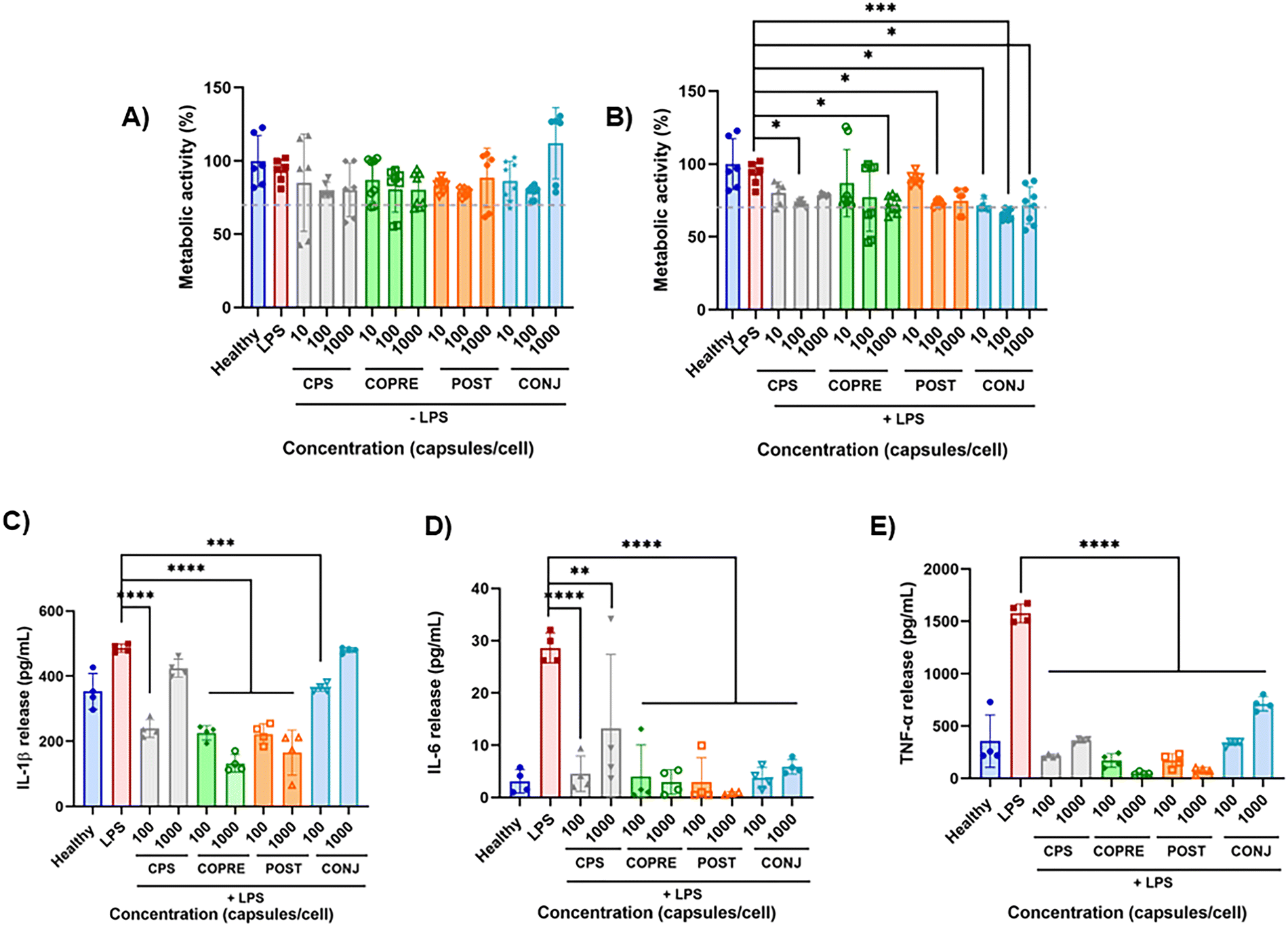 | ||
| Fig. 6 Curcumin (CUR)-loaded capsule performance over THP-1 derived macrophages. (A) and (B) Normalized metabolic activity of THP-1 derived macrophages to the dsDNA content without (A) and with (B) a 100 ng mL−1 lipopolysaccharide (LPS) pre-treatment in the presence of the different formulations at doses from 10 to 1000 capsules per cell. (C)–(E) Effect on the cytokine release (i.e., tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6) from THP-1 derived macrophages treated with 100 or 1000 capsules per cell, measured as the concentration in the supernatant after the treatment. Data obtained from N = 4. The values are reported as mean ± standard deviation (SD). Single ANOVA followed by Dunnet post hoc test comparisons with the controls (healthy or LPS groups) was used (*p < 0.05; **p < 0.01, ***p < 0.0005; ****p < 0.0001). | ||
To investigate any potential anti-inflammatory effect of our capsules, we quantified three pivotal cytokines mostly involved in the pro-inflammatory phase and secreted by the LPS-activated THP-1 derived macrophages, i.e., IL-1β, IL-6, and TNF-α. The effect of the unloaded capsules and CUR-loaded capsules on both resting and LPS-stimulated THP-1 derived macrophages was assessed at 100 and 1000 capsules per cell (Fig. S9C–E, ESI,† and Fig. 6(C)–(E), respectively). As observed in Fig. S9C–E (ESI†), the level of the secreted pro-inflammatory cytokines by resting cells was below the LPS control for all the tested formulations and concentrations, highlighting that our capsules would not induce a further inflammation in a pathological microenvironment. However, it was observed that CONJ-cps promoted a significant release of IL-1β and TNF-α with respect to the healthy cells (Fig. S9C and E, ESI†). We assumed that this phenomenon could be due to the different form of CUR, i.e., CUR–PGlu, which might have a different bioactivity with respect to the free drug loaded into the other formulations. Nonetheless, CONJ-cps exhibited good performance on inflamed cells by significantly reducing the secretion of IL-6 and TNF-α (Fig. 6(D) and (E)). A similar effect was appreciated in the case of the other LbL-capsules (i.e., CPS, COPRE-cps, and POST-cps) with a slightly greater reduction in the cytokine level at 1000 capsules per cell in the case of COPRE-cps and POST-cps compared to CPS (Fig. 6(D) and (E)). Moreover, COPRE-cps and POST-cps at both concentrations (i.e., 100 and 1000 capsules per cell) induced a significant reduction in the secretion of IL-1β with respect to the LPS control, exhibiting a major effect at the highest concentration (Fig. 6(C)). Contrarily, in the case of the CPS and CONJ-cps at 1000 capsules per cell, no significant suppression of IL-1β was detected (Fig. 6(C)). The production of IL-1β by monocytes and macrophages is pivotal to keep the inflammation in immune responses under homeostasis as well as under pathological inflammation.55 Thus, the detected secretion of IL-1β for all the tested formulations in both resting cells and LPS-induced inflammation (Fig. S9C, ESI,† and Fig. 6(C), respectively) was expected due to the intrinsic reactivity of THP-1 cell line.
