
Laser-ablation sampling for inductively coupled plasma distance-of-flight mass spectrometry†
Alexander
Gundlach-Graham‡
a,
Elise A.
Dennis
a,
Steven J.
Ray
a,
Christie G.
Enke
ab,
Charles J.
Barinaga
c,
David W.
Koppenaal
c and
Gary M.
Hieftje
*a
aDepartment of Chemistry, Indiana University, Bloomington, IN 47405, USA. E-mail: hieftje@indiana.edu
bDepartment of Chemistry, University of New Mexico, Albuquerque, NM 87131, USA
cPacific Northwest National Laboratory, Richland, WA 99352, USA
First published on 14th August 2014
Abstract
An inductively coupled plasma distance-of-flight mass spectrometer (ICP-DOFMS) has been coupled with laser-ablation (LA) sample introduction for the elemental analysis of solids. ICP-DOFMS is well suited for the analysis of laser-generated aerosols because it offers both high-speed mass analysis and simultaneous multi-elemental detection. Here, we evaluate the analytical performance of the LA-ICP-DOFMS instrument, equipped with a microchannel plate-based imaging detector, for the measurement of steady-state LA signals, as well as transient signals produced from single LA events. Steady-state detection limits are 1 μg g−1, and absolute single-pulse LA detection limits are 200 fg for uranium; the system is shown capable of performing time-resolved single-pulse LA analysis. By leveraging the benefits of simultaneous multi-elemental detection, we also attain a good shot-to-shot reproducibility of 6% relative standard deviation (RSD) and isotope-ratio precision of 0.3% RSD with a 10 s integration time.
Introduction
Distance-of-Flight Mass Spectrometry (DOFMS) is a form of velocity-based mass spectrometry in which batches of ions are spatially separated based on mass-to-charge (m/z) ratio across a field-free region and then detected at m/z-dependent flight distances. As a velocity-based MS approach, DOFMS shares performance advantages with time-of-flight mass spectrometry (TOFMS), such as unlimited mass range, batch analysis, and simple instrument design.1 In addition, DOFMS demonstrates capabilities that are unique to spatially dispersive MS approaches, including improved precision and accuracy through simultaneous ion detection,2 and the incorporation of ion detector arrays.3 In past research, DOFMS has been characterized with inductively coupled plasma (ICP) and reduced-pressure direct-current glow-discharge ionization sources.3–7 In the present study, we investigate the analytical performance of our ICP-DOFMS instrument for the measurement of transient signals produced by laser-ablation sample introduction.LA-ICPMS was first reported in 1985,8 and has since become a routine analytical method for the elemental analysis of solid samples across a broad range of disciplines and sample types. For example, LA-ICPMS has been used for targeted elemental microanalysis of heterogeneous samples such as geological and anthropological specimens, and also for two- and three-dimensional elemental mapping of a range of samples, from cultural heritage objects to animal tissues.9–14 In addition to applications-driven LA-ICPMS research, continued advances in laser-ablation technology and ICPMS mass-analyzer design have pushed the capabilities of LA-ICPMS analysis and, circularly, enabled the development of new analytical methods.15–18 Modern LA-ICPMS instruments can deliver direct elemental analysis with moderate spatial resolution (∼10 μm), multi-element detection, wide dynamic range (9 orders of magnitude), high sensitivity (detection limits of ng g−1 to μg g−1), and isotope-ratio precision approaching 0.01% with multi-collector ICP instruments.18 Unfortunately, these benchmarks are not currently attainable on any single LA-ICPMS instrument design; differences in laser-ablation sampling, ablated-aerosol transport, and ICP-mass-analyzer design each impact particular (and often competing) aspects of performance.19,20 In this paper, we explore the performance of a DOF mass analyzer as it relates to LA-ICPMS analysis and compare our results to those of other mass analyzers.
Experimental
To perform LA-ICP-DOFMS analysis, we coupled our ICP-DOFMS instrument7 with a commercial laser-ablation system (LSX-200, Cetac Technologies, Omaha, NE); a schematic diagram of this instrumental setup is presented in the ESI (Fig. S1†). The basic operating principles and design of the DOFMS instrument have been previously reported,6,7,21,22 and the analyzer used here is identical to that used for solution-sample-introduction ICP-DOFMS.7 This DOFMS instrument uses a microchannel-plate/phosphor imaging ion detector in combination with a scientific CCD camera (iKon CCD, Andor Technologies plc., UK) to record DOF mass spectra. A description of the design and operation of the ICP-DOFMS instrument, including aspects of DOFMS image collection, is provided in the ESI.† Typical operating conditions of the LA system and ICP are provided in Table 1.| Laser ablation condition | |
|---|---|
| Laser power | 5 mJ |
| Spot size | 300 μm |
| Repetition rate | Variable |
| Cell volume | 2.8 cm3 |
| Helium flow rate | 0.7 L min−1 |
| Argon make-up gas | 0.8 L min−1 |
| ICP conditions | |
|---|---|
| Forward power | 1.3 kW |
| Reflected power | <15 W |
| ICP torch central-channel diameter | 1.5 mm |
| Outer-channel argon flow | 15.7 L min−1 |
| Middle-channel argon flow | 1.2 L min−1 |
| Central-channel argon and helium flow | 1.5 L min−1 |
| ICP sampling depth | 12 mm |
Laser ablation system
The LSX-200 laser-ablation system employs a Q-switched, frequency-quadrupled Nd:YAG laser (266 nm) and produces laser pulses that are about 6 ns long. For steady-state LA experiments, a laser-pulse repetition rate of 10 Hz was used, whereas laser pulses were manually triggered for single-pulse LA studies. For time-delayed CCD-acquisition experiments, the 50 Ω TTL Q-switch sync output from the Cetac LA system was used to trigger a pulse generator (Model 9514, Quantum Composers Inc., Bozeman MT), in order to produce a time-delayed output TTL pulse to externally trigger the CCD shutter. The laser beam was focused onto each sample manually and ablation progress was monitored with the LA system's video camera. Following ablation, an optical-profiling microscope (Zeta-20, Zeta Instruments, San Jose, CA) was used to determine ablation crater width and depth. The ablation chamber was constructed in-house and features a 2.8 cm3 ablation volume.23 Helium (99.9% Purity, Air Gas Inc., Radnor, PA) was used as the ablation sweep gas and mixed with argon (99.9% Purity, Air Gas Inc.) in a glass T-connector before carrying ablated aerosol through approximately 1 m of 64 mm diameter tubing to the ICP torch.Laser ablation standards
Three solid sample standards were used in these studies. The NIST Standard Reference Material (SRM) glass standard 612 (National Institute of Standards and Technology, Washington DC) was used for the steady-state detection-limit (DL) determination of uranium; this standard contains 37.38 μg g−1 uranium.24 The NIST SRM 610 glass standard was used for single-pulse LA-ICP-DOFMS measurement of uranium, and isotope-ratio precision determinations of silver, tin, and lead. The NIST 610 standard has certified concentrations of 268 and 426 μg g−1 for silver and lead, respectively, and is not certified for tin.25 Finally, a BNRM Carp20Cb3 copper-alloy standard (Brammer Standard Company Inc., Houston TX) was used for molybdenum and copper determinations; this standard has assigned values of 3.10% (w/w) for copper and 2.06% for molybdenum.26 Samples underwent no sample preparation apart from placement in the ablation chamber.DOFMS spectrum acquisition
In DOFMS, ions are mass-separated and detected by first sorting groups of ions across a field-free region according to their m/z-dependent velocities, and then directing these spatially separated ion packets onto a spatially selective ion detector. As previously described (and elaborated in the ESI†), DOFMS is operated so ions of all m/z values that are spread across the mass-detection axis are simultaneously brought into energy focus at the energy-focus time (tef). The tef is a function of the electrostatic potentials used in a given DOF experiment and can be adjusted to bring ions of different m/z ranges onto the DOF detector. Moreover, detection of ions at tef reduces the adverse effects of the ions' initial kinetic energies on mass resolution for all groups of ions separated across the DOF axis.
