Nucleic acid therapeutics: basic concepts and recent developments
Vivek K. Sharma
*,
Pallavi Rungta
and
Ashok K. Prasad
Bioorganic Laboratory, Department of Chemistry, University of Delhi, Delhi-110 007, India. E-mail: viveksharmakumar@yahoo.com; Tel: +91-9810562322
First published on 17th March 2014
Abstract
Nucleic acid based therapeutics have an allure for medicinal chemists due to their potential for specific control of gene expression. Persistent efforts have resulted in the FDA approval of the nucleic acid based drugs Vitravene™, Macugen™ and recently Kynamro™, leading to a sudden leap in the number of active clinical trials involving the nucleic acid moieties. Beginning with antigene and antisense technology, nucleic acid therapeutics have recently gained more potent strategies, e.g. RNA interference, ribozymes, decoy oligonucleotides and aptamers. Nucleic acid based therapeutics which have opened up new frontiers in medicinal chemistry research, along with various chemical modifications undertaken to improve their utility and their mechanism of action, will be discussed in this review article.
 Vivek K. Sharma | Mr Vivek Kumar Sharma received his undergraduate degree in chemistry from Hansraj College, University of Delhi in 2007. After receiving his masters in organic chemistry from the Department of Chemistry, University of Delhi in 2009, he joined the same department for a PhD. He has published 6 research papers in internationally reputed journals such as The Journal of Organic Chemistry and Nucleosides, Nucleotides & Nucleic Acids, etc. His research interest lies in nucleic acid chemistry, biotransformations and multicomponent reactions. |
 Pallavi Rungta | Ms Pallavi Rungta received her undergraduate degree in chemistry in 2010 and masters in organic chemistry in 2012 from Hindu College, University of Delhi. She then joined the Department of Chemistry, University of Delhi for a PhD. She has received several academic awards including a Gold Medal in her masters. She has two publications and her research interest lies in biocatalytic transformations, nucleoside chemistry and heterocyclic chemistry. |
 Ashok K. Prasad | Professor Ashok K. Prasad obtained his PhD from University of Delhi in 1990 in the area of synthesis of bioactive polyphenolic natural products. After spending about a decade as a post-doctoral fellow/visiting scientist at the University of Southern Denmark, the Max-Planck-Institute for Molecular Physiology (Germany), Sapienza University Rome (Italy) and the University of Massachusetts Lowell (USA), Professor Prasad joined the Department of Chemistry, University of Delhi as Reader in 2001 and subsequently became Professor in 2009. He has published over 175 research papers in journals of international repute. His research interest lies in the areas of nucleic acid chemistry, biotransformations, natural product chemistry and synthesis of bioactive heterocyclic compounds. |
Introduction
The quest of modern drug research is to develop a therapeutic technology which can selectively and efficiently act only on the specific target responsible for the disease. The majority of currently available drugs interact with proteins which have been identified as being critical for the dysfunction of certain cells or tissues, but they also often bind to non-target proteins or exert an effect through unknown interactions.1 This has led to drugs that may specifically turn off genes by targeting the nucleic acids that code for the proteins instead of targeting the product protein itself. Paterson et al.2 in 1977 first demonstrated the utility of nucleic acids in modulating gene expression, and in the following year Zamecnik and Stephenson3 showed the capacity of antisense oligonucleotides to inhibit viral replication, which further affirmed that the nucleic acids can be exploited as therapeutic agents.4,5 Various classes of oligonucleotides (ONs) have been developed successfully, e.g. antisense and antigene oligonucleotide (AON), small interfering RNA (siRNA), microRNA (miRNA) and ribozymes that specifically inhibit gene expression by Watson–Crick base pairing to a complementary mRNA, whereas decoy ONs and aptamers bind to their target by structural recognition. This review briefly discusses the different nucleic acid based therapeutics, their mechanism of action, various chemical modifications undertaken to improve their stability and their progress in various disease conditions.Antisense and antigene oligonucleotides
The era of antisense therapeutics has been on a roller coaster ride since Zamecnik and Stephenson3 in 1978 reported that the replication of Rous sarcoma virus (RSV) can be inhibited by a synthetic 13mer oligodeoxynucleotide complementary to a specific mRNA of RSV. This novel approach to drug design was more promising than rational drug design in the sense that it can be used for specific control of gene expression at the level of nucleic acid. The first AON to enter the clinic was Vitravene™ for the treatment of cytomegalovirus (CMV) infection6 followed by Kynamro™, which has recently been approved by the FDA for the treatment of hypercholesterolemia (HoFH).7 Antisense agents function in the cytoplasm of the cell, where they interfere with the translation of mRNA via two common pathways (Fig. 1).8 One pathway involves binding of the AON to the target mRNA strand, which sterically hinders ribosome binding and ultimately mRNA translation. The second pathway comprises degradation of the target mRNA by the enzyme RNase H.8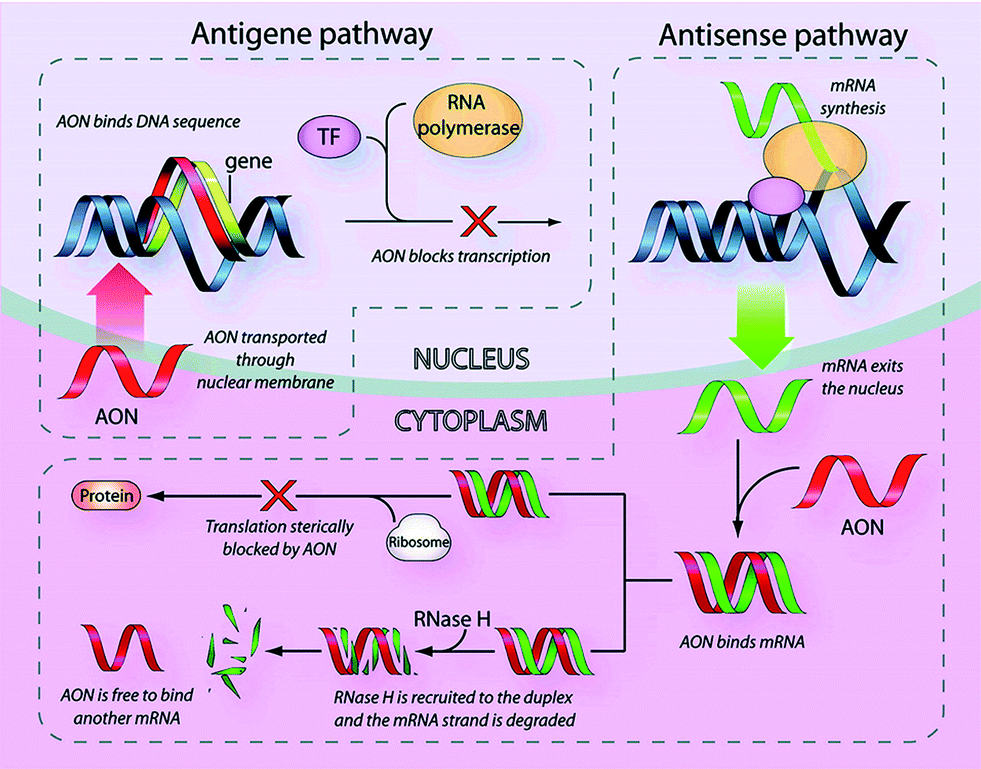 | ||
| Fig. 1 Antisense and antigene pathways [reprinted with permission from (Jain M. L., Bruice P. Y., Szabó I. E. and Bruice T. C., Chem. Rev., 112, 2012, 1284), ©2012 American Chemical Society]. | ||
Antigene agents are nucleic acid analogues that function by entering the nucleus of the cell and interfering with the transcription of DNA (Fig. 1). The introduction of a single-stranded antigene agent results in formation of a double-stranded DNA (dsDNA) antigene triplex that prevents transcription of the blocked DNA sequence into mRNA.9 Successful use of this strategy for blocking transcription and inducing specific mutations, both in vitro and in vivo, has been reported.9,10
In order to be effective, antisense/antigene candidates must have high affinity and specificity for their target mRNA or DNA sequences and confer resistance to degradation by cellular nucleases. Nucleic acids composed of naturally occurring DNA or RNA nucleotides pose some limitations which affect their versatility as therapeutic agents because of their poor binding affinity with complementary ON strands and low degree of nuclease resistance. To overcome these limitations, the ongoing synthetic studies have been focused on chemical modifications of backbone,11 base12 and/or sugar13 functionalities of the natural DNA–RNA and have resulted in significant progress towards establishing ONs as viable therapeutic agents (Fig. 2). Consequently, the AONs have been classified into three generations based upon variation of these modifications.