Our results are in line with studies reported in the literature that show the anti-inflammatory potential of CUR in downregulating TNF-α and IL-6.56–59 For instance, Xu et al. found that CUR could ameliorate heat-killed P. acnes-stimulated THP-1 cells inhibiting the TLR2-NF-kB signaling pathway by suppressing the production of IL-6.56 In a similar study, Ma et al. demonstrated that CUR could inhibit the inflammatory response by targeting miR-155 in THP-1 cells and clearly reduce the expression of TNF-α and IL-6.59 Our findings showed either a downregulation of pro-inflammatory cytokines in a concentration-dependent manner in the presence of the loaded CUR or the potential of our engineered CUR-loaded capsules due to the synergistic effect of the capsule composition and the incorporated CUR. Indeed, although the two polypeptides contained in the multilayer structure promoted a reduction of the pro-inflammatory cytokines by themselves, higher concentrations of loaded CUR could contribute to ameliorate the LPS-triggered inflammatory response. However, this phenomenon was less marked when CUR was included in the capsule structure as the CUR–PGlu conjugate, probably due to either the very low amount of the drug present in the systems compared to the other formulations or the potential reduced bioactivity of CUR after conjugation.
3.7 Anti-inflammatory response and intracellular release on BV-2 cells
As an in vitro proof-of-concept, we used BV-2 cells to evaluate the potential anti-inflammatory properties of our LbL formulations in the context of the central nervous system (CNS). Microglia are resident immune cells of the CNS, which play an essential role in both CNS homeostasis and neuroinflammation.60 Under pathological conditions, activated microglia can polarize into M1-like and M2-like phenotypes and release pro-inflammatory or anti-inflammatory mediators, respectively, to promote cell repair.61,62It has been reported that natural molecules, such as CUR, exert a protective effect on neuroinflammation, reducing the expression of key pro-inflammatory cytokines and chemokines and inhibiting the nitric oxide (NO) production.59,63–65 Herein, we observed that our CUR-loaded capsules did not produce any detrimental effect on the metabolic activity of resting and LPS-activated BV-2 cells at any of the tested concentrations (i.e., 10, 100, and 1000 capsules per cell) with respect to the healthy cells (metabolic activity > 70%) (Fig. S10A and B, ESI†). When BV-2 cells are activated with LPS, their response to the environmental stress induces the release of pro-inflammatory mediators, such as NO, prostaglandin E2, reactive oxygen species, and pro-inflammatory cytokines.66,67 Hence, in this work, we explored the