
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLead-free lanthanide-based Cs3LnCl6 metal halides
Yuanzhe
Ding
ab,
Dongjie
Liu
a,
Peipei
Dang
*a,
Guogang
Li
 *c and
Jun
Lin
*ab
*c and
Jun
Lin
*ab
aState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: ppdang@ciac.ac.cn; jlin@ciac.ac.cn
bSchool of Applied Chemistry and Engineering, University of Science and Technology of China, Hefei 230026, P. R. China
cFaculty of Materials Science and Chemistry, China University of Geosciences, Wuhan 430074, P. R. China. E-mail: ggli@cug.edu.cn
First published on 9th December 2024
Abstract
All-inorganic lead-free luminescent metal halides have attracted considerable interest for their special optical properties in various optoelectronic applications. Among them, lanthanide-based Cs3LnCl6 metal halides, with low phonon energies and suitable crystallographic features, have been in the spotlight due to their appealing optical properties. However, it remains challenging for us to know how to simply and efficiently synthesize lanthanide-based Cs3LnCl6 metal halides, and how to optimize their low absorption and emission efficiencies owing to the parity-forbidden f–f transitions. In this minireview, we summarized several synthesis approaches towards both Cs3LnCl6 polycrystals and nanocrystals. The association of the crystal/electronic structure, optical properties and applications is discussed in three sections: self-trapped emission, down-conversion luminescence, and upconversion luminescence. We provide not only the synthesis strategies and recent progress in the luminescence properties of Cs3LnCl6, but also their promising future as alternatives to their toxic and poorly stable lead-based counterparts.
1. Introduction
In recent years, a large number of studies have shown that replacing lead with non-toxic elements can solve the issue of lead toxicity in metal halide perovskites (MHPs) with excellent photoelectric properties.1–3 Based on the similarities in ion radius, charge, chemical reactivity, and structural tolerance of elements, lead is replaced to form lead-free metal halide perovskite (LFMHP) materials.4,5 The LFMHPs are categorized into equivalent substitution and heterovalent substitution of Pb2+. Equivalent substitution involves replacing Pb2+ with a bivalent cation (Sn2+, Eu2+, and Mn2+) to form AMX3 type perovskites.6–9 Heterovalent substitution mainly includes the following types. First, replacing two Pb2+ lattice positions with a univalent cation (Ag+, Na+, and K+) and a trivalent cation (In3+, Bi3+, and Ln3+), resulting in a double perovskite structure with the structural formula A2B(I)B(III)X6.10–12 Second, substituting two Pb2+ lattice positions with a tetravalent cation (Sn4+, Zr4+, and Hf4+) and a lattice vacancy, leading to a vacancy ordered double perovskite structure with the structural formula A2B(IV)X6.13–15 Third, replacing three Pb2+ lattice positions with two trivalent cations (Bi3+ and Sb3+) and one vacancy, forming a triple perovskite structure with the structural formula A3B(III)2X9.16,17 Fourth, substituting four Pb2+ lattice positions with two trivalent cations (Bi3+ and Sb3+) and one divalent cation (Mn2+, Zn2+, and Ca2+) results in a quadruple perovskite structure, also known as a layered double perovskite structure with the structural formula A4B(II)B(III)2X12.18–21 Among them, lanthanide doping and lanthanide-based halide perovskite luminescent materials have attracted extensive research interest due to their stable characteristic emission.22–25 However, there are a number of difficulties that hinder their further development. First, the optical properties of lanthanide-doped nanocrystals (NCs) and polycrystals (PCs) are very sensitive to the ratios of lanthanide dopants, sensitizers and substrates, and figuring out the optimal ratios is time-consuming and laborious.26,27 Second, the emission of lanthanide ions is usually accompanied by bright broadband self-trapped exciton (STE) emission or sensitizer emission,28–30 which may overlap with the luminescence of the lanthanide ions, thus masking the advantage of the narrowband emission of the lanthanide ions.31,32 STEs refer to excited states in insulators and semiconductors where an electron and a hole become localized in the same region due to their mutual Coulomb attraction. This localization leads to unique optical properties, such as enhanced luminescence efficiency.10 Furthermore, the introduction of lanthanide ions as dopants leads to concentration bursts and increased defects in perovskite NCs. In the field of atomic physics, f–f transitions are the movements of electrons between the f subshells of heavy elements known as rare earth elements. The Laporte rule states that transitions involving orbitals with the same angular momentum quantum number (l) are not allowed, whereas transitions between orbitals of different parity are permitted. Yet, for specific rare earth elements, including lanthanides and actinides, the f–f transitions are prohibited by symmetry because the f orbitals are centrosymmetric. Finally, the luminescence of doped lanthanide ions depends heavily on the energy transfer (ET) from the host to the lanthanide ions, and the energy level mismatch between the host and the lanthanide ions leads to a low ET efficiency, which affects the overall luminescence performance. Therefore, it is crucial to simplify the chemical composition and reduce the interference of other emissions. It is worth mentioning that all-inorganic zero-dimensional (0D) lanthanide-based Cs3LnCl6 (Ln = Sc, Y, La, etc.) halide NCs and SCs, with excellent luminescence performance, have been reported recently.33–35 They have low phonon energies and suitable crystallographic lattices for potential applications in light-emitting diodes (LEDs),36–38 X-ray scintillators,39–41 photodetectors42 and chemical sensors.43–45 Their crystal and electronic structure can be easily adjusted without changing the chemical elements. 0D lanthanide halide NCs and PCs are some of the best research objects to discuss and analyze the structure–optical property relationship. However, the low-temperature wet synthesis of Cs3LnCl6 crystals remains challenging due to the tendency of rare-earth halides to crystallize into hydrated phases in solution.27,46 In addition, the luminescence efficiency and stability of lanthanide halides are still relatively poor and need to be improved.47–49 Therefore, it is significant and challenging to find an efficient and stable lanthanide halide luminescent material, to elucidate its luminescence mechanism and excited state dynamics, and to apply it in the field of optoelectronics.50,51In this review, we aim to summarize the recent progress of Cs3LnCl6 (Ln: Sc, Y, La, Ce, Eu, Gd, Tb, Ho, Er, Tm, Yb, Lu) luminescent materials and discuss their synthesis, characterization, and structural and optical properties. More specifically, we summarize the effects of Sb3+ alloying, small organic molecule modification, and lanthanide ion doping on the luminescence properties of Cs3LnCl6. These strategies regulate luminescence of Cs3LnCl6, achieving highly efficient lanthanide emission and multi-mode luminescence based on upconversion and down-conversion luminescence. Finally, the current challenges and future opportunities for the research of Cs3LnCl6 are proposed, and an outlook on the prospects for its further applications is provided.
2. Synthesis methods of Cs3LnCl6
The control of the synthesis of either PCs or NCs plays an important role in the design optimization of the luminescent properties of the material, which certainly applies to Cs3LnCl6.52–54 In this section, we summarize several synthesis methods for Cs3LnCl6 polycrystals and nanocrystals.2.1. Cs3LnCl6 polycrystals
The solid-state reaction route is usually reported to synthesize Cs3LnCl6 PCs. As shown in Fig. 1a, a starting material consisting of stoichiometric amounts of CsCl and LnCl3·6H2O was ground in an agate mortar under an inert atmosphere. The mixture was then placed in an alumina crucible and compressed into granular form using a hydraulic press. Finally, the samples were fired at 450 °C for 24 h. Cs3GdCl6 and Cs3LuCl6 were synthesized by solid-state reaction, respectively.50,55 The solid-state reaction method is characterized by its simplicity in operation and the controllability of the synthesis process; however, it is associated with high energy consumption and the requirement of high temperatures. Recently, Im et al. reported an innovative mechanochemical approach for the synthesis of Cs3LnCl6. This method involves the continuous grinding of precursors, which reduces their particle size and exposes reactive sites on the material surface, thereby facilitating the reaction of the precursors to yield the desired products. This mechanochemical synthesis is characterized by its straightforward reaction conditions and a relatively low generation of by-products, which simplifies the purification process and enhances the overall efficiency of the synthesis. Compared to solution-phase methods, this approach allows for a larger reaction scale, which is advantageous for scaling up the production process. The mechanochemical method not only reduces the use of solvents but also minimizes environmental impact, aligning with the principles of green chemistry.56,57 Cs3LnCl6 single crystals can be prepared through a facile solvothermal method, which involves the direct reaction of CsCl and LnCl3·6H2O or Ln2O3 in a concentrated HCl solution. The requisite quantities of CsCl and LnCl3·6H2O or Ln2O3, in accordance with the specified stoichiometric ratio and with the addition of an appropriate amount of concentrated HCl, were loaded into a polytetrafluoroethylene (PTFE) container. The aforementioned PTFE container was then sealed within a stainless-steel Parr autoclave, which was heated to a temperature of 180 °C for a period of 10 hours. Upon cooling to room temperature, crystals were observed to precipitate. This synthesis method is simple and at low temperature, but it produces unknown impurity phases, so the synthesis process of the promising Cs3LnCl6 needs to be optimized.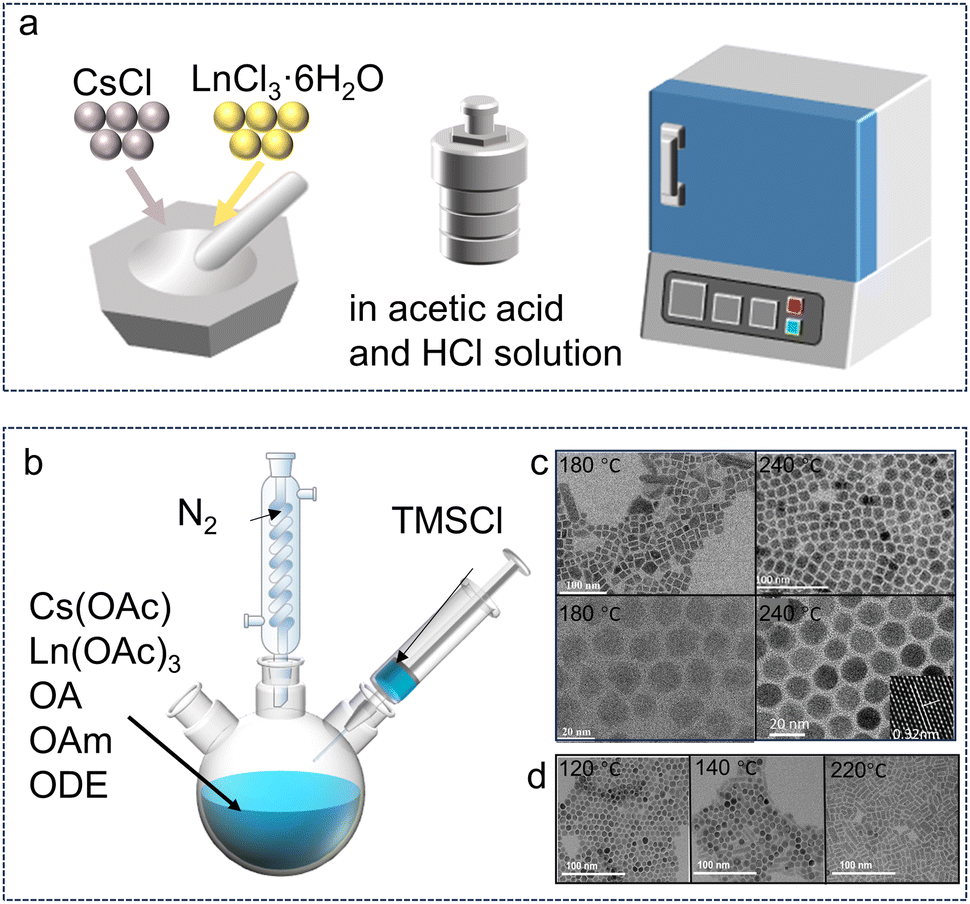 | ||
| Fig. 1 Synthesis methods for Cs3LnCl6 polycrystals and nanocrystals. (a) Synthesis of Cs3LnCl6 single crystals or microcrystals. (b) Hot-injection synthesis using metal chlorides as well as oleic acid, oleylamine, and 1-octadecene. (c) Cs3TbCl6 with different synthesis temperatures (180–240 °C). Reproduced with permission from ref. 58. Copyright 2023, Wiley. Reproduced with permission from ref. 59. Copyright 2024, Wiley. Reproduced with permission from ref. 60. Copyright 2022, Wiley. (d) TEM images of Cs3ScCl6:Sb3+ with the different reaction temperatures (120, 140, and 220 °C). Reproduced with permission from ref. 61. Copyright 2023, Wiley. | ||
2.2. Cs3LnCl6 nanocrystals
The decrease in crystal size leads to a large number of attractive properties of the material, which are also exhibited by Cs3LnCl6.62 They were prepared by thermal injection and solvothermal methods. Through different synthesis processes and conditions, different Cs3LnCl6 crystal structures can be prepared and the product can be rapidly obtained at low temperature. Liu et al. synthesized Cs3TbCl6 quantum dots using a thermal injection method, in which Tb atoms occupy the B site in the cube and are bonded to Cl atoms.60 Cs3TbCl6 exhibit a cubic crystal structure with the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. As shown in Fig. 1b, Cs3TbCl6 synthesized using a modified thermal injection method adopts a monoclinic crystal structure with the C2/c space group.25 In both structures, each Tb atom is coordinated with six neighboring Cl atoms to form an isolated [TbCl6]3− octahedron.60 The Cs atoms are filled in the interlayer formed by the [TbCl6]3− octahedral structure. The prepared cesium precursors are usually injected into oleic acid and TbCl3 solutions at high synthesis temperatures (180–240 °C) and under a nitrogen atmosphere (Fig. 1c).
m space group. As shown in Fig. 1b, Cs3TbCl6 synthesized using a modified thermal injection method adopts a monoclinic crystal structure with the C2/c space group.25 In both structures, each Tb atom is coordinated with six neighboring Cl atoms to form an isolated [TbCl6]3− octahedron.60 The Cs atoms are filled in the interlayer formed by the [TbCl6]3− octahedral structure. The prepared cesium precursors are usually injected into oleic acid and TbCl3 solutions at high synthesis temperatures (180–240 °C) and under a nitrogen atmosphere (Fig. 1c).