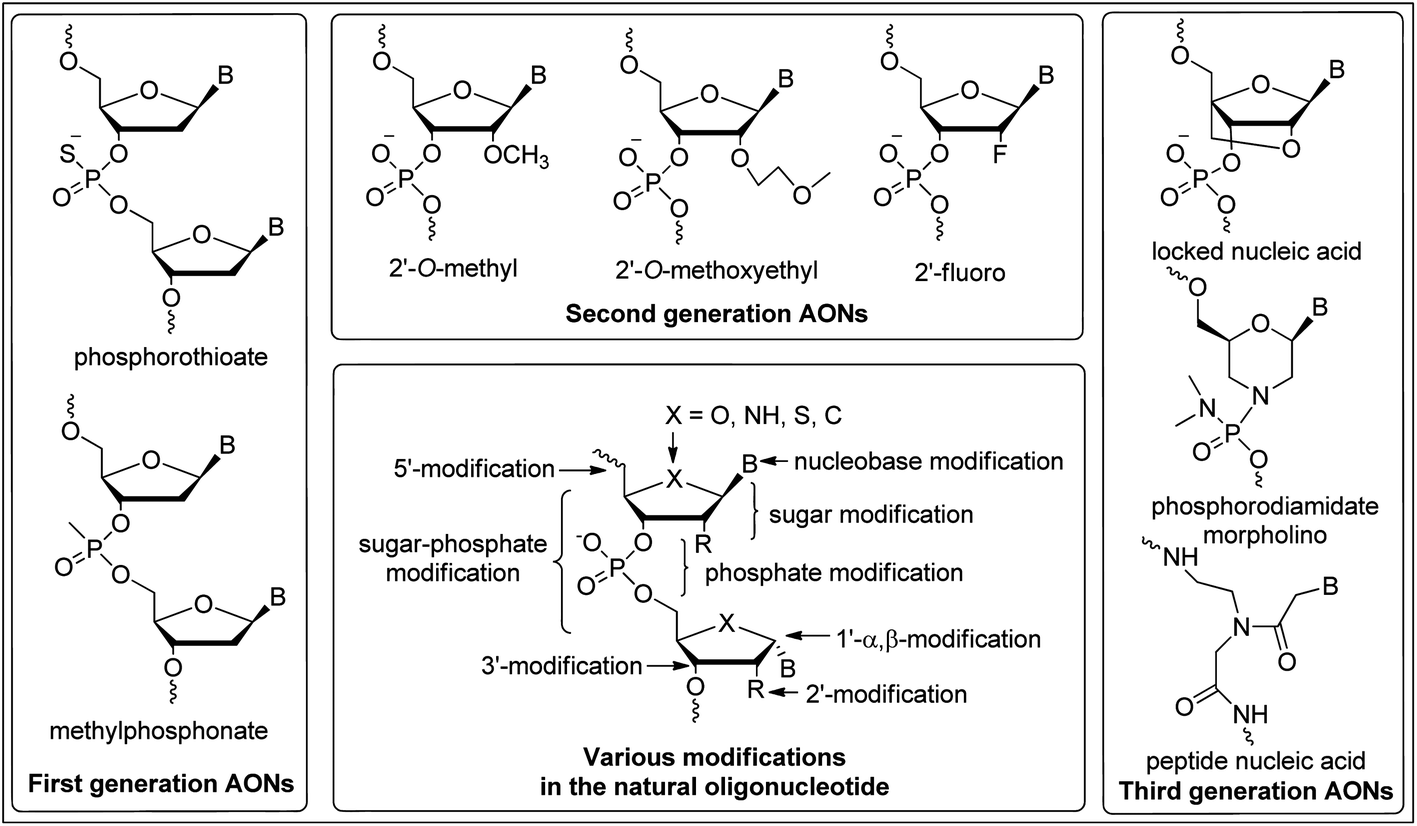 | ||
| Fig. 2 An overview of general modifications and different generations of antisense oligonucleotides (AONs). | ||
The backbone modification of natural ONs such as replacement of an oxygen atom of the phosphate linkage by sulfur (phosphorothioate)14 or a methyl group (methylphosphonate)15 are referred to as first-generation AONs (Fig. 2). Phosphorothioate oligonucleotides (PS-ONs) have been used most successfully for gene silencing and are the major representative of this. Both the FDA-approved antisense drugs, i.e. Vitravene™ and Kynamro™, contains PS-backbone. The PS modification imparts chirality at phosphorus present in the internucleoside linkage. Among the two diastereomers, it is the Sp phosphorothioate diastereomer which has high stability to nucleolytic degradation but has low binding affinity towards the complementary ON relative to the natural phosphodiester linkage.16 In contrast, the Rp phosphorothioate diastereomer confers high binding affinity but is found to be nuclease sensitive. Generally, PS-AONs are synthesized as diastereomeric mixtures as these can form regular Watson–Crick base pairs, carry negative charge for cell delivery, display attractive pharmacokinetic properties and activate RNase H.17,18 This modification also facilitates the binding to plasma proteins, prevents rapid renal excretion and enhances binding to other receptor sites.5 However, their hybridization affinity to the complementary target sequences is less satisfactory.19 Despite this, the FDA approved the antisense drug Vitravene™, a first-generation PS-modified AON for the treatment of AIDS-related CMV retinitis6 (Fig. 3).
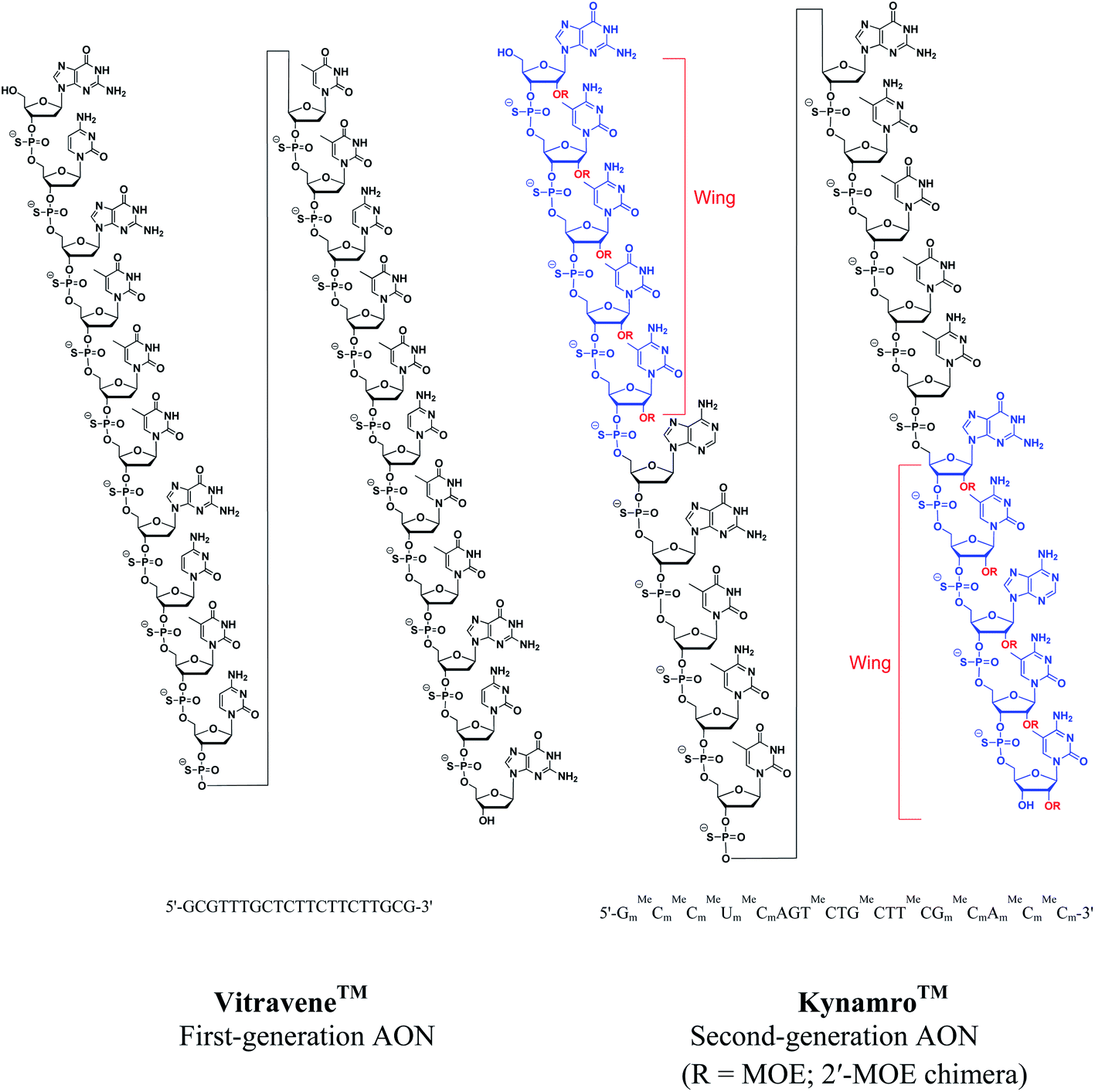 | ||
| Fig. 3 Structures of the FDA-approved antisense drugs Vitravene™ and Kynamro™. | ||
In order to conquer the limitations associated with PS-ONs, further modifications to PS oligos, e.g. introduction of C-5 propyne modified pyrimidines,20 C-7 modified (7-deaza) purines21 and 2′-O-alkyl sugar substitution, lead to substantial increases in binding affinity and nuclease stability.22 2′-O-Methyl (2′-OMe) and 2′-O-methoxyethyl (2′-OMOE) RNAs are the major representatives of second-generation ONs (Fig. 2).23 The designed AONs containing these building blocks are found to be less toxic than PS-AONs and have demonstrated enhanced affinity towards their complementary RNAs, but their efficiency to induce RNase H cleavage of the target RNA is a matter of concern.24 The 2′-OMOE modification has demonstrated up to 2 °C increase in melting temperature (Tm) per modification as compared to PS-ON.25 The conformationally restricted sugar moiety (N-type) and extensive hydration of the 2′-substituent are the probable reason for the observed thermal stability of 2′-OMOE-ONs.26 Further, this modification in natural ONs containing the phosphodiester linkage leads to stability against nucleolytic degradation comparable to that of PS-ON.5 Plasma pharmacokinetics of 2′-OMOE-AONs have been demonstrated in rats and compared to PS-AON.27 The tissue half-life of these modified AONs was found to be tenfold higher than PS modified AONs, which allowed weekly dosing of 2′-OMOE AONs compared to daily dosing of the PS-AONs.27 Substantial reduction in immune stimulation has been observed in mice even in high doses of 2′-OMOE modified ON compared to PS modified ON, which are toxic towards immune stimulation.28 2′-OMOE-AON has also shown 10–20 fold improved potency relative to PS-AON both in cell culture and in animal models.29
Since RNase H cleavage is the most common mechanism for the antisense effect and 2′-O-alkyl modifications are desirable for nuclease resistance and binding affinity, a ‘gapmer’ AON construct can be designed by incorporation of multiple modifications.30 A gapmer contains a central ‘gap’ of deoxynucleotides, sufficient to induce RNase H cleavage, and terminal blocks containing 2′-O-modified ribonucleotide ‘wings’ that protect the central gap from nuclease degradation, e.g. 2′-OMOE sugar modified nucleosides have been combined with phosphorothioate-linkage (PS) in the recently approved antisense drug Kynamro™ (Fig. 3).7 A vast number of AONs undergoing multiple clinical trials are exploring this ‘gapmer’ chimera to have improved therapeutic properties.31
Locked nucleic acid (LNA), phosphorodiamidate morpholino oligomer (PMO) and peptide nucleic acid (PNA) are the three most studied third-generation AONs and mainly contains modification of the furanose ring of the nucleotide (Fig. 2).32
Locked nucleic acid (LNA), contains a ribose ring which is locked in an N-type (C3′-endo) sugar puckering by the introduction of a methylene bridge between 2′-O and 4′-C atoms.13,33 LNA offers key properties needed for successful therapeutic exploitation of ONs. Introduction of LNA monomer unit(s) into ONs confers extraordinary thermal stability when hybridized with either DNA, RNA or with LNA itself.34 Like other 2′-O ribose modifications, LNA is unable to activate RNase H. In order to restore RNase H-mediated cleavage of mRNA, LNA monomers can be freely incorporated into RNA and DNA to form gapmer ONs. It has been reported that the chimeric LNA/DNA/LNA gapmer with 7–10 PS-modified DNA central gaps flanked by three to four LNA nucleotides on both ends provides highly efficient mRNA cleavage and nuclease resistance.35 A number of variations in the LNA skeleton have been employed in the search for improved biological properties, e.g. the α-L-LNA diastereomer of LNA shows superior stability against a 3′-exonuclease. The α-L-LNA chimeras with an interrupted DNA stretch recruit RNase H and show good levels of antisense activity, while the same design with LNA results in a drop in antisense potency.36 Carbocyclic analogues of LNA, such as carbocyclic-LNA-T (five membered) and carbocyclic-ENA-T and carbocyclic-aza-ENA-T (six membered), have shown enhanced target affinity and unprecedented nuclease stability in blood serum (stable >48 h) as compared to the natural (<3 h) and LNA modified AON (<9 h).37 Gymnosis, a process of delivering AONs to cells in the absence of any carriers or conjugation was originally demonstrated using LNA-PS gapmers. The gene silencing can be continuously maintained with little or no toxicity for more than 240 days. Also the pattern of gene silencing of in vitro gymnotically delivered ONs correlates particularly well with in vivo silencing.18