potential anti-inflammatory effect of our CUR-loaded capsules by quantifying the amount of nitrites (NO2−) and TNF-α released in the supernatant (Fig. S10C and D, ESI†). NO is a small membrane-permeable gas, which plays a crucial role in inflammation, because, if secreted at high levels, it induces oxidative stress and cell death.68 Due to the instability of NO, the Griess reagent assay allows the detection of the concentration of NO2− that is proportional to NO.69 A significant inhibition of nitrites was observed when LPS-activated cells were incubated with COPRE-cps and POST-cps at 1000 capsules per cell (Fig. S10C, ESI†), whereas no production of nitrites was detected in resting cells for any of the tested formulations and concentrations (data not shown). Furthermore, the secreted TNF-α by LPS-activated cells was clearly inhibited in the case of the formulations containing CUR (Fig. S10D, ESI†). Interestingly, compared to our previous study on THP-1 derived macrophages, the anti-inflammatory properties associated with CUR were more evident in BV-2 cells with the effect being more attributable to CUR than to the capsule composition itself. Indeed, CPS at both concentrations (i.e., 100 and 1000 capsules per cell) did not induce any significant suppression of the pro-inflammatory cytokine, while COPRE-cps and POST-cps at both concentrations and CONJ-cps at the highest concentration significantly reduced TNF-α (Fig. S10D, ESI†). Therefore, CUR played a decisive role in the anti-inflammatory properties of the capsules, inducing an inhibition in the production of cytokines in a dose-dependent manner. This phenomenon is in accordance with reported studies, where NO and TNF-α produced by activated BV-2 cells were suppressed by CUR in a concentration-dependent manner of both free drug or the one included in delivery systems.64,66
Overall, we could corroborate that our CUR formulations might show promising therapeutic performance in BV-2 cells in the presence of a higher concentration of the drug. However, as our formulations are on the micrometer scale, they may not reach the CNS through systemic administration due to the presence of biological barriers, such as the blood–brain barrier. As an alternative, they could be considered for local administration, such as intrathecal or intraparenchymal. Thus, we explored the interaction of microglia with the LbL-capsules, and the capability of the cells to uptake them. We demonstrated that LPS-activated BV-2 cells were able to uptake the microcapsules compared to the resting cells (Fig. 7 and 8, respectively). Due to the inherent fluorescence of CUR,70 the LbL-capsules were not further labeled with a fluorescent dye, and this allowed us to observe the distribution of the drug inside the capsules and the intracellular release by means of confocal microscopy. Very little uptake was observed by the resting cells, where the capsules were mostly adhering to the cellular membrane (Fig. 7). In