The injection temperature plays a crucial role in obtaining pure phase materials and determining their luminescent properties. For example, the phase stability of lead halide chalcogenides can be enhanced by simply changing the reaction temperature. The reaction temperature for the synthesis of Cs3ScCl6:Sb3+ NCs was thus determined. When the reaction temperature increased from 120 to 220 °C, the NCs changed from spherical to cylindrical rods as shown in Fig. 1d.61 Through this thermal injection route, the synthesis parameters, including temperature, precursor, ligand, and vacuum or not, can be easily adjusted to produce different photoluminescence (PL) properties.
During the heating process, the amount of oleylamine (OAm) and oleic acid (OA) in the reaction had a significant effect on the crystal structure and size distribution of the products. Taking Cs3YbCl6 as an example, monoclinic Cs3YbCl6 NCs with a size larger than several hundred nanometers were formed when the amount of OA was more than 2 mL, suggesting that OA induces the growth or agglomeration of Cs3YbCl6 NCs. In contrast, when the relative content of OAm is larger than that of OA, the byproducts form colloidal CsCl NCs with a size of ∼6 nm. The relative content of CsCl NCs in the final product increases with the relative volume amount of OAm, indicating that OAm promotes the formation of colloidal CsCl NCs. The size and surface properties of these surfactant-covered CsCl NCs were similar to those of Cs3LnCl6 NCs, which made it difficult to separate these NCs during purification. Therefore, optimization of the reaction is crucial for the selective synthesis of phase-pure Cs3LnCl6 NCs. The reaction temperature is also a key factor in controlling its size and phase purity. For example, large polydisperse Cs3YbCl6 NCs with nanoflake impurities were formed when the reaction temperature was higher than 300 °C, whereas CsCl NCs were formed as by-products with Cs3YbCl6 NCs when it was lower than 230 °C.63
3. Crystal/electronic structure of Cs3LnCl6
Cs3LnCl6 has suitable crystal positions and is an efficient host material with good luminescence properties. The lead-free zero-dimensional (0D) Cs3LnCl6 mostly belongs to the monoclinic C2/c space group. Each Ln atom is coordinated to six neighboring Cl atoms to form an isolated [LnCl6]3− octahedron (Fig. 2a). Three Cs atoms occupy the interstices of the structure, and each Cl atom shares a corner with the other octahedra. In addition, Cs3LnCl6 has two distinct [LnCl6]3− octahedra with different bond lengths, as shown in Fig. 2b. Taking Cs3YbCl6 as an example, its crystal structure, bond lengths, and bond angles were calculated by Lee et al.63 Yb1 and Yb2 are each coordinated to six adjacent Cl atoms to form distorted octahedra with Yb–Cl bond lengths ranging from 2.548–2.881 Å (Fig. 2c). The distance between the Yb centers ranges from 7.374 to 7.879 Å, which is more than twice as long as the Yb–Cl bond. In addition, the closest Cl–Cl distance between neighboring octahedra is 3.556 Å, which is much longer than the covalent bond distance of the Cl2 molecule. Thus, the [YbCl6]3− octahedra are spatially isolated from each other within the Cs3YbCl6 NCs, forming a 0D metal halide structure. To understand the electronic structure of Cs3LnCl6, their partial charge densities, orbital projected state densities, and electronic energy band structures have been studied. Han et al. reported the electronic structures of Cs3TbCl6 and Rb3TbCl6, giving the charge of the Cl 3p orbital and the charges of the Tb 5d and 4f orbitals.25 The former is mainly involved in the valence band states and are distributed at the edges of the [TbCl6]3− octahedron (Fig. 2d–f). For the latter, the 5d orbitals mainly contribute to the conduction band state and are distributed around the [TbCl6]3− octahedron, and the 4f orbitals are distributed around the Tb atoms in [TbCl6]3− between the band gaps. As shown in Fig. 2g and h, the effect of Cs and Rb cations on the frontier orbitals is negligible.61,64,65 The direct bandgap of both Cs3TbCl6 and Rb3TbCl6 at the Γ-point is 5.1 eV. In addition, the Cs3LnCl6 also exhibit a cubic crystal structure with an octahedral [LnCl6]3−. For example, Liu et al. reported different Cs3TbCl6 with a cubic crystal structure with the Fmm space group (Fig. 2i). The Tb atoms occupy the B sites interacting with the Cs atoms.60 For this cubic Cs3TbCl6, the valence band mainly consists of the d and f orbitals of Tb and the p orbitals of Cl by first-principles density functional theory (DFT) calculations, and the band gap of Cs3TbCl6 was calculated to be 3.21 eV (Fig. 2j and k).
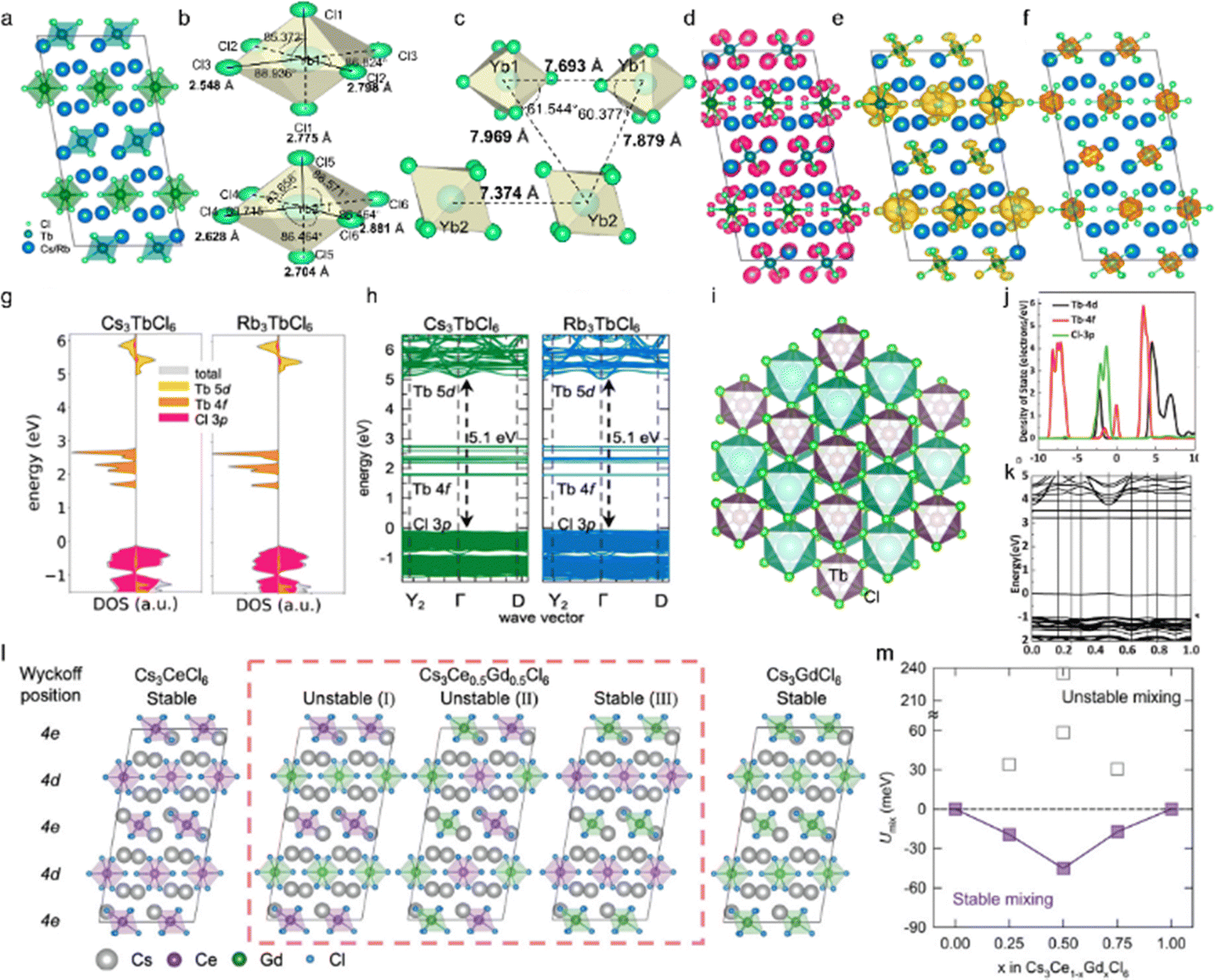 | ||
| Fig. 2 Structural and morphological properties of Cs3LnCl6 metal halides. (a) Crystal structure of 0D Cs3TbCl6. Reproduced with permission from ref. 25. Copyright 2023, American Chemical Society. (b) and (c) Distinct crystallographic sites of Yb with sixfold coordination. (d)–(f) Partial charge density of Cl 3p, Tb 5d, and 4f orbitals in Cs3TbCl6, respectively. (g) and (h) Density of states and calculated electronic band structures of Cs3TbCl6 and Rb3bCl6, respectively. Reproduced with permission from ref. 25. Copyright 2023, American Chemical Society. (i) The crystalline structure of Cs3TbCl6, Cs3TbCl6 and Rb3TbCl6. (j) and (k) The first-principles calculations (PDOS) of Cs3TbCl6 and the electronic band structure obtained by theoretical calculation. Reproduced with permission from ref. 60. Copyright 2022, Wiley. (l) The optimized unit cells of Cs3Ce1−xGdxCl6 for x are 0, 0.5 (with red dashed box), and 1, where different Wyckoff positions for Ce/Gd sites are notated. (m) Internal energy of mixing as a function of the alloying concentration of Gd3+ ions in Cs3Ce1−xGdxCl6. Reproduced with permission from ref. 66. Copyright 2024, Wiley. | ||
By partially replacing Ln(1)3+ with Ln(2)3+ ions, a significant improvement in their optical properties and stability might be achieved. Fig. 2l illustrates the optimized unit cells of Cs3Ce1−xGdxCl6, which exhibit two potential Ce/Gd sites, 4d and 4e, in accordance with the Wyckoff position.66 In this case, x is equal to 0 or 1, and all Ce3+ (or Gd3+) ions occupy both the 4d and 4e sites. In the case of x being neither 0 nor 1, the 4d and 4e sites are found to be partially occupied by the Ce3+ and Gd3+ ions. For example, three potential configurations of Ce3+ and Gd3+ ions are observed in Cs3Ce0.5Gd0.5Cl6: I (in which all 4e sites are occupied by Ce3+ and all 4d sites are occupied by Gd3+). The second configuration (half of the 4d and 4e sites are occupied by Ce3+ while the remaining sites are occupied by Gd3+) and the third configuration (all 4d sites are occupied by Ce3+ and all 4e sites are occupied by Gd3+) are also possible. The aforementioned site-preference can be evaluated by calculating the internal energy of mixing (Umix) using the following equation:
| Umix = E1 − (1 − x)E2 − xE3 |
4. Optical properties, optimized strategies and applications
Here are several effective strategies to endow functional materials with new and improved optical properties. By doping or alloying Sb3+ with ns2 electrons, Cs3LnCl6 can become a new type of highly emissive lanthanide halide for various luminescence applications. In addition, Cs3LnCl6 NCs can achieve impressive PLQYs through the introduction of multifunctional small organic molecules as intermediate states. All the above strategies mainly focus on the down-conversion luminescence. At the same time, Cs3LnCl6 can realize the upconversion luminescence regulation of the material based on the stable characteristic emission of different lanthanide ions. It is meaningful to explore the luminescence regulation mechanism and multi-functional applications.4.1. STE emission in Sb3+-doped Cs3LnCl6 (Ln: Sc, Y, La, Gd, Lu)