Phosphorodiamidate morpholino oligomer (PMO) is a non-charged AON agent in which the ribose sugar is replaced by a six-membered morpholino ring and the phosphodiester bond is replaced by a phosphorodiamidate-linkage (Fig. 2).38 This modification also does not activate RNase H and can be used only as a steric blocker to inhibit gene expression in biological systems. This chemical modification also confers excellent stability against nucleases and has similar target affinity to that of the isosequential unmodified AON. Since the backbone in PMOs is uncharged, they are unlikely to have unwanted interactions with proteins but on the other hand it affects their cellular uptake.39 Conjugation of arginine-rich peptides to PMO shows improved cellular uptake and antisense potency by increasing the binding affinity of the PMO to complementary RNA.40 The scrape loading technique has been used to facilitate the delivery of PMOs into the cytosol of cultured anchorage-dependent animal cells.41 Phosphoroamidate morpholino oligomers have also demonstrated antisense efficacy in animal models in vivo and in human clinical trials.40,42
In peptide nucleic acid (PNA), the phosphodiester backbone is replaced with a flexible pseudopeptide polymer N-(2-aminoethyl)glycine and nucleobases are attached to the backbone via a methylenecarbonyl-linkage (Fig. 2).43 PNA is a non-charged ON analogue and hybridizes with complementary DNA or RNA with higher affinity and specificity than unmodified DNA–DNA and DNA–RNA duplexes. Peptide nucleic acid exerts its antisense effect by forming a sequence-specific duplex with mRNA, which mainly causes steric hindrance of translational machinery leading to protein knockdown because it is not a substrate for RNase H.44 In order to obtain desired biological properties, use of charged amino acids and conjugation of short peptides to PNAs has been employed.45 Improved in vivo bioavailability and sequence specific antisense activity was observed for PNA-ON conjugated to four lysine molecules at the C-terminus.46
A number of approaches have been explored for efficient in vitro and in vivo delivery of AONs such as development of delivering vehicle, e.g. liposomes and charged lipids, and ON conjugates to increase circulation time and improve cellular and targeted tissue uptake.47 Both local and systemic delivery has been investigated with some successful clinical trials.48 The two marketed ON drugs, the AON Vitravene™ and the aptamer Macugen™, both treat ophthalmologic diseases and are delivered locally by injecting directly into the eye. Kynamro™ represents the first approved AON drug which is systemically delivered. The drug targets the liver, without the need of any delivery vehicle. To the beginning of the antisense therapeutics, two drugs have been approved by FDA, and with ongoing promising clinical trials more new antisense drugs are expected to come in the near future.31
RNA interference (RNAi)
RNA interference (RNAi) is an evolutionary conserved mechanism by which small non-coding double-stranded RNA (dsRNA) silences gene expression, either by inducing the sequence-specific degradation of complementary mRNA or by inhibiting translation via endogenous cellular machinery. The concept of RNAi was introduced in 1998 when dsRNA molecules were found to inhibit expression of homologue genes in C. elegans.49 Craig Mello and Andrew Fire were awarded the Nobel Prize in 2006 for this discovery. Research in ON-based approaches dramatically increased when Elbashir et al.50 were able to specifically silence genes in mammalian cells with short dsRNA molecules – small interfering RNAs (siRNAs). This new technology not only revolutionized life-science research but also opened the road for new therapeutic strategies, e.g. miRNAs, which are endogenous small non-coding RNAs, although discovered first in 1993 as post-transcriptional regulators of gene expression,51 caught the attention of the scientific community eight years later only after the discovery of siRNAs. Chemical relevance of siRNA and miRNA suggested that the mechanism of gene silencing by these non-coding dsRNAs were correlated, with miRNAs being an endogenous form of siRNAs. In the ensuing years, miRNAs and siRNAs continued to be at the forefront of the research world-wide.RNAi is a fundamental pathway in eukaryotic cells by which sequence-specific siRNA is able to target and cleave complementary mRNA.52 RNAi is triggered by long pieces of dsRNA, which are cleaved into fragments known as siRNA (21–23 nucleotides long) by the enzyme Dicer.53 In practice, siRNA can be synthetically produced and then directly introduced into the cell, thus circumventing Dicer mechanics (Fig. 4). This technique reduces the potential for an innate immune interferon response and the shutdown of cellular protein expression that can occur following the interaction of long pieces (>30 nucleotides) of dsRNA with intracellular RNA receptors.54
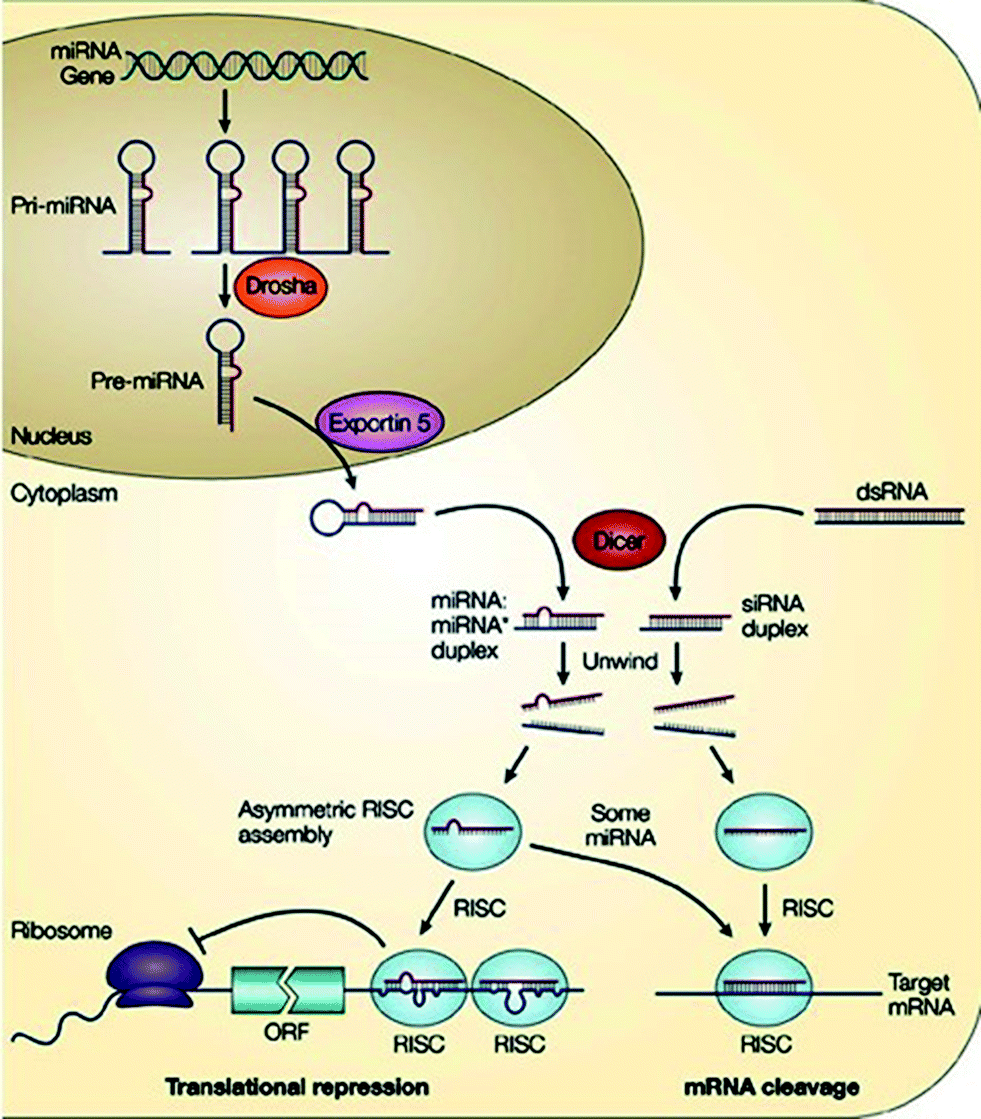 | ||
| Fig. 4 RNA interference pathway [reprinted with permission from Macmillan Publishers Ltd. (He L. and Hannon G. J., Nat. Rev. Genet., 5, 2004, 522), ©2004]. | ||
In the cytoplasm of the cell, siRNA is incorporated into a protein complex called the RNA induced silencing complex (RISC),55 which unwinds the siRNA, after which the sense strand (or passenger strand) of the siRNA is cleaved.56 The activated RISC containing the antisense strand (or guide strand) of the siRNA then selectively degrades mRNA that is complementary to the antisense strand (Fig. 4).57 The cleavage of mRNA occurs at a position between nucleotides 10 and 11 on the complementary antisense strand, relative to the 5′-end.58 The activated RISC complex can further move on to destroy additional mRNA targets.
The early applications of siRNA used long dsRNA which was not effective in most mammalian cells because it induced the antiviral interferon (IFN) response,59 which usually leads to cell death. This view changed in 2001, with the crucial insight of Elbashir et al.50 and Caplen et al.60 They showed that chemically synthesized dsRNA molecules of 21–22 nucleotides known as small interfering RNAs could be used to target mammalian genes by RNAi while evading the interferon response in vivo.