contrast, in the case of LPS-activated cells, the confocal images with an orthogonal view showed that the CUR-loaded capsules were engulfed by cells (Fig. 8). This phenomenon could be associated with the increased phagocytic activity of the activated cells into M1 phenotype, which has been demonstrated in the literature.61,62 Although the study of the polarization of microglia is beyond the scope of our work, the demonstrated preferential uptake by classically activated microglia might be very interesting for several neuroinflammatory diseases (e.g., Alzheimer's diseases, Parkinson's diseases, traumatic brain injury, and stroke). In fact, although the M1-like microglia are crucial in eliciting the innate immune response against pathogens, the persistence of the inflammatory stimulus may induce chronic neuroinflammation, where the M1 phenotype exerts its neurotoxic nature.71 Therefore, modulators of the microglia phenotype may represent a potential therapeutic approach for the treatment of several diseases and disorders occurring in the CNS. Interestingly, we observed that when POST-cps were taken up by the LPS-activated cells, an intracellular release was evident (Fig. 8(C) and (F)). Contrarily, in the case of COPRE-cps and CONJ-cps, very low intracellular release was appreciated, and CUR appeared homogeneously localized in the capsules (Fig. 8(B), (E) and (D), (G)). Zoomed confocal images are reported in Fig. S11 of the ESI.† This finding correlates with the release studies showed in Fig. 5, and might justify the diverse in vitro release profiles of CUR between the three different formulations.
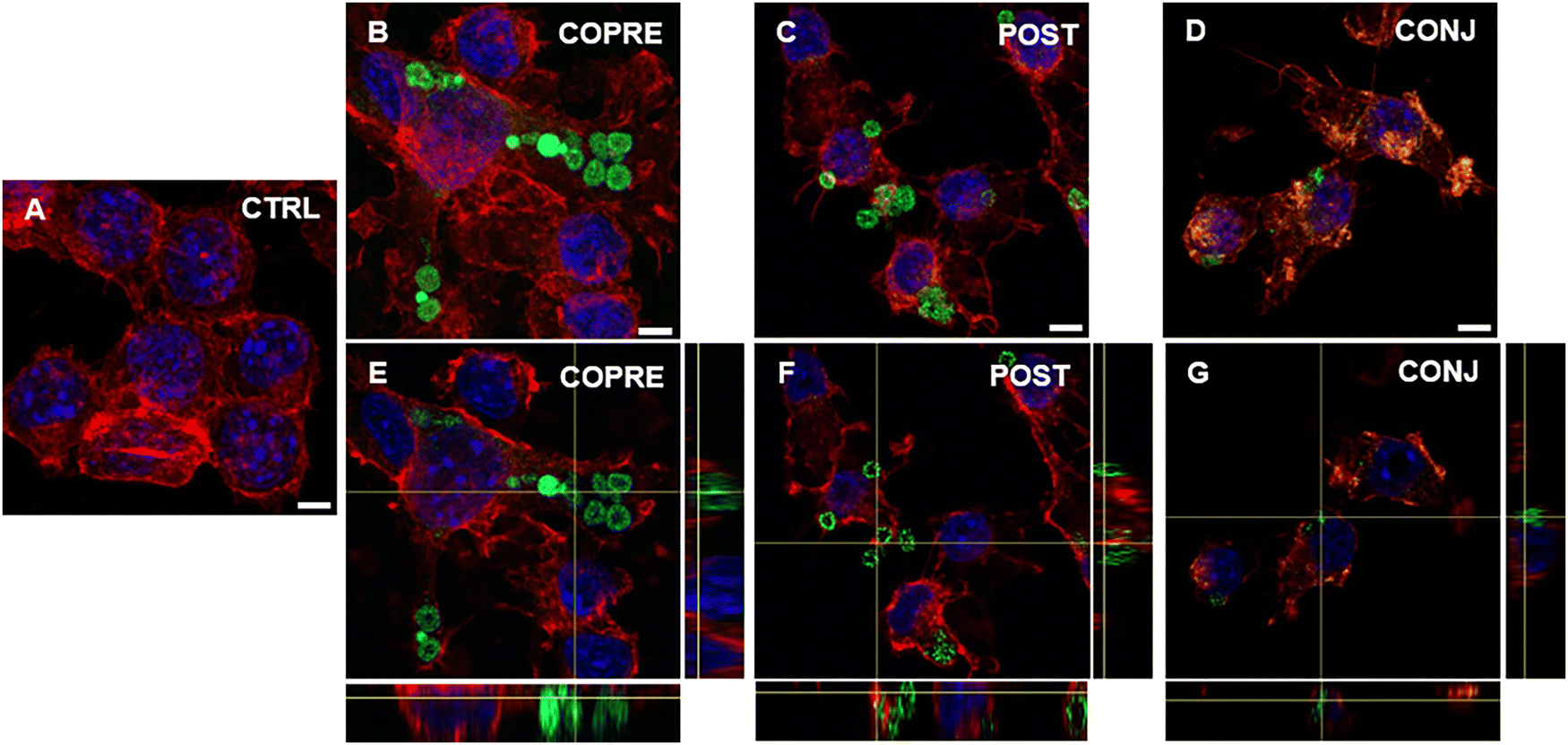 | ||
| Fig. 7 Confocal images showing the uptake of COPRE (B), POST (D), and CONJ (D) capsules by resting BV-2 cells after 24 h of incubation with respect to the control (CTRL) (A). An orthogonal view of the uptake of COPRE (E), POST (F), and CONJ (G) capsules by resting BV-2 cells after 24 h of incubation. The nuclei are stained in blue (DAPI), F-actin is stained in red (tetramethylrhodamine isothiocyanate (TRITC)-phalloidin), and the green is attributed to the presence of curcumin. The scale bar is 5 μm. | ||
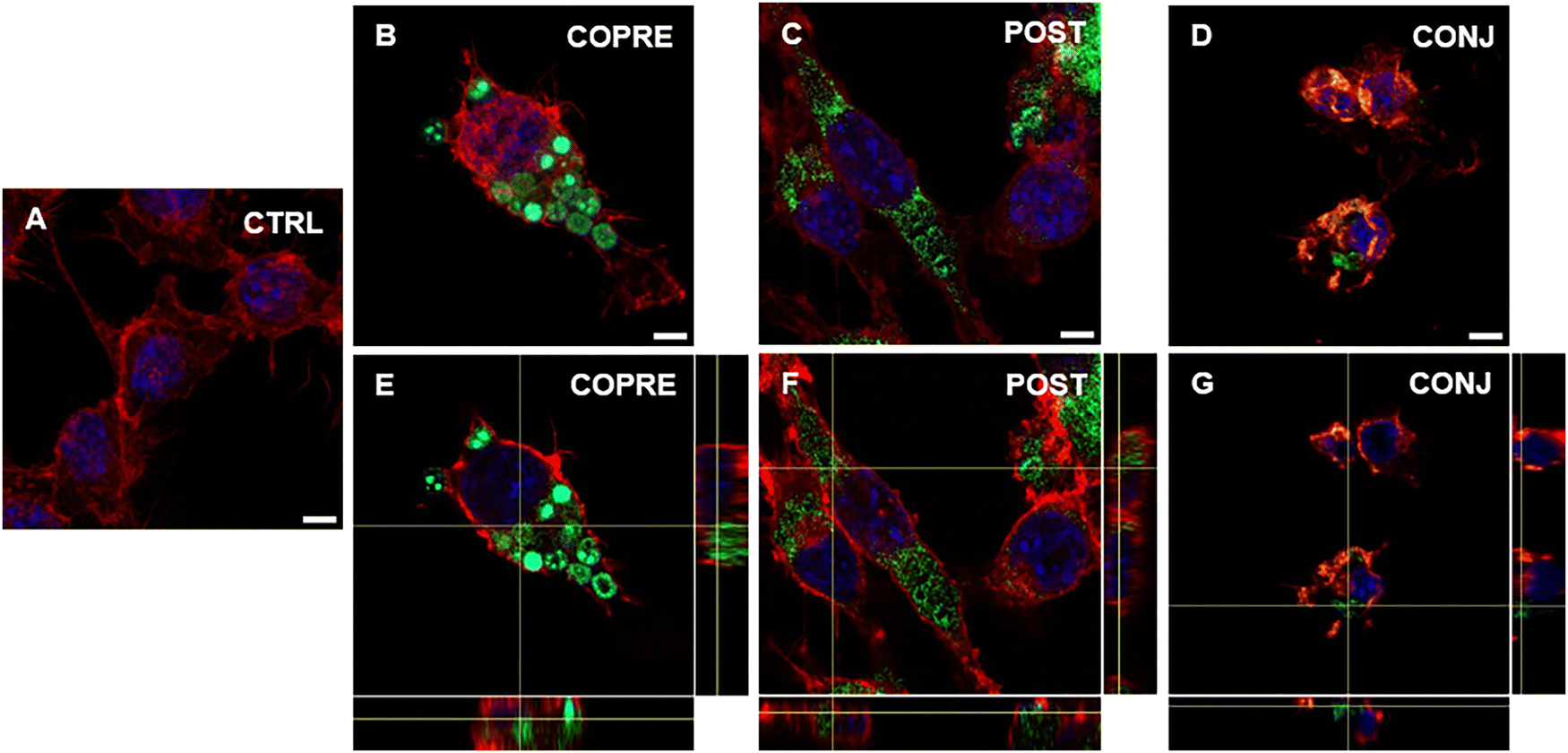 | ||
| Fig. 8 Confocal images showing the uptake of COPRE (B), POST (C), and CONJ (D) capsules by activated BV-2 cells by 20 ng mL−1 LPS after 24 h of incubation with the capsules with respect to the control (CTRL) (A). An orthogonal view of the uptake of COPRE (E), POST (F), and CONJ (G) capsules by activated BV-2 cells after 24 h of incubation. The nuclei are stained in blue (DAPI), F-actin is stained in red (tetramethylrhodamine isothiocyanate (TRITC)-phalloidin), and the green is attributed to the presence of curcumin (CUR). The scale bar is 5 μm. | ||
Therefore, we believe that the facilitated release of CUR into the cells and the downregulation of pro-inflammatory mediators could be very promising to treat pathological neuroinflammation.