Sb3+ dopant occupies the position of octahedral Ln3+ sites in Cs3LnCl6 to form a series of new efficient 0D luminescent materials. Due to the spin orbitals of Sb3+ allowing 3P1 → 1S0 leaps, these materials not only exhibit broadband photoluminescence, but also have excellent resistance to thermal burst and photostability.71–73 Liang et al. developed Sb3+-doped 0D Cs3GdCl6 PCs.74 The lattice contraction of Cs3GdCl6 PCs with the increase in Sb3+ concentration indicates that the smaller Sb3+ successfully substitutes the larger Gd3+ (Fig. 3a). Samanta et al. investigated the luminescence properties of Sb3+-doped Cs3ScCl6 NCs.61 Upon illumination with 345 nm ultraviolet (UV) light, the Cs3ScCl6:Sb3+ NCs exhibit asymmetric yellow emission, which is characteristic of STE-assisted emission (Fig. 3b). The broadband emission is attributed to the 3P1 → 1S0 and 1P1 → 1S0 transitions of Sb3+ ions, followed by STE levels. Typically, STEs are observed in 0D MH structures due to lattice deformation, and similar STE-assisted emission is observed in Cs3ScCl6:Sb3+ through Sb3+ doping. Sb3+-activated MHs typically emit in the blue, green, or red regions, with emission unlikely to extend across the entire visible spectrum.75 Interestingly, the Cs3ScCl6:Sb3+ NCs exhibit bright white emission across the entire visible spectrum when excited with 365 nm UV light (Fig. 3c). The emissions from Cs3ScCl6:Sb3+ NCs have different origins from their corresponding photoluminescence excitation (PLE) spectra (Fig. 3d). Upon illumination with 365 nm UV light, electrons in the ground state of 1S0 are simultaneously excited to the higher- and lower-energy excited states of the carbon dots, 1P1 and 3P1, respectively (Fig. 3e). Excitons present in the energy states of carbon dots radiatively decay with luminescent centers. Subsequently, the higher and lower energy states of 1P1 and 3P1 transfer the energy to the STE1 and STE2 levels, respectively, and radiatively decay to 1S0, thereby emitting white light. From the absolute photoluminescence quantum yield (PLQY) of Cs3ScCl6:Sb3+ NCs at different doping levels, the optimal Sb3+ ion concentrations under excitation wavelengths of 345 and 365 nm were ∼1.5 and ∼3 mol%, respectively (Fig. 3f). The difference between the two optimal concentrations may be attributed to excessive carbon dot emission. Furthermore, upon illumination with 365 nm UV light, Cs3ScCl6:Sb3+ NCs exhibit higher PLQY values than those observed with 345 nm UV light. This is because of a reduction in the Stokes shift (i.e., energy loss) from 1.34 to 0.72 eV. The PL lifetime of Cs3ScCl6:Sb3+ NCs, as measured as a function of Sb3+ concentration (Fig. 3g), is consistent with the concentration-dependent PLQY results (Fig. 3f, yellow emission). Moreover, a typical broadband STE-assisted emission upon doping of Sb3+ ions into the host lattice of Cs3LnCl6 (Ln: La, Gd, Y, Lu) can be observed with the emission wavelength shift from 570 to 525 nm (Fig. 3h) and similar PLE spectral shape.76 In general, the emission color observed in Sb3+-doped zero-dimensional metal halides is dependent on the degree of asymmetry inherent to the Sb3+-doped lattice site. To illustrate, Rb3InCl6:Sb3+ exhibits green emission, while Cs2InCl5·H2O:Sb3+ displays red emission. The diverse emission colors can be attributed to variations in the degree of asymmetry within each Sb3+-doped lattice site.77–79 Furthermore, the distinct crystal structures of these materials contribute to different excitation spectra. However, in similar crystal structure systems, the emission color is primarily governed by the degree of asymmetry present in the Sb3+-doped lattice site. Upon changing the Ln ion type from Lu to La, the PLQY progressively increased (Fig. 3i). A mechanism for tuning the emission energies in Cs3LnCl6:Sb3+ NCs is proposed in the structure systems shown in Fig. 3j, the emission color is primarily governed by the degree of asymmetry present in the Sb3+-doped lattice site. Upon changing the Ln ion type from Lu to La, the PLQY progressively increased (Fig. 3i). A mechanism for tuning the emission energies in Cs3LnCl6:Sb3+ NCs is proposed in Fig. 3j. Irradiation of the Cs3LnCl6:Sb3+ NCs with 320 nm light results in the excitation of electrons in the ground state of 1S0 into the 1P1 and 3P1 states. Subsequently, electrons from the higher energy excited state, 1P1, are transferred to the STE1 state and radiatively recombine with 1S0, thereby causing the 450 nm emission band. In contrast, a lower energy excited state transfers the energy to STE2 and radiatively recombines with 1S0, resulting in emissions in the green and yellow regions, respectively.80 This is dependent on the degree of asymmetry present in the LnCl6 octahedra. The doping of the host material with Sb3+ ions typically results in the excitation of two distinct types of STE states. The first type corresponds to a higher energy STE state, resulting from the electronic transition from 1P1 to 1P0, which is a spin-allowed transition. The second type of transition involves the formation of lower-lying STE states, which arise from the electronic transition from 3Pn (where n can take values of 0, 1, or 2) to 1P0. This transition is classified as a parity-spin-allowed transition. Additionally, the spin-allowed transition from 1P0 to 1P1 is comparatively weak in comparison to the parity-allowed transition from 3P1 to 1P0 in zero-dimensional metal halides. This disparity in transition strengths contributes to the observation of a broad emission band within the blue-green-yellow spectral regions.81–83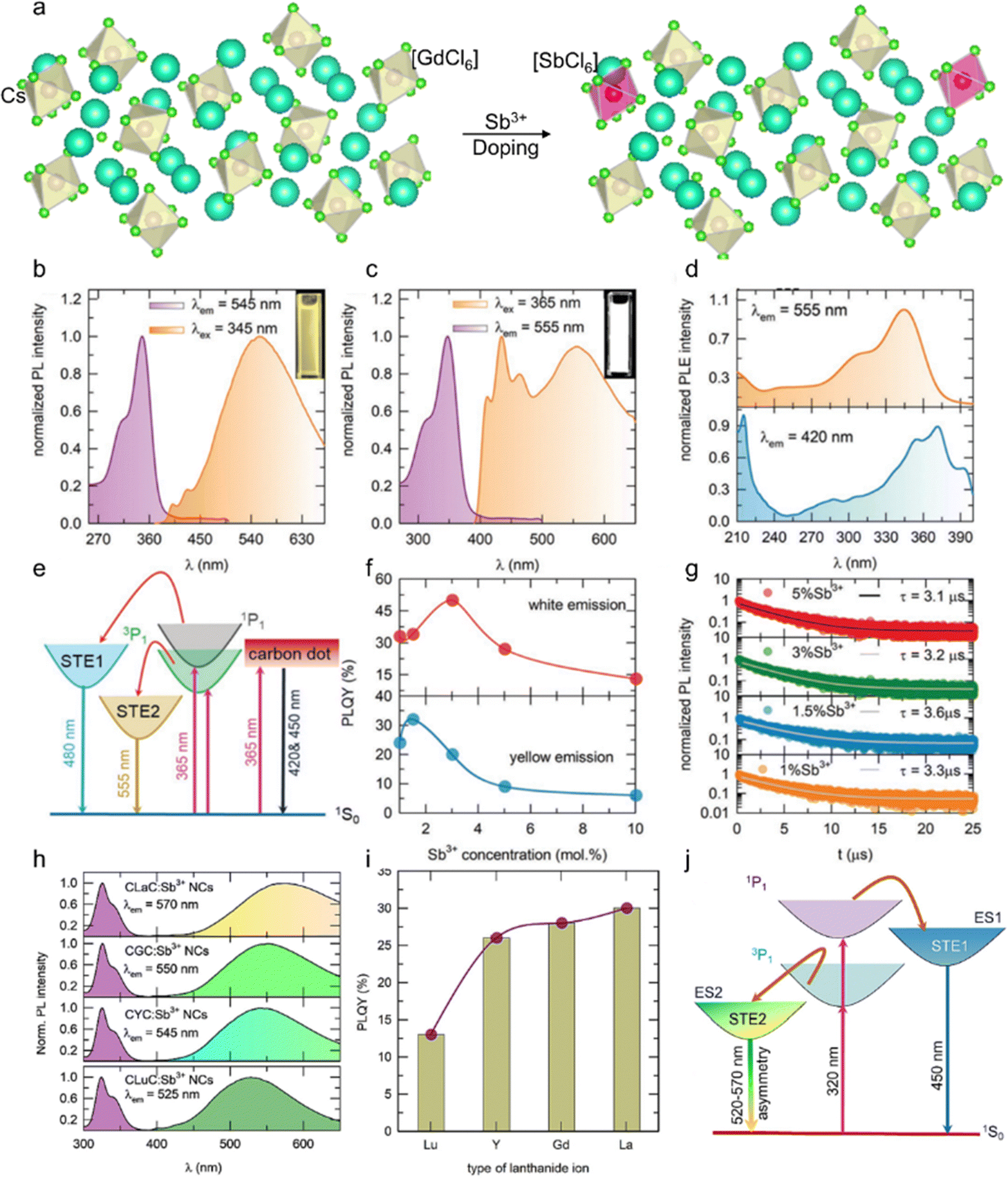 | ||
| Fig. 3 (a) Schematic of the crystal structure of orthorhombic Cs3GdCl6 and the crystallographic site for the Sb3+ dopant. PL and PLE spectra of (b) yellow-emitting and (c) white-emitting Cs3ScCl6:Sb3+ NCs, and the insets show the bright yellow and white light under 345 nm UV light excitation and 365 nm UV light excitation, respectively. (d) PLE spectra of Cs3ScCl6:Sb3+ NCs by monitoring the 420 and 555 nm emission peaks. (e) Schematic for the proposed mechanism of the origin of white-light-emission. (f) PLQY values and (g) PL lifetime of Cs3ScCl6:Sb3+ NCs as a function of Sb3+ concentration. Reproduced with permission from ref. 61. Copyright 2023, Wiley. (h) PL and PLE spectra and (i) PLQY values of Cs3LnCl6:Sb3+ (Ln: La, Gd, Y, Lu) NCs under 320 nm UV light excitation. (j) Schematic for the proposed mechanism for the origin of tuning the emission wavelengths in Cs3LnCl6:Sb3+ NCs. Reproduced with permission from ref. 80. Copyright 2024, Elsevier. | ||
4.2. Down-conversion luminescence in Cs3LnCl6 (Ln: Ce, Tb, Eu)
Cs3LnCl6 (Ln: Ce, Tb, Eu) usually exhibits blue, green and red emission, and their luminescence properties can be improved through Sb3+ doping and the introduction of multifunctional small organic molecules.59 Recently, Zhou et al. synthesized Sb3+-doped Cs3TbCl6 NCs and investigated their PL properties and compared them with undoped Cs3TbCl6 NCs.58 For the Sb3+-doped NCs, a new absorption peak appeared, which was attributed to the 5s2 → 5s15p1 (S → P) transition of Sb3+ in the [SbCl6]3+ octahedral unit (Fig. 4a). The optimal PLQY of Cs3TbCl6:2.5% Sb NCs is about three times higher than that of the undoped NCs (Fig. 4b), which is a new record for the PLQY of Tb-emissive halide perovskite and variant NCs. To further explore this luminescence process, they provide two photophysical pathways for radiative recombination of Tb3+-PL emission in this system. The charge carrier dynamics model is shown in Fig. 4c. Carriers generated by band-edge photons are directly trapped by [TbCl6]3− under UV light excitation, leading to PL emission, and carriers generated by photons can also be directly trapped by [SbCl6]3− octahedra via the 5s2 → 5s15p1 (S → P) transition of Sb3+. Subsequently, the thermally enhanced energy transfer pathway assisted by the more matched [SbCl6]3− induced STE states play a dominant role, which is more favorable for the narrower and more efficient intrinsic f-energy level PL emission of Tb3+.25 Moreover, Im's group demonstrated the optical properties of Cs3TbCl6 and Rb3TbCl6 polycrystals (PCs). They show bright green emission (λem = 548 nm, 5D4 → 7F5 transition) of Tb3+ ions under 280 nm UV light excitation (Fig. 4d and e). The broad absorption band ranges from 250 to 300 nm, corresponding to the charge transfer band (CTB) of Cl− → Tb3+, with the maximum peak at 280 nm.84,85 The narrow bands ranging from 300 to 400 nm are assigned to the spin-forbidden 4f–4f transitions of the Tb3+ ions.86 The scintillation process of Cs3TbCl6 and Rb3TbCl6 can be divided into three phases, namely, the conversion, the transport and the luminescence (Fig. 4f). | ||
| Fig. 4 (a) Normalized steady-state absorption spectra of undoped and Sb-doped Cs3TbCl6 NCs. (b) PL spectra of undoped and Sb-doped Cs3TbCl6 NCs. (c) The charge-carrier dynamics model and photophysical processes of Cs3TbCl6:Sb NCs. (1) Transfer process assisted by Cl−–Tb3+ charge transition in [TbCl6]3− and (2) thermally boosting energy transfer assisted by the [SbCl6]3−-induced STE state. Reproduced with permission from ref. 58. Copyright 2023, Wiley. (d) PL and PLE spectra of Cs3TbCl6 and Rb3TbCl6. (e) Schematic illustration of the luminescence mechanism of Cs3TbCl6 and Rb3TbCl6. (f) Schematic diagram of the scintillator conversion mechanism in Cs3TbCl6 and Rb3TbCl6. The process is divided into conversion, transport, and luminescence. Reproduced with permission from ref. 25. Copyright 2023, American Chemical Society. (g) PL and PLE spectra of Cs3CeCl6 NCs along with the schematic luminescence mechanism. The inset shows the image of Cs3CeCl6 NC colloids in hexane under white light (left) and 365 nm UV irradiation (right). (h) The PLQYs as a function of the Gd3+ concentration of Cs3CeGdCl6 NCs. Reproduced with permission from ref. 66. Copyright 2024, Wiley. (i) PL spectra of Cs3CeCl6 NCs, Cs3TbCl6 NCs, and Cs3TbxCe1−xCl6 (x = 0.1, 0.3, 0.5, 0.7, 0.9) alloy NCs. (j) Ce3+ → Tb3+ energy transfer mechanism in Cs3TbxCe1−xCl6 alloy NCs. (k) PL decay curves for Cs3TbxCe1−xCl6 (x = 0, 0.1, 0.3, 0.5, 0.7, 0.9, 0.99) alloy NCs. Reproduced with permission from ref. 51. Copyright 2024, Wiley. | ||