Chemically unmodified siRNAs are not optimal drug-like molecules as they are highly prone to be degraded by serum nucleases, leading to a short half-life in serum. Because of their anionic nature, these have poor cell penetration and are unlikely to interact with albumin and other serum proteins, resulting into rapid elimination.61 Chemical modification of siRNAs is an arduous task since the modification must not interfere with the silencing activity. The two strands of the siRNA duplex have different functions: the antisense strand is of major functional relevance since it will guide RISC to the target RNA, while the sense strand (passenger strand) will be discarded after the loading of RISC. Hence the sense strand is likely to be more tolerant to the introduction of modifications. The modification approaches to mitigate enzymatic degradation include the introduction of boranophosphate siRNAs which are consistently found to be more effective than siRNAs with the widely used PS modification. The boranophosphate siRNAs are at least ten times more nuclease resistant than unmodified siRNAs and are also found to increase siRNA potency, and they are promising candidates for RNAi-based therapeutics.62 2′-OMe or 2′-OMOE sugar modification into the selected nucleotides within both the sense and antisense strands have been shown to confer resistance to endonucleases.63 2′-OMOE phosphorothioate modification in the 3′-overhangs of 21 nucleotide siRNA perfectly complementary to pain-related cation-channel P2X3 resulted in successful gene targeting in vivo.64 It is also possible to incorporate alternative 2′-sugar modifications to increase resistance to endonucleases.65 2′-F-RNA is among the best-known siRNA modifications, and is well tolerated in both the sense and antisense strands.66 2′-F pyrimidine modified siRNA duplexes have shown significantly increased serum and nuclease stability.67 The 2′-deoxy-2′-fluoro-β-D-arabino nucleic acid (2′-F-ANA) modification is structurally similar to 2′-F-RNA, except for the sugar which is arabinose instead of ribose. 2′-F-ANA enhances binding affinity with complementary RNA68 and enhances nuclease stability in siRNA constructs.69,70 Modified siRNAs gapmer contructs by combining 2′-F-ANA with LNA have demonstrated enhanced gene silencing potency, increased nuclease stability, and significantly reduced immunostimulatory properties.70 4′-S-F-ANA have also been found to be compatible with siRNA-mediated gene silencing.71
LNA has also shown compatibility with siRNAs72,73 and confers improved nuclease resistance74 and reduction in siRNA immunostimulation.75 North bicyclo methanocarba thymidine nucleosides were substituted into siRNAs to investigate the effect of bicyclo[3.1.0]hexane-2′-deoxy-pseudosugars on RNA interference activity. It was found that these modified siRNAs are compatible with the intracellular RNAi machinery and lead to enhanced thermal stability of duplexes and enhanced siRNA serum stability.76 Introduction of PNAs has also been found to be compatible with the siRNA intracellular machinery, because all the PNA-modified siRNAs can efficiently mediate specific gene silencing in mammalian cells. Furthermore, modification on the sense strand of siRNA results in increased RNAi activity, whereas modification on both strands results in significant resistance against serum-derived nucleases without the loss of RNAi activity.77
Interestingly, the duplex architecture of siRNAs can also be modified, e.g. siRNAs made of three strands (an intact AON with two 9–13 nucleotide sense strands), called small internally segmented interfering RNA (sisiRNA), can reduce off-target effects and increase potency.72 Single-stranded hairpin-type duplexes (shRNA) have also been introduced exogenously. These shRNA have been designed or expressed within a cell in two possible orientations: left- or right-hand loop, designated L-sshRNAs and R-sshRNAs, respectively.78 Closing the end of the shRNA results in a dumbbell, which retains RNAi activity along with stability against exonucleases.79
While improvement in nuclease stability and immunogenicity can be achieved by chemical modification of the siRNA itself, delivery materials are required to surmount other barriers in the body.61 The first targeted systemic delivery of siRNA drug candidate CALAA-01 in humans has been reported with cyclodextrin polymer (CDP)-based nanoparticles.80 Delivery efficacy can also be improved by end-capping of the polymer termini with imidazole functional groups.81 For stabilization and efficacy in vivo, additional formulation components are required for these nanocomplexes, e.g. adamantane–PEG (AD–PEG) and adamantane–PEG–transferrin (AD–PEG–Tf).82 More than five RNAi drugs utilising lipid nanoparticle (LNP) as a delivering system have entered into clinical trials targeting hypercholesterolemia, transthyretin-mediated amyloidosis and cancer.83 Directly conjugating delivery systems such as cholesterol84 and other lipophilic molecules85 have shown efficacy in vivo. Other conjugate delivery systems include polymers, peptides, antibodies, aptamers and small molecules.86 Despite the recent discovery of RNAi, several siRNA molecules are already undergoing various human clinical trials.83,87
MicroRNAs (miRNAs) are another class of small non-coding RNAs that have been found to regulate gene expression at the post-transcriptional level. Mature miRNAs originate from longer transcripts, called primary-miRNAs (pri-miRNAs) that are transcribed in the cell nucleus by polymerase II.88 These pri-miRNAs comprise hairpins which are recognized and cleaved by the enzyme complex called microprocessor, the core component of which is an RNase III type endonuclease, called Drosha. Drosha cuts the hairpin within the pri-miRNA to release a shorter hairpin of 70–100 nucleotides, called the precursor-miRNA (pre-miRNA).89 The pre-miRNAs are then transported from the nucleus to the cytoplasm via Exportin 5 complex.90 Like siRNAs, pre-miRNAs are processed by Dicer from imperfect RNA hairpins of 18–24 nucleotides (miRNA:miRNA*) containing the mature miRNA strand and its complementary strand miRNA* (Fig. 4).91 Subsequently, miRNA* is discarded on the basis of its thermodynamic stability, and the mature miRNA is incorporated into a RISC-like complex, which, depending on its extent of complementarity to the target mRNA, can either lead to mRNA cleavage (near perfect complementarity) or translational repression (imperfect complementarity).92 Hence, a single miRNA can regulate multiple genes due to its successful gene silencing ability even with partial complementarity between the miRNA and its target.
Fundamentally, siRNAs and miRNAs are similar regarding their biogenesis, molecular characteristics and functions, e.g. both are processed by RNase III enzyme and Dicer, and utilise RISCs for post-transcriptional repression (Fig. 4). However, miRNAs differ from siRNAs in three primary ways. First, miRNAs are purposefully expressed endogenously from an organism's own genome, whereas siRNAs are introduced exogenously. Second, miRNAs are processed from stem-loop precursors with incomplete double-stranded character, while siRNAs form fully complementary dsRNAs.93 Third, miRNAs usually bind to target mRNA through imperfect complementarity at multiple sites and hence negatively regulate gene expression at the translational level, whereas siRNAs often form perfect duplexes with target mRNA due to nearly perfect complementarity.92
Over 1000 miRNAs have been identified in humans, and these can target >30% of the human genome.94 miRNAs are important post-transcriptional regulators of nearly every biological process in the cell, e.g. early development,95 cellular differentiation,96 proliferation,97 apoptosis,98 developmental timing99 and hematopoiesis,100 etc. Dysregulation of miRNAs has been associated with the pathogenesis of human diseases101 and a number of disease-implicated miRNAs have been identified,102 leading to the development of miRNA targeting ONs known as anti-miRNA ONs (AMOs).103 Generally, exogenously introduced AMOs function via a steric block mechanism where they hybridize strongly with the targeted miRNA in order to inhibit its loading to RISC. Progress in the elucidation of RNAi pathways and design of chemically modified AONs have resolved several issues for AMOs in advance, such as cell permeability, nuclease stability and hybridization affinity, leading to a sudden leap in the clinical development of miRNA therapeutics.
Sugar modifications (2′-F, 2′-OMe, 2′-OMOE, LNA)104 and backbone modifications (PS and PMO)105,106 have been successfully applied to inhibit miRNA function in vitro and in vivo.107 Krutzfeldt et al. reported the silencing of miR-122 in the mouse liver using 3′ cholesterol-conjugated, 2′-OMe ONs, known as “antagomirs.”108 Dextran-conjugated AONs injected into the C. elegans germ line efficiently and specifically inhibit the function of a specific miRNA function in the progeny.109 PMO modification has also proved to be specific and non-toxic inhibitors of both pri-miRNA and mature miRNA in zebrafish and Xenopus laevis.106 Miravirsen (SPC3649), currently undergoing a phase II clinical trial, is an inhibitor of miR-122, a liver specific miRNA that the hepatitis C virus (HCV) requires for replication. Miravirsen consist of LNA and PS modified 15-nucleotide sequence complementary to mature miR-122 (Fig. 5).105
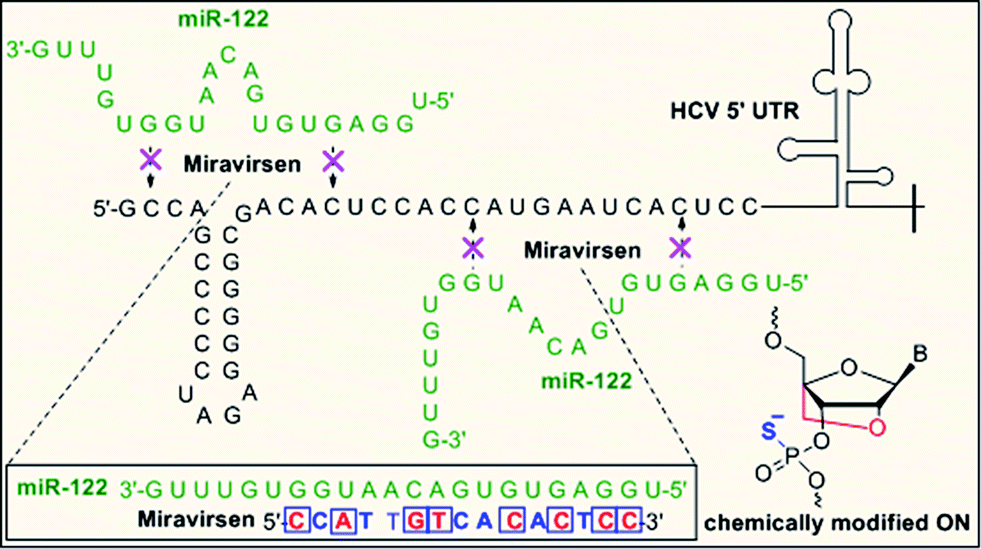 | ||
| Fig. 5 Mechanism of action of PS-backbone modified drug miravirsen (nucleobases modified with LNA are shown red). | ||
Ribozymes
Another strategy for combating diseases by targeting the transcriptional process is the use of ribozymes, a unique class of RNA molecules that not only store information but also possess catalytic activity.110 Ribozymes catalytically cleave specific target RNA (Fig. 6), whereas AONs inhibit translation by binding to mRNA sequences on a stoichiometric basis (Fig. 1). Theoretically, ribozymes are more effective for inhibiting targeted gene expression.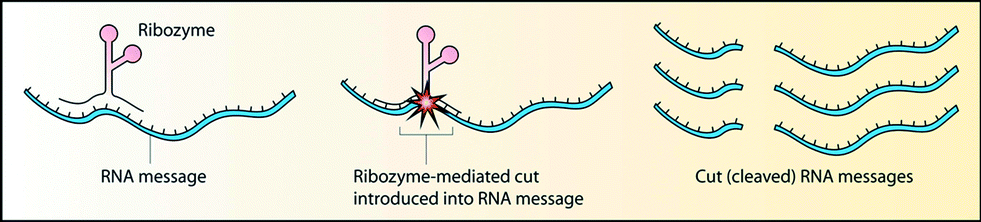 | ||
| Fig. 6 Mechanism of action of ribozymes [reprinted from Robinson R., PLoS Biol., 2, 2004, e28; © 2004 Richard Robinson]. | ||
All known enzymes were considered to be proteins until the discovery of some RNA molecules that can act as enzymes.111 In the early 1980s, Cech and coworkers discovered that RNA molecules are capable of catalysing reactions even in the absence of any protein component.112 These ribonucleic acids with enzyme-like activity were named ribozymes. Later on, Altman et al.113 showed that the RNA component of RNase P, M1 RNA, from Escherichia coli was likewise able to process tRNA precursors without any protein co-factors. Cech and Altman shared the Nobel Prize in Chemistry in 1989 for this discovery.