4. Conclusions
In this work, we developed biodegradable multilayer microcapsules by using the LbL approach. PLys and PGlu were used as positively and negatively charged building blocks due to their biocompatibility and biodegradability. In particular, we built high engineered carriers, establishing three methodologies to include a poor water-soluble drug (i.e., CUR) in the core by co-precipitation or post-encapsulation or in the multilayer membrane as polymer-conjugate.An initial optimization procedure was carried out for the encapsulation of the free CUR into the CaCO3 microparticles to achieve high encapsulation efficiencies and systems with a spherical-like shape. In the case of the co-precipitation strategy, the ethanol played a crucial role in the morphology of the resulting particles, promoting the formation of a flower-like shape at high volume and a spherical-like shape when the volume used to dissolve CUR was minimized compared to the salt solutions. Furthermore, in the case of the post-encapsulation strategy, the dropwise addition of CUR to the pre-synthesized CaCO3 microparticles pursued high encapsulation efficiencies compared to the direct addition. Interestingly, the two selected approaches proved to be decisive either in terms of the location of the drug in the LbL-capsules or in the subsequent release of CUR, which was accelerated in POST-cps compared to the COPRE-cps.
We also presented an alternative manner to include CUR in our LbL systems, which consisted of increasing the solubility of the drug by synthesizing drug-conjugates and then incorporating them into the multilayer membrane. This approach is very promising in the field of drug delivery as it would allow to include responsive entities in the multilayer membrane, control the amount of the drug loaded in the nanostructure, and have an ideal system for the controlled release of the drug. However, the lack of selectivity in the reaction we performed did not allow us to control the conjugation of CUR, which further exhibits two potential OH groups that could react with PGlu. Therefore, a further improvement of this methodology might be needed for future studies.
Moreover, the study of the release profile of CUR from the three formulations under acidic and neutral conditions and in the presence of proteolytic enzymes revealed that several parameters may influence the release of the drug (e.g., the pH, the ionic strength, the nature of the layers and the drug, or the typology of loading). Noteworthily, the LbL technique offered a valid tool to fabricate drug delivery systems that allowed a controlled and sustained release of the drug, preventing its initial burst release.
Due to the documented pleiotropic properties of CUR as therapeutics, in vitro studies on two different models of inflamed cell lines (i.e., THP-1 derived macrophages and BV-2 cells) were performed to evaluate whether CUR was still bioactive after the loading process, the conjugation, and in the presence of the multilayer membrane. Our CUR-loaded capsules did not induce any detrimental effect regardless of the explored concentration. Notably, although the capsule composition showed an anti-inflammatory effect on THP-1 derived macrophages, the highest concentrations of CUR loaded into the capsules could ameliorate the inflammation by suppressing the release of pro-inflammatory cytokines, such as TNF-α and IL-6. This suggested that the capsules and the conjugation preserved the bioactivity of the drug, which resulted clearer in the treatment of inflamed microglia. Furthermore, the uptake studies of the capsules by LPS-activated microglia compared to the corresponding resting cells highlighted the tendency of the M1-like phenotype to phagocytize the capsules when an inflammation occurs. In particular, the intracellular release of CUR confirmed the different release profiles of CUR obtained in vitro. In fact, the uptake studies via confocal microscopy indicated that the release of CUR was facilitated in POST-cps, probably due to a mechanism of diffusion, whereas, in the other two systems, CUR looked more localized inside the capsules.
Overall, regardless of the methodology applied to include the hydrophobic drug into the biodegradable multilayer carriers, our LbL formulations could have a great potential as modulators of the microglia phenotype towards the reduction of the acute neuroinflammation. Furthermore, the possibility to apply the LbL technique on nanoparticles (e.g., CaCO3 nanoparticles, silica nanoparticles, and nanocrystals) and functionalize the external layer of the multilayer membrane may lead to exploration of alternative administration routes for specific neuronal diseases. Hence, we believe that this study could pave the way to extended investigations on novel drug delivery systems in both the micro- and nano-meter scales to treat the inflammation associated with different diseases.