Fig. 1 shows a representative LA-ICP-DOFMS spectrum and an averaged CCD image of the DOFMS lines obtained with our MCP/phosphor-CCD detection system. For this spectrum, ion packets were injected into the DOFMS analyzer, and subsequently mass separated and pushed onto the DOF detector, at a rate of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz. Here, instrument conditions were adjusted to bring ions from m/z 172 to 190.5 into focus on the 40 mm-long DOF detector at a flight time of 44.7 μs. This repetition rate and DOF flight time are typical for experiments presented here.
000 Hz. Here, instrument conditions were adjusted to bring ions from m/z 172 to 190.5 into focus on the 40 mm-long DOF detector at a flight time of 44.7 μs. This repetition rate and DOF flight time are typical for experiments presented here.
 | ||
| Fig. 1 (a) LA-ICP-DOFMS spectrum from NIST 610 multi-element glass standard. (b) Image of DOFMS lines used to generate the mass spectrum in (a). The vertical lines in the image correspond to ion strikes from m/z-separated ion packets; the regular spacing of these lines is characteristic of the unit-mass spacing of the singly charged elemental ions generated in the ICP. The isotopic signals in the DOFMS image are aligned with the above spectrum. In DOFMS, flight distance is proportional to (m/z)−1. | ||
Fig. 1 illustrates that the range of simultaneously detectable masses is limited on our DOFMS instrument. In DOFMS, a constant high-to-low m/z ratio of ions is simultaneously detectable and is controlled by the total flight distance of ions through the analyzer and by the length of the DOF detector. In previous work, we found that the high-to-low mass range of our current DOFMS setup is 1.11;7 this range is evident in Fig. 1. While the high-to-low mass range is fixed on our DOFMS instrument, ions of any m/z value can be brought into focus on the DOFMS detector by simple voltage adjustments, so any desired region of the DOF mass spectrum can be analyzed.3
Fig. 1b displays a typical CCD image collected during the LA-ICP-DOFMS analysis of NIST SRM 610. This image was collected with a CCD exposure time of 1.5 s, so around 15000 DOFMS experiments (ion-packet injections) are averaged in this image. For studies reported here, CCD images with exposure times from 0.1 s to 5 s were collected; and exposure times were adjusted depending on analyte concentration to achieve best S/N ratio with as little temporal resolution loss as possible. As shown in Fig. 1b, the CCD images are two-dimensional: the horizontal axis is the distance-of-flight mass-separation axis, and the positions at which ions strike along this axis are proportional to (m/z)−1. The vertical axis of the CCD image is parallel to the initial ion-beam axis; the height of the DOFMS lines corresponds to the portion of the input ion beam that is sampled in each DOFMS experiment. Importantly, the tall DOFMS lines obtained are advantageous because the detection of a large portion of the initial ion beam with each DOFMS experiment increases the analyzer's efficiency.4 The brightness of each DOFMS line across the vertical axis of the CCD image is proportional to ion abundance, and conventional mass spectra were generated by vertically binning (i.e. summing) pixel signals normal to the mass-separation axis. Spectra were also blank-subtracted to correct for background illumination of the CCD. As is highlighted in Fig. 1a, our DOFMS with the MCP/phosphor-CCD detection assembly provides at least unit-mass resolution across the elemental mass range.
Results and discussion
LA-ICP-DOFMS detection limits
An often-cited advantage of LA-ICPMS is the extreme sensitivity and absolute detection limits that it can provide; in fact, with commercial systems, detection limits (DLs) below 0.01 μg g−1 are achievable for over 20 elements across the periodic table.27,28 However, detection limits reported for LA-ICPMS must be considered cautiously because many instrumental and experimental variables influence them. For instance, such factors as laser-sample interaction, ablation volume, aerosol-transport efficiency, atomization and ionization efficiency of the ICP, and mass-analyzer sensitivity all impact attained DLs.29,30 Here, we report the performance of LA-ICP-DOFMS with a 266 nm, ns-pulsed laser ablation system for the analysis of glass standards. We compare our results to those obtained for the analysis of glass with related mass-analyzer types, with an emphasis on LA-ICP-Mattauch–Herzog Mass Spectrograph (MHMS) and LA-ICP-TOFMS systems.We investigated the performance of our LA-ICP-DOFMS setup for two types of LA analysis: steady-state and single-shot. In the steady-state approach, a laser-repetition rate of 5–10 Hz is used so aerosol ablated from several laser pulses is mixed within the ablation chamber to produce a time-averaged analyte signal. (For the analysis of a homogenous sample, this approach produces a steady stream of sample aerosol into the ICP.) Steady-state LA is a typical ablation approach with scanning-type mass analyzers, such as quadrupole MS and single-collector sector-field MS instruments because these analyzers benefit from averaged and relatively stable multi-isotopic signal levels to minimize spectral skew and improve isotope-ratio precision and accuracy.11
Fig. 2 provides the averaged DOF mass spectrum of uranium and thorium obtained from the steady-state laser ablation of a NIST SRM 612 standard, and the time trace for integrated DOFMS signals. For this spectrum, CCD images were collected with 1 s exposures and summed offline to create 3 s-equivalent exposures. As seen in Fig. 2b, the uranium signal is not constant, but gradually declines as the experiment progresses; this decline is most likely caused by non-stoichiometric LA sampling (i.e. elemental fractionation) and is consistent with previous studies.31–33 No attempt was made to optimize ICP or LA conditions for a unity U/Th signal ratio; the much higher uranium signal seen here compared to thorium is a consequence of plasma and laser-ablation conditions, not the DOF mass analyzer. From this behavior, it is clear that Th and U do not constitute a good internal-standard pair under these conditions.