PL and PLE spectra of Cs3CeCl6, along with its luminescence mechanism, are illustrated in Fig. 4g. Upon UV excitation, Cs3CeCl6 NCs exhibited intense luminescence in the UV to visible region, characterized by asymmetric luminescence bands at 376 and 408 nm. In the case of the Ce3+ ion, the 4f1 energy level undergoes a splitting into 2F5/2 and 2F7/2 as a consequence of spin–orbit coupling. Additionally, the 5d1 (2D) energy level experiences a splitting into T2g and Eg, which is attributed to the influence of the crystal field effect.87,88 The principal luminescence bands at 379 nm and 408 nm are derived from the transition from the lowest 5d1 (T2g Γ8) to the 4f1 (2F5/2 and 2F7/2) multiple levels.89 The PLE spectra of the Cs3CeCl6 NCs were determined by monitoring the 376 nm emission wavelength. Excitation bands were observed at 264 and 336 nm, due to the 4f–5d electronic transition.90 A salient feature of Ce3+-based luminescent materials is the parity-allowed f–d transition exhibited by Ce3+ ions, which typically manifests as a broad emission band, a short PL lifetime (commonly on the nanosecond scale), and a tunable emission spectrum. These PL properties render Ce3+-based materials promising candidates for optoelectronic devices. The [CeCl6]3− octahedra, comprising Cl− ions as ligands, exert a dominant influence on the crystal field splitting strength. With a sixfold coordination number, the octahedra exhibit strong splitting in the 5d of Ce3+ ions.91,92 A gradual increase in the PLQY of the Cs3CeCl6 NCs was observed with an increase in the Gd3+ concentration, as illustrated in Fig. 4h. This represents a significant enhancement in comparison to the initial PLQY of 57% in the absence of Gd3+. This improvement in PLQY is ascribed to the substitution of Ce3+ ions with Gd3+ ions in [CeCl6]3− octahedra, which indicates the suppression of concentration quenching by providing higher distances between Ce3+ ions. Furthermore, this process reduces radiation reabsorption.
However, following the complete substitution of Ce3+ with Gd3+ ions, the PLQY of the Cs3GdCl6 NCs decreased to 6.2%, which is because of the surfactant ligands. It can be postulated that the only possible reason why Cs3GdCl6 NCs emit in the UV region would be oleic acid and oleylamine on the surface of NCs. Fig. 4i illustrates the PL spectra of the Cs3TbxCe1−xCl6 (x = 0, 0.1, 0.3, 0.5, 0.7, 0.9, 1) NCs. Upon excitation in the CTB band (290 nm), the Cs3TbCl6NCs emit a pronounced green luminescence at 546 nm. Furthermore, additional peaks were observed at 489, 588, and 621 nm. The emission peaks observed at 489, 546, 588, and 621 nm are assigned to 5D4 → 7F6, 5D4 → 7F5, 5D4 → 7F4, and 5D4 → 7F3 transitions, respectively.93 PLE spectra of the Cs3TbCl6 NCs were obtained by monitoring the 546 nm emission wavelength. A broad PLE band was observed in the 250–300 nm range, which is attributed to the charge transfer from Cl− to Tb3+. The PLE bands corresponding to the 4f–4f transition in the UV-A region were observed to be extremely weak. It is noteworthy that the Cs3TbxCe1−xCl6 (x = 0.1, 0.3, 0.5, 0.7, and 0.9) NCs exhibited intense blue-green luminescence upon excitation at 340 nm. In addition to the emission peaks at 489, 546, 588, and 621 nm from Tb3+ ions, blue-violet emission peaks (360–410 nm) were observed. The blue-violet peaks of Ce3+ ions are also characterized by asymmetric luminescent bands at 375 and 308 nm, which originate from the transition from the lowest 5d1 (T2g Γ8) to the 4f1 (2F5/2 and 2F7/2) multiple.94 The 4f1 energy level of Ce3+ ions undergoes a splitting into 2F5/2 and 2F7/2, while the 5d1 (2D) energy level undergoes a splitting into T2g and Eg as a result of the crystal field effect. It is noteworthy that an increase in the concentration of Ce3+ resulted in an enhancement in the intensity of the blue-violet peak and a concomitant reduction in the intensity of the Tb3+ ion luminescence. In the case of Cs3TbCl6 NCs, a broad excitation band was observed in the UV-B region (260–310 nm). Upon the incorporation of Ce3+ ions into the Cs3TbCl6 (Cs3TbxCe1−xCl6) NCs, an additional peak emerged in the UV-A region (340–370 nm), while the UV-B PLE peak of Cs3TbCl6 was no longer discernible for the Cs3CeCl6 NCs. A schematic representation of the energy transfer process from Ce3+ to Tb3+ is presented in Fig. 4j. Upon excitation at 340 nm, the Ce3+ ions absorbed photons and were excited to the excited state (5d1). A proportion of the excitons underwent radiative decay from the 5d1 state to the 2F5/2 and 2F7/2 states, resulting in the emission of blue-violet luminescence. The excited state of the Ce3+ ions exhibited an overlap with a 5d1 energy level of Tb3+ ions. Consequently, efficient energy transfer occurs from the 5d1 state to the 5G1 (Tb3+) energy level. Subsequently, a non-radiative energy transfer occurs from the 5D1 (Tb3+) to 5D4 (Tb3+) energy level. The hot excitons were radiatively transferred from the 5D4 level to the 7FJ (J = 0, 1, 2, 3, 4, 5, and 6) levels. Fig. 4k illustrates the time-correlated PL (TRPL) Cs3TbxCe1−xCl6 (x = 0, 0.1, 0.3, 0.5, 0.7, and 0.9) alloy NCs. The average lifetime τave of Cs3CeCl6 NCs was calculated. As a result, the average lifetime decreased from 36.80 ns (Cs3CeCl6 NCs) to 7.27 ns (Cs3Tb0.1Ce0.9Cl6 NCs), which can be attributed to the Ce3+ → Tb3+ energy transfer. Recently, a new type of Ce3+-based metal halide Cs3CeCl6·3H2O was presented. Single-crystal XRD measurements reveal that the material exhibits a distinctive one-dimensional crystalline structure, characterized by H2O–Ce–Cl tetrakaidecahedral units linked together in a linear chain. The synthesized compound exhibits the characteristic luminescence of Ce3+ ions, featuring a near-unity PLQY.95
In the conversion stage, the scintillator converts the incident radiant energy into energetic electrons and holes due to the photoelectric effect. The generated electrons and holes induce secondary electrons and holes. In the transport phase, the generated electrons and holes are transferred to the luminescent center. In the luminescence stage, the transferred electrons and holes from the excited and ground states recombine at the luminescence center to produce light. The high radioluminescence (RL) performance of Cs3TbCl6 and Rb3TbCl6 can be explained due to their low exciton binding energy to produce more free carriers by thermalization, which helps Cs3TbCl6 and Rb3TbCl6 to produce strong radiation at their Tb3+ luminescence centers. This allows them to be applied to homemade X-ray imaging systems. Thick films of Cs3TbCl6 and Rb3TbCl6 were fabricated as shown in Fig. 5a. Cs3TbCl6 and Rb3TbCl6 PCs were mixed with a polydimethylsiloxane (PDMS) binder, and then large area (25 cm2) films were fabricated using a spin-coating method. The bottom images in Fig. 5a are photographs of a scintillator film in daylight and under a 254 nm UV light in the dark. Due to the high ductility of PDMS, the scintillator films could be well stretched and bent while maintaining homogeneous green emission under a 254 nm UV light (Fig. 5b and c). Fig. 5d shows the schematic device structure of the indirect X-ray detector constructed using Cs3TbCl6 and Rb3TbCl6 scintillator films. The indirect X-ray detector consists of a carbon fiber reinforced polymer (CFRP) film/scintillator film/arrayed CMOS photodiode, which acquires X-ray images via a data interface with a digital video output and a frame grabber. Another important indicator of the performance of an X-ray detector is its spatial resolution, which can be determined using the modulation transfer function (MTF). The spatial resolution of the X-ray detector is affected by the spatial resolution of the photodiode array and the thickness and performance of the scintillator film. The MTF was measured using the oblique edge method and the spatial resolution of the commercial GOS film was measured in the same system as the Cs3TbCl6 and Rb3TbCl6 scintillator films for comparison (Fig. 5e). The spatial resolutions of the Cs3TbCl6 and Rb3TbCl6 films at MTF values of 0.2 were 3.3 and 3.9 lp mm−1 respectively. These values are slightly higher than the spatial resolution of the commercial GOS film reported in a previous study (2.7 lp mm−1). Static X-ray imaging was then performed using Cs3TbCl6 and Rb3TbCl6 scintillator films. As shown in Fig. 5f, normal and bent clips cannot be identified with the naked eye when under a key ring. However, they could be clearly distinguished in the radiographs obtained, indicating the application potential in homemade X-ray imaging systems.