Author contributions
M. A. M.: conceptualization, investigation, methodology, formal analysis, and writing – original draft. S. M.-S.: investigation, formal analysis, writing – original draft, and writing – review and editing. A. B.: writing – review and editing, supervision, and funding acquisition. M. C.: conceptualization, writing – review and editing, supervision, and funding acquisition. A. L.: conceptualization, writing – review and editing, supervision, and funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The authors acknowledge the financial support from the Basque Government (projects 2023333010, 2023333023, PIBA2023-1-0043, IT-1766-22, IKUR Strategy), the University of the Basque Country (projects COLLAB22/05 and GIU21/033), the IKERBASQUE-Basque Foundation for Science, the Ministry of Science and Innovation of the Government of Spain (grant PID2022-142739OB-I00 funded by MICIU/AEI/10.13039/501100011033 and by FEDER, UE; 'María de Maeztu' Programme for Center of Excellence in R&D, grant CEX2023-001303-M funded by MICIU/AEI/10.13039/501100011033; PID2022-142128NB-I00 funded by MCIN/AEI/10.13039/501100011033/and by the “European Union NextGenerationEU/PRTR”; RYC2018-025923-I from RyC program – MCIN/AEI/10.13039/501100011033 and FSE “invierte en tu futuro”). SGIker technical services (UPV/EHU) are gratefully acknowledged for the support in ATR-FTIR, confocal microscopy, XRD, and SEM. Open Access funding provided by University of the Basque Country.References
- S. K. Prajapati, A. Jain, A. Jain and S. Jain, Eur. Polym. J., 2019, 120, 109191 CrossRef.
- H. O. Alsaab, F. D. Alharbi, A. S. Alhibs, N. B. Alanazi, B. Y. Alshehri, M. A. Saleh, F. S. Alshehri, M. A. Algarni, T. Almugaiteeb, M. N. Uddin and R. M. Alzhrani, Pharmaceutics, 2022, 14, 2728 CrossRef.
- T. Melnyk, S. Đorđević, I. Conejos-Sánchez and M. J. Vicent, Adv. Drug Delivery Rev., 2020, 160, 136–169 CrossRef.
- I. Ekladious, Y. L. Colson and M. W. Grinstaff, Nat. Rev. Drug Discovery, 2019, 18, 273–294 CrossRef PubMed.
- T. Ramasamy, Z. S. Haidar, T. H. Tran, J. Y. Choi, J. H. Jeong, B. S. Shin, H. G. Choi, C. S. Yong and J. O. Kim, Acta Biomater., 2014, 10, 5116–5127 CrossRef PubMed.
- T. Ramasamy, H. B. Ruttala, N. Chitrapriya, B. K. Poudal, J. Y. Choi, S. T. Kim, Y. S. Youn, S. K. Ku, H. G. Choi, C. S. Yong and J. O. Kim, Acta Biomater., 2017, 48, 131–143 CrossRef.
- E. Marin, N. Tiwari, M. Calderón, J. R. Sarasua and A. Larrañaga, ACS Appl. Mater. Interfaces, 2021, 13, 18511–18524 CrossRef.
- S. W. Morton, Z. Poon and P. T. Hammond, Biomaterials, 2013, 34, 5328–5335 CrossRef CAS.
- S. Roy, N. M. Elbaz, W. J. Parak and N. Feliu, ACS Appl. Bio Mater., 2019, 2, 3245–3256 CrossRef CAS PubMed.
- J. Pang, Z. Gao, H. Tan, X. Mao, J. Xu, J. Kong and X. Hu, Front. Chem., 2019, 7, 620 CrossRef CAS PubMed.
- G. B. Sukhorukov, A. L. Rogach, M. Garstka, S. Springer, W. J. Parak, A. Muñoz-Javier, O. Kreft, A. G. Skirtach, A. S. Susha, Y. Ramaye, R. Palankar and M. Winterhalter, Small, 2007, 3, 944–955 CrossRef CAS.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS.
- C. Peng, Q. Zhao and C. Gao, Colloids Surf., A, 2010, 353, 132–139 CrossRef CAS.
- M. A. Motta, L. Mulko, E. Marin, A. Larrañaga and M. Calderón, Adv. Colloid Interface Sci., 2024, 331, 103248 CrossRef.
- D. V. Volodkin, N. I. Larionova and G. B. Sukhorukov, Biomacromolecules, 2004, 5, 1962–1972 CrossRef PubMed.
- D. Sp and R. Rmg, J. Nanomed. Biother. Discov., 2017, 7, 1000150 Search PubMed.
- Z. She, C. Wang, J. Li, G. B. Sukhorukov and M. N. Antipina, Biomacromolecules, 2012, 13, 2174–2180 CrossRef PubMed.
- P. Zhao, Y. Tian, J. You, X. Hu and Y. Liu, Bioengineering, 2022, 9, 691 CrossRef PubMed.
- S. K. Kim, M. B. Foote and L. Huang, Cancer Lett., 2013, 334, 311–318 CrossRef CAS PubMed.
