Frustrated Lewis pairs: a new strategy to small molecule activation and hydrogenation catalysis
Douglas W.
Stephan
*
Department of Chemistry, University of Toronto, 80 St. George St., Toronto, Ontario, Canada M5S 3H6. E-mail: dstephan@chem.utoronto.ca; Tel: +1 416 946 3294
First published on 18th February 2009
Abstract
The combination of Lewis acids and bases that are sterically precluded from forming Lewis acid–base adducts, termed Frustrated Lewis pairs provide a unique route to the activation of small molecules and applications in catalysis.
 Douglas W. Stephan Douglas W. Stephan | Doug Stephan attended McMaster University and the University of Western Ontario earning a PhD in 1980. He was a NATO postdoctoral Fellow at Harvard University (R. H. Holm) and in 1982 joined the faculty at Windsor, and was subsequently promoted through the ranks to University Professor in 2002. Author of over 200 scientific papers, his research interest focused on early transition metal and main group chemistry and catalysis. His work in olefin polymerization resulted in a number of patents and has been commercialized. Most recently Stephan's group uncovered “frustrated Lewis pairs” and developed the first main group hydrogenation catalysts. Stephan received the 2001 Alcan Award (Canadian Society for Chemistry), a 2003 NSERC of Canada Synergy Award, a 2003 Humboldt Research Award (Germany), the 2004 Ciapetta Lectureship (North American Catalysis Society) and the 2005 LeSueur Memorial Award from the Society for Chemical Industry and in 2005 was named a Canada Research Chair at Windsor. In January 2008, he took up a position as Professor of Chemistry and Canada Research Chair in Inorganic Materials and Catalysis at the University of Toronto. |
Introduction
The concept of Lewis acids and bases is fundamental in inorganic chemistry.1 Indeed, this notion is a guiding principle for the understanding of both bonding and reactivity interactions in both main group and transition metals chemistry. For example, Lewis donor–acceptor interactions account for the formation of acid–base adducts and transition metal–ligand complexes. The principles of Lewis acidity and basicity also extend to and help shape our understanding of organic, organometallic, solid state chemistry and surface science. Clearly, the influence of this simple rationale for bonding involving filled and vacant molecular orbitals put forward by Lewis some 90 years ago impacts across the discipline. Indeed it continues to be one of the most powerful tools for both understanding and predicting a broad spectrum of chemical reactivity.In our explorations of olefin polymerization catalysts we have extensively exploited Lewis acid activators. As part of these efforts we began to explore simple donor–acceptor adducts involving such activators and the impact of steric bulk. We asked could steric congestion preclude Lewis acid–base adduct formation? If the answer was yes, then could such sterically frustrated Lewis pairs (FLPs) prompt alternate and previously unknown reactivity as a result of the unquenched of Lewis acidity and basicity?
Historical perspective
In pursuing this avenue of thinking, we note that H. C. Brown and co-workers commented in 1942 that steric demands can intervene in the formation of donor–acceptor adducts of pyridines with BMe3.2 They showed that while adduct formation was seen for pyridine, lutidine failed to form an adduct. Molecular models provided a rational for this observation based on steric interactions of methyl groups of lutidine and BMe3 (Fig. 1). Some years later in 1950, the research group of Wittig described the reaction of Ph3CNa with THF(BPh3).3 In this case, displacement of THF was not observed as might be anticipated on the basis of Lewis acid–base chemistry; rather the trityl anion effected THF ring opening affording the anion [Ph3C(CH2)4OBPh3]−. These findings by Nobel laureates did not garner much attention at the time, but in some sense these works were prophetic for us, as these represent to our knowledge, the first references to the curious and unique behaviour of sterically frustrated Lewis pairs.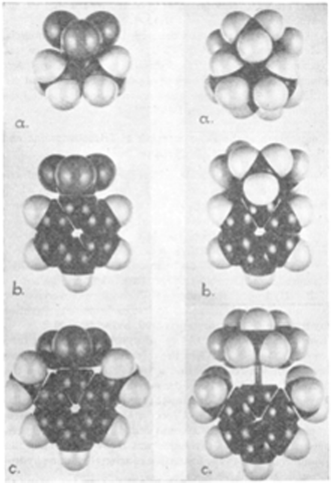 | ||
| Fig. 1 Molecular models of left (a) NMe3BF3, (b) C5H5NBF3, (c) C5H3Me2NBF3; right (a) NMe3BMe3, (b) C5H5NBMe3, (c) C5H3Me2NBMe3. Reprinted with permission from ref. 2. © 2006 American Chemical Society. | ||
In the 1960's non-classical reaction of sterically encumbered amines with trityl cation were shown to result not in amine-quaternization but rather proton abstraction to give an iminium cation.4 Subsequently reactions of pyridine with trityl cation was reported to give rise to attack at the carbonpara to the carbocation,5 although this report was subsequent disputed.6 (Scheme 1).
