
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing coiled-coil assembly by paramagnetic NMR spectroscopy†
TingTing
Zheng
,
Aimee
Boyle
,
Hana
Robson Marsden
,
Dayenne
Valdink
,
Giuliana
Martelli
,
Jan
Raap
and
Alexander
Kros
 *
*
Dept. Supramolecular & Biomaterials Chemistry, Leiden Institute of Chemistry, Leiden University, PO Box 9502, 2300RA, Leiden, The Netherlands. E-mail: A.kros@chem.leidenuniv.nl
First published on 18th November 2014
Abstract
Here a new method to determine the oligomeric state and orientation of coiled-coil peptide motifs is described. Peptides K and E, which are designed to form a parallel heterodimeric complex in aqueous solution, were labeled with the aromatic amino acids tryptophan and tyrosine on the C-terminus respectively as ‘fingerprint’ residues. One of the peptides was also labeled with the paramagnetic probe MTSL. One dimensional proton NMR spectroscopy was used to study the peptide quaternary structure by monitoring the signal suppression of the aromatic labels due to proximity of the nitroxyl radical. 1D-NMR confirmed that the peptides K and E form a heterodimeric coiled coil with a parallel orientation. In addition, fluorescence emission quenching of the aromatic labels due to electron exchange with a nitroxyl radical confirmed the parallel coiled coil orientation. Thus, paramagnetic nitroxide and aromatic fluorophore labeling of peptides yields valuable information regarding the quaternary structure from 1D-NMR and steady-state fluorescence measurements. This convenient method is useful not only to investigate coiled coil assembly, but can also be applied to any defined supramolecular assembly.
Introduction
Coiled coils are a protein motif comprised of two to nine α-helices folded around each other in a superhelical fashion, with the helices aligned either parallel or antiparallel to one another.1–8 In nature, this protein folding motif assembles into a wide range of structures with a variety of functions, such as gene expression regulation (e.g. transcription factors) and molecular membrane trafficking (e.g. SNARE proteins).7,9–12 The simplicity of the individual peptides coupled with the functionality of the complexes has led to the inclusion of coiled-coil complexes in many synthetic supramolecular systems.5,13–16To fully utilize coiled-coil motifs as building blocks in supramolecular systems it is important to obtain a detailed view of the quaternary structure of the assemblies. Knowledge of the number of peptides in an assembly, their stoichiometry and their relative orientation are required for the effective design of functional systems that incorporate coiled-coil elements.17–28 Coiled-coil peptide complexes are usually identified by circular dichroism, which can also determine the peptide stoichiometry. However, elucidating the number of peptides in the complexes and their orientation is often time-intensive and requires expensive and complex equipment. Typically, analytical ultracentrifugation, X-ray diffraction, disulfide exchange, electron paramagnetic resonance (EPR), two-dimensional nuclear magnetic resonance (2D-NMR), 3D-NMR and even 4D-NMR techniques are employed to study these aspects of the quaternary structure. These measurements are not always practical, therefore it is valuable to develop methods that are simple to prepare, execute, and analyze.
In this paper we demonstrate the simple and easy use of paramagnetic 1D-NMR to investigate coiled-coil peptide assembly and orientation. We also show that the results can be confirmed with fluorescence measurements.
For the 1D-NMR measurements, the paramagnetic nitroxyl radical MTSL (S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate) is introduced at specific positions in the peptides. Magnetic dipole interactions between the unpaired electron of MTSL and nuclei that are within 13 Å of it result in the drastic suppression of their NMR signals.29–37
The amino acids tryptophan and tyrosine are introduced as the ‘fingerprint’ functional groups in this method. Tryptophan and tyrosine have aromatic side chains, whose signals in 1D-proton NMR spectra are well-separated from all other proton signals, making the signal-quenching effect of the spin label unambiguous. Peptides which already contain an aromatic group would not require labeling with an additional aromatic amino acid. Indeed, the effect of the spin label on any fully resolved proton signal could be analyzed. However, another advantage of using tryptophan and tyrosine as the fingerprint groups is that these amino acids have an intrinsic fluorescence, which is quenched when the fluorophores are within 12 Å of the nitroxide radical,38–42 meaning that the NMR results can be confirmed with fluorescence quenching measurements.
To demonstrate our approach, the well-known heterodimeric coiled coil E/K was used as a model system.18,43,44 This coiled coil has been used by several groups in the field of supramolecular chemistry, polymers and membrane fusion.45–51
The peptides, which we denote Coil-K and Coil-E, were labeled at the C-terminus with tryptophan and tyrosine respectively. Variants of the peptides were also labeled with MTSL at the C- or the N-terminus. An equimolar mixture of Coil-K and Coil-E results in the formation of a heterodimeric coiled coil motif, CC-K/E. The C-termini of both peptides will be in close proximity when a parallel orientation is adopted; but will be at opposite ends of the coiled coil complex if an antiparallel orientation is adopted (Scheme 1).
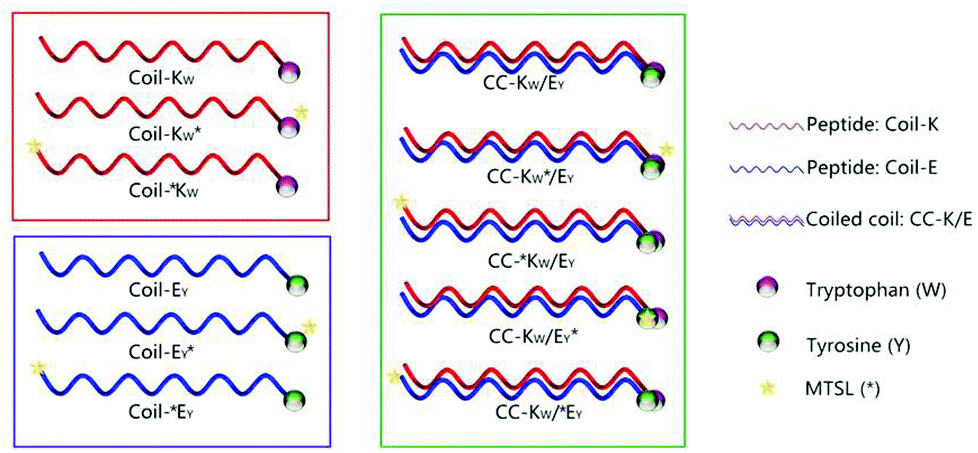 | ||
| Scheme 1 Schematic illustration of designed peptides and coiled coil motifs. Coil-K peptides are labeled with a tryptophan (W) residue while Coil-E peptides are decorated with a tyrosine (Y) residue. In this paper W and Y are always at the C-terminus of the peptides. A yellow star represents the MTSL- (*) nitroxyl radical spin label, which is added to either the C- or the N-terminus, to investigate the possibility of CC-K/E parallel and antiparallel orientations respectively. See Table 1 for primary sequences of all peptides. | ||
The paramagnetic 1D-proton NMR and steady state fluorescence studies demonstrate that the peptides Coil-K and Coil-E form a parallel heterodimeric coiled coil CC-K/E in phosphate buffered saline (PBS). This is the first example of a paramagnetic nitroxide spin label being used to determine the orientation and assembly of peptide strands in a coiled-coil motif. Furthermore, the peptide assembly ratio can be determined within a heteromeric-coiled-coil complex. This method does not disturb self-assembly and can be applied to any discrete supramolecular structure.