Among different types of ribozymes in experimental usage, the hammerhead and hairpin ribozymes have been extensively studied due to their small size and high cleavage efficiency.114,115 As they are susceptible to nucleolytic degradation, chemical modification of the RNA molecules is required. The task here is a tougher challenge than the modification of AONs, since the modified nucleotides may interfere with the three dimensional structure and can lead to reduction of catalytic activity. However, ribozymes have been chemically modified by incorporation of 2′-O-methyl RNA monomers, terminal protection by an inverted 3′-3′ deoxy-abasic sugar and phosphorothioate-linkage. Hammerhead ribozymes with these modification patterns were tested in clinical trials to treat cancer and HCV.116,117 Several ribozymes have been tested in clinical trials to treat viral infections and cancer. The first clinical trial using a ribozyme targeted human immunodeficiency virus 1 (HIV-1) and showed that an anti-HIV-l gag ribozyme can interfere with both pre-integration and post-integration events of the HIV replication cycle, by cleaving incoming viral RNA and transcribed mRNAs.118 Various clinical trials are in progress to evaluate the potential of ribozymes to fight cancer, e.g. angiozyme,117 which targets mRNA that encodes vascular endothelial growth factor (VEGF), is being examined in a phase II trial for treatment of metastatic colorectal cancer, while herzyme (Zinzyme)119 targets the mRNA that encodes human epidermal growth factor-2 and is in a phase I clinical trial for treatment of breast and ovarian cancer.
Decoy oligonucleotides
Regulation of gene expression is a complex biological process involving transcription factor–DNA interaction that initiates gene transcription.120 These transcription factor proteins bind specific sequences found in the promoter regions of genes (target genes) whose expression is regulated thereafter (switch on or off). These binding sequences are generally 6–10 base pairs in length and are occasionally found in multiple copies within the promoter regions of target genes. The transcription factor ON decoy approach involves flooding the cell with competing synthetic, transcription factor-specific consensus sequences. These synthetic decoys compete for binding to the transcription factor with consensus sequences in target genes. If delivered into the cell in sufficient concentrations these “decoys” thus have the potential to diminish the binding of the transcription factor to promoter regions of target genes and thus attenuate the function of the transcription factor to regulate the expression of its target gene(s).Decoy ONs are conceptually related to aptamers and are short, double-stranded DNA molecules which mimic the binding ligand of their target transcription factor, thereby altering the expression of the target gene.121 Their transfer mechanism may involve receptor-mediated uptake or a saturable carrier mediated component with subsequent intracellular release. The first pathway seems to operate primarily at low pH conditions, while the second pathway gains importance at the neutral pH of physiological conditions. After delivery of decoy-ON into the target cell, they can bind to the transcription factors with a promoter of the target genes (Fig. 7).122,123
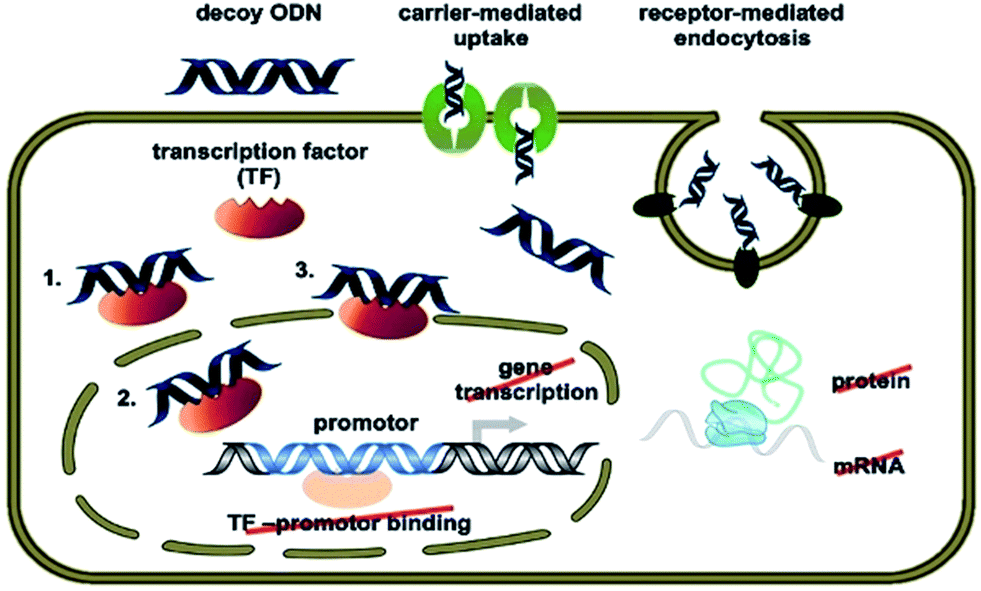 | ||
| Fig. 7 Decoy ON mechanism of action for neutralization of the targeted transcription factor (TF) [© Stojanovic T., Wagner A. H., Schöndube F. A., Hecker M.; originally published in ref. 123 under CC BY 3.0 license]. | ||
Different chemical modifications and/or delivery systems have been investigated in order to harness the therapeutic potential of the decoy ONs, which is usually hampered by their poor bioavailability and short half-life.124 Phosphorothioate (PS) modified double-stranded ONs were reported to specifically bind to transcription nuclear factor kappa beta (NF-kB). These PS modified ONs have increased serum stability and modulate gene expression in a specific manner.125 Transcription factor decoy molecules based on a PNA–DNA–PNA (PDP) chimera mimicking Sp1 binding sites have been investigated. These PDP-based decoy molecules are more resistant than DNA–DNA hybrids towards enzymatic degradation. PDP chimeric decoy molecules also effectively inhibit Sp1–DNA interactions.126 Decoy ONs that bind to the transcription NF-kB have been stabilized successfully by the introduction of LNA and α-L-LNA and were protected against degradation by exonucleases.127 Augmentation of the therapeutic properties of decoy ONs has been accomplished by delivery systems. Cationic liposomes, fusogenic nanoparticles and biodegradable microspheres have been investigated as delivery systems for decoy ONs to impart stability in biological fluids and to increase cellular uptake and target specificity.122,128
Aptamers
Aptamers are single-stranded ONs (DNA–RNA) that form stable three-dimensional structures capable of binding with high affinity and specificity to a variety of molecular targets. Aptamers bind to protein targets and can modulate protein function. In 1990, Tuerk et al. and Ellington et al. simultaneously observed in vitro selection of ONs with high affinity to a target molecule referred to as aptamers (a Latin term that means ‘to fit’).129 This therapeutic principle has now been developed into one marketed product, Macugen™,130 an anti-VEGF aptamer approved for the treatment of age-related macular degeneration in 2004 and several other aptamers are at various stages of clinical development.The most common way to obtain these nucleic acids with high target affinity is by an in vitro selection procedure called SELEX (systematic evolution of ligands by exponential enrichment).131 This technology comprises an iterative process of selection and amplification of target-binding ONs, while unbound ONs are discarded. The SELEX procedure starts with a large library consisting of 1014 to 1015 different DNA or RNA species that fold into distinct three-dimensional structures, depending on their sequence. The library is incubated with the target molecule to separate the DNA or RNA molecules that bind to the ligand from those that do not. The retained ONs are amplified and undergo further rounds of enrichment, until ONs with high affinity to the target molecule can be isolated, cloned, sequenced and further optimized (Fig. 8).132
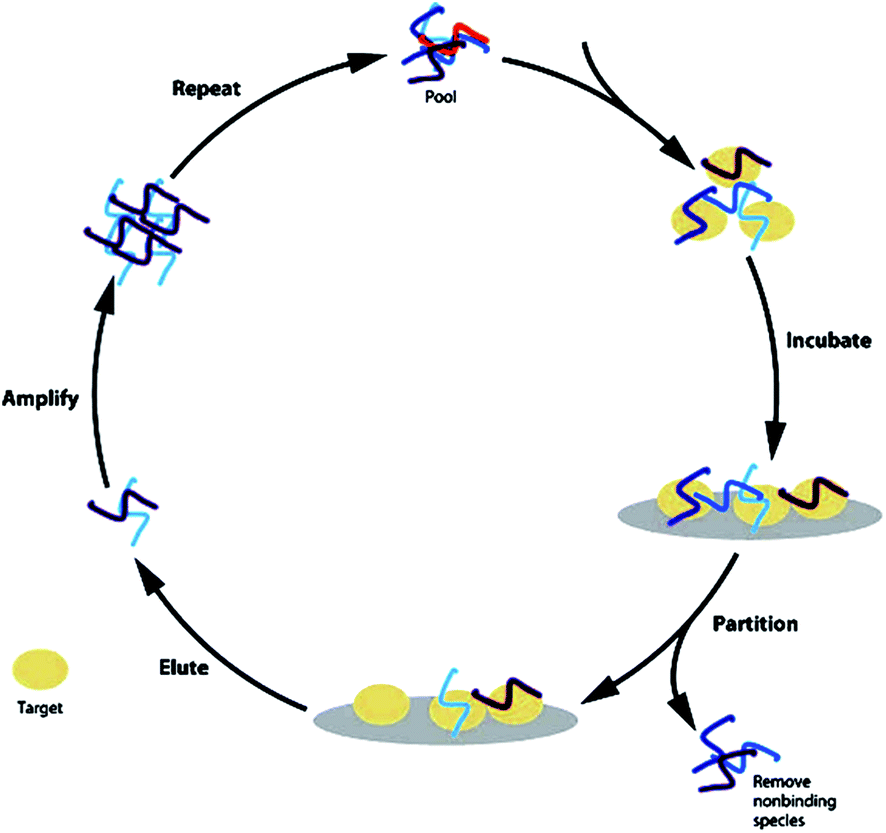 | ||
| Fig. 8 The general SELEX strategy. Starting with combinatorial libraries, the specific binders are isolated by an iterative process of ligand binding, elution and amplification [reprinted with permission from Bouchard P. R., Hutabarat R. M. and Thompson K. M., Annu. Rev. Pharmacol. Toxicol., 50, 2010, 237; © 2010 by Annual Reviews]. | ||
Since the targets are either in solution in the blood plasma or are displayed on the surface of cells, aptamers in this milieu are prone to degradation by serum nucleases and quick renal filtration. Unmodified aptamers may have half-lives in the blood as short as 2 minutes.133 Capping of ONs at the 3′-terminus, often followed by inverting the nucleotide at the 3′-terminus, increases stability to endogenous serum nucleases.134 Another elegant approach to improve the stability of aptamers is the ‘Spiegelmer’ concept, which relies on the high enantioselectivity of nucleases. Spiegelmers135 are aptamers in which the sugars are the enantiomers (non-identical mirror images) of wild-type nucleic acid sugars. In the case of RNA spiegelmers, the ON backbone is composed entirely of L-riboses linked by phosphodiesters. Because nucleases are highly stereoselective, these molecules are much more nuclease-resistant than the corresponding wild-type RNA (D-ribose) sequences.