- E. Larrañeta, S. Stewart, M. Ervine, R. Al-Kasasbeh and R. F. Donnelly, J. Funct. Biomater., 2018, 9, 13 CrossRef.
- S. Ghosh and M. Banerjee, Sci. Rep., 2021, 11, 7030 CrossRef CAS PubMed.
- M. Kolter, M. Wittmann, M. Köll-Weber and R. Süss, Eur. J. Pharm. Biopharm., 2019, 140, 20–28 CrossRef CAS.
- M. Ghezzi, S. Pescina, C. Padula, P. Santi, E. Del Favero, L. Cantù and S. Nicoli, J. Controlled Release, 2021, 332, 312–336 Search PubMed.
- V. Mohanta, G. Madras and S. Patil, ACS Appl. Mater. Interfaces, 2014, 6, 20093–20101 Search PubMed.
- N. A. Feoktistova, A. S. Vikulina, N. G. Balabushevich, A. G. Skirtach and D. Volodkin, Mater. Des., 2019, 185, 108223 CrossRef.
- A. M. Ferreira, A. S. Vikulina and D. Volodkin, J. Controlled Release, 2020, 328, 470–489 CrossRef CAS.
- D. B. Trushina, T. V. Bukreeva, M. V. Kovalchuk and M. N. Antipina, Mater. Sci. Eng., C, 2014, 45, 644–658 Search PubMed.
- A. S. Vikulina, N. A. Feoktistova, N. G. Balabushevich, A. G. Skirtach and D. Volodkin, Phys. Chem. Chem. Phys., 2018, 20, 8822–8831 RSC.
- N. G. Balabushevich, A. V. Lopez De Guerenu, N. A. Feoktistova, A. G. Skirtach and D. Volodkin, Macromol. Biosci., 2016, 16, 95–105 CrossRef CAS.
- E. Abdollahi, A. A. Momtazi, T. P. Johnston and A. Sahebkar, J. Cell. Physiol., 2018, 233, 830–848 CrossRef CAS PubMed.
- A. Larrañaga, C. Bello-Álvarez and E. Lizundia, Biomacromolecules, 2023, 24, 5737–5748 CrossRef.
- D. B. Trushina, T. N. Borodina, S. Belyakov and M. N. Antipina, Mater. Today Adv., 2022, 14, 100214 CrossRef CAS PubMed.
- G. B. Sukhorukov, D. V. Volodkin, A. M. Günther, A. I. Petrov, D. B. Shenoy and H. Möhwald, J. Mater. Chem., 2004, 14, 2073–2081 RSC.
- D. V. Volodkin, A. I. Petrov, M. Prevot and G. B. Sukhorukov, Langmuir, 2004, 20, 3398–3406 CrossRef CAS PubMed.
- B. V. Parakhonskiy, A. M. Yashchenok, S. Donatan, D. V. Volodkin, F. Tessarolo, R. Antolini, H. Möhwald and A. G. Skirtach, ChemPhysChem, 2014, 15, 2817–2822 CrossRef CAS PubMed.
- S. Donatan, A. Yashchenok, N. Khan, B. Parakhonskiy, M. Cocquyt, B. El Pinchasik, D. Khalenkow, H. Möhwald, M. Konrad and A. Skirtach, ACS Appl. Mater. Interfaces, 2016, 8, 14284–14292 CrossRef CAS.
- F. Manoli and E. Dalas, J. Cryst. Growth, 2000, 218, 359–364 CrossRef CAS.
- L. Zhang, L. H. Yue, F. Wang and Q. Wang, J. Phys. Chem. B, 2008, 112, 10668–10674 CrossRef CAS PubMed.
- M. Hegde, S. Girisa, B. BharathwajChetty, R. Vishwa and A. B. Kunnumakkara, ACS Omega, 2023, 8, 10713–10746 CrossRef CAS.
- S. Pillarisetti, S. Maya, S. Sathianarayanan and R. Jayakumar, Colloids Surf., B, 2017, 159, 809–819 CrossRef CAS.
- S. B. Wan, H. Yang, Z. Zhou, Q. C. Cui, D. Chen, J. Kanwar, I. Mohammad, Q. P. Dou and T. H. Chan, Int. J. Mol. Med., 2010, 26, 447–455 CAS.
- G. Córdoba-David, A. Duro-Castano, R. C. Castelo-Branco, C. González-Guerrero, P. Cannata, A. B. Sanz, M. J. Vicent, A. Ortiz and A. M. Ramos, Sci. Rep., 2020, 10, 2056 CrossRef.
- A. Duro-Castano, C. Borrás, V. Herranz-Pérez, M. C. Blanco-Gandía, I. Conejos-Sánchez, A. Armiñán, C. Mas-Bargues, M. Inglés, J. Miñarro, M. Rodríguez-Arias, J. M. García-Verdugo, J. Viña and M. J. Vicent, Sci. Adv., 2021, 7, eabf9180 CrossRef CAS PubMed.
- A. V. Dubrovskii, A. L. Kim, E. V. Musin and S. A. Tikhonenko, Sci. Rep., 2022, 12, 4032 CrossRef CAS PubMed.
- P. Kittitheeranun, W. Sajomsang, S. Phanpee, A. Treetong, T. Wutikhun, K. Suktham, S. Puttipipatkhachorn and U. R. Ruktanonchai, Int. J. Pharm., 2015, 492, 92–102 CrossRef CAS PubMed.