Peptide design and synthesis
In this study the feasibility of using paramagnetic NMR and fluorescence spectroscopy to investigate the orientation of the complementary peptides in a coiled-coil motif was explored. For this, the heterodimeric coiled-coil pair Coil-K and Coil-E was used as a model system. Coil-K and Coil-E have three heptad repeats, with the sequences (Ac-(KIAALKE)3-CONH2) and (Ac-(EIAALEK)3-CONH2), respectively. These sequences were modified with an aromatic amino acid, either a tryptophan (W) or tyrosine (Y) at the C-terminus. A glycine residue was added between the aromatic fluorophore and the original peptide sequence to minimize any potential influence on the coiled-coil assembly. The paramagnetic nitroxyl radical MTSL was introduced at either the C- or N-terminus of the peptides as the sensitive ‘signal suppression’ functional group (Table 1).18,43,44 Facile site-directed spin labeling was achieved via a disulfide bond to a cysteine residue.52 The distance between the aromatic fluorophore and the nitroxide determines whether the signal is suppressed or not. Initially, the spin label was conjugated to the C-terminus of the peptide to probe the parallel orientation in the coiled-coil heterodimer (peptides denoted KW* and EY*). For comparison, the spin label was conjugated at the N-terminus of the peptide to probe whether the antiparallel orientation would (co)exist as well (peptides denoted *KW and *EY) (Table 1).| Peptide name | Sequence | 1H-NMR signala | Fluorescence signalb |
|---|---|---|---|
| a 1D-proton NMR chemical signal for aromatic protons of tryptophan (W) and tyrosine (Y) in the range of 6–8 ppm. b Fluorescence emission spectra from 285–445 nm with excitation at 275 nm. Both 1H-NMR and fluorescence measurements were performed in pH = 7.4 PBS buffer. ‘+’ Indicates there is signal observed while ‘−’ indicates there is no signal observed. ‘ * ’ Denotes the position of the nitroxide spin label. | |||
| Coil-KW | Ac-(KIAALKE)3GW-NH2 | + | + |
| Coil-KW* | Ac-(KIAALKE)3GWC*-NH2 | − | − |
| Coil-*KW | Ac-C*(KIAALKE)3GW-NH2 | + | + |
| Coil-EY | Ac-(EIAALEK)3GY-NH2 | + | + |
| Coil-EY* | Ac-(EIAALEK)3GYC*-NH2 | − | − |
| Coil-*EY | Ac-C*(EIAALEK)3GY-NH2 | + | + |
Circular dichroism spectroscopy
As single mutations in amino acid sequences can alter the propensity of the peptides to form coiled coils, the secondary structures and binding properties of the Coil-K and Coil-E peptides and their derivatives were studied using circular dichroism (CD) spectroscopy (Fig. 1 and S4†). These results showed that in PBS buffer (pH = 7.4) there is no significant change in the secondary structure of Coil-K after introduction of the aromatic amino acids and the spinlabel (Fig. 1A), while the introduction of the labels causes Coil-E to adopt a more α-helical conformation (Fig. 1B).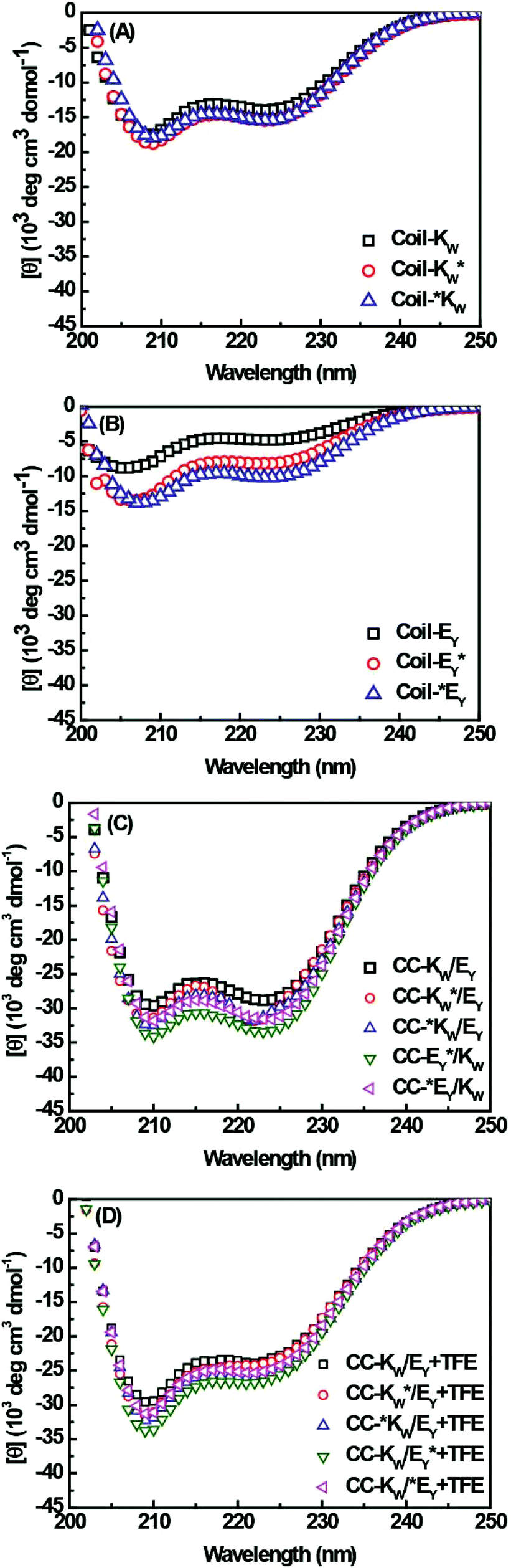 | ||
Fig. 1 (A) CD spectra showing secondary structure comparisons of the peptide Coil-K and its derivatives. (B) Secondary structure comparison of CD spectra from Coil-E and its derivatives (C) Comparison of CD spectra of equimolar mixtures of Coil-K and Coil-E peptides and their derivatives in PBS, and in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) PBS:TFE (D) [Total peptide] = 200 μM, PBS pH = 7.4, 25 °C. :1 (v/v) PBS:TFE (D) [Total peptide] = 200 μM, PBS pH = 7.4, 25 °C. | ||
Next, coiled-coil formation of equimolar mixtures of coil-E and coil-K and their derivatives, were studied. All peptide pairs exhibited spectra typical of coiled-coils, with the α-helical content higher than 90% and the ratio of [θ]222/[θ]208 close to 1 (Fig. 1C).53,54 Trifluoroethanol (TFE) is known to enhance the intramolecular α-helicity while disrupting intermolecular interactions.55,56 As expected, addition of TFE resulted in a lower [θ]222/[θ]208 ratio and decreased α-helicity, consistent with the transition from a coiled-coil to monomeric α-helical peptides (Fig. 1D and Table 2 in ESI†).1
CD spectroscopy was then used to confirm that the labels added for the purpose of this study did not affect the stoichiometry of the coiled-coil complexes. A job-plot of [θ]222 as a function of the mole fraction of Coil-E yields the binding stoichiometry.57,58 For all of the coiled coil complexes studied, a minimum of [θ]222 was always observed at an equimolar ratio of Coil-K (or its derivative) and Coil-E (or its derivative), indicating that all peptide pairs bind in a 1:1 stoichiometry (Fig. 2A).
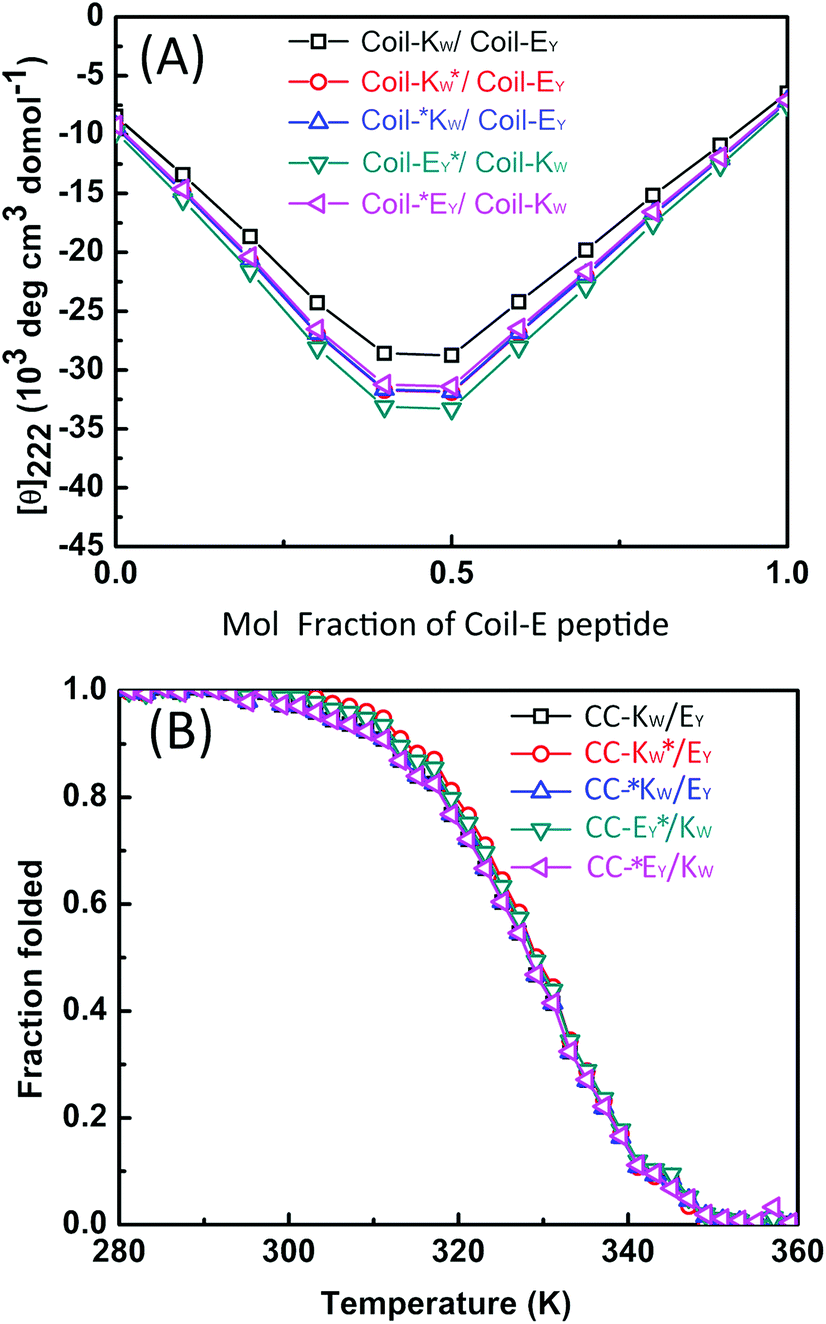 | ||
| Fig. 2 (A) Mean residue molar ellipticities at 222 nm for mixtures of the Coil-K and Coil-E peptides as a function of the mole fraction of the Coil-E peptide. All the measurements were carried out at a total peptide concentration of 200 μM at 25 °C, in 1 mm quartz cuvettes. (B) Thermal unfolding curves based on [θ]222 as a function of temperature. 1 cm quartz cuvette with stirring at 900 rpm was used. [Total peptide] = 40 μM, PBS, pH = 7.4. | ||
The thermodynamic stability of the all of the CC-K/E pairs was then determined. The molar ellipticity at 222 nm is directly proportional to the amount of helical structure and therefore thermal denaturation curves provide information as to the stability of the coiled coils.59–61 Temperature-dependent CD measurements showed that all the peptide complexes used in this study have an identical two-state transition denaturation process, dissociating from coiled coils to random coil (Fig. 2B). The dissociation constants for all of the coiled coils are in the same order of magnitude (10−8 M). The binding parameters are summarized in Table 3 in the ESI.†
Thus, while the introduction of the additional amino acids and the MTSL label does adjust the secondary structure of Coil-E, the labels do not significantly alter coiled-coil assembly. All of the labelled peptide pairs formed coiled-coil complexes with the same stoichiometry and with similar binding energies to the original peptides Coil-K and Coil-E.
1H-NMR spectroscopy
1H-nuclear magnetic resonance spectroscopy was used to study the coiled-coil complex formation of the Coil-K and Coil-E peptide derivatives, including their orientation and binding stoichiometry. Tryptophan (W) and tyrosine (Y) residues show characteristic aromatic signals with a chemical shift in the range of 6 to 8 ppm. The N–H signals were suppressed by H–D exchange to prevent overlap with the aromatic region. Typical individual NMR signals of Coil-KW and Coil-EY in the 6–8 ppm range are shown in Fig. 3 A/C (blue lines).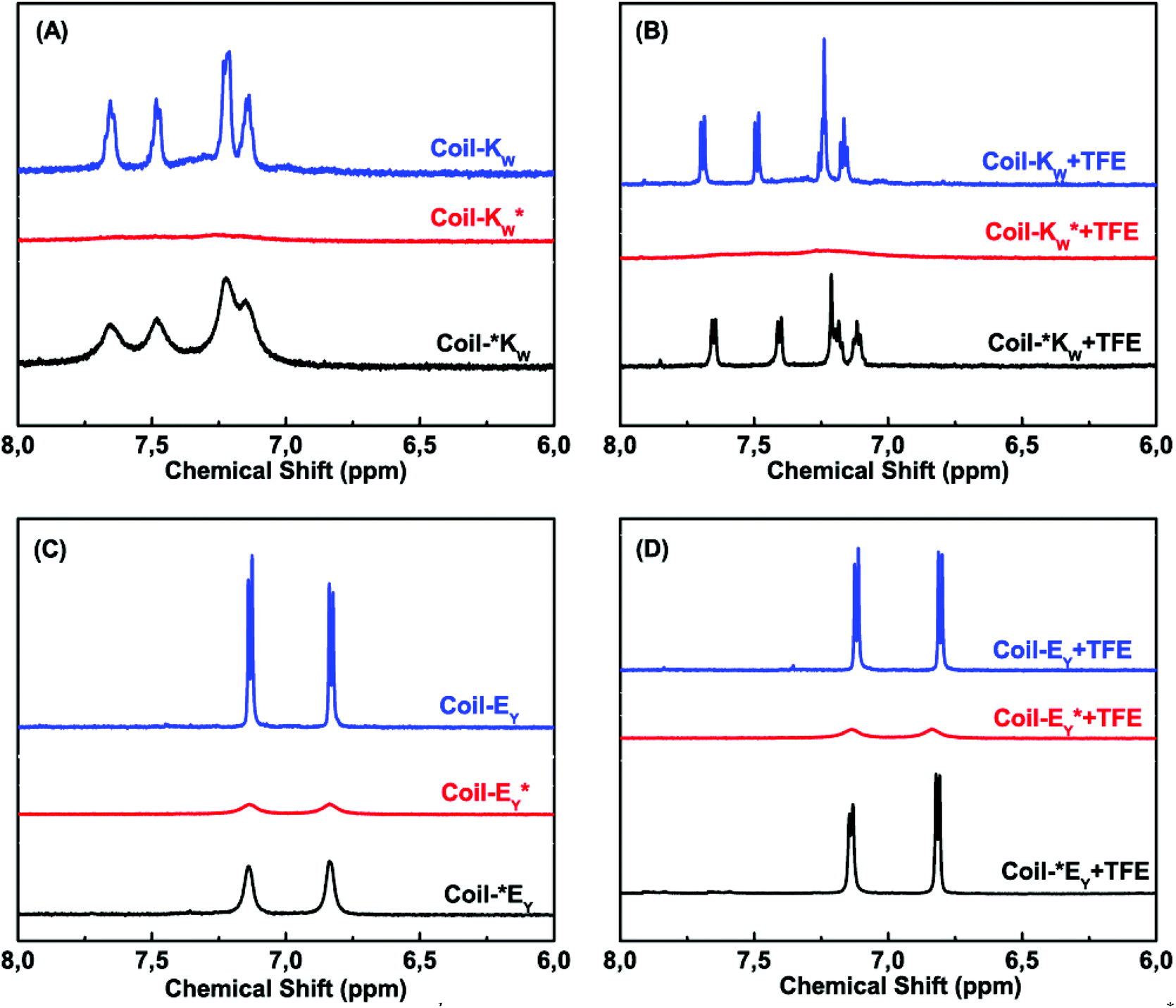 | ||
| Fig. 3 Aromatic region (6–8 ppm) of 600 MHz 1H-nuclear magnetic resonance spectra showing the tryptophan indole and tyrosine hydroxyphenyl functional groups of Coil-K and Coil-E derivatives respectively. (A) Aromatic signals of Coil-KW, Coil-*KW and Coil-KW* in PBS. (B) Aromatic signals of Coil-KW, Coil-*KW and Coil-KW* in 1:1 (v/v) PBS:TFE solution. (C) Aromatic signals of Coil-EY, Coil-*EY and Coil-EY* in PBS. (D) Aromatic signal of Coil-EY, Coil-*EY and Coil-EY* in 1:1 (v/v) PBS:TFE solution. [Total peptide] = 0.8 mM (see ESI Fig. S8† for 0–8 ppm chemical shift). | ||
When a spin label is located close to a proton, magnetic dipole interactions lead to paramagnetic relaxation enhancement (PRE),31–36 increasing the relaxation rate of the nuclear magnetization and resulting in the suppression of the NMR signal of the neighboring nucleus.29,33,34,37,62–65 The line width of a proton signal will get significantly perturbed when the proton is within 13.0 Å of the paramagnetic MTSL probe, and fully suppressed if the distance is less than 10.5 Å due to its fast transverse relaxation rate.36 Theoretical calculations using Hyperchem software showed that for Coil-KW* the average distance between the MTSL nitroxide radical and the aromatic protons of the tryptophan group is 6.6 Å, while in Coil-EY* the distance between MTSL and the aromatic tyrosine group is 13.0 Å (see Part 3 of the ESI†). As expected, significant suppression of the aromatic proton signals is observed in spectra of Coil-KW* and Coil-EY* (Fig. 3 A/C, red lines).
In contrast, when the MTSL label is at the N-terminus of the peptides, the distance between the nitroxyl radical and the tyrosine or tryptophan residues is too large to cause a PRE effect. In Coil-*KW, the calculated average distance between the radical and W is 36.7 Å, while in Coil-*EY the distance between the radical and Y is 40.1 Å. In the NMR spectra, however, there is some degree of line broadening (Fig. 3 A/C, black lines). This line broadening was caused by peptide aggregation, as evidenced by its elimination from spectra when the peptides were in a monomeric α-helical form as measured in 1:1 (v/v) TFE:PBS solution (Fig. 3 B/D, black lines).18,66–69 For the peptides Coil-KW* and Coil-EY* in which the MTSL label is adjacent to the aromatic amino acids, suppression of the aromatic protons was retained in 1:1 (v/v) TFE:PBS solution, confirming that the NMR signal suppression is due to intramolecular paramagnetic relaxation enhancement (Fig. 3 B/D, red lines).
Next, the coiled-coil assembly of all the peptide pairs was investigated. In coiled coils, complementary peptides fold together in close proximity resulting in a tight peptide complex. When the MTSL label was positioned at the C-terminus of either Coil-KW or Coil-EY, adjacent to the aromatic amino acids, it effectively suppressed the aromatic signals from both of the peptides (Fig. 4 A/C, compare blue and red traces). This indicates that Coil-KW and Coil-EY assemble into a parallel coiled-coil complex. In contrast, when the MTSL label was positioned at the N-terminus, no PRE effect was observed, indicating that all of the peptides assemble into parallel coiled coils, without the coexistence of antiparallel complexes (Fig. 4 A/C, compare blue and black traces).
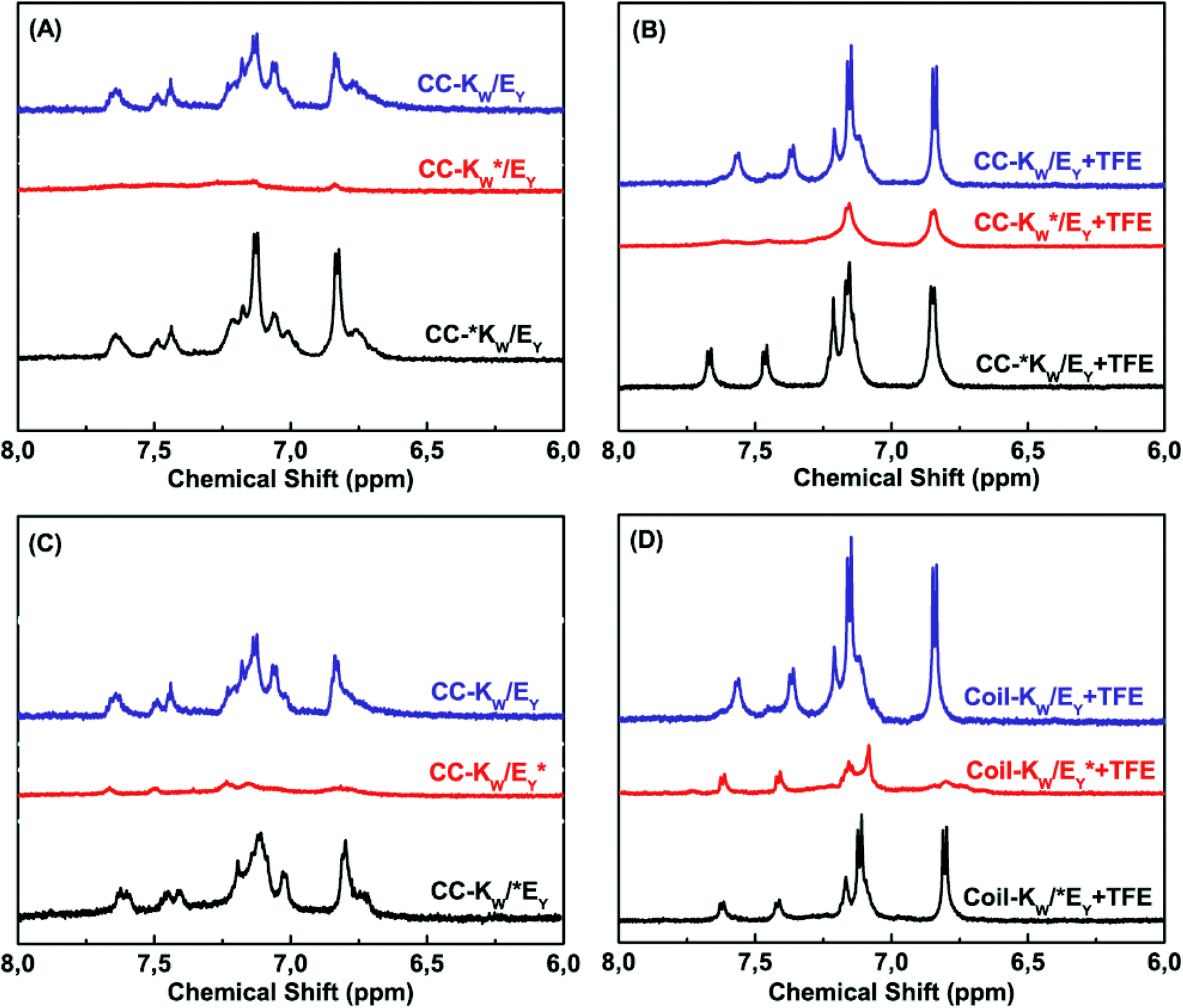 | ||
| Fig. 4 Aromatic region (6–8 ppm) of 600 MHz 1H-nuclear magnetic resonance spectra showing tryptophan indole and tyrosine hydroxyphenyl functional groups of equimolar mixtures of Coil-K and Coil-E (short name CC-K/E). (A) Aromatic signals of peptide CC-K/E complex in PBS. (B) Aromatic signals of CC-K/E in 1:1 (v/v) TFE:PBS. (C) Aromatic signals of CC-K/E in PBS. (D) Aromatic signals of CC-K/E in 1:1 (v/v) TFE:PBS [Total peptide] = 0.8 mM, PBS, pH = 7.4. (see ESI Fig. S9† for 0–8 ppm chemical shift). | ||
Measuring the same peptide mixtures in 1:1 (v/v) TFE:PBS revealed the dissociation of the coiled coil complexes as observed by the reappearance of the aromatic protons of the non-MTSL labeled peptide (Fig. 4 B/D red traces).
These results show that the PRE effect can be utilized to probe coiled coil formation and the relative orientation of the peptides within the complex. This is the first time that the PRE effect has been used to study intermolecular interactions in coiled coils.
Next, 1H-NMR measurements were used to study the coiled-coil binding stoichiometry. The molar ratio of the complementary peptides was varied from 2:1, 1:1 and 1:2 using peptides Coil-KW* and Coil-EY (Fig. 5A). The tryptophan NMR signals from Coil-KW* are always silent, while parallel coiled coil formation results in PRE suppression of the tyrosine NMR signals from Coil-EY as well, due to its close proximity with MTSL. The measurements show that at Coil-KW*: Coil-EY ratios of 2:1 and 1:1, the aromatic NMR region is silent. However, at a 1:2 ratio of Coil-KW* to Coil-EY, the tyrosine signals were visible. This shows that peptides Coil-KW and Coil-EY indeed form a 1:1 coiled coil complex as the excess of Coil-EY is not bound to Coil-KW* and thus the aromatic proton signal is no longer suppressed. Measuring the 1H-NMR spectrum of MTSL labeled peptide Coil-EY* and non-labeled Coil-KW mixtures confirmed this finding (Fig. 5B).
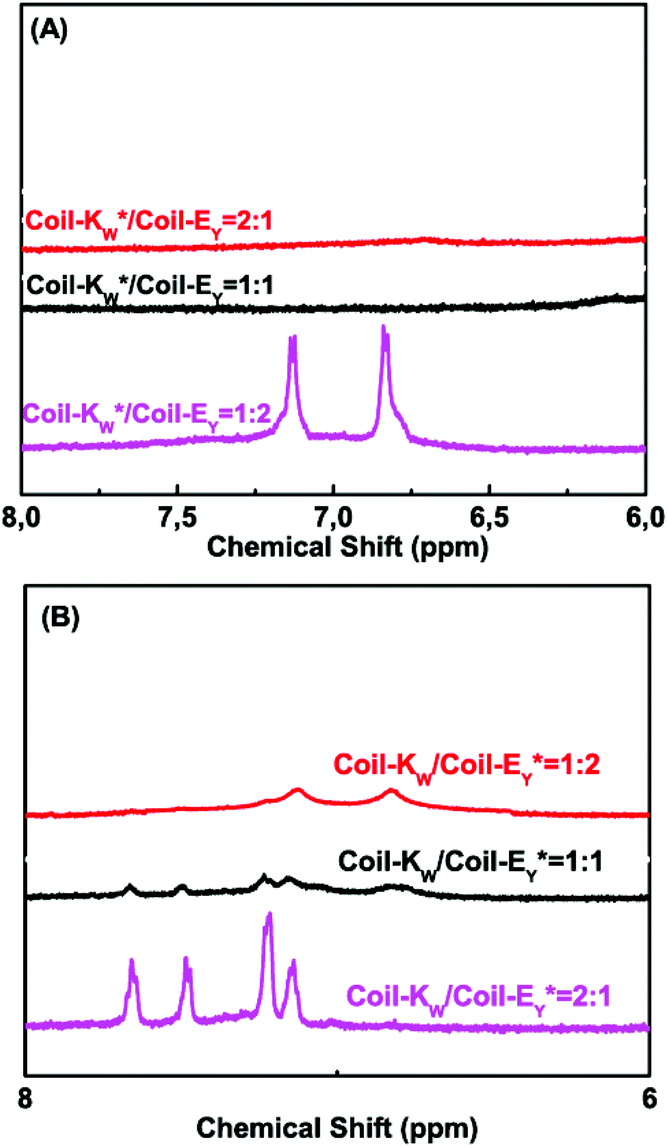 | ||
| Fig. 5 Aromatic region (6–8 ppm) of 600 MHz 1H-nuclear magnetic resonance spectra showing tryptophan indole and tyrosine hydroxyphenyl functional groups of different molar ratio mixtures of Coil-K and Coil-E. (A) Aromatic signals of peptide Coil-KW* and Coil-EY mixtures. From top to bottom, the molar ratio between Coil-KW* and Coil-EY is 2:1, 1:1 and 1:2 respectively. (B) Aromatic signals of peptide Coil-KW and Coil-EY* mixtures. From top to bottom, the molar ratio between Coil-KW and Coil-*EY is 1:2, 1:1 and 2:1. [Total peptide] = 0.8 mM (see ESI Fig. S10† for 0–8 ppm chemical shift). | ||
Nitroxyl radical PRE ‘signal suppression’ in 1H-NMR experiments is a fast and reliable method to determine the peptide folding in a coiled coil motif, allowing the determination of the peptide orientation and stoichiometry within the complex.
Fluorescence spectroscopy
To support the 1H-NMR measurements, steady-state fluorescence spectroscopy was used to probe the orientation of peptides Coil-KW and Coil-EY in the coiled coils by monitoring fluorophore electron excited singlet state quenching.41 Within a 12 Å radius, fluorescence emission quenching occurs due to electron exchange interaction between the MTSL nitroxyl radical and a tryptophan (W) or a tyrosine (Y) fluorophore.39,70–75 The degree of quenching is proportional to the electron exchange interaction, which is inversly proportional to the distance.76,77Excitation at a wavelength of 275 nm results in fluorescence of both tryptophan and tyrosine residues. When an MTSL group is present at the C-terminus, significant fluorescence quenching was observed for individual measurements of Coil-KW* and Coil-EY* (Fig. 6, blue line). However, when the MTSL label is positioned at the N-terminus, the quenching is absent (Fig. 6, red line).
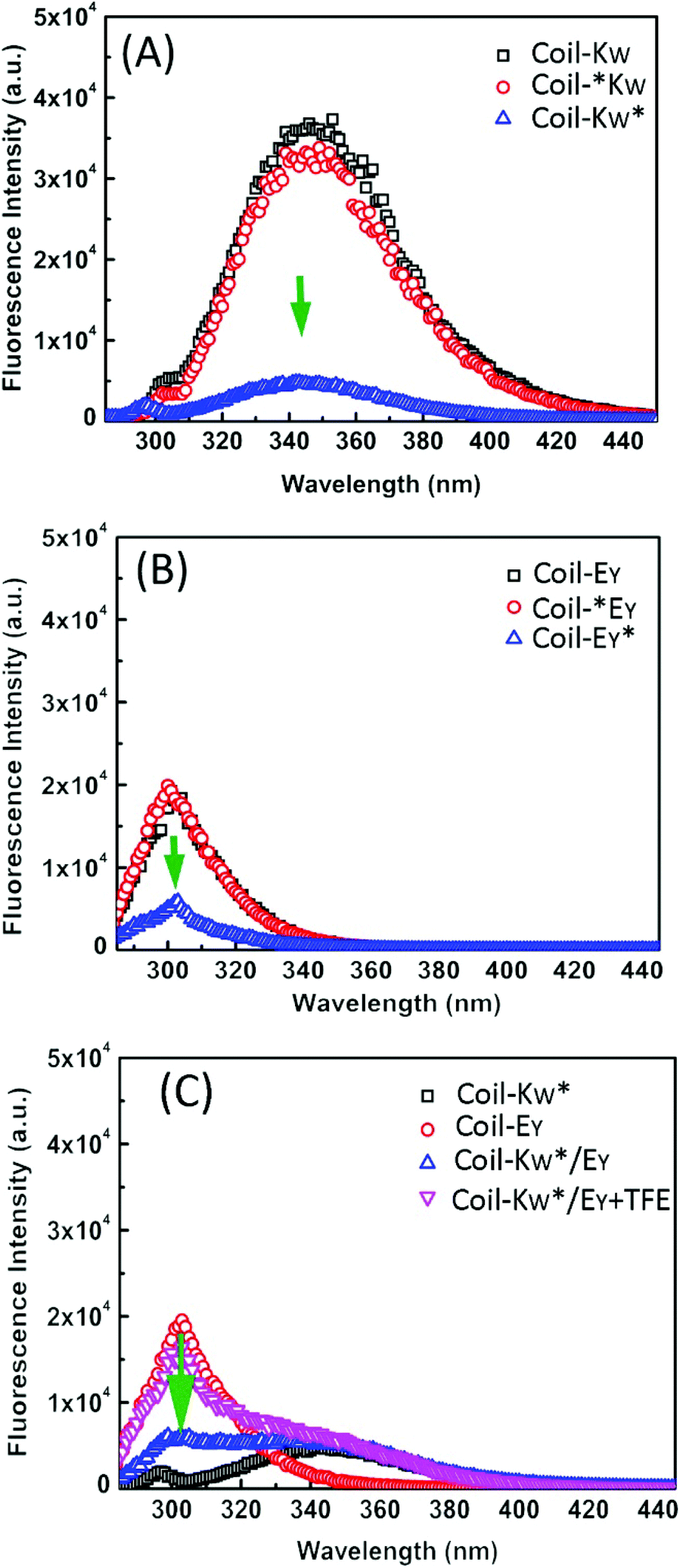 | ||
| Fig. 6 Fluorescence emission spectra of (A) Coil-KW and its spin labeled derivatives, and (B) Coil-EY and its spin labeled derivatives. (C) equimolar mixture of coil-Kw*/Ey in the absence and presence of TFE. Coil-Ey and coil-Kw* are shown for comparison. [Peptide concentration] = 50 μM in pH = 7.4 PBS buffer solution at 25 °C. Excitation at 275 nm. The green arrows indicate the fluorescence quenching positions. | ||
In equimolar mixtures of the peptides with a C-terminal MTSL label, the fluorescence signals of both of the peptides are quenched (Fig. 6C). This means that the fluorophores of both of the peptides are within 12 Å of the MTSL label. For example, in Coil-KW*, the MTSL quenches the tryptophan signal. In an equimolar mixture of Coil-KW* and Coil-EY, the tyrosine is also quenched, indicating that the tyrosine is in the vicinity of MTSL due to coiled coil formation. This can only occur when Coil-KW* and Coil-EY assemble into a parallel heterodimer. Addition of TFE results in separation of the peptides and the appearance of the tyrosine signal. These results again prove the parallel orientation of the K/E coiled coil, supporting the findings of the paramagnetic NMR studies.
Conclusions
In this paper we applied paramagnetic 1D proton-NMR to study peptide–peptide interactions for the first time. This new approach is used to investigate the supramolecular assembly of a well-known coiled coil pair. Labeling the peptides with tryptophan, tyrosine, or the spin label did not influence the binding properties of the peptides. The nitroxide spin label induced the suppression of specific NMR signals, enabling the determination of the orientation and stoichiometry of the peptides in the coiled-coil motifs. Fluorescence quenching by MTSL, using the same labeled peptides, confirmed the finding of the NMR studies. In this study aromatic amino acids were used as the proton signals are well-separated from the other peptide signals. In principle however any unambiguous and diagnostic ‘fingerprint’ proton signal could be used for this purpose.34In comparison to existing methods used to study coiled coil assembly, this method does not change the environment of the peptide (e.g. crystallization is necessary for X-ray diffraction) and avoids intermolecular interaction competition between chemical bonds and the hydrophobic core (e.g. disulfide exchange). The field of paramagnetic NMR spectroscopy is rapidly developing, and in this contribution it is used to study coiled coil assembly. In addition, it is compatible with two- or multi-dimensional NMR, and the same peptides can be used for further studies, for example for EPR measurements.78
All the required peptide manipulations are easily performed with high efficiency. The careful choice in labeling combined with fast 1H-NMR and fluorescence measurements significantly simplifies the study of non-covalent interactions in coiled coils or other supramolecular assemblies. Further development of this approach will aid in the investigation of not only peptide quaternary structure, but also many other self-assembly systems.
Experimental section
Materials
Fmoc-protected amino acids and Rink amide resin (0.53 mmol g−1) were purchased from NovaBiochem. HCTU (O-(1H-6-Chlorobenzotriazole-1-yl)-1,1,2,2-tetramethyluronium hexafluorophosphate), HOBT (1-Hydroxybenzotriazole) and DIPEA (N,N-Diisopropylethylamine) were from IRIS Biotech GmbH. NMP (N-methyl-2-pyrrolidone) and DMF (N,N-dimethylformamide) were from Biosolve. DCM (dichloromethane), TFE (2,2,2-Trifluoroethanol), TFE-D3 (2,2,2-Trifluoroethanol-d3), and deuterium oxide were obtained from Sigma-Aldrich. Acetic anhydride, piperidine, MeCN (acetonitrile), TFA (trifluoroacetic acid), and TIS (triisopropylsilane) were obtained from Fluka Chemie GmbH. MTSL (S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate) was obtained from Toronto Research Chemicals Inc. PBS buffer contains: 30 mM K2HPO4, 19 mM KH2PO4, 150 mM NaCl, pH = 7.4. The pH value was adjusted with either 0.1 M HCl or 0.1 M NaOH. Tris buffer contains 1 M tris (2-amino-2-hydroxymethyl-propane-1,3-diol), pH = 7.0.Peptide synthesis
Peptides were synthesized on a CEM-Liberty 1 Single Channel Microwave Peptide Synthesizer using standard Fmoc chemistry.79 Fmoc-protected Rink amide resin (0.53 mmol g−1) was used to synthesize the peptides on a 0.25 mmol scale. The resin was swollen in DMF for 30 min before use. Fmoc deprotection was performed using 20% (v/v) piperidine in DMF for 3 min at 50 W with a maximum temperature of 80 °C. Four equivalents of a Fmoc-amino acid, four equivalents of HCTU and five equivalents of DIPEA in DMF were used for amino acid coupling for 5 min at 40 W with a maximum temperature of 80 °C. For each amino acid coupling cycle, a deprotection and coupling time of 5 and 30 min were used respectively. For cysteine coupling, a cycle comprising 2 min at 0 W followed by 4 min at 40 W with a maximum temperature of 50 °C was used. Two wash steps (1.5 mL DMF) were performed between every amino acid coupling cycle. All peptides were acetylated manually at the N-terminus after completion of the synthesis using 20% (v/v) acetic anhydride in DMF for 1.5 hours. Peptides without a cysteine residue were cleaved from the resin and side-chain deprotected using a mixture of TFA–water–TIS = 95:2.5:2.5 (v/v) for 1 hour.80 Peptides with a Trt (trityl-) protected cysteine residue were cleaved from the resin with simultaneous side-chain deprotection using TFA–thioanisole–ethandithiol–phenol–H2O = 8.4:0.7:0.5:0.2:0.2 (v/v) for 3 hours at room temperature.81 The resulting solution was added drop-wise into an excess of 50 ml cold diethyl ether to precipitate the deprotected peptide, followed by centrifugation and the liquid supernatant was removed. This procedure was repeated 3 times with the addition of fresh cold diethyl ether. All the peptides were dried under vacuum, dissolved in MilliQ water and lyophilized yielding a white powder.
MTSL nitroxyl radical labelling
MTSL was conjugated to the peptide via a disulfide bond with the cysteine residue. One equivalent peptide (1 mM) was dissolved in 1 M tris buffer (pH = 7.0) and five equivalents of MTSL in DMF (50 mM) were added slowly under an argon atmosphere and the final mixture was stirred for 3 hours at room temperature.82 Next, the samples were lyophilized and stored at −20 °C before purification. All peptides were characterized by both MALDI-TOF and LC-MS mass spectrometry.Peptide purification
The crude peptides were purified by RP-HPLC, using a Shimadzu HPLC system with two LC-8A pumps, and an SPD-10A VP UV-VIS detector. Sample elution was monitored by UV detection at 214 nm and 254 nm. Purification of peptides was performed on a Vydac C18 reversed phase preparative column with a flow rate 15 mL min−1. Peptides were dissolved at a concentration of 5 mg ml−1 in a mixture of acetonitrile–H2O–tert-butanol = 1:1:1 (v/v) and eluted with a linear gradient from B to A. Solvent A = acetonitrile, while solvent B = 0.1% TFA in H2O. Acetylated peptides were purified using a 20 min gradient from 90% to 10% B, with a yield of 30%. MTSL labeled peptides were purified using a 25 min gradient elution from 80% to 20% B, with a typical yield of 20%. Purified peptides were lyophilized and characterized by LC-MS using a Vydac C18 analytical column with a 1 mL min−1 flow rate. Analytical HPLC confirmed the purity of the peptide to be 99%, while UV measurements showed a purity of at least 95% (see ESI†).
Circular dichroism spectroscopy
CD (circular dichroism) spectra were obtained using a Jasco J-815 spectropolarimeter equipped with a peltier controlled thermostatic cell. The ellipticity is given as mean residue molar ellipticity, [θ] (103 deg cm2 dmol−1), calculated by eqn (1).43,61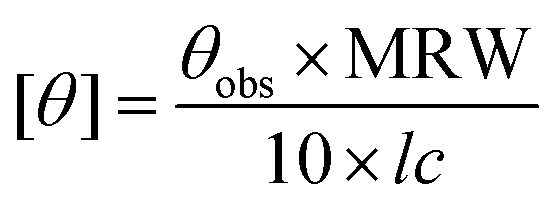 | (1) |
A 1.0 mm quartz cuvette was used, with a final peptide concentration of 200 μM in PBS (pH = 7.4). Spectra were recorded from 250 nm to 200 nm at 25 °C. Unless stated otherwise data points were collected with a 0.5 nm interval with a 1 nm bandwidth and scan speed of 1 nm per second. Each spectrum was an average of 5 scans. For analysis each spectrum had the appropriate background spectrum (buffer or 50% TFE) subtracted.
For determination of the coiled coil thermal dissociation constant, temperature dependent CD spectra were obtained using an external temperature sensor immersed in the sample.83,84 The temperature was controlled with the internal sensor and measured with the external sensor. A 10 mm quartz cuvette was used, and the solutions were stirred at 900 rpm. Spectra were recorded from 250 nm to 200 nm, with data collected at 0.5 nm intervals with a 1 nm bandwidth and a scan speed of 1 nm per second. The temperature range was 6 °C to 96 °C with a temperature gradient of 2.0 °C min−1 and a 60 s delay after reaching the set temperature. The spectrum of PBS at 6 °C (average of 5 scans) was subtracted from each spectrum. All the thermal unfolding curves were analyzed using a two-state conformation transition model.85,86
The data was analyzed using a two-state unfolding model to determine the fraction folded using eqn (2),
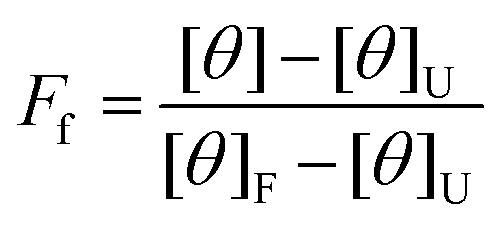 | (2) |
The fraction unfolded, FU, was calculated by eqn (3),
| FU = 1 − Ff | (3) |
The dimer dissociation constant in the transition zone was calculated using eqn (4),
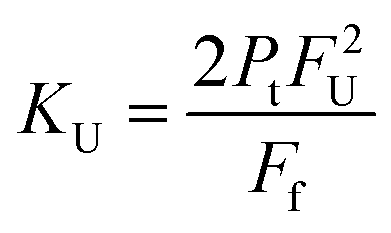 | (4) |
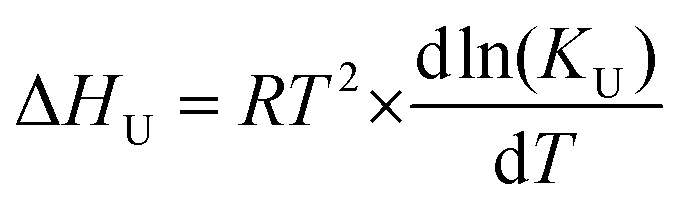 | (5) |
The gradient of the enthalpy vs. Temperature plot ΔCp, is the difference in heat capacity between the folded and unfolded forms, and can be used in the Gibbs–Helmholtz equation adapted to monomer–dimer equilibrium, eqn (6), to obtain the Gibbs free energy of unfolding as a function of temperature by least-squares fitting,
| ΔGU = ΔHm (1 – T/Tm) + ΔCp [T – TmTln(T/Tm)] – RTln[Pt] | (6) |
1H-magnetic resonance spectroscopy
To monitor the aromatic region 1H-NMR signals in the range from 8 ppm to 6 ppm of the amino acids W and Y, the proton signals of the peptide amide bonds were suppressed by proton–deuterium exchange using D2O. Lyophilized peptide samples were dissolved at a concentration of 0.5 mg ml−1 and incubated in D2O for one hour, followed by lyophilization. This procedure was repeated three times. PBS (10 ml, pH = 7.4) was lyophilized and redissolved in D2O to prepare a PBS/D2O buffer solution. Peptide samples were prepared with a final concentration of 0.8 mM in PBS/D2O buffer solution. All 1H-NMR spectra were recorded at 298 K on a Bruker Avance III 600 MHz spectrometer with 32 scans for each sample.Fluorescence spectroscopy
Fluorescence experiments were conducted on a TECAN Infinite M1000 PRO fluorometer using a 96 well plate. The Z-position was 12500 μm, and the gain was optimized according to the amount of fluorophore in the sample. Excitation and emission slits were set at 5 nm. Emission spectra were measured from 290 nm to 450 nm in 1 nm steps at a fixed excitation wavelength of 275 nm. The temperature was set at 25 °C. For consistent mixing, the plate was shaken inside the fluorometer for 30 seconds (2 mm linearly, 70 × per minute). The spectra were corrected by subtraction of PBS or PBS–TFE = 1:1 (v/v) spectra as a background spectrum. The total peptide concentration was 50 μM in each measurement, with 250 μL volume of peptide solution in each well.
Acknowledgements
T.Z. acknowledges the support of the CSC. A.B. acknowledges the support of the FOM. A.K. acknowledges the support of the European Research Council (ERC) via an ERC starting grant (240394).Notes and references
- B. Apostolovic and H. A. Klok, Biomacromolecules, 2008, 9, 3173–3180 CrossRef CAS PubMed.
- P. Burkhard, J. Stetefeld and S. V. Strelkov, Trends Cell Biol., 2001, 11, 82–88 CrossRef CAS PubMed.
- J. M. Mason and K. M. Arndt, Chembiochem, 2004, 5, 170–176 CrossRef CAS PubMed.
- A. M. Slovic, J. D. Lear and W. F. DeGrado, J. Pept. Res., 2005, 65, 312–321 CrossRef CAS PubMed.
- A. L. Boyle and D. N. Woolfson, Chem. Soc. Rev., 2011, 40, 4295–4306 RSC.
- M. Spinola-Amilibia, J. Rivera, M. Ortiz-Lombardia, A. Romero, J. L. Neira and J. Bravo, J. Mol. Biol., 2011, 411, 1114–1127 CrossRef CAS PubMed.
- J. Liu, Q. Zheng, Y. Q. Deng, C. S. Cheng, N. R. Kallenbach and M. Lu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15457–15462 CrossRef CAS PubMed.
- G. Grigoryan and A. E. Keating, Curr. Opin. Struct. Biol., 2008, 18, 477–483 CrossRef CAS PubMed.
- A. Lupas, Trends Biochem. Sci., 1996, 21, 375–382 CrossRef CAS PubMed.
- S. H. White and W. C. Wimley, Annu. Rev. Biophys. Biomol. Struct., 1999, 28, 319–365 CrossRef CAS PubMed.
- A. K. Gillingham and S. Munro, Biochim. Biophys. Acta, Mol. Cell Res., 2003, 1641, 71–85 CrossRef CAS.
- J. R. S. Newman and A. E. Keating, Science, 2003, 300, 2097–2101 CrossRef CAS PubMed.
- H. Gradisar, S. Bozic, T. Doles, D. Vengust, I. Hafner-Bratkovic, A. Mertelj, B. Webb, A. Sali, S. Klavzar and R. Jerala, Nat. Chem. Biol., 2013, 9, 362–366 CrossRef CAS PubMed.
- J. M. Fletcher, R. L. Harniman, F. R. H. Barnes, A. L. Boyle, A. Collins, J. Mantell, T. H. Sharp, M. Antognozzi, P. J. Booth, N. Linden, M. J. Miles, R. B. Sessions, P. Verkade and D. N. Woolfson, Science, 2013, 340, 595–599 CrossRef CAS PubMed.
- H. R. Marsden and A. Kros, Angew. Chem., Int. Ed., 2010, 49, 2988–3005 CrossRef PubMed.
- S. Cavalli, F. Albericio and A. Kros, Chem. Soc. Rev., 2010, 39, 241–263 RSC.
- M. N. Oda, T. M. Forte, R. O. Ryan and J. C. Voss, Nat. Struct. Biol., 2003, 10, 455–460 CrossRef CAS PubMed.
- D. A. Lindhout, J. R. Litowski, P. Mercier, R. S. Hodges and B. D. Sykes, Biopolymers, 2004, 75, 367–375 CrossRef CAS PubMed.
- C. Landon, F. Barbault, M. Legrain, L. Menin, M. Guenneugues, V. Schott, F. Vovelle and J. L. Dimarcq, Protein Sci., 2004, 13, 703–713 CrossRef CAS PubMed.
- M. Huber, S. Hiller, P. Schanda, M. Ernst, A. Bockmann, R. Verel and B. H. Meier, ChemPhysChem, 2011, 12, 915–918 CrossRef CAS PubMed.
- D. Sheppard, C. Y. Guo and V. Tugarinov, J. Am. Chem. Soc., 2009, 131, 1364–1365 CrossRef CAS PubMed.
- R. P. Meadows, D. Nettesheim, R. X. Xu, E. T. Olejniczak, A. M. Petros, T. F. Holzman, J. Severin, E. Gubbins, H. Smith and S. W. Fesik, J. Cell. Biochem., 1993, 279–279 Search PubMed.
- M. Huber, A. Bockmann, S. Hiller and B. H. Meier, Phys. Chem. Chem. Phys., 2012, 14, 5239–5246 RSC.
- D. M. Engelman and P. B. Moore, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 1997–1999 CrossRef CAS.
- E. K. Oshea, J. D. Klemm, P. S. Kim and T. Alber, Science, 1991, 254, 539–544 CAS.
- K. Nagai and H. Hori, FEBS Lett., 1978, 93, 275–277 CrossRef CAS PubMed.
- M. G. Oakley and P. S. Kim, Biochemistry, 1998, 37, 12603–12610 CrossRef CAS PubMed.
- U. I. M. Gerling, E. Brandenburg, H. von Berlepsch, K. Pagel and B. Koksch, Biomacromolecules, 2011, 12, 2988–2996 CrossRef CAS PubMed.
- L. J. Berliner, J. Grunwald, H. O. Hankovszky and K. Hideg, Anal. Biochem., 1982, 119, 450–455 CrossRef CAS PubMed.
- G. L. Kenyon and T. W. Bruice, in Methods in Enzymology, ed. C. H. W. Hirs and N. T. Serge, Academic Press, 1977, vol. 47, pp. 407–430 Search PubMed.
- I. Solomon, Phys. Rev., 1955, 99, 559–565 CrossRef CAS.
- N. Bloembergen and L. O. Morgan, J. Chem. Phys., 1961, 34, 842–850 CrossRef CAS.
- J. Iwahara and G. M. Clore, Nature, 2006, 440, 1227–1230 CrossRef CAS PubMed.
- C. Peggion, M. Jost, W. M. De Borggraeve, M. Crisma, F. Formaggio and C. Toniolo, Chem. Biodiversity, 2007, 4, 1256–1268 CAS.
- K. A. Bolin, P. Hanson, S. J. Wright and G. L. Millhauser, J. Magn. Reson., 1998, 131, 248–253 CrossRef CAS PubMed.
- Z. O. Shenkarev, A. S. Paramonov, T. A. Balashova, Z. A. Yakimenko, M. B. Baru, L. G. Mustaeva, J. Raap, T. V. Ovchinnikova and A. S. Arseniev, Biochem. Biophys. Res. Commun., 2004, 325, 1099–1105 CrossRef CAS PubMed.
- H. E. Lindfors, P. E. de Koning, J. W. Drijfhout, B. Venezia and M. Ubbink, J. Biomol. NMR, 2008, 41, 157–167 CrossRef CAS PubMed.
- S. A. Green, D. J. Simpson, G. Zhou, P. S. Ho and N. V. Blough, J. Am. Chem. Soc., 1990, 112, 7337–7346 CrossRef CAS.
- N. V. Blough and D. J. Simpson, J. Am. Chem. Soc., 1988, 110, 1915–1917 CrossRef CAS.
- P. L. Privalov, E. I. Tiktopulo and V. M. Tischenko, J. Mol. Biol., 1979, 127, 203–216 CrossRef CAS PubMed.
- S. E. Herbelin and N. V. Blough, J. Phys. Chem. B, 1998, 102, 8170–8176 CrossRef CAS.
- B. Pispisa, A. Palleschi, L. Stella, M. Venanzi and C. Toniolo, J. Phys. Chem. B, 1998, 102, 7890–7898 CrossRef CAS.
- J. R. Litowski and R. S. Hodges, J. Pept. Res., 2001, 58, 477–492 CrossRef CAS PubMed.
- J. R. Litowski and R. S. Hodges, J. Biol. Chem., 2002, 277, 37272–37279 CrossRef CAS PubMed.
- B. Apostolovic and H. A. Klok, Biomacromolecules, 2010, 11, 1891–1895 CrossRef CAS PubMed.
- B. Apostolovic, M. Danial and H. A. Klok, Chem. Soc. Rev., 2010, 39, 3541–3575 RSC.
- J. Voskuhl, C. Wendeln, F. Versluis, E. C. Fritz, O. Roling, H. Zope, C. Schulz, S. Rinnen, H. F. Arlinghaus, B. J. Ravoo and A. Kros, Angew. Chem., Int. Ed., 2012, 51, 12616–12620 CrossRef CAS PubMed.
- F. Versluis, J. Dominguez, J. Voskuhl and A. Kros, Faraday Discuss., 2013, 166, 349–359 RSC.
- A. S. Lygina, K. Meyenberg, R. Jahn and U. Diederichsen, Angew. Chem., Int. Ed., 2011, 50, 8597–8601 CrossRef CAS PubMed.
- G. Martelli, H. R. Zope, M. Brovia Capell and A. Kros, Chem. Commun., 2013, 49(85), 9932–9934 RSC.
- T. Zheng, J. Voskuhl, F. Versluis, H. R. Zope, I. Tomatsu, H. R. Marsden and A. Kros, Chem. Commun., 2013, 49, 3649–3651 RSC.
- E. J. Hustedt and A. H. Beth, Annu. Rev. Biophys. Biomol. Struct., 1999, 28, 129–153 CrossRef CAS PubMed.
- N. E. Zhou, C. M. Kay and R. S. Hodges, Biochemistry, 1992, 31, 5739–5746 CrossRef CAS PubMed.
- N. E. Zhou, C. M. Kay and R. S. Hodges, J. Biol. Chem., 1992, 267, 2664–2670 CAS.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2527–2535 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed.
- C. Y. Huang, Methods Enzymol., 1982, 87, 509–525 CAS.
- Z. D. Hill and P. Maccarthy, J. Chem. Educ., 1986, 63, 162–167 CrossRef CAS.
- Y. H. Chen, J. T. Yang and K. H. Chau, Biochemistry, 1974, 13, 3350–3359 CrossRef CAS PubMed.
- J. A. Boice, G. R. Dieckmann, W. F. DeGrado and R. Fairman, Biochemistry, 1996, 35, 14480–14485 CrossRef CAS PubMed.
- H. Robson Marsden, A. V. Korobko, T. Zheng, J. Voskuhl and A. Kros, Biomater. Sci., 2013, 1(10), 1046–1054 RSC.
- T. Gruene, M. K. Cho, I. Karyagina, H. Y. Kim, C. Grosse, K. Giller, M. Zweckstetter and S. Becker, J. Biomol. NMR, 2011, 49, 111–119 CrossRef CAS PubMed.
- J. Y. Guan, P. H. J. Keizers, W. M. Liu, F. Lohr, S. P. Skinner, E. A. Heeneman, H. Schwalbe, M. Ubbink and G. Siegal, J. Am. Chem. Soc., 2013, 135, 5859–5868 CrossRef CAS PubMed.
- S. Scanu, J. M. Foerster, G. M. Ullmann and M. Ubbink, J. Am. Chem. Soc., 2013, 135, 7681–7692 CrossRef CAS PubMed.
- S. P. Skinner, M. Moshev, M. A. S. Hass and M. Ubbink, J. Biomol. NMR, 2013, 55, 379–389 CrossRef CAS PubMed.
- J. Davies and L. Riechmann, FEBS Lett., 1994, 339, 285–290 CrossRef CAS PubMed.
- T. Sugiki, C. Yoshiura, Y. Kofuku, T. Ueda, I. Shimada and H. Takahashi, Protein Sci., 2009, 18, 1115–1120 CrossRef CAS PubMed.
- R. Page, W. Peti, I. A. Wilson, R. C. Stevens and K. Wuthrich, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1901–1905 CrossRef CAS PubMed.
- S. Y. M. Lau, A. K. Taneja and R. S. Hodges, J. Biol. Chem., 1984, 259, 3253–3261 Search PubMed.
- F. Mito, T. Yamasaki, Y. Ito, M. Yamato, H. Mino, H. Sadasue, C. Shirahama, K. Sakai, H. Utsumi and K. Yamada, Chem. Commun., 2011, 47, 5070–5072 RSC.
- G. I. Likhtenstein, K. Ishii and S. Nakatsuji, Photochem. Photobiol., 2007, 83, 871–881 CrossRef CAS PubMed.
- E. Gatto, G. Bocchinfuso, A. Palleschi, S. Oncea, M. De Zotti, F. Formaggio, C. Toniolo and M. Venanzi, Chem. Biodiversity, 2013, 10, 887–903 CAS.
- S. K. Chattopadhyay, P. K. Das and G. L. Hug, J. Am. Chem. Soc., 1983, 105, 6205–6210 CrossRef CAS.
- C. V. Kumar, S. K. Chattopadhyay and P. K. Das, J. Am. Chem. Soc., 1983, 105, 5143–5144 CrossRef CAS.
- W. A. Yee, V. A. Kuzmin, D. S. Kliger, G. S. Hammond and A. J. Twarowski, J. Am. Chem. Soc., 1979, 101, 5104–5106 CrossRef CAS.
- J. Karpiuk and Z. R. Grabowski, Chem. Phys. Lett., 1989, 160, 451–456 CrossRef CAS.
- S. Atik and L. A. Singer, J. Am. Chem. Soc., 1978, 100, 3234–3235 CrossRef CAS.
- X. Han, J. H. Bushweller, D. S. Cafiso and L. K. Tamm, Nat. Struct. Biol., 2001, 8, 715–720 CrossRef CAS PubMed.
- S. A. Palasek, Z. J. Cox and J. M. Collins, J. Pept. Sci., 2007, 13, 143–148 CrossRef CAS PubMed.
- H. Robson Marsden and A. Kros, Angew. Chem., Int. Ed., 2010, 49, 2988–3005 CrossRef CAS PubMed.
- T. Kaiser, G. J. Nicholson, H. J. Kohlbau and W. Voelter, Tetrahedron Lett., 1996, 37, 1187–1190 CrossRef CAS.
- A. Y. Kornilova, J. F. Wishart, W. Z. Xiao, R. C. Lasey, A. Fedorova, Y. K. Shin and M. Y. Ogawa, J. Am. Chem. Soc., 2000, 122, 7999–8006 CrossRef CAS.
- S. M. Kelly and N. C. Price, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1997, 1338, 161–185 CrossRef CAS.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1751, 119–139 CrossRef CAS PubMed.
- P. Lavigne, M. P. Crump, S. M. Gagne, R. S. Hodges, C. M. Kay and B. D. Sykes, J. Mol. Biol., 1998, 281, 165–181 CrossRef CAS PubMed.
- P. Lavigne, L. H. Kondejewski, M. E. Houston, F. D. Sonnichsen, B. Lix, B. D. Sykes, R. S. Hodges and C. M. Kay, J. Mol. Biol., 1995, 254, 505–520 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Mass spectra, additional CD spectra, hyperchem modelling of distances. See DOI: 10.1039/c4ob02125h |
| This journal is © The Royal Society of Chemistry 2015 |