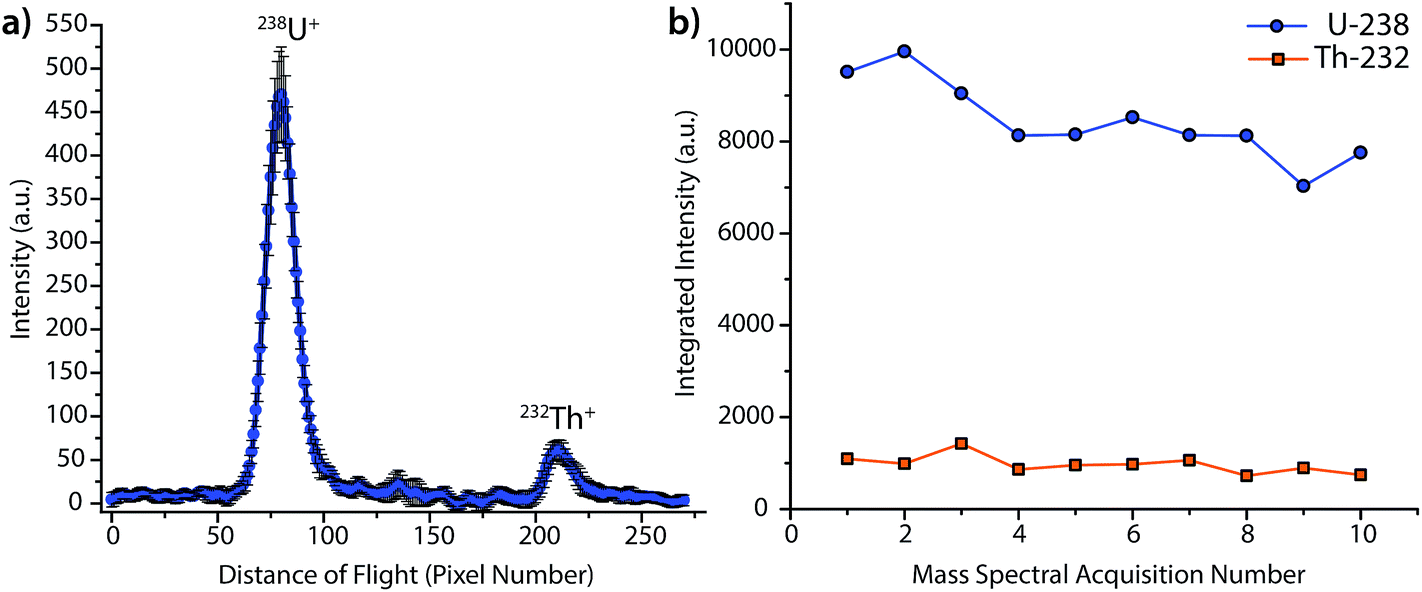 | ||
| Fig. 2 (a) Averaged LA-ICP-DOFMS mass spectrum of the uranium and thorium isotopes ablated from a NIST SRM 612 glass standard. The laser was fired at a repetition rate of 10 Hz to create a steady-state ICP-DOFMS signal; DOFMS images were collected at 1 s exposures and summed off-line to generate spectra with 3 s exposures. (b) Time traces of integrated signals of 238U+ and 232Th+. | ||
The detection limit (DL) for uranium by steady-state LA was determined to be 1 μg g−1, and was calculated as three times the σblank, which was obtained at same mass-spectral position as uranium, but with the laser off (i.e. as a gas blank). For reference, this DL is about two orders of magnitude higher than results obtained in our laboratory with a LA-ICP-MHMS system equipped with the same LA system used here.34 On the LA-ICP-MHMS instrument, researchers obtained a detection limit of 50 ng g−1 for uranium, with a 20 Hz laser repetition rate and a 10 s integration.34 The better DL with the MHMS is most likely due to higher sample throughput, unity duty factor, and a longer integration period than in the DOFMS experiments. While the ICP-MHMS operates at a 100% duty factor, DOFMS, as a batch-analysis MS approach, has a duty factor that depends on the frequency of DOF mass-separation experiments and the portion of the ion beam extracted in each experiment.4 For results presented here, we estimate the duty cycle to be about ∼10%, based on geometric considerations and a DOFMS repetition rate of 10 kHz.4,7 This estimated duty factor accounts for an order of magnitude loss in sensitivity for DOFMS compared to the MHMS. Ion-throughput from the ICP to mass analyzer is most likely the other prominent sensitivity-limiting factor in the DOFMS instrument, and, in the future, will be overcome by modification of the MS-interface and ion-transport optics. Though the current sensitivity of LA-ICP-DOFMS is not competitive with alternative, established LA-ICPMS systems, the detection limit of ∼1 μg g−1, demonstrates a proof-of-principle for trace elemental analysis with LA-ICP-DOFMS.
Single-pulse LA offers advantages over steady state LA, including better spatial and depth resolution, the elimination of pulse-to-pulse mixing, the correction of shot-to-shot laser-power fluctuations for improved quantitation, and reduced sample consumption.11,14,35 For single-pulse LA analysis, we reduced the laser repetition rate so the ablated aerosol from each LA event is transferred as a discrete aerosol cloud into the ICP-DOFMS instrument. This approach produces time-dependent signal peaks rather than steady-state signals. Moreover, for this analysis, the mass analyzer must have sufficient speed or simultaneous spectral coverage to detect ion signals of interest at the same point along each LA peak, and thus eliminate spectral skew.2 Currently, there are three commercially available ICPMS instrument designs that deliver simultaneous (or quasi-simultaneous) multi-isotope and/or multi-element detection: multi-collector (MC)-ICPMS, ICP-MHMS, and ICP-TOFMS. Of these approaches, both MC-ICPMS and ICP-MHMS continuously and simultaneously collect ion signals across space with an array of ion detectors. In contrast, ICP-TOFMS instruments rapidly (every ∼50 μs) generate ion packets and then separate and detect all ion signals from these packets based on flight time; ICP-TOFMS offers a duty cycle approaching 30%.36 DOFMS combines features of MC-ICPMS and TOFMS by offering simultaneous, spatially dispersive multi-isotope detection of batches of ions, and has the potential to produce simultaneously acquired spectra on the timescale of a single DOFMS experiment (50 μs, up to 20 kHz).