- N. M. Elbaz, A. Owen, S. Rannard and T. O. McDonald, Int. J. Pharm., 2020, 574, 118866 CrossRef CAS.
- R. Medzhitov, Nature, 2008, 454, 428–435 CrossRef CAS.
- E. R. Brannon, M. V. Guevara, N. J. Pacifici, J. K. Lee, J. S. Lewis and O. Eniola-Adefeso, Nat. Rev. Mater., 2022, 7, 796–813 CrossRef CAS.
- U. Ikoba, H. Peng, H. Li, C. Miller, C. Yu and Q. Wang, Nanoscale, 2015, 7, 4291–4305 RSC.
- W. Chanput, J. J. Mes and H. J. Wichers, Int. Immunopharmacol., 2014, 23, 37–45 CrossRef CAS PubMed.
- T. Liu, T. Huang, J. Li, A. Li, C. Li, X. Huang, D. Li, S. Wang and M. Liang, PLoS One, 2023, 18, e0286056 CrossRef CAS PubMed.
- Y. K. Kim, J. H. Hwang and H. T. Lee, Innate Immun., 2022, 28, 122–129 CrossRef CAS.
- C. M. Mulvey, L. M. Breckels, O. M. Crook, D. J. Sanders, A. L. R. Ribeiro, A. Geladaki, A. Christoforou, N. K. Britovšek, T. Hurrell, M. J. Deery, L. Gatto, A. M. Smith and K. S. Lilley, Nat. Commun., 2021, 12, 5773 Search PubMed.
- O. Sharif, V. N. Bolshakov, S. Raines, P. Newham and N. D. Perkins, BMC Immunol., 2007, 8, 1 CrossRef.
- E. Hadadi, B. Zhang, K. Baidzajevas, N. Yusof, K. J. Puan, S. M. Ong, W. H. Yeap, O. Rotzschke, E. Kiss-Toth, H. Wilson and S. C. Wong, Sci. Rep., 2016, 6, 39035 CrossRef CAS PubMed.
- C. Xu, R. Zeng, Q. Chen, L. Du, J. Tong, Y. He, H. Xu and M. Li, Br. J. Dermatol., 2019, 181, 1320–1322 CrossRef CAS.
- C. Wang, Z. Han, Y. Wu, X. Lu, X. Tang, J. Xiao and N. Li, Food Chem. Toxicol., 2021, 151, 112123 CrossRef CAS PubMed.
- C. Liu, X. Yan, Y. Zhang, M. Yang, Y. Ma, Y. Zhang, Q. Xu, K. Tu and M. Zhang, J. Nanobiotechnol., 2022, 20, 206 CrossRef CAS.
- F. Ma, F. Liu, L. Ding, M. You, H. Yue, Y. Zhou and Y. Hou, Pharm. Biol., 2017, 55, 1263–1273 CrossRef CAS PubMed.
- A. Tawbeh, Q. Raas, M. Tahri-Joutey, C. Keime, R. Kaiser, D. Trompier, B. Nasser, E. Bellanger, M. Dessard, Y. Hamon, A. Benani, F. Di Cara, T. Cunha Alves, J. Berger, I. Weinhofer, S. Mandard, M. Cherkaoui-Malki, P. Andreoletti, C. Gondcaille and S. Savary, Front. Mol. Neurosci., 2023, 16, 1299314 CrossRef CAS PubMed.
- S. Li, I. Wernersbach, G. S. Harms and M. K. E. Schäfer, Front. Immunol., 2022, 13, 945485 CrossRef CAS PubMed.
- D. J. Loane and A. Kumar, Exp. Neurol., 2016, 275, 316–327 CrossRef CAS PubMed.
- Z. Yang, T. Zhao, Y. Zou, J. H. Zhang and H. Feng, Immunol. Lett., 2014, 160, 89–95 CrossRef CAS.
- P. Ganesan, B. Kim, P. Ramalaingam, G. Karthivashan, V. Revuri, S. Park, J. S. Kim, Y. T. Ko and D. K. Choi, Molecules, 2019, 24, 1170 CrossRef.
- F. Gao, J. Lei, Z. Zhang, Y. Yang and H. You, RSC Adv., 2019, 9, 38397–38406 RSC.
- C. Y. Jin, J. D. Lee, C. Park, Y. H. Choi and G. Y. Kim, Acta Pharmacol. Sin., 2007, 28, 1645–1651 CrossRef CAS.
- M. A. Lynch, Mol. Neurobiol., 2009, 40, 139–156 CrossRef CAS PubMed.
- N. Cho, E. H. Moon, H. W. Kim, J. Hong, J. A. Beutler and S. H. Sung, Molecules, 2016, 21, 459 CrossRef PubMed.
- N. S. Bryan and M. B. Grisham, Free Radicals Biol. Med., 2007, 43, 645–657 CrossRef CAS.
- N. A. N. Hanafy, Int. J. Biol. Macromol., 2021, 182, 1981–1993 CrossRef CAS PubMed.
- G. J. Song and K. Suk, Front. Aging Neurosci., 2017, 9, 139 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01906g |
| This journal is © The Royal Society of Chemistry 2025 |