The conjugation of high molecular mass poly(ethylene glycol) (PEG) appeared to facilitate distribution of aptamer to tissues, particularly those of highly perfused organs, and showed considerable drop in the rate of renal filtration.136 In the mouse circulatory system, conjugation of 40 kDa PEG to a fully 2′-OMe modified aptamer was found to have a circulating half-life as long as 1 day compared to unconjugated aptamers having a half-life of 5–10 minutes only.137
Interestingly, aptamers have been developed as “escort” molecules for the delivery of appended cargoes to disease targets,138 e.g. targeted delivery of doxorubicin (Dox) to cancer cells has been accomplished using A10 RNA aptamer that binds to the prostate-specific membrane antigen (PSMA) protein on the surface of prostate cancer cells.139 Another cornerstone application of aptamers is the delivery of ONs such as siRNAs, which otherwise faces tremendous hurdles.140
When an anti-PSMA aptamer was coupled to siRNAs via a modular streptavidin bridge, the resulting siRNA-mediated inhibition of gene expression was found to be as efficient as observed with conventional lipid-based reagents.141 Zhou et al. demonstrated cell type-specific delivery of anti-human immunodeficiency virus (anti-HIV) siRNAs through fusion to an anti-gp120 aptamer. The anti-gp120 aptamer–siRNA chimera was specifically taken up by cells expressing HIV-1 gp120, and the appended siRNA was processed by Dicer, releasing an anti-tat/rev siRNA which, in turn, inhibited HIV replication. This construct can inhibit HIV replication through both the aptamer and the siRNA components.142 Apart from the aptamer–siRNA chimera, various types of chimerization strategies have been employed to construct aptamer–aptamer, aptamer–proteins and aptamer–nanoparticle chimeras. Introduction of chemical modification, e.g. LNA, to these chimeric aptamers has improved their stability, biodistribution and targeting efficiency, and have facilitated accurate targeting in preclinical trials.143 Magalhães et al.144 have recently reported a general RNA motif that could internalize into the cells without the need of any transfection reagents. When this aptamer was applied to the vaginal mucosa of mice, staining and uptake patterns were found to be similar to those with the administration of siRNAs using transfection agents145 or following the administration of cholesterol-derivatized siRNAs.146
In December 2004, the FDA approved Macugen™ (pegaptanib sodium), an anti-VEGF RNA aptamer, for the treatment of all types of neovascular age-related macular degeneration (Fig. 9).130 Pegaptanib's approval represents a milestone in drug development since it is the first aptamer to be successfully developed as a therapeutic agent in humans, and it has led to six further therapeutic aptamers which are under active clinical development. All the six aptamers in clinical development possess favourable safety profiles and promising clinical activity.147
 | ||
| Fig. 9 Structure of the aptamer-based FDA-approved drug Macugen™ (pegaptanib sodium). | ||
Conclusion
Antisense technology and more recent advances in ON based drugs, such as siRNAs, miRNAs, ribozymes, decoy ONs and aptamers, have strengthened nucleic acid therapeutics and opened up unlimited scope for the research and development of new and highly specific drugs. This allows researchers to focus on a broad range of disease targets once considered “undruggable”. Carefully designed chemically modified nucleic acid moieties have demonstrated specific modulation of gene expression of selected targets compared with rational drugs, which rather exert non-specific effects. The last three decades of effort have resulted in a promising deep pipeline of clinical and preclinical drug candidates based on nucleic acid therapeutics. Current research is focused on the RNA based technologies, which are very near to achieving their clinical potential and are envisioned as the future of nucleic acid based therapeutics.Acknowledgements
We are thankful to the University of Delhi for providing financial support under DU-DST Purse and R&D Grants. VKS and PR thank CSIR, New Delhi, for the award of JRF/SRF Fellowships.References
-
(a) Y. Guan, C. Hao, D. R. Cha, R. Rao, W. Lu, D. E. Kohan, M. A. Magnuson, R. Redha, Y. Zhang and M. D. Breyer, Nat. Med., 2005, 11, 861 CrossRef CAS PubMed
; (b) H. Zhang, A. Zhang, D. E. Kohan, R. D. Nelson, F. J. Gonzalez and T. Yang, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9406 CAS
- B. M. Paterson, B. E. Roberts and E. L. Kuff, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 4370 CrossRef CAS
- P. C. Zamecnik and M. L. Stephenson, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 280 CrossRef CAS
-
(a) S. R. Zheng, G. L. Guo, Q. Zhai, Z. Y. Zou and W. Zhang, Asian Pacific Journal of Cancer Prevention, 2013, 14, 2361 CrossRef
- C. F. Bennett and E. E. Swayze, Annu. Rev. Pharmacol. Toxicol., 2010, 50, 259 CAS
- G. B. Mulamba, A. Hu, R. F. Azad, K. P. Anderson and D. M. Coen, Antimicrob. Agents Chemother., 1998, 42, 971 CAS
- M. P. McGowan, J. Tardif, R. Ceska, L. J. Burgess, H. Soran, I. Gouni-Berthold, G. Wagener and S. Chasan-Taber, PLoS One, 2012, 7, e49006 CAS
- M. L. Jain, P. Y. Bruice, I. E. Szabó and T. C. Bruice, Chem. Rev., 2012, 112, 1284 CrossRef CAS PubMed
- B. A. Armitage, Nat. Chem. Biol., 2005, 1, 185 CrossRef CAS PubMed
- H. A. Quigley, Lancet, 2011, 377, 1367 CrossRef
- J. Mickelfield, Curr. Med. Chem., 2001, 8, 1157 CrossRef
- D. R. Corey, J. Clin. Invest., 2007, 117, 3615 CrossRef CAS PubMed
-
(a) T. P. Prakash, A. M. Kawasaki, E. V. Wancewicz, L. Shen, B. P. Monia, B. S. Ross, B. Bhat and M. Manoharan, J. Med. Chem., 2008, 51, 2766 CrossRef CAS PubMed
-
(a) X. Xie, J. Liang, T. Pu, F. Xu, F. Yao, Y. Yang, Y. L. Zhao, D. You, X. Zhou, Z. Deng and Z. Wang, Nucleic Acids Res., 2012, 40, 9115 CrossRef CAS PubMed
-
(a) S. T. Monn and S. Schürch, J. Am. Soc. Mass Spectrom., 2007, 18, 984 CrossRef CAS PubMed
- F. Eckstein, Biochimie, 2002, 84, 841 CrossRef CAS
- S. Agrawal and E. R. Kandimalla, Mol. Med. Today, 2000, 6, 72 CrossRef CAS
- C. A. Stein, J. B. Hansen, J. Lai, S. Wu, A. Voskresenskiy, A. Høg, J. Worm, M. Hedtjärn, N. Souleimanian, P. Miller, H. S. Soifer, D. Castanotto, L. Benimetskaya, H. Ørum and T. Koch, Nucleic Acids Res., 2010, 38, e3 CrossRef CAS PubMed
-
(a) S. T. Crooke, Therapeutic applications of oligonucleotides, ed. R. G. Landes, Austin, vol. 79, 1995 Search PubMed
-
(a) C. Moulds, J. G. Lewis, B. C. Froehler, D. Grant, T. Huang, J. F. Milligan, M. D. Matteucci and R. W. Wagner, Biochemistry, 1995, 34, 5044 CrossRef CAS
- C. A. Buhr, R. W. Wagner, D. Grant and B. C. Froehler, Nucleic Acids Res., 1996, 24, 2974 CrossRef CAS PubMed
-
(a) R. A. McKay, L. L. Cummins, M. J. Graham, E. A. Lesnik, S. R. Owens, M. Winniman and N. M. Dean, Nucleic Acids Res., 1996, 24, 411 CrossRef CAS PubMed
- T. P. Prakash, Chem. Biodiversity, 2011, 8, 1616 CAS
- B. S. Sproat, A. I. Lamond, B. Beijer, P. Neuner and U. Ryder, Nucleic Acids Res., 1989, 17, 3373 CrossRef CAS PubMed
- S. M. Freier and K. H. Altmann, Nucleic Acids Res., 1997, 25, 4429 CrossRef CAS PubMed
-
(a) M. Teplova, G. Minasov, V. Tereshko, G. B. Inamati, P. D. Cook, M. Manoharan and M. Egli, Nat. Struct. Biol., 1999, 6, 535 CrossRef CAS PubMed