 | ||
| Fig. 5 X-Ray imaging of Cs3TbCl6 and Rb3TbCl6 scintillator films. (a) Fabrication method of Cs3TbCl6 and Rb3TbCl6 scintillator films and photographs of a scintillator film under daylight and a 254 nm UV lamp. (b) and (c) Photographs of stretched and bent scintillator films, respectively. (d) Device schematic of the X-ray imaging system. (e) Spatial resolution measurement of Cs3TbCl6, Rb3TbCl6, and GOS scintillator films. (f) Photographs of normal (left) and bent (right) clips and a key ring. Reproduced with permission from ref. 25. Copyright 2023, American Chemical Society. | ||
Recently, Sun et al. achieved highly efficient red emission by introducing multifunctional coumarin small molecules into Cs3EuCl6 NCs as intermediate states.59 The coumarin-modified Cs3EuCl6 exhibits pure red emission with 92.4% PLQY and excellent energy transfer efficiency. The energy transfer pathway in the Cs3EuCl6 system is shown in Fig. 6a. Upon excitation at 365 nm UV light, the electron leaps from the ground state to the excited state of the Cs3EuCl6 NCs, and the incident photon is absorbed by the ethyl 7-hydroxycoumarin-3-carboxylate (EHC), which facilitates the electron leaps from the ground state to S1.96–98 Subsequently, strong electron–phonon coupling induces lattice deformation, leading to free exciton capture by the self-trapped excited state.99–101 At the same time, energy transfer occurs between (i) the self-trapped exciton and Eu3+, (ii) the self-trapped exciton and S1 of the EHC, and (iii) the EHC and S1 of Eu3+. This complex network of energy transfer pathways achieves efficient multichannel energy transfer to Eu3+, leading to emission at 593 and 617 nm (5D0 → 7F1 and 5D0 → 7F2). Remarkably, the STE emission was largely suppressed due to the efficient energy transfer from the STE to Eu3+ and S1 of the EHC. The PL intensity was maximized when 4% of EHC molecules were added, and the PLQY was increased from 51% to 92% (Fig. 6b). The coumarin molecules improved the PLQY of Cs3LnCl6 NCs by passivating the halogen vacancy defects and increasing the crystallinity of the materials. By UV photoelectron spectroscopy (UPS) testing, they found that the coumarin molecule post-treated films had a low figure of merit (3.6 eV) and a high energy shift between the figure of merit and valence band (2.3 eV) (Fig. 6c). To extend the investigation, EHC molecules were introduced during the synthesis of Cs3LnCl6 (Ln: La, Pr, Nd, Sm, Eu, Tb, Er, Yb) NCs, resulting in lead-free perovskite materials with tunable emission wavelengths from the visible to the near-infrared range, together with a high PLQY (Fig. 6d). When excited by 365 nm light, Cs3LnCl6 NCs exhibit exciton emissions at ∼438 nm, consistent with previous studies in a similar context.102 In addition, the PL spectra show transitions related to the intrinsic electron transitions of lanthanide ions. These include the 1D2–3F4 and 1G4–3H5 transitions for Pr3+, 4F3/2–4I11/2 and 4F3/2–4I3/2 transitions for Nd3+, 4G5/2–6HJ(5,7,9)/2 transition for Sm3+, 5D0–7FJ(1,2,3,4) transition for Eu3+, 5D4–7FJ(6,5,4,3) transition for Tb3+, 4I13/2–4I15/2 transition for Er3+ and the 2F5/2–2F7/2 transition for Yb3+.103–105 Moreover, the introduction of EHC molecules significantly increased the overall PLQY of Cs3LnCl6 NCs compared to Cs3LnCl6 NCs without EHC (Fig. 6e).
 | ||
| Fig. 6 (a) Schematic diagram of the luminescence mechanism of Cs3EuCl6. (b) PLQY of Cs3EuCl6-NCs and Cs3EuCl6-NC-treated EHC. (c) Energy level diagram of the pristine film and EHC post-treated film obtained from parameters derived from UPS spectra (EVAC, vacuum level; EF, Fermi level). (d) Normalized PL spectra and (e) PLQY values of 4% EHC-treated Cs3LnCl6 (λex = 365 nm). Reproduced with permission from ref. 59. Copyright 2023, Wiley. (f) Luminescence photographs of Cs3EuCl6 and Cs3TbCl6 NC films under excitation wavelengths from 260 to 360 nm with a 10 nm interval. (g) Direct writing using brush transfer printing under ambient light, 254 nm UV light, and 365 nm UV light. Reproduced with permission from ref. 35. Copyright 2023, The Royal Society of Chemistry. | ||
In addition, Cs3EuCl6 and Cs3TbCl6 NC films also show the excitation-wavelength dependent emission colors, attributed to the presence of two different emission mechanisms. For instance, Cs3EuCl6 NCs exhibit blue emission under the excitation range of 310 to 360 nm and red emission under the excitation range from 260 to 330 nm, both of which depend on the excitation wavelength. This blue emission may be from the nanocrystals’ organic components or the d–f transition of Eu2+, and red emission is from the f–f transition of Eu3+. Consequently, the relative PL intensity ratio of blue and red PL can be systematically varied by changing the excitation wavelength, which enables the emission color to be fine-tuned over a wide spectrum range between blue and red, as illustrated in Fig. 6f. The dynamic color changes observed under conventional UV light suggest that Cs3EuCl6 and Cs3TbCl6 NCs may be suitable for use as writeable and printable security inks for high-level anti-counterfeiting applications. Fig. 6g illustrates the direct writing of a flower image on printing paper using Cs3EuCl6 and Cs3TbCl6 NCs and a writing brush. The marked image was invisible under ambient light, indicating that the images can be effectively hidden. Upon 365 nm UV light irradiation, a blue flower image emerges due to the luminescence of both the NCs. This image color changes to red and green under 254 nm UV light irradiation. Notably, this multicolor image is clearly visible under UV irradiation using a conventional hand UV lamp, and not using a high-power laser or time-gating equipment. The stamped images clearly show blue/red (Eu) or blue/green (Tb) dual-mode emission, depending on the excitation wavelength.106–108
4.3. Upconversion luminescence in Cs3LnCl6 (Ln: Ho, Er, Tm, Yb)
Upconversion luminescent materials have attracted significant interest in the field of sensors due to their ability to penetrate deep into light-absorbing materials through the use of infrared excitation sources, while causing minimal damage to living tissues.Cs3LnCl6 (Ln: Ho, Er, Tm, Yb) upconversion luminescent material has potential in the field of water detection and information encryption. Fig. 7a shows the upconversion luminescence (UCL) of different Er3+-doped Cs3Gd0.8−yYb0.2EryCl6 (y = 0.005, 0.01, 0.02 and 0.04) PCs. In particular, the 2% Er3+-doped sample (Cs3Gd0.78Yb0.2Er0.02Cl6) showed the most intense luminescence compared to the other Er3+ doped samples. The UCL intensities of the 525, 550, and 660 nm peaks (green and red peak) were determined at varying Er3+ dopant concentrations. Consequently, green luminescence is generated from the 4H11/2 and 4S3/2 manifolds. For red luminescence, the 4F9/2 (Er3+) manifold is populated via multiple phonon relaxations from the 4H11/2/4S3/2 manifold. In general, green luminescence and red luminescence occur via a two-photon process. Under 980 nm laser excitation, Cs3Gd(0.8−y)Yb0.2HoyCl6 NCs (y is 0.005, 0.01, 0.02 and 0.04, respectively) exhibit the typical UCL of Ho3+ ions. The characteristic emission peaks are 540 nm (5S2, 5F4 → 5I8) and 650 nm (5F5 → 5I8), which are capable of emitting green and red light, respectively. Fig. 7b shows Ho3+ doped Cs3Gd(0.8−y)Yb0.2HoyCl6 NCs, the 0.5%Ho3+ sample (Cs3Gd0.795Yb0.2Ho0.005Cl6) has the highest luminescence intensity. Excitation of Cs3Gd(0.8−x)Yb0.2TmxCl6 (x = 0.001, 0.003, 0.005, 0.01, and 0.02) PCs with a 980 nm laser resulted in the emission of intense near infrared (NIR) UCL at 815 nm (3H4 → 3H6) of Tm3+. In addition to the 815 nm peak, a weak peak at 770 nm was also observed. However, the Cs3Gd0.797Yb0.2Tm0.003Cl6 PCs exhibited the most intense UCL compared with the other Tm3+-doped samples (Fig. 7c). The UCL mechanism in Cs3Gd0.78Yb0.2Er0.02Cl6 NCs has been the subject of extensive research and is now well understood (Fig. 7d). In accordance with this mechanism, the Yb3+ ion, acting as a sensitizer, absorbs incoming NIR photons (with a wavelength of 980 nm) and efficiently transfers energy to the 4I11/2 energy level of the Er3+ ion. Subsequently, the 4F7/2 level of the Er3+ ion absorbs another photon from the 4I11/2 level through an energy transfer up-conversion process. Consequently, green luminescence is generated by the 4H11/2 and 4S3/2 energy levels. With regard to red emission, excitation of the 4F9/2 level of the Er3+ ion occurs as a result of relaxation of the 4H11/2/4S3/2 level of multiple phonons.42,109 Analogous to the aforementioned example, the sensitizer Yb3+ absorbed NIR photons at 980 nm, leading to the excitation of the 5S2 and 5F4 energy levels of Ho3+ ions and the 3H4 of Tm3+. The 5S2 and 5F4 energy levels of Ho3+ ions as well as the 3H4 energy levels of Tm3+ ions could be populated either through energy transfer up-conversion or via energy transfer followed by excited state absorption.110–113
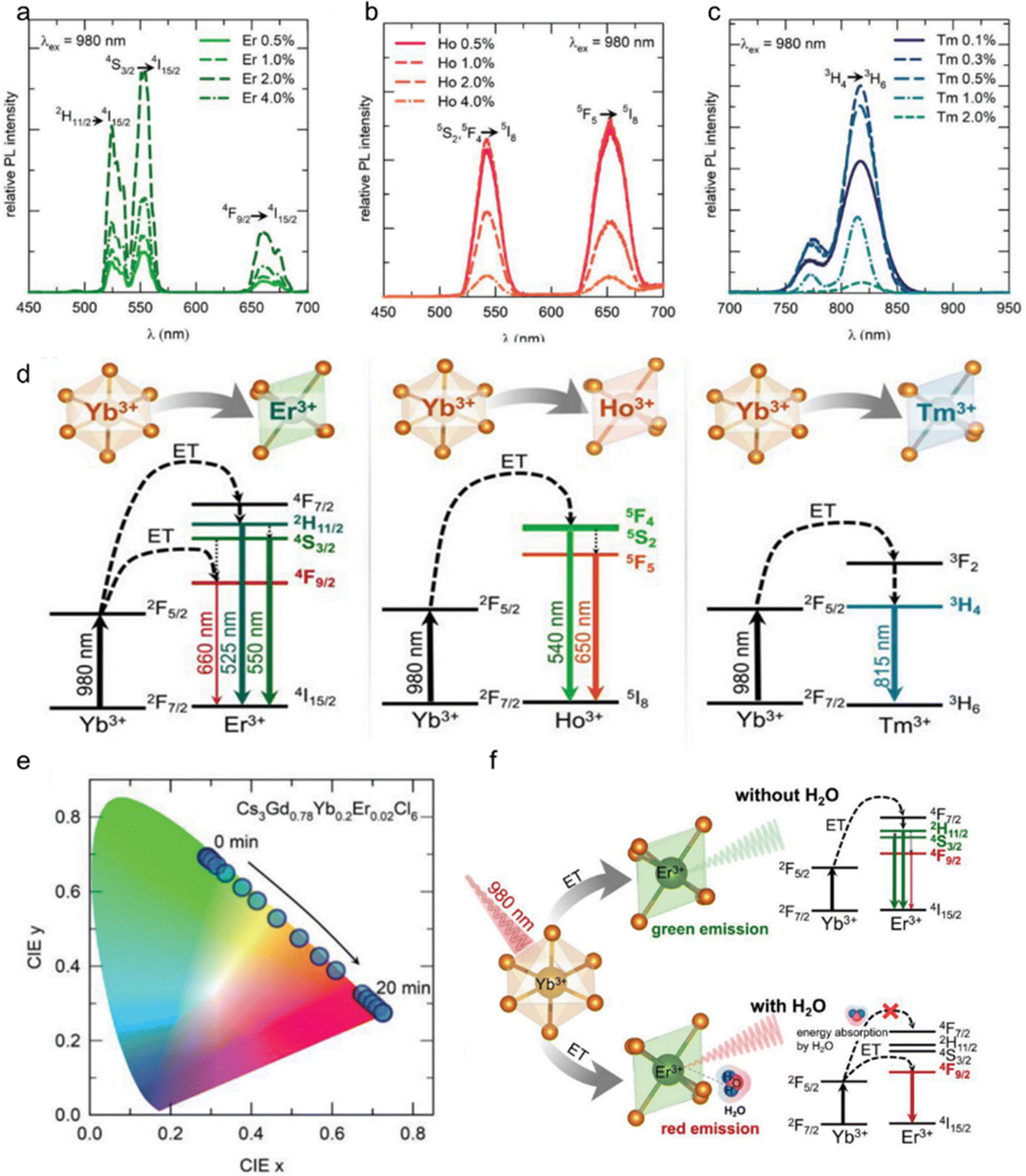 | ||
| Fig. 7 (a) Different Er3+ dopant concentrations in Cs3Gd1−x−yYbxEryCl6 PCs under 980 nm laser excitation; (b) different Ho3+ dopant concentrations in Cs3Gd(1−x−y)YbxHoyCl6 PCs upon 980 nm laser excitation; (c) different Tm3+ dopant concentrations in Cs3Gd(1−x−y)YbxTmyCl6 PCs upon 980 nm laser excitation. (d) Mechanism of energy transfer from Yb3+ to Er3+, Ho3+, and Tm3+ ions for UCL. (e) CIE coordinates of UCL from Er3+, Ho3+, and Tm3+. (f) The UCL mechanism of Cs3GdCl6:Yb3+,Er3+ PCs in the presence and absence of water molecules. Reproduced with permission from ref. 55. Copyright 2023, Wiley. | ||
Interestingly, upon exposure to atmospheric moisture, these PCs absorbed water molecules to influence their UCL properties. Taking Cs3Gd0.78Yb0.2Er0.02Cl6 as a case, with exposure time, the PL intensity of the red peak (λem = 660 nm) increased, and the green peak (λem = 550 nm) intensity decreased gradually. Thus, the UCL color changed from green to red with increasing exposure time (Fig. 7e). In addition, in order to understand the amount of moisture absorption of Cs3GdCl6:Yb3+,Er3+, the change in the intensity of green and red luminescence peaks with the change in the mass of Cs3GdCl6:Yb3+,Er3+ was measured. Also note that the vibrational energy of water (–OH group) overlaps with the 2F5/2 energy level of the Yb3+ ions; consequently, the absorption of water molecules near the 980 nm wavelength causes the excitation photons to be attenuated. Thus, the overall UCL is quenched by the absorption of more water molecules. The role of water molecules in the upconversion process is shown in Fig. 7f.