Fig. 3 shows an example single-pulse LA-ICP-DOFMS time trace for the measurement of uranium and thorium isotopes from a NIST SRM 610 standard. In this experiment, LA pulses were manually triggered, and CCD images were collected at a rate of 4.16 images per s with exposure times of 100 ms. This CCD acquisition speed was chosen to enable several DOFMS spectral images to be captured along the transient profile, and also to maintain a high enough S/N to collect meaningful spectra at the tail of the profiles. Fig. 3a shows the time trace for the mass-spectral signals of 238U+ and 232Th+; transient signal profiles were generated by integrating across the peak for each isotope of interest. Not surprisingly, the pulse-to-pulse variation is substantial: the average peak area is 5000 ± 700 a.u. and the average full-width half-maximum (FWHM) temporal peakwidth is 1.1 ± 0.3 s. This variance in single-pulse LA data is a product of the LA system used here and matches well with previous studies.23Fig. 3b provides a zoomed-in view of signals from the first two laser pulses and demonstrates that our system has sufficient speed to sample each transient LA signal and to indicate the shape of each transient peak. To determine the single-pulse LA detection limits, we measured the average ablation crater dimensions with an optical-profiling microscope. With an average laser energy of 5.4 mJ per pulse and a spot size of 300 μm, the average ablation depth is 300 nm per pulse, which corresponds to an ablation rate of about 100 ng per pulse of SiO2 (major sample constituent).23 It should be noted that the large laser spot size used here enables single-shot element determination with the ICP-DOFMS instrument. This spot size could be useful for depth profiling of materials, but is too large for LA imaging applications.
 | ||
| Fig. 3 (a) Integrated signals from the mass-spectral peaks of 238U and 232Th isotopes (shown at different scales) for single-pulse LA-ICP-DOFMS. In this experiment, the CCD was operated with an exposure time of 100 ms and a frequency of 4.16 Hz. (b) Zoomed-in view of two transient laser-ablation peaks. The single-pulse detection limit for uranium is 200 fg. | ||
From these data, we determine the detection limit for uranium to be 2 μg g−1 pulse and the absolute detection limit to be 200 fg. Our single-pulse LA-ICP-DOFMS detection limits are about an order of magnitude poorer than those previously obtained with TOFMS (18 fg U), but are comparable to those obtained with the MHMS (272 fg U).23,34
Time-gated mass-spectral acquisition
As seen in Fig. 3, the peak heights and widths produced by aerosol plugs from single laser-ablation pulses vary significantly from event to event. In our experiments, the relative standard deviation (RSD) of peak heights for single laser-ablation events was 20%, while that of the time-integrated peak area was 14%. This variation is a product of the laser-pulse irreproducibility, ICP drift, and mass-spectral sampling bias. Mass-spectral sampling bias, or aliasing,37 occurs when the signals from individual laser-ablation events are not measured at the same relative positions along their transient ablation profiles. With DOFMS detection by a CCD detector, the CCD camera exposure and readout times act like a comb filter to modulate the DOFMS signal and produce aliased LA signals (cf. ESI†). Here, the CCD exposure was 100 ms and the camera readout time was 138 ms. If a signal maximum occurs during the detector readout, this information is lost and cannot be recovered mathematically because of the unknown contribution of laser-induced signal variation. Importantly, mass-spectral sampling bias is different from commonly encountered “spectral skew”2,38 in scanning-based MS instruments; because all DOFMS signals are detected simultaneously, the MS signals (and isotope ratios) within each ablation event are internally consistent. To overcome mass-spectral sampling bias and also improve the S/N ratio of spectra of single-pulse LA events, we time-delayed the CCD acquisition from the Q-switch of the Nd:YAG laser. By using a long CCD exposure time of 250 ms and time-delaying the CCD acquisition, we collected DOFMS spectra at a consistent point along each transient LA profile, captured the majority of the ion signals generated with each LA pulse, and minimized pulse-to-pulse signal variation.Fig. 4a shows averaged DOF spectra for CCD delay times from 100 to 1000 ms after each ablation event; each displayed spectrum is the average of 10 original spectra. As seen in Fig. 4a, the time of CCD image collection dramatically affects the magnitudes and S/N ratios of the acquired mass spectra. Fig. 4b highlights the correlation of isotopic signals to CCD delay time for 96Mo and 98Mo. At a delay time of 800 ms, both the S/N ratio and the signal itself are highest because the highest concentration portion of each LA aerosol plug is detected with each CCD exposure. Further experiments have shown that the CCD-time-delayed determination of other elements (see Fig. S2†) also produces a maximum mass-spectral signal at 800 ms. However, because the optimal delay time is dependent on the aerosol transit time from the ablation chamber to the mass analyzer, adjustment of ablation-gas or transfer-tube-gas flow rates will affect the optimal CCD delay time.
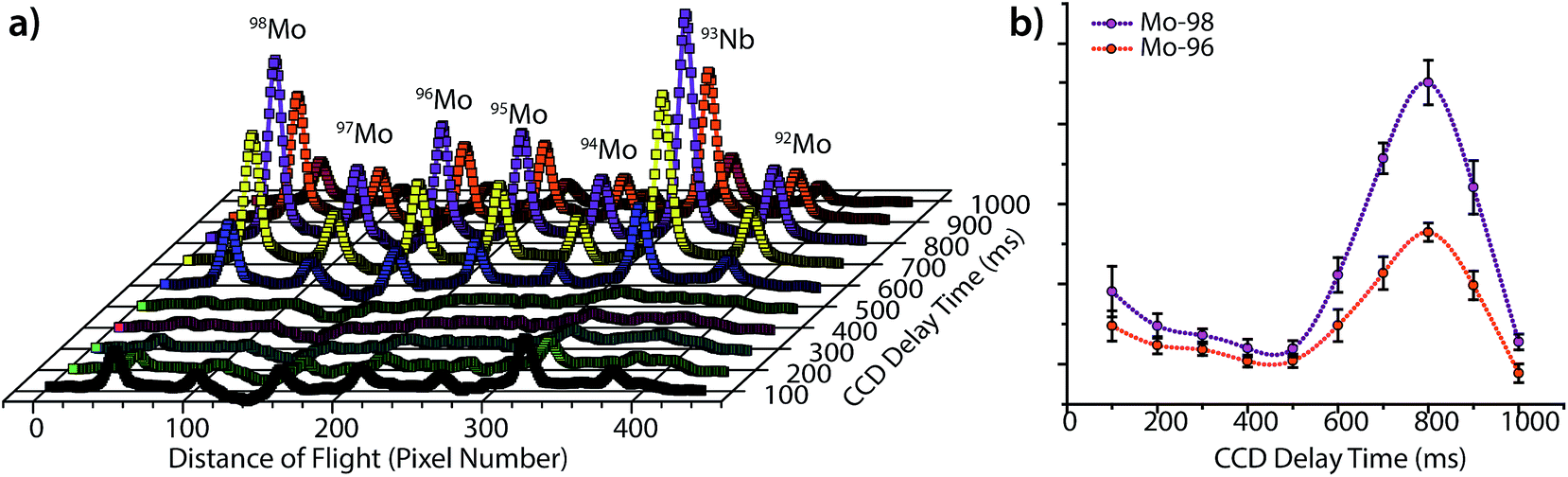 | ||
| Fig. 4 (a) DOFMS spectra of molybdenum isotopes ablated from a BNRM Carb20Cb3 copper-alloy standard and detected at increasing CCD delay times from the onset of the laser pulse. (b) The variation in integrated signals of 96Mo+ and 98Mo+ with CCD delay time shows that both isotopes give a maximum signal at a delay time of 800 ms; the integrated peak intensity in arbitrary units is plotted on the y-axis. The 800 ms delay corresponds to the average transit time of aerosol from ablation chamber to the DOFMS analyzer. | ||
In addition to increasing S/N, time-delayed CCD acquisition improves shot-to-shot precision. Fig. 5a displays the integrated signals of 96Mo and 98Mo for the single-pulse LA experiment when the CCD is operated at a collection rate of 5.25 Hz and an exposure of 100 ms, rather than the time-delayed CCD approach. For a side-by-side comparison of mass spectra with 100 ms and 250 ms exposures, see Fig. S3.† As expected, the RSD of peak height and peak area for the single-pulse LA signals collected at 5.25 Hz is quite large. For comparison, in Fig. 5b, we graph the normalized time-integrated signals of 98Mo from the single-pulse LA data given in Fig. 5a against those from replicate measurements at the 800 ms CCD delay. In both experiments, mass-spectral signals are obtained from single ablation events; however, the precision of the time-delayed CCD acquisition is substantially better than the single-pulse experiments with fast spectral acquisition—6% RSD vs. 25% RSD. While fast image collection allows the LA transient profile to be visualized, it introduces a mass-spectral sampling bias and requires the camera to be read out (and not exposed) when signals are high, which reduces the total ion signal collected and worsens precision.