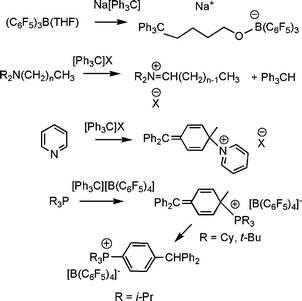 | ||
| Scheme 1 Non-classical Lewis acid–base reactions of trityl anion and cation with various Lewis donors. | ||
In 1998, Erker and his co-workers in their continuing interest in the chemistry of Lewis acids, described a related example involving the combination of the Lewis acid B(C6F5)3 and the sterically encumbered ylide, Ph3PCHPh. While the simple ylide/borane adduct (Ph3PCHPh)B(C6F5)3 was shown to form at room temperature, this species was also shown to undergo thermal rearrangement resulting in the attack by ylide at the para-position of the one of the fluorinated arene rings to give the species [Ph3PCHPh(C6F4)BF(C6F5)2]. (Scheme 2).7 Again, these curious reactions were attributed to both the steric congestion in the Lewis acid–base adduct and the canonical form of the borane that places considerable positive charge on the para-carbon of the fluorinated arene rings.
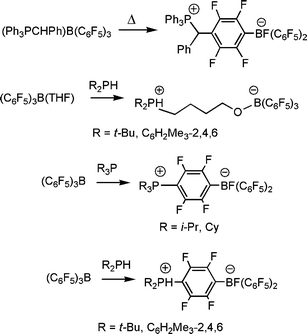 | ||
| Scheme 2 Non-classical Lewis acid–base reactions of B(C6F5)3 with phosphines. | ||
Uncovering FLPs
We began to explore the concept of FLPs as a result of our interest in the use of boranes and borate salts as activators for olefin polymerization. We were examining the simple and corresponding reaction of phosphines with Lewis acid activators.In a related study we also examined the reactions of trityl borates with Lewis donors, such as amine, pyridines and phosphines.8 While classical Lewis acid–base adduct formation yielding species of the form [LCPh3][B(C6F5)4] was observed in most cases, it was noted that the reactions of sterically encumbered phosphines such as PR3, (R = i-Pr, Cy, t-Bu) proceeded differently. In these cases, reactions with [CPh3][B(C6F5)4] prompted nucleophilic aromatic substitution at the carbonpara- to the carbocation, affording either [(R3PC6H5)CPh2][B(C6F5)4] (R= Cy, t-Bu) or [(i-Pr3PC6H4)Ph2CH][B(C6F5)4] (Scheme 1).8 Indeed the former species were well characterized by NMR spectroscopy, while X-ray crystallography support the formation of the latter salts.
In studying reactions of (THF)B(C6F5)3 with phosphines, we fully anticipated a straightforward reaction in which a phosphine displaces THF from the coordination sphere of B, and indeed this was observed in most cases. However, for sterically encumbered phosphines, that are incapable of formation of a classic Lewis adduct with B(C6F5)3 the reactions follows an alternate reaction pathway effecting nucleophilic ring opening of THF to form the butoxy-tethered phosphonium-borates of the form R′R2PC4H8OB(C6F5)3 (Scheme 2).9 These reactions were reminiscent of the results reported by Wittig and Ruckert some 55 years prior. Nonetheless, these results offered a unique strategy to simply generate a library of zwitterionic phosphonium- and anionic phosphine-borates.
In the corresponding reactions of the Lewis acid B(C6F5)3 with phosphines, we found a similar dependence on steric demands. While the formation of simple phosphine-borane adducts was well established, we found that sterically hindered tertiary or secondary phosphines R3P (R = i-Pr, Cy) or R2PH (R′ = t-Bu, C6H2Me3-2,4,6) react with B(C6F5)3via nucleophilic aromatic substitution at the carbonpara to B with concurrent F migration to B, to give the white, air and moisture stable zwitterions formulated as [R3P(C6F4)BF(C6F5)2] (Scheme 2) or [R′2PH(C6F4)BF(C6F5)2].10 This latter reactivity parallels that previously reported for sterically encumbered phosphines and ylides with trityl and B(C6F5)3, respectively.
The synthesis of these para-substituted derivatives of B(C6F5)3 provides facile access to a series of species in which the nature of the substituents alters the Lewis acidity at B. The salts of the form [R2PH(C6F4)BF(C6F5)2] react with Grignard reagents to give the phosphino-boranes R2P(C6F4)B(C6F5)2.10 Alternatively, reaction of the species [R2PH(C6F4)BH(C6F5)2] with [Ph3C][B(C6F5)4] afforded compounds of the form [(R3P)(C6F4)B(C6F5)2][B(C6F5)4] and [(R2PH)(C6F4)B(C6F5)2] [B(C6F5)4] in which the cations are phosphonium-boranes (Scheme 3).10 Collectively, these synthetic pathways afford a family of borane derivatives with both electron donating phosphine or electron withdrawing phosphonium groups in the para-position to a Lewis acidic B center. The variations in the Lewis acidity were assessed employing the Gutmann–Beckett and Childs–Lewis acidity tests. These data confirm that the cationic boranes are significantly more Lewis acidic than B(C6F5)3. Conversely the phosphine-boranes give rise to B centers with significantly lower Lewis acidity.10 Thus, these synthetic routes provide an facile method to tune Lewis acidity of B(C6F5)3 for applications in reactivity.
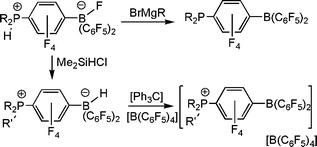
H2Activation
In further pursuing the chemistry of these unusual B/P systems, exchange of the boron bound fluoride for hydride in [R′2PH(C6F4)BF(C6F5)2] was readily achieved by treatment with Me2Si(H)Cl, yielding [R′2PH(C6F4)BH(C6F5)2] This species was targeted as it would be a unusual zwitterion containing both a proton and hydride at the phosphonium and borate centers respectively. It was perhaps not surprising that thermolysis of the species [(C6H2Me3-2,4,6)2PH(C6F4)BH(C6F5)2] resulted in the liberation of H2 and the formation of the phosphino-borane (C6H2Me3-2,4,6)2P(C6F4)B(C6F5)2.11 The B and P centers in this retain sufficient steric bulk to preclude aggregation. However, perhaps the most astounding observation is seen upon exposure of a solution of this phosphino-borane to H2 at 25 °C. This led to the rapid and facile reformation of the zwitterionic salt [(C6H2Me3-2,4,6)2PH(C6F4)BH(C6F5)2] (Scheme 4). This system represents the first non-transition metal system that releases and takes up hydrogen. It is also noteworthy that the species tBu2PH(C6F4)BH(C6F5)2 does not release H2 even on heating to 150 °C presumably a result of the significantly higher basicity of the P center.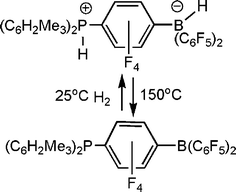 | ||
| Scheme 4 Reversible activation of H2. | ||
It is noteworthy that the phosphine-borane, (C6H2Me3-2,4,6)2P(C6F4)B(C6F5)2 shows no sign of aggregation via Lewis acid–base interactions, presumably a result of the steric demands of the substituents on the donor and acceptor atoms respectively. Thus, the unique reactivity with H2 was ascribed to the presence of this unquenched Lewis acidity and basicity arising from the FLP.
Simple phosphine-boraneFLP activation of H2
Our observation of the linked phosphine-borane systems described above prompted the question: could H2 be activated by a simple combinations of phosphines and boranes?12 Indeed, this is readily achieved for phosphine-borane pairs where the Lewis acidity and basicity is not quenched by donor–acceptor adduct formation. For example, 1 : 1 mixtures of R3P (R = tBu, C6H2Me3) with B(C6F5)3 show no evidence of adduct formation by NMR methods, even on cooling to −50 °C. Exposure of such mixtures to 1 atm. H2 however, prompts immediate reaction to form the salt [R3PH][HB(C6F5)3] (Scheme 5).12 Similarly, no adduct is formed on combination of tBu3P and BPh3, however this system also reacts with H2 to give [tBu3PH][HBPh3] albeit more slowly and in only 33% isolated yield. The analogous reactions of (C6H2Me3)3P and BPh3, (C6F5)3P and B(C6F5)3 or tBu3P and B(C6H2Me3)3 do not form adducts and in that sense are FLPs. However these FLPs do not react with H2 at 25 °C. This suggests that modifications to the net acidity and basicity may be used to favour or preclude the reaction of FLPs with H2. It is noteworthy that the conceptually related reactions of organometallic alkali metal species with H2 were first shown to generate metal hydrides and alkanes,13,14 some 70 years ago. However in these cases, it is the inherent polarity of the metal alkyls that prompts reactivity, whereas, the polarity of FLP systems is a result of steric conflicts.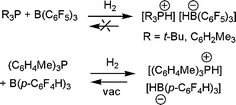 | ||
| Scheme 5 Activation of H2 by phosphine-borane combinations. | ||
It is noteworthy that the salts [R3PH][HB(C6F5)3], do not liberate H2 upon heating to above 100 °C. Given the ability to adjust the phosphine and borane and shut down reactivity with H2, it seemed reasonable to pursue appropriate modifications to generate a simple salt capable of reversible H2activation. To that end, we noted that a borane that avoided nucleophilic aromatic para-attack would broaden the range of phosphines that could be utilized. At the same time, the borane should be less Lewis acidic than B(C6F5)3 to facilitate loss of hydride in the reverse reaction, but be Lewis acidic enough to activate H2 in the first place. For these reasons we prepared the borane B(p-C6F4H)3.15 In selecting the phosphine, we sought a sterically encumbered ligand, yet one that was of similar basicity to P(C6H2Me3)3. Here again a simple modification was selected, namely P(o-C6H4Me)3. Indeed, the combination of B(p-C6F4H)3/P(o-C6H4Me)3 activates H2 at 25 °C as expected to give [HP(o-C6H4Me)3][HB(p-C6F4H)3] (Scheme 5).15 Interestingly placement of this salt under vacuum at 25 °C resulted in the loss of H2, albeit slow, requiring 9 d to effect 85% loss. Nonetheless, 85% loss of H2 was observed on heating this salt to 80 °C for 12 h.
The intimate details of the mechanism of both H2activation and H2 evolution are certainly questions of interest. While early mechanistic study of the loss of H2 from (C6H2Me3-2,4,6)2PH(C6F4)BH(C6F5)2 were reported to exhibit first order kinetics, the nature of the process remains the subject of speculation. The activation of H2 by the simple phosphine-borane system was proposed to involve initial Lewis acid activation of H2 followed by protonation of the Lewis base. This view is supported for early computations for BH3–H2 adducts. It is noteworthy that matrix isolation work demonstrates the interaction of phosphine with H2, suggesting the possibility of an alternative mechanism. Computational studies by Papai and co-workers16 suggest the generation of an “encounter complex” between the phosphine and borane, stabilized by H⋯F interactions, where these centres approach but fail to form a dative bond as a result of steric congestion. It is further proposed that H2 reacts with this ‘species’ (Fig. 2). A related mechanism has been envisioned for R2P(C6F4)B(C6F5)2.17
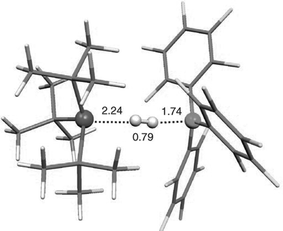 | ||
| Fig. 2 Computational model of phosphine-borane activation of H2. Reproduced with permission from ref. 13. © 2008 Wiley-VCH Verlag GmbH & Co. KGaA | ||
Interest in the potential of ferrocene backbone for system design prompted Erker and co-workers to probe ferrocenophane-based FLPs. The reaction of a phosphino-ferrocenophane, B(C6F5)3 and H2 affords an unfunctionalized ferrocenophane product and a phosphine-borane adduct. This reaction is thought to proceed via a transient phosphonium hydridoborate, however the phosphonium fragment is readily displaced by hydride transfer from anion (Scheme 6).18
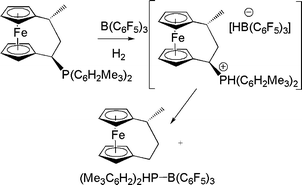 | ||
| Scheme 6 Reactions of phosphinioferrocenophane-B(C6F5)3 with H2. | ||
In very much related work we have recently described related reaction of mono- and bis-ferrocenylphosphines.19 In cases where steric demands about P preclude Lewis acid–base adducts with B(C6F5)3, nucleophilic aromatic substitution is observed. Alternatively, when steric congestion is more severe, a FLP results and reaction with H2 is observed to give ferrocenylphosphonium borates. In the case of (η5-C5H4PtBuR2)2Fe these two reaction pathways can be used in tandem to give [(η5-C5H4PR2)Fe(η5-C5H4PR2(C6F4)BF(C6F5)2] and subsequently [(η5-C5H4PHtBu2)Fe(η5-C5H4PtBu2(C6F4)-BH(C6F5)2][HB(C6F5)3] (Scheme 7).19
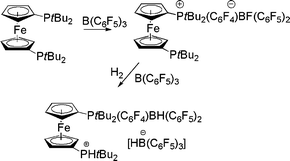 | ||
| Scheme 7 Reactions of bisphosphinoferrocene with B(C6F5)3 with H2. | ||
Carbene based FLP systems
Bertrand and co-workers20 conceived a elegant extension to the notion of FLPs. They recognized that a carbene is a unique form of FLP where the donor and acceptor sites reside on the same atom yet the orthogonal relationship of the lone pair and the vacant p-orbital results in unquenched Lewis acidity and basicity. Based on this notion, these researchers showed that some carbene derivatives react both with H2 or NH3 resulting on the H–H and N–H heterolytic bond cleavage converting the carbene carbon to a methylene (CH2) or a amino-methine (CH(NH2)) fragments (Scheme 8).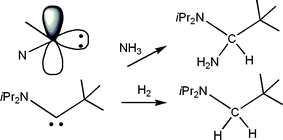 | ||
| Scheme 8 Reactions of amino-carbenes with H2 and NH3. | ||
In very recent related work, we21 and the research group of Tamm22 simultaneously showed that sterically hindered N-heterocyclic carbenes (NHCs) can act as the base component of an FLP. Thus, reactions of tBuNHC do not bind to B(C6F5)3, yet upon addition of H2 the mixture promptly forms the imidazolium hydridoborate salt (Scheme 9). Tamm et al. showed computationally that the transition state in the activation of H2 by carbene and borane was similar to that proposed for the corresponding reaction with phosphine as the base. We also explored the addition of aryl amines to the NHC/B(C6F5)3 and showed that a rapid reactions effect N–H activation and formation of imidazolium amidoborates. While alkylamines were expected to react in a similar sense, the observed products are C6F5H and the amidoboranes R2NB(C6F5)2 (Scheme 10). Indeed, this latter reaction proceeds rapidly and in the process of catalytic amounts of NHC.21
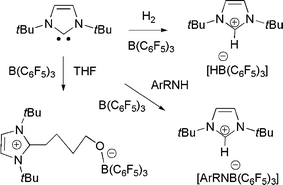 | ||
| Scheme 9 Reactions of N-heterocyclic carbenes with H2, THF and amines. | ||
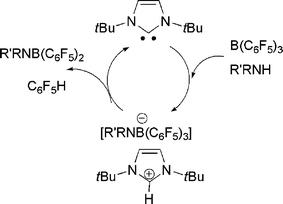 | ||
| Scheme 10 Carbene catalyzed conversion of alkylamine-borane to amidoborane. | ||
Activation of other small molecules
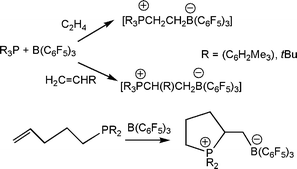
The observation of the activation of H2 prompted questions regarding the ability of FLP to react with other small molecules. Addition of olefins to the combination of tBu3P and B(C6F5)3 resulted in the formation of the zwitterionic species [tBu3P(C2H4)B(C6F5)3] (Scheme 9).23 In the same manner, the analogous products derived from the reactions of propylene and 1-hexene with tBu3P and B(C6F5)3, afforded the salts, [tBu3P(CH(R)CH2B(C6F5)3] (R = CH3, C4H9), respectively. Subsequent deprotonation of these latter materials offers a facile route to a series of anionic phosphines. In addition, this reaction can also be effected in an intramolecular manner and thus reaction of CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(CH2)3PR2 (R = tBu, C6H2Me3) with B(C6F5)3 generates the cyclic phosphonium borate [R2PCH(C2H4)CH2B(C6F5)3] (R = tBu, C6H2Me3) (Scheme 11).23 In each of these products derived from olefins, the regiochemistry of addition is selective for B addition to the terminal methylene unit while the substituted carbon adds to P. These reactions are also thought to be initiated by interaction of the Lewis acid with the olefin, prompting attack by the phosphine. This view is supported by IR studies which demonstrate the formation of BF3-ethylene and BF3-propylene complexes in an Argon matrix at 93–125 K.24 Similarly, computational studies have also suggested weak π-donation complexes for ethylene-alane and borane adducts.25,26
CH(CH2)3PR2 (R = tBu, C6H2Me3) with B(C6F5)3 generates the cyclic phosphonium borate [R2PCH(C2H4)CH2B(C6F5)3] (R = tBu, C6H2Me3) (Scheme 11).23 In each of these products derived from olefins, the regiochemistry of addition is selective for B addition to the terminal methylene unit while the substituted carbon adds to P. These reactions are also thought to be initiated by interaction of the Lewis acid with the olefin, prompting attack by the phosphine. This view is supported by IR studies which demonstrate the formation of BF3-ethylene and BF3-propylene complexes in an Argon matrix at 93–125 K.24 Similarly, computational studies have also suggested weak π-donation complexes for ethylene-alane and borane adducts.25,26
In a closely relate series of reactions, combination of an FLPs with dienes also results in addition reactions. In the case of the reactions of 1,3-cyclohexadiene and 2,3-dimethylbutadiene, 1,4 addition products result (Scheme 12).27 While these reaction mixtures also contain other species that may arise from other stereoisomers or 1,2 addition products, the 1,4 addition products are the major species and are isolated in typically 50–60% yield.
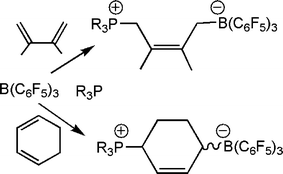 | ||
| Scheme 12 Reactions of phosphine-borane and dienes. | ||
Exploring the activation of other small molecules led us to examine the reactions of the FLP, tBu2RP (R = tBu, C6H4Ph) and B(C6F5)3 with catechol-borane. In these cases, the products [tBu2RPBO2C6H4)][HB(C6F5)3] were isolated (Scheme 13).28 DFT calculations were used to address the nature of this cations as they could be viewed either as a phosphine stabilized borenium cation or alternatively as a borylphosphonium cation. The computations support the localization of the positive charge on P and thus the latter formulation. Nonetheless, this cation is the first, three coordinate B cation, ligated by O-donors.29
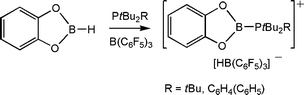 | ||
| Scheme 13 Activation of B–H bond by phosphine-borane. | ||
Metal-free catalytic hydrogenation
While some of the above chemistry may be viewed as idiosyncratic reactivity resulting from steric congestion, the ability to activate H2 by a simple main group system is a significant landmark finding. Indeed, the obvious extension of this discovery is the extension to metal-free hydrogenation catalysis. Conceptually this notion is reminiscent of the Noyori-systems,30–32 which effect the heterolytic cleavage of H2 to form a metal hydride and a protonated ligand. The present FLP systems behave in much the same fashion, although the metal is replaced with a Lewis acid. Nonetheless, the potential for catalysis resides in the prospect of proton and hydride transfer from for example a phosphonium borate to a substrate. This would regenerate the phosphine-borane, which would then further activate H2 to propagate the catalytic reduction.In our initial exploration of this notion, the phosphonium borates (R2PH)(C6F4)BH(C6F5)2 (R = C6H2Me3, tBu) were shown to react stoichiometrically with an imine to afford the amine adduct (R2P)(C6F4)B(C6F5)2(NHRCH2R′). This initial result augured well for catalysis as it infers that both proton and hydride were transferred to the substrate molecule. To achieve a catalytic process, all that was required was to simply break the B–N dative bond, as this would provide the phosphino-borane species which could subsequently activate H2, forming the phosphonium borate, which would react further with substrate. This was readily achieved by simple heating the solutions to between 80–120 °C. In this fashion, thermal dissociation of amine occurs and catalytic reduction of imines occurs under 1–5 atm. of H2 (Scheme 14).33 These reductions were achieved readily when the imine itself incorporated bulky substituents on N. This presumably facilitates dissociation of the amine from the B center. Where this was not the case, only stoichiometric reduction was observed. In addition, it has been demonstrated that these systems are “living” in the sense that one can monitor the conversion of the starting material to hydrogenated product, and subsequently add more substrate and reduction resumes immediately. Also, catalytic reductive ring opening of a non-activated N-aryl aziridine functionality is readily achieved under similar conditions. The mechanism of these reductions was shown to begin with protonation of the imine or aziridine by the phosphonium centre followed by borohydride attack of the iminium salt (Scheme 14).33
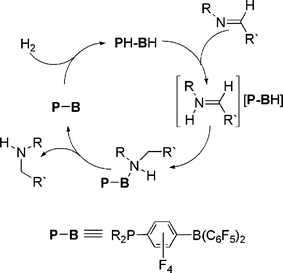 | ||
| Scheme 14 Catalytic reduction of imines by phosphonium borate. | ||
Sterically less encumbered imines as well as nitriles can be effectively reduced by employing B(C6F5)3 as a protecting group. Thus, reduction of adducts such as (PhCHNCH2Ph)B(C6F5)3 or (RC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N)B(C6F5)3 employing the phosphonium borate catalysts, proceeds smoothly to give the corresponding amine-B(C6F5)3 adducts. This strategy is effective as B(C6F5)3 is a much strong Lewis acid than the phosphino-boranes generated from the catalysts.33
N)B(C6F5)3 employing the phosphonium borate catalysts, proceeds smoothly to give the corresponding amine-B(C6F5)3 adducts. This strategy is effective as B(C6F5)3 is a much strong Lewis acid than the phosphino-boranes generated from the catalysts.33
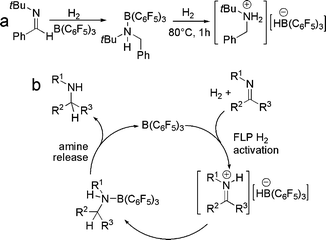
We have also probed the question: can the substrate act as the base of an FLP to catalyze reductions? Indeed, hindered imines themselves can act as the Lewis base partner of an FLP to effect the catalytic reduction of imine. Thus, the simple combination of an imine substrate, B(C6F5)3 and H2 results in reduction of imine to amine under conditions similar to those described for the phosphino-borane catalysts. Related reductions were subsequently reported by Chen and Klankermayer.34 Mechanistic studies of such reactions show that the resulting amine in combination with B(C6F5)3 acts to split H2, as the resting form of the catalyst in these imine reductions is the ammonium borate [R′RNH2][H B(C6F5)3] (Scheme 15).35 Repo et al. subsequently published a related result in which sterically encumbered amines and B(C6F5)3 were shown to heterolyically split H2.36 Interestingly, Chen and Klankermayer also reported an attempt at the asymmetric reduction of PhNCPh(Me) employing the chiral borane37 derived from α-pinene, (α-pinenyl)B(C6F5)2. In this a 13% enantiomeric excess was observed.34
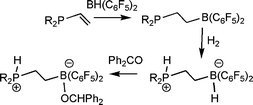
In developing new FLP systems, the Erker group has developed a facile route to the ethylene linked (C6H2Me3)2P(C2H4)B(C6F5)2viahydroboration of the vinylphosphine.38 This species was shown to activation H2 in a similar fashion, to give the corresponding phosphonium borate. Interestingly this reaction was not thermally reversible, although this salt does react with benzaldehyde to give [(C6H2Me3)2PH(C2H4)B(OR)(C6F5)2] (Scheme 16).
 | ||
| Scheme 16 Synthesis and reactivity of alkyl-linked phosphino-boranes. | ||
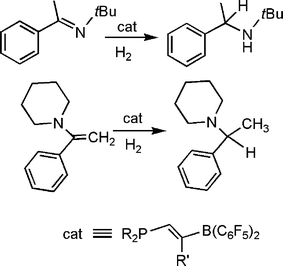
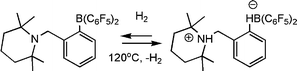
In more recent efforts, Erker and co-workers have prepared the related olefinic linked phosphine borane (C6H2Me3)2P(CHCR)B(C6F5)2viahydroboration of the akynylphosphine (Scheme 17).39 These species also activate H2 and were employed to effect the catalytic reduction of imines and enamines at room temperature. In a similar fashion, the research groups of Repo and Rieger have very recently reported the catalytic hydrogenation of imines and enamines, using a linked amine-borane species derived from tetramethylpiperidine (Scheme 18).40
 | ||
| Scheme 18 Synthesis and reactivity of alkyl-linked amino-boranes. | ||
The Erker research group has most recently developed a interesting new FLP based on the 1,8 diphosphino-napthalene phosphine, 1,8(Ph2P)2(C10H6) (Scheme 19).41 In combination with B(C6F5)3, this diphosphine has been shown to activate H2. At 60 °C the resulting phosphonium borate loses H2 to regenerate the FLP. This system has also been shown to catalytically reduce silyl-enol ethers at 25 °C under H2 pressure of 2 bar.
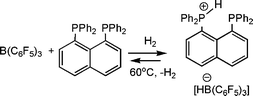 | ||
| Scheme 19 Synthesis and reactivity of alkyl-linked phosphino-boranes. | ||
Future considerations
The discovery of FLP reactivity brings a new perspective to the realm of small molecule activation and applications in catalysis. In particular, the potential for metal-free hydrogenation catalysis has been demonstrated. This approach holds the potential to replace costly precious metal catalysts and thus diminish the environmental impact from heavy metal pollutants. It is noteworthy however other researchers have and continue to explore other avenues to metal-free transformations. For example, previous studies have achieved metal-free hydrogenation of benzophenone, however rather forcing conditions were required (20 mol% KOt-Bu, 200 °C and >100 bar H2).42 Similarly, trialkylboranes and H2 afford hydroboration/hydrogenolysis reactions at >200 °C and 15 atm.43–46 Efforts toward organocatalysts for the hydrogenation of enones and imines have utilized a Hantzsch ester as the stoichiometric source of H2.47–51In addition to the obvious applications in catalysis, the chemistry of FLPs with H2 may be useful in hydrogen transport. The requirement of steric congestion is a serious limitation for applications in hydrogen storage where high H2 content by mass is required. Nonetheless, the discovery does offer new catalysts to consider for use in the liberation of H2 or the regeneration of hydrogen storage materials. Although only in their infancy, FLPs also offer new strategies to the reactivity of other small molecules. The chemistry of the resulting species may also provide unique routes to new synthons and processes. Despite the fact that FLPs were only described 2 years ago, it is clear that the data to date demonstrate that a window of opportunity for new reactivity and catalysis has been opened.
Acknowledgements
The contributions and helpful discussions with a number of very talented students and postdoctoral fellows in my group is gratefully acknowledged In particular, the contributions and discussions of Dr Preston Chase, Dr Greg Welch, Dr Jenny McCahill, Lourisa Cabrera, Dr Emily Hollink, Dr Matthias Ullrich, Meghan Dureen, Rebecca Neu, Xiaoxi Zhao, Dr Alberto Ramos, Kelvin Seto, Dr Zach Heiden, Dr Consuelo Herrera and Dr Edwin Otten were invaluable. Financial support from NSERC of Canada is gratefully acknowledged.Notes and references
- G. N. Lewis, Valence and the Structure of Atoms and Molecules, Chemical Catalogue Company, Inc., New York, 1923 Search PubMed.
- H. C. Brown, H. I. Schlesinger and S. Z. Cardon, J. Am. Chem. Soc., 1942, 64, 325 CrossRef CAS.
- G. Wittig and A. Ruckert, Liebigs Ann. Chem., 1950, 566, 101–113 CrossRef CAS.
- R. Damico and C. D. Broadus, J. Org. Chem., 1966, 31, 1607 CrossRef CAS.
- H. Lankamp, W. T. Nauta and C. MacLean, Tetrahedron Lett., 1968, 249 CrossRef CAS.
- Y. Okamoto and Y. Shimakawa, J. Org. Chem., 1970, 35, 3752 CrossRef CAS.
- S. Doering, G. Erker, R. Fröhlich, O. Meyer and K. Bergander, Organometallics, 1998, 17, 2183 CrossRef.
- L. Cabrera, G. C. Welch, J. D. Masuda, P. Wei and D. W. Stephan, Inorg. Chim. Acta, 2006, 359, 3066–3071 CrossRef CAS.
- G. C. Welch, J. D. Masuda and D. W. Stephan, Inorg. Chem., 2006, 45, 478–480 CrossRef CAS.
- G. C. Welch, L. Cabrera, P. A. Chase, E. Hollink, J. D. Masuda, P. Wei and D. W. Stephan, Dalton Trans., 2007, 3407 RSC.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS.
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS.
- H. Gillman, A. L. Jacoby and H. Ludeman, J. Am. Chem. Soc., 1938, 60, 2336 CrossRef CAS.
- P. A. A. Klusener, L. Brandsma, H. D. Verkruijsse, P. v. R. Schleyer, T. Freidl and R. Pi, Angew. Chem., Int. Ed. Engl., 1986, 25, 465 CrossRef.
- M. Ullrich, A. J. Lough and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 52–53 CrossRef CAS.
- T. A. Rokob, A. Hamza, A. Stirling, T. Soós and I. Pápai, Angew. Chem., Int. Ed., 2008, 47, 2435 CrossRef CAS.
- Y. Guo and S. Li, Inorg. Chem., 2008, 47, 6212 CrossRef CAS.
- D. P. Huber, G. Kehr, K. Bergander, R. Fröhlich, G. Erker, S. Tanino, Y. Ohki and K. Tatsumi, Organometallics, 2008, 27, 5279 CrossRef CAS.
- A. Ramos, A. J. Lough and D. W. Stephan, Chem. Commun., 2009, 1118–1120 RSC.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439 CrossRef CAS.
- P. A. Chase and D. W. Stephan, Angew. Chem., Int. Ed., 2008, 47, 7433 CrossRef CAS.
- D. Holschumacher, T. Bannenberg, C. G. Hrib, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2008, 47, 7428 CrossRef CAS.
- J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 4968 CrossRef CAS.
- W. A. Herrebout and B. J. v. d. Veken, J. Am. Chem. Soc., 1997, 119, 10446 CrossRef CAS.
- P. Tarakeshwar and K. S. Kim, J. Phys. Chem. A., 1999, 103, 9116 CrossRef CAS.
- P. Tarakeshwar, S. J. Lee, J. Y. Lee and K. S. Kim, J. Phys. Chem. A., 1999, 103, 184 CAS.
- K. Seto, M. Ullrich, A. J. Lough and D. W. Stephan, submitted for publication, 2008 Search PubMed.
- M. A. Dureen and D. W. Stephan, Chem. Commun., 2008, 4303 RSC.
- W. E. Piers, S. C. Bourke and K. D. Conroy, Angew. Chem., Int. Ed., 2005, 44, 5016–5036 CrossRef CAS.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS.
- R. Noyori, M. Kitamura and T. Ohkuma, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5356 CrossRef CAS.
- C. P. Casey, G. A. Bikzhanova and I. A. Guzei, J. Am. Chem. Soc., 2006, 128, 2286 CrossRef CAS.
- P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 49, 8050 CrossRef.
- D. Chen and J. Klankermayer, Chem. Commun., 2130 Search PubMed.
- P. A. Chase, T. Jurca and D. W. Stephan, Chem. Commun., 2008, 1701 RSC.
- V. Sumerin, F. Schulz, M. Nieger, M. Leskela, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001 CAS.
- D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492–5503 CrossRef CAS.
- P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fröhlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072–5074 RSC.
- P. Spies, S. Schewendemann, S. Lange, G. Kehr, R. Frohlich and G. Erker, Angew. Chem., Int. Ed., 2008, 47, 7543 CrossRef CAS.
- V. Sumerin, F. Schulz, M. Atsumi, C. Wang, M. Nieger, M. Leskela, T. Repo, P. Pyykko and B. Rieger, J. Am. Chem. Soc., 2008, 130, 14117 CrossRef CAS.
- H. Wang, R. Fröhlich, G. Kehr and G. Erker, Chem. Commun., 2008, 5966 RSC.
- A. Berkessel, T. J. S. Schubert and T. N. Mueller, J. Am. Chem. Soc., 2002, 124, 8693 CrossRef CAS.
- E. J. DeWitt, F. L. Ramp and L. E. Trapasso, J. Am. Chem. Soc., 1961, 83, 4672 CrossRef CAS.
- M. W. Haenel, J. Narangerel, U. B. Richter and A. Rufińska, Angew. Chem., Int. Ed., 2006, 45, 1061 CrossRef CAS.
- R. Koester, G. Bruno and P. Binger, Justus Liebigs Ann. Chem., 1961, 644, 1 CrossRef CAS.
- F. L. Ramp, E. J. DeWitt and L. E. Trapasso, J. Org. Chem., 1962, 27, 4368 CrossRef CAS.
- H. Adolfsson, Angew. Chem., Int. Ed., 2005, 44, 3340 CrossRef CAS.
- P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS.
- M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 3683 CrossRef.
- J. B. Tuttle, S. G. Ouellet and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 12662 CrossRef CAS.
- J. W. Yang, M. T. Hechavarria Fonseca and B. List, Angew. Chem., Int. Ed., 2004, 43, 6660 CrossRef.
| This journal is © The Royal Society of Chemistry 2009 |