Fig. 8a shows the variation of red-green UCL ratio with increasing water content. The UCL color of the solution starts to change from green to red, and when the water content of the tetrahydrofuran (THF) solution is 0.08%, the UCL color changes completely from green to red. Subsequently, a film in the form of a seven-segment display was fabricated, as shown in Fig. 8b, applying Cs3GdCl6:Yb3+,Er3+ PCs in a form that allows for information encryption and wet sensing applications. The prepared film is white in daylight, as shown in Fig. 8c, however, at 980 nm, the UCL reversibly turns red upon hydration and green upon dehydration. As shown in Fig. 8d, the pattern could not be distinguished under daylight conditions. The seven-segment display film was fabricated into a separable shape and separated in the desired pattern. By partial dehydration and hydration, a dial pattern image of 0–9 was obtained at 980 nm, confirming the information encryption effect of the film. In addition, the mechanical stability of the film was measured by folding and unfolding the film. This confirmed the mechanical stability of the film. In addition, Cs3GdCl6:Yb3+,Er3+ PC was cipher encrypted using a 10 mW cm−1 laser. The Cs3GdCl6:Yb3+,Er3+ PC was processed into a plate and the ciphertext was written with the laser. As shown in Fig. 8e, when writing with the laser, the water in the area where the laser passes through will locally evaporate, after which the hidden information will no longer be visible to the naked eye but will be examined with 980 nm light. In addition, when the material is again hydrated by the moisture in the air, the message naturally disappears to remain secure. In order to confirm the possibility of encrypting the ciphertext, the initials “HY” of Hanyang University were laser-engraved here. A heart-shaped ciphertext was also drawn on the board as shown in Fig. 8f. After 15 minutes, the ciphertext disappeared. This confirms the feasibility of encrypting and re-encrypting the ciphertext using a Cs3GdCl6:Yb3+,Er3+ pc machine and a laser.
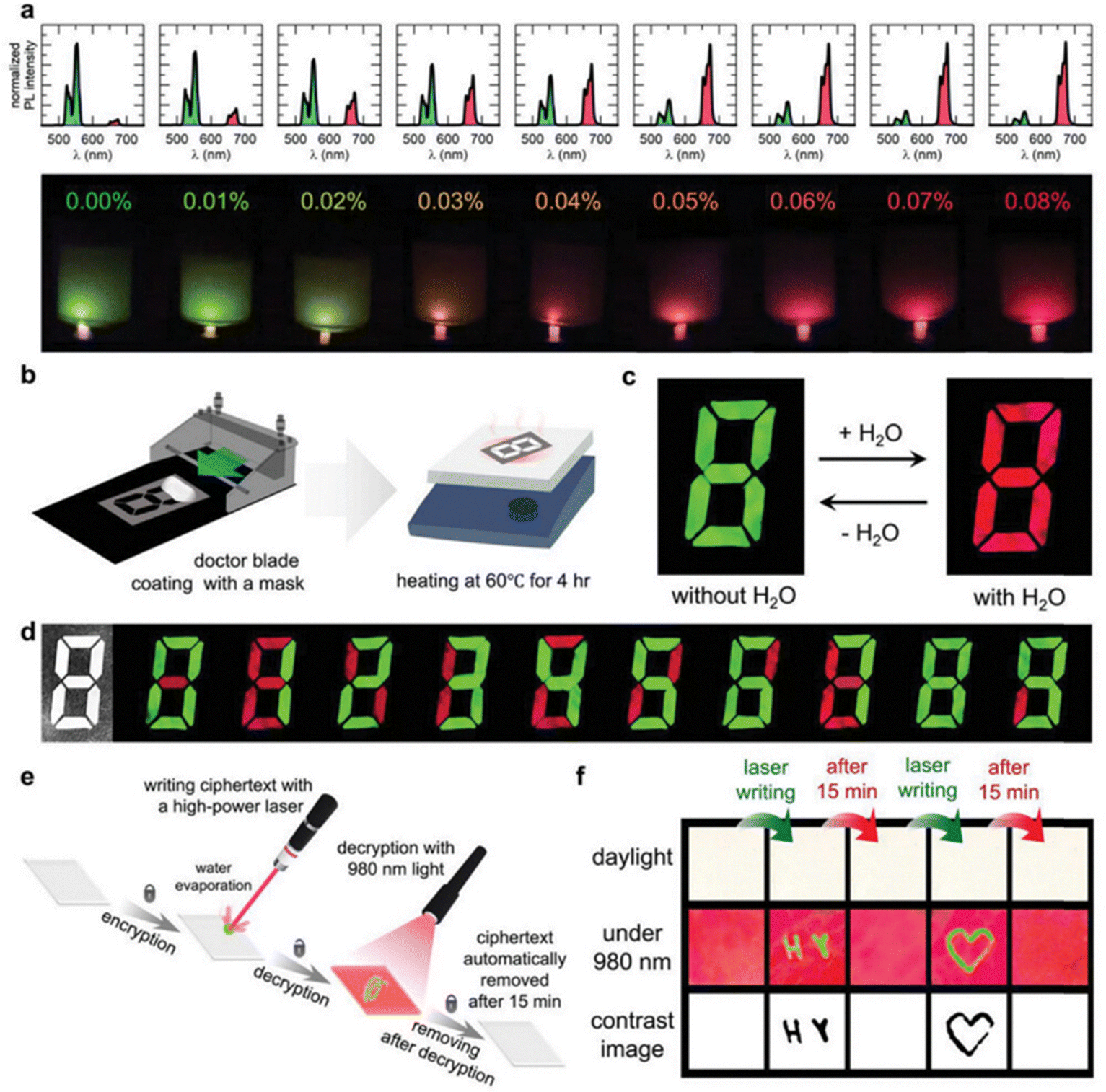 | ||
| Fig. 8 Applications of Cs3GdCl6:Yb3+,Er3+ for sensing moisture and encrypting information. (a) Hydrochromic UCL in Cs3GdCl6:Yb3+,Er3+ nanoparticles in THF varies with water concentration. (b) Diagrams of the creation of a 7-segment display-like moisture-sensing film. (c) Color changes in the moisture-sensing film due to hydrochromic UCL when exposed to 980 nm light. (d) Pattern changes in the moisture-sensing film under both daylight and 980 nm irradiation. (e) A schematic illustrating the encryption of ciphertext using laser writing. (f) Visual representation of the encrypted information using 980 nm laser excitation to decrypt the ciphertext. Reproduced with permission from ref. 55. Copyright 2023, Wiley. | ||
5. Conclusion and outlook
In this review, we briefly introduced several synthetic methods and optical properties of Cs3LnCl6 PCs and NCs. In order to better understand the intrinsic mechanism, luminescence modulation and optimization of Cs3LnCl6 luminescence, the crystal and electronic structures of Cs3LnCl6 are summarized. In particular, alloying and modification strategies are proposed to improve the PLQY and tunable emission. Challenges and perspectives are briefly summarized as follows.(1) The synthesis of Cs3LnCl6 is complex, mainly due to the underdeveloped synthesis conditions and incomplete understanding of optimal reaction parameters. The lack of established experimental conditions and a thorough investigation of these parameters is a key challenge. Addressing these is crucial for research progress. The scarcity of literature on Gibbs free energy changes during the reaction underscores the need for a deeper exploration of the synthesis's thermodynamics and kinetics. Combining chemical reaction thermodynamics with kinetics can establish a robust theoretical framework to guide systematic condition exploration. This approach may identify optimal parameters for efficient Cs3LnCl6 synthesis, overcoming challenges and enabling more effective research methods.
(2) For Cs3LnCl6, the luminescence efficiency is too low for practical application, especially for nanocrystals. Therefore, various luminescence modulation or luminescence optimization strategies are used, such as doping with ns2-cationic metals/lanthanides or modifying the crystal surface with small organic molecules. Therefore, future research should focus on further exploring these methods to enhance luminescence performance and delving into their mechanisms.
(3) In addition to the existing lead-free metal halide application scenarios, the luminescent properties of Cs3LnCl6 can be more suitable for application. Cs3LnCl6 has a high degree of compatibility with rare earth elements, which extends beyond traditional visible light research. It can be used to study near-infrared luminescence and imaging applications, as well as for multi-mode anti-counterfeiting through up-conversion luminescence. In addition, Cs3LnCl6 undergoes a phase change when in contact with water, a property that suggests potential applications in anti-counterfeiting and environmental moisture monitoring. Referring to the current application methods and combining with practical needs, it is better to make people's life better while enriching the application scenes.
In conclusion, the lead-free lanthanide metal halide Cs3LnCl6 has garnered increasing scholarly interest due to its superior chemical stability, commendable lattice adaptability, and distinctive characteristics within the realms of optics and photovoltaics. Despite the existence of extant methodologies for the modification and enhancement of PLQYs, the domain necessitates the engagement of a broader research community to further refine the intrinsic properties of these materials or to diversify their applications. Additionally, computational approaches like ICSD data or high-throughput screening DFT for phase competition can be utilized to support the analysis of synthetic routes and reaction conditions, in the pursuit of determining the most efficient synthesis pathway for Cs3LnCl6. Employing such methods to systematically explore reaction conditions and identify suitable synthetic routes can streamline the experimental process and prevent resource wastage, making this approach highly worthwhile to pursue. Furthermore, Cs3LnCl6 exhibits a favourable interaction with water. Materials such as Cs2YbCl5·HO and Cs3CeCl6·3H2O are progressively garnering the attention of researchers.95,114 These materials frequently demonstrate high PLQY and robust stability, undergo phase transition in the presence of water, and can be more effectively utilised in anti-counterfeiting applications.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2023YFB3506600), the National Natural Science Foundation of China (NSFC No. 12374386, 12374388, 12304461, 52172166, 52072349 and 51932009), and the Project for Science and Technology Development Plan of Jilin Province (20240101316JC).References
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed.
- T.-H. Han, K. Y. Jang, Y. Dong, R. H. Friend, E. H. Sargent and T.-W. Lee, Nat. Rev. Mater., 2022, 7, 757–777 CrossRef.
- M. T. Hoang, A. S. Pannu, Y. Yang, S. Madani, P. Shaw, P. Sonar, T. Tesfamichael and H. Wang, Nano-Micro Lett., 2022, 14, 69 CrossRef CAS PubMed.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- Y.-H. Kim, Y. Zhai, H. Lu, X. Pan, C. Xiao, E. A. Gaulding, S. P. Harvey, J. J. Berry, Z. V. Vardeny, J. M. Luther and M. C. Beard, Science, 2021, 371, 1129 CrossRef CAS PubMed.