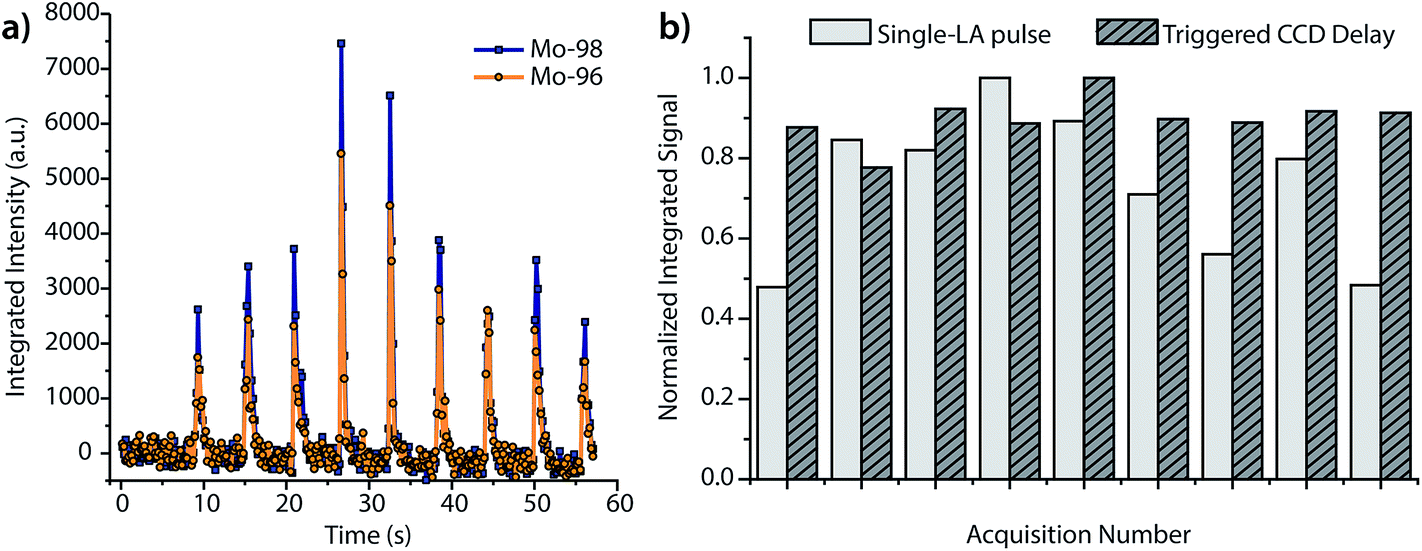 | ||
| Fig. 5 (a) Single-pulse analysis of the BNRM Carb20Cb3 copper-alloy standard with DOFMS detection set at 5.25 Hz and a 100 ms CCD exposure. (b) Normalized time-integrated signals of 98Mo+ for both time-delayed CCD acquisition (cf.Fig. 4) and fast DOFMS detection. The CCD-delayed method offers better precision: 6% vs. 25% RSD. | ||
The temporal resolution of our instrument is currently limited by the MCP/phosphor-CCD detection system, due mostly to the long readout time of the camera. The fastest spectral acquisition rate we have achieved with this detection arrangement is 6.15 Hz; see ESI† for details. The CCD-delay-time approach satisfactorily overcomes the time-resolution limitations of our current detection setup, and improves detection efficiency through collection of ion signal across the majority of each LA peak and reduces the number of spectra (amount of data) required for single-pulse LA analysis. Because the CCD delay allows collection of high-S/N mass spectra from each LA pulse, it should prove especially useful in LA-ICPMS applications that benefit from the analysis of single LA events, such as in elemental mapping and depth profiling.9,11,12,39,40 Of course, incorporation of alternative high-sensitivity, high-speed DOFMS detection strategies would also eliminate mass-spectral sampling bias and improve instrument performance; this is a major direction of current research.
Isotope-ratio analysis by LA-ICP-DOFMS
In addition to the improved shot-to-shot performance obtained with time-gated DOFMS detection, DOFMS also offers the benefits of simultaneous multichannel (multi-isotope) detection. Through ratioing simultaneously acquired ion signals, multichannel detection can overcome correlated noise caused by shot-to-shot variation between laser pulses and drift (flicker noise) from the ICP, and also to mitigate spectral-skew error.30,38,41 These advantages are not available on single-channel ICPMS instruments, such as quadrupole or sector-field mass spectrometers (QMS or SFMS) because, on such instruments, ion signals that fluctuate in concert must be measured sequentially rather than simultaneously.In recent years, the advantages of multichannel LA-ICPMS analysis have been well demonstrated with several instrument designs, including multi-collector sector-field mass spectrometers,12,18,41 Mattauch–Herzog mass spectrographs equipped with charge-detector arrays,34,42–45 and time-of-flight mass spectrometers.35,46–49 With these instruments and ns-LA systems, isotope-ratio precisions of 0.01%,50 0.02%,34 and 0.4%,30 respectively, have been reported. However, a direct comparison of these approaches to LA-ICP-DOFMS is problematic because the performance of LA-ICPMS is dependent on many instrumental factors. Results from a controlled ablation setup, sample type, and mass-analysis approach are not available to compare mass-analyzer performance objectively. Rather, we investigate here the isotope-ratio performance of our LA-ICP-DOFMS instrument and compare it to previous results obtained with solution sample introduction ICP-DOFMS.