- R. S. Geary, T. A. Watanabe, L. Truong, S. Freier, E. A. Lesnik, N. B. Sioufi, H. Sasmor, M. Manoharan and A. A. Levin, J. Pharmacol. Exp. Ther., 2001, 296, 890 CAS
- S. Henry, K. Stecker, D. Brooks, D. Monteith, B. Conklin and C. F. Bennett, J. Pharmacol. Exp. Ther., 2000, 292, 468 CAS
- C. F. Bennett, Pharmacological properties of 2′-O-methoxyethyl modified oligonucleotides in antisense drug technology, in Principles, strategies and applications, ed. S. T. Crooke, CRC Press, Boca Raton, 2nd edn, 2007, pp. 273–303 Search PubMed
-
(a) P. P. Seth, A. Jazayeri, J. Yu, C. R. Allerson, B. Bhat and E. E. Swayze, Mol. Ther.–Nucleic Acids, 2012, 1, e47 CrossRef PubMed
-
(a) V. K. Sharma, R. Kumar, P. Rungta, V. S. Parmar and A. K. Prasad, Trends Carbohydr. Res., 2013, 3, 1 Search PubMed
-
(a) J. Kurreck, Eur. J. Biochem., 2003, 270, 1628 CrossRef CAS
- E. S. Hildebrandt-Eriksen, V. Aarup, R. Persson, H. F. Hansen, M. E. Munk and H. Orum, Nucleic Acid Ther., 2012, 22, 152 CAS
-
(a) R. N. Veedu and J. Wengel, Chem. Biodiversity, 2010, 7, 536 CrossRef CAS PubMed
- J. Kurreck, E. Wyszko, C. Gillen and V. A. Erdmann, Nucleic Acids Res., 2002, 30, 1911 CrossRef CAS PubMed
- M. Frieden, S. M. Christensen, N. D. Mikkelsen, C. Rosenbohm, C. A. Thrue, M. Westergaard, H. F. Hansen, H. Ørum and T. Koch, Nucleic Acids Res., 2003, 31, 6365 CrossRef CAS PubMed
-
(a) P. Srivastava, J. Barman, W. Pathmasiri, O. Plashkevych, M. Wenska and J. Chattopadhyaya, J. Am. Chem. Soc., 2007, 129, 8362 CrossRef CAS PubMed
- F. J. Schnell, S. L. Crumley, D. V. Mourich and P. L. Iversen, BioRes. Open Access, 2013, 2, 61 CrossRef CAS PubMed
- A. Amantana and P. L. Iversen, Curr. Opin. Pharmacol., 2005, 5, 550 CrossRef CAS PubMed
- M. H. Nelson, D. A. Stein, A. D. Kroeker, S. A. Hatlevig, P. L. Iversen and H. M. Moulton, Bioconjugate Chem., 2005, 16, 959 CAS
- M. Partridge, A. Vincent, P. Matthews, J. Puma, D. Stein and J. Summerton, Antisense Nucleic Acid Drug Dev., 1996, 6, 169 CrossRef CAS
- P. L. Iversen, T. K. Warren, J. B. Wells, N. L. Garza, D. V. Mourich, L. S. Welch, R. G. Panchal and S. Bavari, Viruses, 2012, 4, 2806 CrossRef CAS PubMed
-
(a) P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497 CAS
- Y. Ji and T. Lei, Sci. Prog., 2013, 96, 43 CrossRef CAS
- P. E. Nielsen, Q. Rev. Biophys., 2005, 38, 345 CrossRef CAS PubMed
- P. Sazani, F. Gemignani, S. H. Kang, M. A. Maier, M. Manoharan, M. Persmark, D. Bortner and R. Kole, Nat. Biotechnol., 2002, 20, 1228 CrossRef CAS PubMed
-
(a) O. Nakagawa, X. Ming, L. Huang and R. L. Juliano, J. Am. Chem. Soc., 2010, 132, 8848 CrossRef CAS PubMed
- X. Yuan, S. Naguib and Z. Wu, Expert Opin. Drug Delivery, 2011, 8, 521 CrossRef CAS PubMed
- A. Fire, S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806 CrossRef CAS PubMed
- S. M. Elbashir, W. Lendeckel and T. Tuschl, Genes Dev., 2001, 15, 188 CrossRef CAS
-
(a) R. C. Lee, R. L. Feinbaum and V. Ambros, Cell, 1993, 75, 843 CrossRef CAS
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494 CrossRef CAS PubMed
- E. Bernstein, A. A. Caudy, S. M. Hammond and G. J. Hannon, Nature, 2001, 409, 363 CrossRef CAS PubMed
- D. Grimm, K. L. Streetz, C. L. Jopling, T. A. Storm, K. Pandey, C. R. Davis, P. Marion, F. Salazar and M. A. Kay, Nature, 2006, 441, 537 CrossRef CAS PubMed
- T. A. Rand, K. Ginalski, N. V. Grishin and X. Wang, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14385 CrossRef CAS PubMed
- C. Matranga, Y. Tomari, C. Shin, D. P. Bartel and P. D. Zamore, Cell, 2005, 123, 607 CrossRef CAS PubMed
- http://www.alnylam.com.
- T. A. Rand, S. Petersen, F. Du and X. Wang, Cell, 2005, 123, 621 CrossRef CAS PubMed
- G. R. Stark, I. M. Kerr, B. R. Williams, R. H. Silverman and R. D. Schreiber, Annu. Rev. Biochem., 1998, 67, 227 CrossRef CAS PubMed
- N. J. Caplen, S. Parrish, F. Imani, A. Fire and R. A. Morgan, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9742 CrossRef CAS PubMed
- J. K. Watts, G. F. Deleavey and M. J. Damha, Drug Discovery Today, 2008, 13, 842 CrossRef CAS PubMed
- A. H. Hall, J. Wan, E. E. Shaughnessy, B. Ramsay Shaw and K. A. Alexander, Nucleic Acids Res., 2004, 32, 5991 CrossRef CAS PubMed
- A. D. Judge, G. Bola, A. C. Lee and I. MacLachlan, Mol. Ther., 2006, 13, 494 CrossRef CAS PubMed
- G. Dorn, S. Patel, G. Wotherspoon, M. Hemmings-Mieszczak, J. Barclay, F. J. Natt, P. Martin, S. Bevan, A. Fox, P. Ganju, W. Wishart and J. Hall, Nucleic Acids Res., 2004, 32, e49 Search PubMed
- D. Bumcrot, M. Manoharan, V. Koteliansky and D. W. Sah, Nat. Chem. Biol., 2006, 2, 711 CrossRef CAS PubMed
-
(a) Y. L. Chiu and T. M. Rana, RNA, 2003, 9, 1034 CrossRef CAS
- J. M. Layzer, A. P. McCaffrey, A. K. Tanner, Z. Huang, M. A. Kay and B. A. Sullenger, RNA, 2004, 10, 766 CrossRef CAS
- C. J. Wilds and M. J. Damha, Nucleic Acids Res., 2000, 28, 3625 CAS
- T. Dowler, D. Bergeron, A. L. Tedeschi, L. Paquet, N. Ferrari and M. J. Damha, Nucleic Acids Res., 2006, 34, 1669 CrossRef CAS PubMed
- G. F. Deleavey, J. K. Watts, T. Alain, F. Robert, A. Kalota, V. Aishwarya, J. Pelletier, A. M. Gewirtz, N. Sonenberg and M. J. Damha, Nucleic Acids Res., 2010, 38, 4547 CrossRef CAS PubMed
- J. K. Watts, N. Choubdar, K. Sadalapure, F. Robert, A. S. Wahba, J. Pelletier, B. M. Pinto and M. J. Damha, Nucleic Acids Res., 2007, 35, 1441 CrossRef CAS PubMed
- J. B. Bramsen, M. B. Laursen, C. K. Damgaard, S. W. Lena, B. R. Babu, J. Wengel and J. Kjems, Nucleic Acids Res., 2007, 35, 5886 CrossRef CAS PubMed
- O. R. Mook, F. Baas, M. B. de Wissel and K. Fluiter, Mol. Cancer Ther., 2007, 6, 833 CrossRef CAS PubMed
- C. Wahlestedt, P. Salmi, L. Good, J. Kela, T. Johnsson, T. Hökfelt, C. Broberger, F. Porreca, J. Lai, K. Ren, M. Ossipov, A. Koshkin, N. Jakobsen, J. Skouv, H. Oerum, M. H. Jacobsen and J. Wengel, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5633 CrossRef CAS
- K. A. Whitehead, J. E. Dahlman, R. S. Langer and D. G. Anderson, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 77 CrossRef CAS PubMed
- M. Terrazas, S. M. Ocampo, J. C. Perales, V. E. Marquez and R. Eritja, ChemBioChem, 2011, 12, 1056 CrossRef CAS PubMed
- N. Potenza, L. Moggio, G. Milano, V. Salvatore, B. Di Blasio, A. Russo and A. Messere, Int. J. Mol. Sci., 2008, 9, 299 CrossRef
-
(a) D. Siolas, C. Lerner, J. Burchard, W. Ge, P. S. Linsley, P. J. Paddison, G. J. Hannon and M. A. Cleary, Nat. Biotechnol., 2005, 23, 227 CrossRef CAS PubMed
-
(a) N. Abe, H. Abe, C. Nagai, M. Harada, H. Hatakeyama, H. Harashima, T. Ohshiro, M. Nishihara, K. Furukawa, M. Maeda, S. Tsuneda and Y. Ito, Bioconjugate Chem., 2011, 22, 2082 CrossRef CAS PubMed
-
(a) M. E. Davis, J. E. Zuckerman, C. H. Choi, D. Seligson, A. Tolcher, C. A. Alabi, Y. Yen, J. D. Heidel and A. Ribas, Nature, 2010, 464, 1067 CrossRef CAS PubMed
- D. W. Bartlett and M. E. Davis, Bioconjugate Chem., 2007, 18, 456 CAS
-
(a) S. Hu-Lieskovan, J. D. Heidel, D. W. Bartlett, M. E. Davis and T. J. Triche, Cancer Res., 2005, 65, 8984 CrossRef CAS PubMed
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967 CrossRef CAS PubMed
- J. Soutschek, A. Akinc, B. Bramlage, K. Charisse, R. Constien, M. Donoghue, S. Elbashir, A. Geick, P. Hadwiger, J. Harborth, M. John, V. Kesavan, G. Lavine, R. K. Pandey, T. Racie, K. G. Rajeev, I. Röhl, I. Toudjarska, G. Wang, S. Wuschko, D. Bumcrot, V. Koteliansky, S. Limmer, M. Manoharan and H. P. Vornlocher, Nature, 2004, 432, 173 CrossRef CAS PubMed
- C. Wolfrum, S. Shi, K. N. Jayaprakash, M. Jayaraman, G. Wang, R. K. Pandey, K. G. Rajeev, T. Nakayama, K. Charrise, E. M. Ndungo, T. Zimmermann, V. Koteliansky, M. Manoharan and M. Stoffel, Nat. Biotechnol., 2007, 25, 1149 CrossRef CAS PubMed
- J. H. Jeong, H. Mok, Y. K. Oh and T. G. Park, Bioconjugate Chem., 2009, 20, 5 CrossRef CAS PubMed
-
(a) A. De Fougerolles, H. P. Vornlocher, J. Maraganore and J. Lieberman, Nat. Rev. Drug Discovery, 2007, 6, 443 CrossRef CAS PubMed
- Y. Lee, M. Kim, J. Han, K. H. Yeom, S. Lee, S. H. Baek and V. N. Kim, EMBO J., 2004, 23, 4051 CAS
- Y. Lee, C. Ahn, J. Han, H. Choi, J. Kim, J. Yim, J. Lee, P. Provost, O. Rådmark, S. Kim and V. N. Kim, Nature, 2003, 425, 415 CrossRef CAS PubMed
- E. Lund, S. Güttinger, A. Calado, J. E. Dahlberg and U. Kutay, Science, 2004, 303, 95 CrossRef CAS PubMed
-
(a) A. Grishok, A. E. Pasquinelli, D. Conte, N. Li, S. Parrish, I. Ha, D. L. Baillie, A. Fire, G. Ruvkun and C. C. Mello, Cell, 2001, 106, 23 CrossRef CAS
- L. He and G. J. Hannon, Nat. Rev. Genet., 2004, 5, 522 CrossRef CAS PubMed
- Y. Tomari and P. D. Zamore, Genes Dev., 2005, 19, 517 CrossRef CAS PubMed
- S. Griffiths-Jones, H. K. Saini, S. van Dongen and A. J. Enright, Nucleic Acids Res., 2008, 36, 154 CrossRef PubMed
- J. Brennecke, D. R. Hipfner, A. Stark, R. B. Russell and S. M. Cohen, Cell, 2003, 113, 25 CrossRef CAS
- J. Dostie, Z. Mourelatos, M. Yang, A. Sharma and G. Dreyfuss, RNA, 2003, 9, 180 CAS
- Y. L. Wang, D. N. Keys, J. K. Au-Young and C. F. J. Chen, J. Cell Physiol., 2009, 218, 251 CrossRef CAS PubMed
- P. Xu, S. Y. Vernooy, M. Guo and B. A. Hay, Curr. Biol., 2003, 13, 790 CAS
- V. Ambros, Cell, 2003, 113, 673 CrossRef CAS
- C. Z. Chen, L. Li, H. F. Lodish and D. P. Bartel, Science, 2004, 303, 83 CrossRef CAS PubMed
-
(a) J. T. Mendell and E. N. Olson, Cell, 2012, 148, 1172 CrossRef CAS PubMed
-
(a) J. Stenvang, A. Petri, M. Lindow, S. Obad and S. Kauppinen, Silence, 2012, 3, 1 CrossRef CAS PubMed
- K. A. Lennox and M. A. Behlke, Gene Ther., 2011, 18, 1111 CrossRef CAS PubMed
- J. Weiler, J. Hunziker and J. Hall, Gene Ther., 2006, 13, 496 CAS
- L. F. Gebert, M. A. Rebhan, S. E. Crivelli, R. Denzler, M. Stoffel and J. Hall, Nucleic Acids Res., 2014, 42, 609 CrossRef CAS PubMed
-
(a) G. Martello, L. Zacchigna, M. Inui, M. Montagner, M. Adorno, A. Mamidi, L. Morsut, S. Soligo, U. Tran, S. Dupont, M. Cordenonsi, O. Wessely and S. Piccolo, Nature, 2007, 449, 183 CrossRef CAS PubMed
-
(a) S. Davis, B. Lollo, S. Freier and C. Esau, Nucleic Acids Res., 2006, 34, 2294 CrossRef CAS PubMed
- J. Krützfeldt, N. Rajewsky, R. Braich, K. G. Rajeev, T. Tuschl, M. Manoharan and M. Stoffel, Nature, 2005, 438, 685 CrossRef PubMed
- G. Zheng, V. Ambros and W. H. Li, Silence, 2010, 1, 9 CrossRef PubMed
- M. Kiehntopf, E. L. Esquivel, M. A. Brach and F. Herrmann, Lancet, 1995, 345, 1027 CrossRef CAS
- N. Lehman, Ribozymes and evolution, in Encyclopedia of Biol. Chem., 2013, p. 142 Search PubMed
- K. Kruger, P. J. Grabowski, A. J. Zaug, J. Sands, D. E. Gottschling and T. R. Cech, Cell, 1982, 31, 147 CrossRef CAS
- S. Altman, M. F. Baer, M. Bartkiewicz, H. Gold, C. Guerrier-Takada, L. A. Kirsebom, N. Lumelsky and K. Peck, Gene, 1989, 82, 63 CrossRef CAS
- R. Robinson, PLoS Biol., 2004, 2, e28 CrossRef PubMed
-
(a) D. Mueller, U. Stahl and V. Meyer, J. Microbiol. Methods, 2006, 65, 585 CrossRef CAS PubMed
- L. Beigelman, J. A. McSwiggen, K. G. Draper, C. Gonzalez, K. Jensen, A. M. Karpeisky, A. S. Modak, J. Matulic-Adamic, A. B. DiRenzo, P. Haeberli, D. Sweedler, D. Tracz, S. Grimm, F. E. Wincott, V. G. Thackaray and N. Usman, J. Biol. Chem., 1995, 270, 25702 CAS
- N. Usman and L. M. Blatt, J. Clin. Invest., 2000, 106, 1197 CrossRef CAS PubMed
-
(a) K. Konopka, J. J. Rossi, P. Swiderski, V. A. Slepushkin and N. Duzqunes, Biochim. Biophys. Acta, 1998, 1372, 55 CrossRef CAS
- A. Peracchi, Rev. Med. Virol., 2004, 14, 47 CAS
- J. Morschhäuser, K. S. Barker, T. T. Liu, J. BlaB-Warmuth, R. Homayouni and P. D. Rogers, PLoS Pathog., 2007, 3, e164 Search PubMed
-
(a) C. Deng, J. Zheng, W. Wan, S. Zhang, Z. Ding, G. Mao and S. Yang, Mol. Med. Rep., 2013, 7, 785 CAS
- M. Z. Ahmad, S. Akhter, N. Mallik, M. Anwar, W. Tabassum and F. J. Ahmad, Curr. Drug Discovery Technol., 2013, 10, 71 CAS
- T. Stojanovic, A. H. Wagner, F. A. Schöndube and M. Hecker, Pre-Transplant Therapy in Experimental Heart Transplantation, in Cardiac Transplantation, ed. S. D. Moffatt-Bruce, InTech, Rijeka, 1st edn, 2012, ch. 8, pp. 143–154. DOI:10.5772/28531
- S. Akhtar, R. Kole and R. L. Juliano, Life Sci., 1991, 49, 1793 CrossRef CAS
- A. Bielinska, R. A. Shivdasani, L. Q. Zhang and G. J. Nabel, Science, 1990, 250, 997 CAS
- M. Borgatti, I. Lampronti, A. Romanelli, C. Pedone, M. Saviano, N. Bianchi, C. Mischiati and R. Gambari, J. Biol. Chem., 2003, 278, 7500 CrossRef CAS PubMed
- R. Crinelli, M. Bianchi, L. Gentilini, L. Palma, M. D. Sørensen, T. Bryld, R. B. Babu, K. Arar, J. Wengel and M. Magnani, Nucleic Acids Res., 2004, 32, 1874 CrossRef CAS PubMed
-
(a) D. De Stefano, G. De Rosa, M. C. Maiuri, F. Ungaro, F. Quaglia, T. Iuvone, M. P. Cinelli, M. I. La Rotonda and R. Carnuccio, Pharmacol. Res., 2009, 60, 33 CrossRef CAS PubMed
-
(a) C. Tuerk and L. Gold, Science, 1990, 249, 505 CAS
- E. W. Ng, D. T. Shima, P. Calias, E. T. Cunningham, Jr, D. R. Guyer and A. P. Adamis, Nat. Rev. Drug Discovery, 2006, 5, 123 CrossRef CAS PubMed
-
(a) S. Ohuchi, BioRes. Open Access, 2012, 1, 265 CrossRef CAS PubMed
- P. R. Bouchard, R. M. Hutabarat and K. M. Thompson, Annu. Rev. Pharmacol. Toxicol., 2010, 50, 237 CrossRef CAS PubMed
- L. C. Griffin, G. F. Tidmarsh, L. C. Bock, J. J. Toole and L. L. Leung, Blood, 1993, 81, 3271 CAS
-
(a) Y. Kasahara, S. Kitadume, K. Morihiro, M. Kuwahara, H. Ozaki, H. Sawai, T. Imanishi and S. Obika, Bioorg. Med. Chem. Lett., 2010, 20, 1626 CrossRef CAS PubMed
-
(a) S. Hoffmann, J. Hoos, S. Klussmann and S. Vonhoff, Current Protocols in Nucleic Acid Chemistry, 2011 Search PubMed
-
(a) J. M. Healy, S. D. Lewis, M. Kurz, R. M. Boomer, K. M. Thompson, C. Wilson and T. G. McCauley, Pharm. Res., 2004, 21, 2234 CrossRef CAS
- P. E. Burmeister, S. D. Lewis, R. F. Silva, J. R. Preiss, L. R. Horwitz, P. S. Pendergrast, T. G. McCauley, J. C. Kurz, D. M. Epstein, C. Wilson and A. D. Keefe, Chem. Biol., 2005, 12, 25 CrossRef CAS PubMed
- B. J. Hicke and A. W. Stephens, J. Clin. Invest., 2000, 106, 923 CAS
- V. Bagalkot, O. C. Farokhzad, R. Langer and S. Jon, Angew. Chem., Int. Ed., 2006, 45, 8149 CrossRef CAS PubMed
-
(a) B. R. Cullen, Nat. Immunol., 2002, 3, 597 CrossRef CAS PubMed
- T. C. Chu, K. Y. Twu, A. D. Ellington and M. Levy, Nucleic Acids Res., 2006, 34, e73 CrossRef PubMed
- J. Zhou, H. Li, S. Li, J. Zaia and J. J. Rossi, Mol. Ther., 2008, 16, 1481 CrossRef CAS PubMed
- J. R. Kanwar, K. Roy and R. K. Kanwar, Crit. Rev. Biochem. Mol. Biol., 2011, 46, 459 CAS
- M. L. Magalhães, M. Byrom, A. Yan, L. Kelly, N. Li, R. Furtado, D. Palliser, A. D. Ellington and M. Levy, Mol. Ther., 2012, 20, 616 CrossRef PubMed
-
(a) S. Y. Wu, H. I. Chang, M. Burgess and N. A. McMillan, J. Controlled Release, 2011, 155, 418 CrossRef CAS PubMed
- Y. Wu, F. Navarro, A. Lal, E. Basar, R. K. Pandey, M. Manoharan, Y. Feng, S. J. Lee, J. Lieberman and D. Palliser, Cell Host Microbe, 2009, 5, 84 CAS
- X. Yang, N. Li and D. G. Gorenstein, Expert Opin. Drug Discovery, 2011, 6, 75 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2014 |