- H. Xiao, P. Dang, X. Yun, G. Li, Y. Wei, Y. Wei, X. Xiao, Y. Zhao, M. S. Molokeev, Z. Cheng and J. Lin, Angew. Chem., Int. Ed., 2021, 60, 25183 CrossRef CAS PubMed.
- Z. Wu, Q. Zhang, B. Li, Z. Shi, K. Xu, Y. Chen, Z. Ning and Q. Mi, Chem. Mater., 2019, 31, 4999–5004 CrossRef CAS.
- J. Huang, T. Lei, M. Siron, Y. Zhang, S. Yu, F. Seeler, A. Dehestani, L. N. Quan, K. Schierle-Arndt and P. Yang, Nano Lett., 2020, 20, 3734–3739 CrossRef CAS PubMed.
- X. Li, X. Chen, H. Jiang, M. Wang, S. Lin, Z. Ma, H. Wang, H. Ji, M. Jia, Y. Han, J. Zhu, G. Pan, D. Wu, X. Li, W. Xu, Y. Liu, C.-X. Shan and Z. Shi, Nano Lett., 2024, 24, 6601–6609 CrossRef CAS PubMed.
- J. Luo, X. Wang, S. Li, J. Liu, Y. Guo, G. Niu, L. Yao, Y. Fu, L. Gao, Q. Dong, C. Zhao, M. Leng, F. Ma, W. Liang, L. Wang, S. Jin, J. Han, L. Zhang, J. Etheridge, J. Wang, Y. Yan, E. H. Sargent and J. Tang, Nature, 2018, 563, 541–545 CrossRef CAS PubMed.
- Y. Zhang, X. Liu, H. Sun, J. Zhang, X. Gao, C. Yang, Q. Li, H. Jiang, J. Wang and D. Xu, Angew. Chem., Int. Ed., 2021, 60, 7587–7592 CrossRef CAS PubMed.
- H. Tang, Y. Xu, X. Hu, Q. Hu, T. Chen, W. Jiang, L. Wang and W. Jiang, Adv. Sci., 2021, 8, 2004118 CrossRef CAS PubMed.
- S. Liu, B. Yang, J. Chen, D. Wei, D. Zheng, Q. Kong, W. Deng and K. Han, Angew. Chem., Int. Ed., 2020, 59, 21925–21929 CrossRef CAS PubMed.
- P. Vishnoi, J. L. Zuo, J. A. Cooley, L. Kautzsch, A. Gómez-Torres, J. Murillo, S. Fortier, S. D. Wilson, R. Seshadri and A. K. Cheetham, Angew. Chem., Int. Ed., 2020, 60, 5184–5188 CrossRef PubMed.
- X.-D. Wang, Y.-H. Huang, J.-F. Liao, Y. Jiang, L. Zhou, X.-Y. Zhang, H.-Y. Chen and D.-B. Kuang, J. Am. Chem. Soc., 2019, 141, 13434–13441 CrossRef CAS PubMed.
- S. Guha, A. Dalui, P. K. Sarkar, S. Roy, A. Paul, S. Kamilya, A. Mondal, I. Dasgupta, D. D. Sarma and S. Acharya, J. Phys. Chem. Lett., 2024, 15, 3061–3070 CrossRef CAS PubMed.
- Z. Guo, Y. Chaowei, L. Xiaofang, Y. Lin, Y. Wenjia, F. Ruimei, S. Yanjuan, S. Jianping and D. Fan, Chem. Eng. J., 2021, 430, 132974 Search PubMed.
- M. B. Gray, J. D. Majher, N. P. Holzapfel and P. M. Woodward, Chem. Mater., 2021, 33, 2165–2172 CrossRef CAS.
- N. P. Holzapfel, J. D. Majher, T. A. Strom, C. E. Moore and P. M. Woodward, Chem. Mater., 2020, 32, 3510–3516 CrossRef CAS.
- T. Cai, W. Shi, S. Hwang, K. Kobbekaduwa, Y. Nagaoka, H. Yang, K. Hills-Kimball, H. Zhu, J. Wang, Z. Wang, Y. Liu, D. Su, J. Gao and O. Chen, J. Am. Chem. Soc., 2020, 142, 11927–11936 CrossRef CAS PubMed.
- D. Liu, Z. Zhang and L. Wang, J. Chin. Rare Earth Soc., 2023, 4, 701 Search PubMed.
- J. Huang, L. Yan, Z. An, H. Wei, C. Wang, Q. Zhang and B. Zhou, Adv. Mater., 2024, 36, 2310524 CrossRef CAS PubMed.
- Y.-J. Yu, H.-S. Wang, J.-L. Pan, S.-N. Li, W.-S. Shen, S.-N. Cheng, L.-J. Jin, Y.-K. Wang, Y.-Y. Li and L.-S. Liao, Adv. Funct. Mater., 2023, 33, 2305125 CrossRef CAS.
- J.-L. Pan, W.-S. Shen, S.-N. Li, Z.-D. Zhang, F. Zhao, H.-W. Duan, Y.-K. Wang and L.-S. Liao, Nano Lett., 2024, 24, 2765–2772 CrossRef CAS PubMed.
- J. H. Han, T. Samanta, Y. M. Park, H. J. Kim, N. S. Manikanta Viswanath, H. W. Kim, B. K. Cha, S. B. Cho and W. B. Im, ACS Energy Lett., 2022, 8, 545–552 CrossRef.
- X. Li, X. Gao, X. Zhang, X. Shen, M. Lu, J. Wu, Z. Shi, V. L. Colvin, J. Hu, X. Bai, W. W. Yu and Y. Zhang, Adv. Sci., 2021, 8, 2003334 CrossRef CAS PubMed.
- Y. Huang, Y. Pan, C. Peng, Y. Ding, H. Lian, L. Li and J. Lin, Inorg. Chem. Front., 2022, 10, 991–1000 RSC.
- W. Zhou, C. Li, T. Wu, R. Liu, Z. Ding, R. Zhang, Y. Yu, P. Han and R. Lu, J. Phys. Chem. Lett., 2023, 14, 8577–8583 CrossRef CAS PubMed.
- W. Huang, H. Peng, Q. Wei, J. Wang, B. Ke, W. Liang, J. Zhao and B. Zou, Chem. Mater., 2024, 36, 2483–2494 CrossRef CAS.
- Z. Rao, M. Cao, Z. Chen, X. Zhao and X. Gong, Adv. Funct. Mater., 2023, 34, 2311568 CrossRef.
- J. Jia, D. Wang, X. Gao, Y. Xu, X. Ren and G. Zou, J. Mater. Chem. C, 2023, 11, 4351–4356 RSC.
- Y. Liu, R. Yun, H. Yang, W. Sun, Y. Li, H. Lu, L. Zhang and X. Li, Mater. Horiz., 2024, 11, 5341–5351 RSC.
- W. Xu, J. Liu, B. Dong, J. Huang, H. Shi, X. Xue and M. Liu, Sci. Adv., 2023, 9, 7931 CrossRef PubMed.
- X. X. Guo, J. H. Chen, J. B. Luo, J. H. Wei, Z. Z. Zhang, Z. L. He, Q. P. Peng and D. B. Kuang, Adv. Opt. Mater., 2024, 12(22), 2400681 CrossRef CAS.
- M. Lee, H. Chung, S. V. Hong, H. Y. Woo, J.-Y. Chae, T. Y. Yoon, B. T. Diroll and T. Paik, Nanoscale, 2022, 15, 1513–1521 RSC.
- L. Xu, Y. Li, Q. Pan, D. Wang, S. Li, G. Wang, Y. Chen, P. Zhu and W. Qin, ACs Appl. Mater. Interfaces, 2020, 12, 18934–18943 CrossRef CAS PubMed.
- Y. Liu, F. Di Stasio, C. Bi, J. Zhang, Z. Xia, Z. Shi and L. Manna, Adv. Mater., 2024, 36, 2312483 Search PubMed.
- H. Li, M. Zhang, Y. Li, X. Fu, J. Feng and H. Zhang, Adv. Opt. Mater., 2023, 11, 2300429 CrossRef CAS.
- X. Liu, R. Li, X. Xu, Y. Jiang, W. Zhu, Y. Yao, F. Li, X. Tao, S. Liu, W. Huang and Q. Zhao, Adv. Mater., 2022, 35, 2206741 CrossRef PubMed.
- H. Li, Y. Li, L. Zhang, E. Hu, D. Zhao, H. Guo and G. Qian, Adv. Mater., 2024, 36, 2405535 CrossRef CAS PubMed.
- Q. Hu, Z. Deng, M. Hu, A. Zhao, Y. Zhang, Z. Tan, G. Niu, H. Wu and J. Tang, Sci. China Chem., 2018, 61, 1581–1586 CrossRef CAS.
- Y. Zhang, T. Zhao and G. Chen, J. Chin. Rare Earth Soc., 2023, 41, 580–586 Search PubMed.
- P. Zhao, Y. Liu, C. He and C. Duan, Inorg. Chem., 2022, 61, 3132–3140 CrossRef CAS PubMed.
- D. Paderni, L. Giorgi, V. Fusi, M. Formica, G. Ambrosi and M. Micheloni, Coord. Chem. Rev., 2020, 429, 213639 CrossRef.
- W. Liu, C. Liu, L. Pilia, H. Zhang, P. Van Der Voort, A. M. Kaczmarek and R. Van Deun, Dalton Trans., 2023, 52, 11949–11957 RSC.
- G. Li, X. Chen, M. Wang, S. Cheng, D. Yang, D. Wu, Y. Han, M. Jia, X. Li, Y. Zhang, C. Shan and Z. Shi, Adv. Mater., 2023, 35, 2305495 CrossRef CAS PubMed.
- Z. Li, B. Zhang, Z. Zhang, J.-C. Bünzli, A. R. B. Mohd Yusoff, Y.-Y. Noh and P. Gao, Mater. Sci. Eng., R, 2023, 152, 100710 CrossRef.
- A. K. Singh, Coord. Chem. Rev., 2022, 455, 214365 CrossRef CAS.
- H. Bahmani Jalali, A. Pianetti, J. Zito, M. Imran, M. Campolucci, Y. P. Ivanov, F. Locardi, I. Infante, G. Divitini, S. Brovelli, L. Manna and F. Di Stasio, ACS Energy Lett., 2022, 7, 1850–1858 CrossRef CAS PubMed.
- H. W. Kim, J. H. Han, H. Ko, T. Samanta, D. G. Lee, D. W. Jeon, W. Kim, Y.-C. Chung, W. B. Im and S. B. Cho, ACS Energy Lett., 2023, 8, 3621–3630 CrossRef CAS.
- T. Samanta, A. N. Yadav, J. H. Han, M. Kim, S. W. Jang, N. S. M. Viswanath and W. B. Im, Adv. Opt. Mater., 2024, 12(23), 2400909 CrossRef CAS.
- Z. Ma, Z. Liu, S. Lu, L. Wang, X. Feng, D. Yang, K. Wang, G. Xiao, L. Zhang and S. A. Redfern, Nat. Commun., 2018, 9, 4506 CrossRef PubMed.
- W. Liu, Q. Lin, H. Li, K. Wu, I. Robel, J. M. Pietryga and V. I. Klimov, J. Am. Chem. Soc., 2016, 138, 14954–14961 CrossRef CAS PubMed.
- C. Coughlan, M. Ibanez, O. Dobrozhan, A. Singh, A. Cabot and K. M. Ryan, Chem. Rev., 2017, 117, 5865–6109 CrossRef CAS PubMed.
- J. H. Han, T. Samanta, H. B. Cho, S. W. Jang, N. S. M. Viswanath, Y. R. Kim, J. M. Seo and W. B. Im, Adv. Mater., 2023, 35, 2302442 CrossRef CAS PubMed.
- T. Samanta, J. H. Han, H. U. Lee, B. K. Cha, Y. M. Park, N. S. M. Viswanath, H. B. Cho, H. W. Kim, S. B. Cho and W. B. Im, Inorg. Chem., 2024, 63, 16483–16490 CrossRef CAS PubMed.
- L. Yang, H. Du, J. Li, Y. Luo, X. Lin, J. Pang, Y. Liu, L. Gao, S. He, J.-W. Kang, W. Liang, H. Song, J. Luo and J. Tang, Nat. Commun., 2024, 15, 6240 CrossRef CAS PubMed.