Fig. 6 highlights the isotope-ratio performance of our LA-ICP-DOFMS instrument. Here, we ablated a copper alloy standard, BNRM Carb20Cb3, at a laser repetition rate of 5 Hz to create a steady-state signal and used a 250 ms CCD exposure time (2.4 Hz spectrum-collection rate). In Fig. 6a, the integrated signals of 63Cu+ and 65Cu+ each show an average RSD of 8.4%, which is consistent with expectations for single-channel LA-ICPMS analysis.28,51Fig. 6b shows the ratio of 65Cu:63Cu and illustrates that the noise between the two mass-spectral windows is correlated; the isotope-ratio precision is 1.3% for 300 consecutively acquired DOFMS signals. In Fig. 6b, we also mimic the performance of DOFMS as a single-channel analyzer by ratioing copper signals from sequentially acquired spectra. With this approach, the isotope-ratio precision worsens to 9.7% RSD, and so demonstrates how simultaneous multi-element detection improves precision. The observed isotope ratio of 65Cu:63Cu is 0.466, 0.02 above the expected isotope ratio of 0.446;52 this discrepancy is most likely due to mass bias in ion sampling from the ICP,53,54 and is consistent with our previous solution-sample-introduction ICP-DOFMS studies.
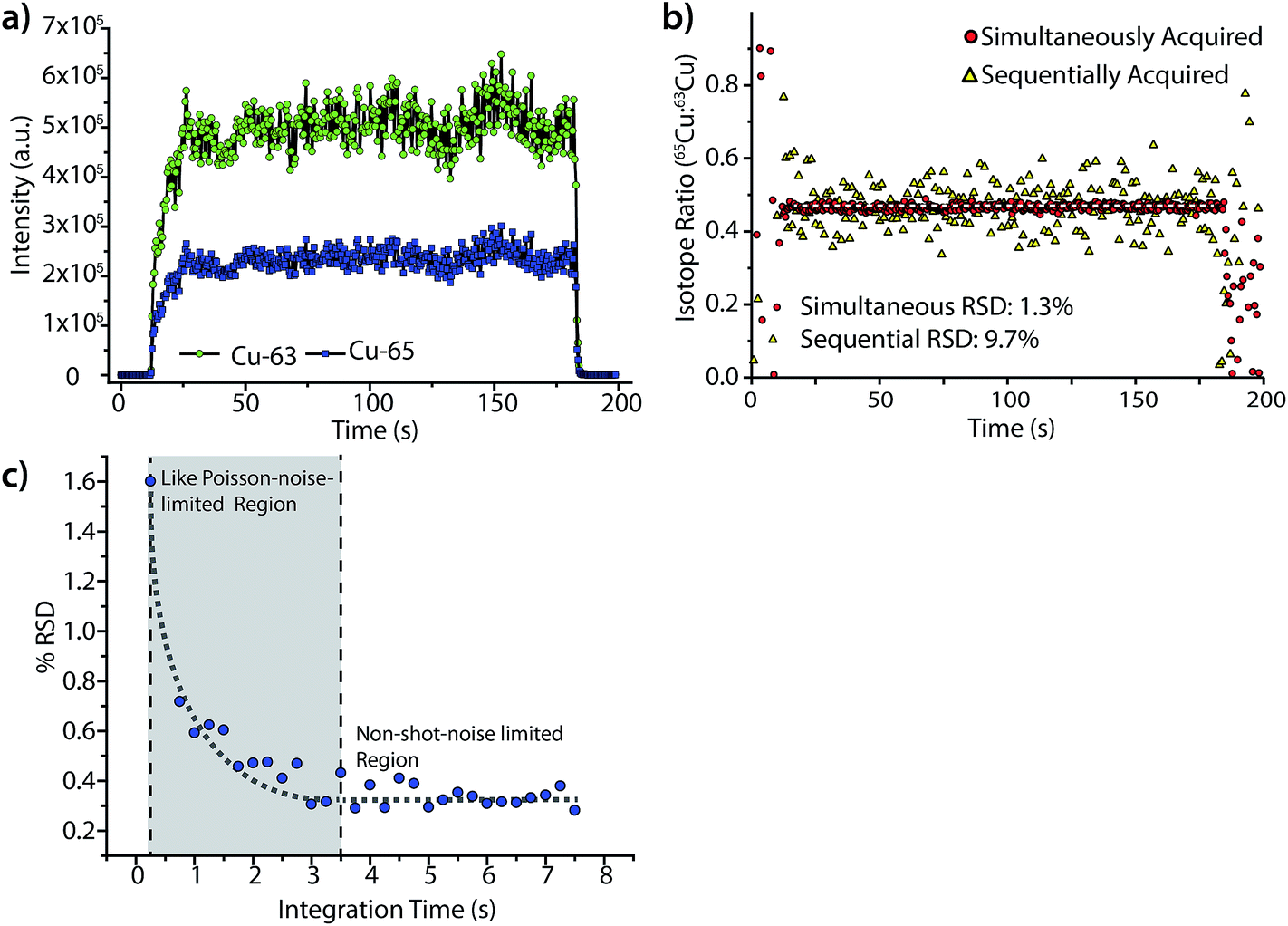 | ||
| Fig. 6 (a) Time trace of integrated mass-spectral peaks of 63Cu+ and 65Cu+ for the steady-state LA of a BNRM Carb20Cb3 copper-alloy standard. (b) Ratio of simultaneously acquired 63Cu+ and 65Cu+ signals gives a relative standard deviation (RSD) of 1.3% over 300 measurements. However, if the ratios of 63Cu+ and 65Cu+ signals are calculated from sequentially taken DOF mass spectra, the RSD worsens to 9.7%. (c) By summing spectra offline to increase integration time, the isotope-ratio precision is shown to improves with integration time from exposure times of 0.25 to 3.5 s, as predicted by Poisson statistics. However, the RSD levels off after a CCD exposure time of about 3.5 s. | ||
To further improve the isotope-ratio precision of LA-ICP-DOFMS analysis, we summed consecutively acquired spectra offline to generate spectra with longer equivalent CCD exposure times. As shown in Fig. 6c, the isotope-ratio RSD initially improves, down to 0.3% RSD, with lengthened integration time as predicted by Poisson statistics.55,56 However, as the integration time grows from 3.5 to 7.5 s, the isotope-ratio precision does not continue to improve; this behavior indicates that another noise source dominates at these long integration times. Studies are ongoing to identify this other noise source; a strong candidate is ion-signal instability and drift across the MCP/phosphor detector. Because the MCP/phosphor detector most likely suffers from inconsistent ion-detection efficiency across its surface, any spatial drift of ion peaks will worsen precision, and mass-analyzer drift is a well-known precision-limiting noise source.18 For example, multi-collector ICPMS instruments designed for isotope-ratio analysis mitigate the negative effects of spectral drift by collecting all mass-separated ions of each isotope with dedicated detectors so ion peaks that drift spatially still equally strike their detector. A narrow scan across the m/z range for a single isotope then produces a characteristic “flat-topped” peak, which demonstrates the small variance of isotopic signals with spatial drift.56 In the future, similar ion-collection strategies and peak shapes could be incorporated into DOFMS.
Table 2 shows that our LA-ICP-DOFMS instrument equipped with an MCP/phosphor-CCD detection system can provide isotope-ratio precision better than 2% RSD, though this experimental approach is not yet usable for isotope-ratio determinations because systematic isotope-ratio errors have not been defined. For comparison, the best isotope-ratio precision results achieved for solution-based ICP-DOFMS trace-element determinations are about one order of magnitude better than that achieved here. Most likely, this poorer isotope-ratio precision is influenced by the laser-ablation process. However, more research is required to calibrate absolute ion signals obtained with DOFMS so isotope-ratio results can be directly compared and evaluated with counting statistics. On the whole, isotope-ratio precision obtained with the DOFMS are worse than those obtained with commercial multichannel ICPMS instruments, but surpass, or are on par with, those of single-collector ICPMS instruments.30 Additionally, it is expected that the future incorporation of solid-state ion detector arrays into the LA-ICP-DOFMS instrument will result in even better precision.
| Isotope Pair | Solution-ICP-DOFMSa | LA-ICP-DOFMSb | ||
|---|---|---|---|---|
| RSD (%) | Integration time (s) | RSD (%) | Integration time (s) | |
| a Analyte concentration was 1 ppm. b Analyte concentration varied from 500 ppm to 3% depending on sample. | ||||
| 65/63Cu | 0.30 | 6 | 0.28 | 3 |
| 109/107Ag | 0.11 | 6 | 1.18 | 10 |
| 120/118Sn | — | — | 1.87 | 8 |
| 98/96Mo | — | — | 1.35 | 7 |
| 98/95Mo | — | — | 1.57 | 11.5 |
| 154/152Sm | 0.14 | 11.5 | — | — |
| 208/207Pb | 0.10 | 8 | 1.01 | 7 |
Conclusions
In this study, we report the first combination of ICP-DOFMS with laser ablation. In addition to evaluating figures of merit such as detection limits and isotope-ratio precision, we tested the temporal resolution of the DOF mass analyzer and demonstrated continuous signal acquisition at 6.15 Hz. This temporal resolution was sufficient to detect and characterize atomic signals from single LA events. We also developed a CCD-delay detection method to improve the S/N of LA-ICP-DOFMS spectra and shot-to-shot precision between LA events. In particular, the 6% RSD between single LA events obtained with our CCD-delay method compares well with single-element precision available with alternative LA-ICPMS systems.30The initial results presented here indicate that DOFMS has promise as a mass analyzer for LA analysis, as well as for other time-dependent measurements, such as online combination with separation methods. Though our current MCP/phosphor-CCD detection system has fundamentally limited measurement speed due to camera readout time, this speed limitation is not inherent to the DOFMS technique. We expect future developments in high-speed ion-detection arrays to facilitate the continued growth of DOFMS for time-sensitive analysis.
Acknowledgements
Alexander Gundlach-Graham thanks the Division of Analytical Chemistry (DAC) of the American Chemistry Society and Agilent Technologies for a DAC graduate-research fellowship. AGG also thanks Indiana University, Department of Chemistry for support under the Briscoe Teacher-Scholar fellowship program. Partial salary support was provided by the U.S. Department of Energy through grant DE-FG02-98ER14890. This research was also supported in part by the National Science Foundation through Grant DBI-1062846 and performed in collaboration with Pacific Northwest National Laboratory, operated for the US DOE by Battelle Memorial Institute under Contract DE-AC06-76RLO-1830op.References
- C. G. Enke., The Unique Capabilities of Time-of-Flight Mass Analyzers, in Adv. Mass Spectrom., Elsevier Science Publishers B. V., Amsterdam, 1998, 14, pp. 197–219 Search PubMed.
- G. D. Schilling, F. J. Andrade, J. H. Barnes, R. P. Sperline, M. B. Denton, C. J. Barinaga, D. W. Koppenaal and G. M. Hieftje, Anal. Chem., 2007, 79(20), 7662–7668 CrossRef CAS PubMed.
- A. W. G. Graham, S. J. Ray, C. G. Enke, J. A. Felton, A. J. Carado, C. J. Barinaga, D. W. Koppenaal and G. M. Hieftje, Anal. Chem., 2011, 83(22), 8552–8559 CrossRef CAS PubMed.
- A. Gundlach-Graham, E. Dennis, S. Ray, C. Enke, C. Barinaga, D. Koppenaal and G. Hieftje, J. Am. Soc. Mass Spectrom., 2013, 24(11), 1736–1744 CrossRef CAS PubMed.
- A. W. Gundlach-Graham, E. A. Dennis, S. J. Ray, C. G. Enke, A. J. Carado, C. J. Barinaga, D. W. Koppenaal and G. M. Hieftje, Rapid Commun. Mass Spectrom., 2012, 26(21), 2526–2534 CrossRef CAS PubMed.
- A. Graham, S. Ray, C. Enke, C. Barinaga, D. Koppenaal and G. Hieftje, J. Am. Soc. Mass Spectrom., 2011, 22(1), 110–117 CrossRef CAS PubMed.
- A. Gundlach-Graham, E. A. Dennis, S. J. Ray, C. G. Enke, C. J. Barinaga, D. W. Koppenaal and G. M. Hieftje, J. Anal. At. Spectrom., 2013, 28(9), 1385–1395 RSC.
- A. L. Gray, Analyst, 1985, 110(5), 551–556 RSC.
- J. Elteren, A. Izmer, M. Sala, E. F. Orsega, V. S. Selih, S. Panighello and F. Vanhaecke, J. Anal. At. Spectrom., 2013, 28, 994–1004 RSC.
- D. Rubatto, Chem. Geol., 2002, 184(1), 123–138 CrossRef CAS.
- C. Heinrich, T. Pettke, W. Halter, M. Aigner-Torres, A. Audétat, D. Günther, B. Hattendorf, D. Bleiner, M. Guillong and I. Horn, Geochim. Cosmochim. Acta, 2003, 67(18), 3473–3497 CrossRef CAS.
- J. Sabine Becker, J. Mass Spectrom., 2013, 48(2), 255–268 CrossRef CAS PubMed.
- M. Resano, E. García-Ruiz, R. Alloza, M. P. Marzo, P. Vandenabeele and F. Vanhaecke, Anal. Chem., 2007, 79(23), 8947–8955 CrossRef CAS PubMed.
- J. D. Woodhead, J. Hellstrom, J. M. Hergt, A. Greig and R. Maas, Geostand. Geoanal. Res., 2007, 31(4), 331–343 CAS.
- D. Gunther, R. Frischknecht, C. A. Heinrich and H.-J. Kahlert, J. Anal. At. Spectrom., 1997, 12(9), 939–944 RSC.
- D. M. McClenathan, S. J. Ray, W. C. Wetzel and G. M. Hieftje, Anal. Chem., 2004, 76(9), 158A–166A CrossRef CAS.
- J. Pisonero and D. Günther, Mass Spectrom. Rev., 2008, 27(6), 609–623 CrossRef CAS PubMed.
- N. Jakubowski, T. Prohaska, L. Rottmann and F. Vanhaecke, J. Anal. At. Spectrom., 2011, 26(4), 693–726 RSC.
- D. Günther and B. Hattendorf, TrAC, Trends Anal. Chem., 2005, 24(3), 255–265 CrossRef PubMed.
- C. C. Garcia, H. Lindner and K. Niemax, J. Anal. At. Spectrom., 2009, 24(1), 14–26 RSC.
- C. G. Enke and G. S. Dobson, Energy Focus for Distance of Flight Mass Spectometry with Constant Momentum Acceleration and an Ion Mirror, US 2008/0017792 A1, Jan. 24 2008.
- C. G. Enke, S. J. Ray, A. W. Graham, E. A. Dennis, G. M. Hieftje, A. J. Carado, C. J. Barinaga and D. W. Koppenaal, Ann. Rev. Anal. Chem., 2012, 5(1), 487–504 CrossRef CAS PubMed.
- A. M. Leach and G. M. Hieftje, Appl. Spectrosc., 2002, 56(1), 62–69 CrossRef CAS.
- National Institute of Standards & Technology, Certificate of Analysis: Standard Reference Material 612 2012.
- National Institute of Standards & Technology, Certificate of Analysis: Standard Reference Material 610 2012.
- Brammer Standard, http://www.brammerstandard.com/certificates/bshas-12.pdf.
- B. Hattendorf and D. Günther, Laser Ablation Inductively Coupled Plasma Mass Spectrometry (LA-ICPMS), in Handbook of Spectroscopy, Wiley-VCH Verlag GmbH & Co. KGaA, 2014, pp 647–698 Search PubMed.
- J. Koch and D. Günther, Appl. Spectrosc., 2011, 65(5), 155A–162A CrossRef CAS PubMed.
- J. Gonzalez, X. Mao, J. Roy, S. Mao and R. Russo, J. Anal. At. Spectrom., 2002, 17(9), 1108–1113 RSC.
- R. E. Russo, X. Mao, J. J. Gonzalez, V. Zorba and J. Yoo, Anal. Chem., 2013, 85(13), 6162–6177 CrossRef CAS PubMed.
- R. E. Russo, X. L. Mao, O. V. Borisov and H. Liu, J. Anal. At. Spectrom., 2000, 15(9), 1115–1120 RSC.
- M. Guillong, I. Horn and D. Günther, J. Anal. At. Spectrom., 2003, 18(10), 1224–1230 RSC.
- J. J. Gonzalez, D. Oropeza, X. Mao and R. E. Russo, J. Anal. At. Spectrom., 2008, 23(2), 229–234 RSC.
- J. H. Barnes IV, G. D. Schilling, G. M. Hieftje, R. P. Sperline, M. B. Denton, C. J. Barinaga and D. W. Koppenaal, J. Am. Soc. Mass Spectrom., 2004, 15(6), 769–776 CrossRef PubMed.
- A. M. Leach and G. M. Hieftje, Anal. Chem., 2001, 73(13), 2959–2967 CrossRef CAS.
- O. Borovinskaya, B. Hattendorf, M. Tanner, S. Gschwind and D. Gunther, J. Anal. At. Spectrom., 2013, 28(2), 226–233 RSC.
- H. A. Wang, D. Grolimund, C. Giesen, C. N. Borca, J. R. Shaw-Stewart, B. Bodenmiller and D. Günther, Anal. Chem., 2013, 85(21), 10107–10116 CrossRef CAS PubMed.
- T. Pettke, C. A. Heinrich, A. C. Ciocan and D. Gunther, J. Anal. At. Spectrom., 2000, 15(9), 1149–1155 RSC.
- D. S. Gholap, A. Izmer, B. De Samber, J. T. van Elteren, V. S. Šelih, R. Evens, K. De Schamphelaere, C. Janssen, L. Balcaen, I. Lindemann, L. Vincze and F. Vanhaecke, Anal. Chim. Acta, 2010, 664(1), 19–26 CrossRef CAS PubMed.
- H. A. O. Wang, D. Grolimund, L. R. Van Loon, K. Barmettler, C. N. Borca, B. Aeschlimann and D. Günther, Anal. Chem., 2011, 83(16), 6259–6266 CrossRef CAS PubMed.
- G. E. Gehrels, V. A. Valencia and J. Ruiz, Geochem., Geophys., Geosyst., 2008, 9(3), Q03017 CrossRef.
- M. Resano, K. S. McIntosh and F. Vanhaecke, J. Anal. At. Spectrom., 2012, 27(1), 165–173 RSC.
- G. D. Schilling, S. J. Ray, A. A. Rubinshtein, J. A. Felton, R. P. Sperline, M. B. Denton, C. J. Barinaga, D. W. Koppenaal and G. M. Hieftje, Anal. Chem., 2009, 81(13), 5467–5473 CrossRef CAS PubMed.
- G. D. Schilling, F. J. Andrade, J. H. Barnes, R. P. Sperline, M. B. Denton, C. J. Barinaga, D. W. Koppenaal and G. M. Hieftje, Anal. Chem., 2006, 78(13), 4319–4325 CrossRef CAS PubMed.
- J. A. Felton, G. D. Schilling, S. J. Ray, R. P. Sperline, M. B. Denton, C. J. Barinaga, D. W. Koppenaal and G. M. Hieftje, J. Anal. At. Spectrom., 2011, 26(2), 300–304 RSC.
- A. M. Leach and G. M. Hieftje, J. Anal. At. Spectrom., 2000, 15(9), 1121–1124 RSC.
- P. P. Mahoney, G. Li and G. M. Hieftje, J. Anal. At. Spectrom., 1996, 11(6), 401–405 RSC.
- S. Willie, Z. Mester and R. E. Sturgeon, J. Anal. At. Spectrom., 2005, 20(12), 1358–1364 RSC.
- M. Tanner and D. Günther, Anal. Bioanal. Chem., 2008, 391(4), 1211–1220 CrossRef CAS PubMed.
- J. Woodhead, S. Swearer, J. Hergt and R. Maas, J. Anal. At. Spectrom., 2005, 20(1), 22–27 RSC.
- J. J. González, D. D. Oropeza, H. Longerich, X. Mao and R. E. Russo, J. Anal. At. Spectrom., 2012, 27(9), 1405–1412 RSC.
- M. Berglund and M. E. Wieser, Pure Appl. Chem., 2009, 83(2), 397–410 Search PubMed.
- K. G. Heumann, S. M. Gallus, G. Radlinger and J. Vogl, J. Anal. At. Spectrom., 1998, 13(9), 1001–1008 RSC.
- T. W. Burgoyne, G. M. Hieftje and R. A. Hites, Anal. Chem., 1997, 69(3), 485–489 CrossRef CAS.
- D. Solyom and G. Hieftje, J. Am. Soc. Mass Spectrom., 2003, 14(3), 227–235 CrossRef CAS.
- F. Vanhaecke and P. Degryse, Isotopic Analysis, Wiley, 2012 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ja00231h |
| ‡ Current address: Department of Chemistry and Applied Biosciences, ETH Zürich, 8093 Zürich, Switzerland. |
| This journal is © The Royal Society of Chemistry 2015 |