- W. Zhou, Y. Yu, P. Han, C. Li, T. Wu, Z. Ding, R. Liu, R. Zhang, C. Luo, H. Li, K. Zhao, K. Han and R. Lu, Adv. Mater., 2024, 36, 2302140 CrossRef CAS PubMed.
- L. Sun, B. Dong, J. Sun, Y. Wang, Y. Wang, S. Hu, B. Zhou, X. Bai, L. Xu, D. Zhou and H. Song, Adv. Mater., 2024, 36, 2310065 CrossRef CAS PubMed.
- S. Liu, J. Lyu, D. Zhou, X. Zhuang, Z. Shi, R. Sun, L. Liu, Y. Wu, B. Liu, D. Liu and H. Song, Adv. Funct. Mater., 2022, 32, 2112647 CrossRef CAS.
- T. Samanta, N. S. M. Viswanath, S. W. Jang, J. W. Min, H. B. Cho, J. H. Han and W. B. Im, Adv. Opt. Mater., 2023, 11, 2202744 CrossRef CAS.
- V. Morad, Y. Shynkarenko, S. Yakunin, A. Brumberg, R. D. Schaller and M. V. Kovalenko, J. Am. Chem. Soc., 2019, 141, 9764–9768 CrossRef CAS PubMed.
- M. Lee, D. H. D. Lee, S. V. Hong, H. Y. Woo, J. Y. Chae, D. W. Lee, M. J. Han and T. Paik, Adv. Opt. Mater., 2022, 10, 2201389 CrossRef.
- Y.-C. Lin, M. Bettinelli and M. Karlsson, Chem. Mater., 2019, 31, 3851–3862 CrossRef CAS.
- H. S. Ra, J. Ahn, J. Jang, T. W. Kim, S. H. Song, M. H. Jeong, S. H. Lee, T. Yoon, T. W. Yoon and S. Kim, Adv. Mater., 2022, 34, 2107468 CrossRef CAS PubMed.
- J. W. Min, T. Samanta, A. Y. Lee, Y. K. Jung, N. S. M. Viswanath, Y. R. Kim, H. B. Cho, J. Y. Moon, S. H. Jang, J. H. Kim and W. B. Im, Small, 2024, 20(43), 2402951 CrossRef CAS PubMed.
- Y.-C. Lin, M. Bettinelli and M. Karlsson, Chem. Mater., 2019, 31, 3851 CrossRef CAS.
- H.-S. Ra, J. Ahn, J. Jang, T. W. Kim, S. H. Song, M.-H. Jeong, S.-H. Lee, T. Yoon, T. W. Yoon, S. Kim, T. Taniguch, K. Watanabe, Y. J. Song, J.-S. Lee and D. K. Hwang, Adv. Mater., 2021, 34, 2107468 CrossRef PubMed.
- Y. H. Kim, P. Arunkumar, B. Y. Kim, S. Unithrattil, E. Kim, S.-H. Moon, J. Y. Hyun, K. H. Kim, D. Lee and J.-S. Lee, Nat. Mater., 2017, 16, 543–550 CrossRef CAS PubMed.
- W. Jang, S. Rasool, B. G. Kim, J. Kim, J. Yoon, S. Manzhos, H. K. Lee, I. Jeon and D. H. Wang, Adv. Funct. Mater., 2020, 30, 2001402 CrossRef CAS.
- B. Su, M. Li, E. Song and Z. Xia, Adv. Funct. Mater., 2021, 31, 2105316 CrossRef CAS.
- S. Saikia, A. Ghosh and A. Nag, Angew. Chem., Int. Ed., 2023, 135, e202307689 CrossRef.
- K. M. McCall, V. Morad, B. M. Benin and M. V. Kovalenko, ACS Mater. Lett., 2020, 2, 1218–1232 CrossRef CAS PubMed.
- X. Liang, W. Zhang, Y. Shi, W. Zhang, H. Yang, P. Huang, L. Li, Q. Zhang, W. Zheng and X. Chen, J. Mater. Chem. C, 2024, 12, 5538–5548 RSC.
- S. X. Bo Liu, J. Chang, Y. Li and B. Chen, Chin. J. Lumin., 2023, 45, 1095 Search PubMed.
- A. Noculak, V. Morad, K. M. McCall, S. Yakunin, Y. Shynkarenko, M. Wörle and M. V. Kovalenko, Chem. Mater., 2020, 32, 5118–5124 CrossRef CAS PubMed.
- Y. Zhou, W. Zhuang, Y. Hu, R. Liu, H. Xu, Y. Liu and Y. Li, Inorg. Chem., 2019, 58, 9108–9117 CrossRef CAS PubMed.
- H. Dong, L.-D. Sun and C.-H. Yan, J. Am. Chem. Soc., 2021, 143, 20546–20561 CrossRef CAS PubMed.
- L. Mao, Y. Wu, C. C. Stoumpos, B. Traore, C. Katan, J. Even, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 11956–11963 CrossRef CAS PubMed.
- T. Samanta, N. S. M. Viswanath, H. W. Kim, S. W. Jang, J. H. Han, S. B. Cho and W. B. Im, Chem. Eng. J., 2024, 484, 149697 CrossRef CAS.
- R. A. Walton, R. W. Matthews and C. K. Jørgensen, Inorg. Chim. Acta, 1967, 1, 355–359 CrossRef CAS.
- A. Vogler, A. Paukner and H. Kunkely, Coord. Chem. Rev., 1990, 97, 285–297 CrossRef CAS.
- H. Nikol and A. Vogler, J. Am. Chem. Soc., 1991, 113, 8988–8990 CrossRef CAS.
- E. Cavalli, P. Boutinaud, R. Mahiou, M. Bettinelli and P. Dorenbos, Inorg. Chem., 2010, 49, 4916–4921 CrossRef CAS PubMed.
- J. J. Joos, D. Van der Heggen, L. I. D. J. Martin, L. Amidani, P. F. Smet, Z. Barandiarán and L. Seijo, Nat. Commun., 2020, 11, 1–11 CrossRef PubMed.
- D. J. Starkenburg, P. M. Johns, J. E. Baciak, J. C. Nino and J. Xue, J. Appl. Phys., 2017, 122, 225502 CrossRef.
- Y. H. Kim, B. Y. Kim, N. S. M. Viswanath, P. Arunkumar and W. B. Im, J. Korean Ceram. Soc., 2017, 54, 422 CrossRef CAS.
- S. Afrin and P. Bollini, J. Phys. Chem. C, 2022, 127, 234 CrossRef.
- P. A. Tanner, C. S. K. Mak, N. M. Edelstein, K. M. Murdoch, G. Liu, J. Huang, L. Seijo and Z. Barandiarán, J. Am. Chem. Soc., 2003, 125, 13225 CrossRef CAS PubMed.
- N. S. M. Viswanath, G. Krishnamurthy Grandhi, H. Tran Huu, H. Choi, H. Jun Kim, S. Min Kim, H. You Kim, C.-J. Park and W. Bin Im, Chem. Eng. J., 2020, 420, 127664 CrossRef.
- S. Wang, Z. Song, Y. Kong, Z. Xia and Q. Liu, J. Lumin., 2018, 194, 461 CrossRef CAS.
- X. Qin, X. Liu, W. Huang, M. Bettinelli and X. Liu, Chem. Rev., 2017, 117, 4488 CrossRef CAS PubMed.
- J. Liu, M.-A. Morikawa and N. Kimizuka, J. Am. Chem. Soc., 2011, 133, 17370–17374 CrossRef CAS PubMed.
- P. A. Tanner, C. S. K. Mak, N. M. Edelstein, K. M. Murdoch, G. Liu, J. Huang, L. Seijo and Z. Barandiarán, J. Am. Chem. Soc., 2003, 125, 13225–13233 CrossRef CAS PubMed.
- H. Sun, X. Yang, P. Li, Y. Bai, Q. Meng, H. Zhao, Q. Wang, Z. Wen, L. Huang, D. Huang, W. W. Yu, H. Chen and F. Liu, Nano Lett., 2024, 24, 10355–10361 CrossRef CAS PubMed.
- Y. Shen, Y. Q. Li, K. Zhang, L. J. Zhang, F. M. Xie, L. Chen, X. Y. Cai, Y. Lu, H. Ren, X. Gao, H. Xie, H. Mao, S. Kera and J. X. Tang, Adv. Funct. Mater., 2022, 32, 2206574 CrossRef CAS.
- Y. Xia, Y. H. Lou, Y. H. Zhou, Y. R. Shi, K. L. Wang, L. Cai, C. H. Chen, F. Hu, Z. K. Wang and L. S. Liao, Adv. Funct. Mater., 2022, 32, 2208538 CrossRef CAS.
- R. Sun, D. Zhou, Y. Ding, Y. Wang, Y. Wang, X. Zhuang, S. Liu, N. Ding, T. Wang, W. Xu and H. Song, Light: Sci. Appl., 2022, 11, 340 CrossRef CAS PubMed.
- J. Guo, J. Sun, L. Hu, S. Fang, X. Ling, X. Zhang, Y. Wang, H. Huang, C. Han, C. Cazorla, Y. Yang, D. Chu, T. Wu, J. Yuan and W. Ma, Adv. Energy Mater., 2022, 12, 2200537 CrossRef CAS.
- J. Wang, J. Zhang, Y. Zhou, H. Liu, Q. Xue, X. Li, C.-C. Chueh, H.-L. Yip, Z. Zhu and A. K. Y. Jen, Nat. Commun., 2020, 11, 177 CrossRef CAS PubMed.
- W. Xu, Q. Hu, S. Bai, C. Bao, Y. Miao, Z. Yuan, T. Borzda, A. J. Barker, E. Tyukalova, Z. Hu, M. Kawecki, H. Wang, Z. Yan, X. Liu, X. Shi, K. Uvdal, M. Fahlman, W. Zhang, M. Duchamp, J.-M. Liu, A. Petrozza, J. Wang, L.-M. Liu, W. Huang and F. Gao, Nat. Photonics, 2019, 13, 418 CrossRef CAS.
- L. Sun, B. Dong, J. Sun, Y. Wang, R. Sun, S. Hu, B. Zhou, W. Xu, X. Bai and L. Xu, Laser Photonics Rev., 2023, 17, 2300045 CrossRef CAS.
- M. Plonska, J. Pisarska and W. A. Pisarski, J. Am. Ceram. Soc., 2017, 100, 1295 CrossRef CAS.
- G. Pan, X. Bai, D. Yang, X. Chen, P. Jing, S. Qu, L. Zhang, D. Zhou, J. Zhu, W. Xu, B. Dong and H. Song, Nano Lett., 2017, 17, 8005 CrossRef CAS PubMed.
- Q. Yang, Opt. Commun., 2022, 527, 128969 CrossRef.
- X. Li, B. Lou, X. Chen, M. Wang, H. Jiang, S. Lin, Z. Ma, M. Jia, Y. Han, Y. Tian, D. Wu, W. Xu, X. Li, C. Ma and Z. Shi, Mater. Horiz., 2024, 11, 1294–1304 RSC.
- Y. Wang, P. Dang, G. Li, H. Lian and J. Lin, J. Chin. Rare Earth Soc., 2023, 41, 39–53 Search PubMed.
- Y. G. Ping Chen, Y. He and N. Dai, Chin. J. Lumin., 2023, 44, 2128 Search PubMed.
- Y. Y. Guotao Xiang, Y. Zhang, M. Xiong, Q. Xu, H. Chen, Y. Chang and L. Yao, Chin. J. Lumin., 2024, 45, 952 Search PubMed.
- C. Sun, S. Su, Z. Gao, H. Liu, H. Wu, X. Shen and W. Bi, ACS Appl. Mater. Interfaces, 2019, 11, 8210 CrossRef CAS PubMed.
- Y. Jiang, J. Ma, Z. Ran, H. Zhong, D. Zhang and N. Hadjichristidis, Angew. Chem., Int. Ed., 2022, 61, 202208516 CrossRef PubMed.
- D. Xu, C. Liu, J. Yan, S. Yang and Y. Zhang, J. Phys. Chem. C, 2015, 119, 6852 CrossRef CAS.
- R. Z. Yucong Zhao, J. Wang, G. Gong and J. Huang, J. Chin. Rare Earth Soc., 2024, 005, 041 Search PubMed.
- X.-X. Guo, J.-H. Chen, J.-B. Luo, J.-H. Wei, Z.-Z. Zhang, Z.-L. He, Q.-P. Peng and D.-B. Kuang, Adv. Opt. Mater., 2024, 12, 2400681 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |