
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A radical rebound mechanism for the methane oxidation reaction promoted by the dicopper center of a pMMO enzyme: a computational perspective†
Júlio C. S.
Da Silva
a,
Robert C. R.
Pennifold
b,
Jeremy N.
Harvey
*c and
Willian R.
Rocha
*d
aBioMat: Biomaterial Modeling Group, Departamento de Química Fundamental, CCEN, Universidade Federal de Pernambuco, Cidade Universitária, 50670-901, Recife, PE, Brazil
bCentre for Computational Chemistry, School of Chemistry, University of Bristol, Cantock's Close, Bristol, UK
cDepartment of Chemistry, KU Leuven, Celestijnenlaan 200F, B-3001 Heverlee, Belgium. E-mail: jeremy.harvey@chem.kuleuven.be
dLQC-MM: Laboratório de Química Computacional e Modelagem Molecular, Departamento de Química, ICEX, Universidade Federal de Minas Gerais, Campus Universitário Pampulha, 31270-901, Belo Horizonte, MG, Brazil. E-mail: wrocha@ufmg.br
First published on 30th October 2015
Abstract
In this article, we investigated the hydroxylation of methane catalyzed by the binuclear copper site of a pMMO enzyme, through a radical rebound mechanism. All intermediates and transition states along the reaction coordinate were located and the energies involved in the mechanism calculated using the B3LYP functional including dispersion effects. Our B3LYP-D2 results show that the singlet state of the (μ-1,2-peroxo)Cu(II)2 complex plays an important role as the lowest energy species prior to C–H bond activation. A crossing between the singlet and triplet PES is suggested to occur before the cleavage of the C–H bond of methane, where the triplet (bis-μ-oxo)Cu(III)2 is very reactive towards activation of the strong C–H bond of methane. The C–H bond activation is the rate-determining step of the reaction, with an activation energy of 18.6 kcal mol−1 relative to the singlet (μ-1,2-peroxo)Cu(II)2 species. Comparison with previous theoretical results for a non-synchronous concerted mechanism suggests the radical rebound mechanism as a possible alternative pathway.
1. Introduction
The activation and functionalization of an “unreactive” C–H bond of alkane molecules is currently a theme of great interest in both scientific and industrial communities due to the fact that control of this chemical process can lead to the generation of compounds with direct economic impact, since alkanes are the main constituents of natural gas.1–3 Nevertheless, the current chemical processes employed to activate the strong C–H bond (∼100 kcal mol−1) in alkanes require extreme conditions, are costly and inefficient, and produce a considerable amount of waste.3 Therefore, alternative ways to activate and functionalize alkanes are highly desirable. In this regard, natural catalysts found in the methanotrophic bacteria emerge as promising candidates.4–6Methanotrophic bacteria are organisms that are capable of selectively activating alkane molecules under mild conditions using Methane Monooxygenase (MMO) enzymes.7 The discovery of the catalytic function of MMO enzymes has led to renewed interest in the alkane activation chemistry aiming at understanding the fundamental steps of the catalytic process promoted by MMO. Detailed knowledge about how these enzymes promote the cleavage of C–H bonds of alkanes could lead to the development of new synthetic catalysts with improved activity and selectivity.
Two types of MMO enzymes are found in these methanotrophic organisms:8–10 the soluble form (sMMO), present in the form of a cytoplasmic complex, and the more abundant membrane-associated form, particulate MMO (pMMO). In fact, pMMO represents the most abundant catalyst for methane oxidation found in nature.7 These two metalloenzymes differ significantly in their structures and in the nature of the active sites. It is now well established that in sMMO the activation of methane takes place at a diiron active site and, based on spectroscopic measurements and computational studies, several possible mechanisms have been proposed for hydroxylation of methane catalyzed by sMMO.10 In contrast, little is known about the methane oxidation process catalyzed by the particulate form of the MMO enzyme, which can partially be attributed to a long-standing lack of detailed information regarding its structure.
The first crystal structure of pMMO was determined ten years ago, at a resolution of 2.8 Å, by Rosenzweig and co-workers.11 The results obtained from this investigation revealed a trimeric structure spatially organized in a α3β3γ3 polypeptide arrangement. In total three distinct types of metallic centers were included in this structure: (i) a mononuclear copper center in the soluble domain of the β subunit, also called pmoB, located 25 Å above the membrane. The coordination sphere of this copper center is composed of the His 48, His 72 and Gln 404 residue sidechains (Fig. 1a). The pmoB subunit also contains a binuclear copper center with the residues His 33, His 137 and His 139 composing the first coordination sphere (Fig. 1b), with the His 33 forming what is called a histidine brace. The third metallic center was modeled as a mononuclear zinc complex containing the residues Asp 156, His 160, His 173 and Glu 195 as ligands (Fig. 1c). This zinc center is situated in the subunit γ, also called pmoC, which is located in the domain composed of the α-helices bound to the membrane. The presence of some water molecules close to each of these metallic centers is also possible based on the crystal structure. The detailed description of the coordination environment of the metallic sites in metalloenzymes is crucial, since the chemical nature of the ligands directly bound to the metal ions can change their oxidation number, electronic states and, consequently, their reactivity.12–14
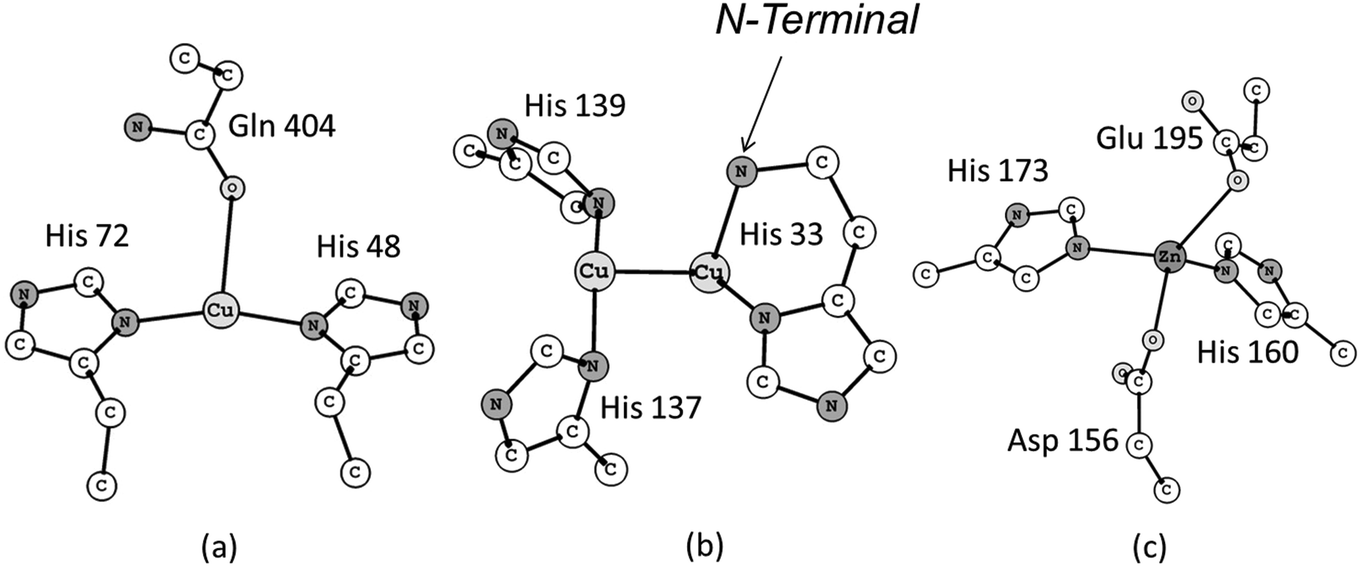 | ||
| Fig. 1 The three metallic centres revealed from the crystal structure of the pMMO enzyme. | ||
The structure determined within 2.8 Å of resolution, together with the set of spectroscopic data obtained by Rosenzweig et al. and other research groups, represented a significant step towards elucidating the pMMO structure.11,15–17 Nevertheless, at 2.8 Å it was not possible to determine precisely the main residues and possible water molecules close to the active sites, which can be important to the global catalytic process. Experimentally it is very complicated to work with integral membrane proteins due to difficulties in crystallization.18,19 Hence, the main insights obtained about the methane oxidation reaction catalyzed by pMMO have been obtained from experimental and theoretical studies involving either recombinant fragment proteins of the soluble part of pMMO, or other copper-containing proteins (tyrosinase, catechol oxidase and hemocyanin),10,20–23 or the soluble form sMMO, which has a well-established structure and reactivity.10,24,25
Despite the considerable progress in the structure determination and spectroscopic studies, the nature of the pMMO active site remains controversial. In principle, the three metal sites revealed from the pMMO crystal structure are possible candidates for the catalytic site. A single trinuclear copper cluster was also proposed by Chan and co-workers.26 Mössbauer spectroscopic results led Martinho et al.27 to propose a dinuclear iron center for the pMMO active site. Nevertheless, biochemical and spectroscopic studies provide some evidence in favour of the binuclear copper center of pMMO as the most likely catalytic site.28–31 Recently, a new class of Lytic Polysaccharide Monooxygenase (LPMO) enzymes involved in the oxidative cleavage of C–H bonds in polysaccharides, including cellulose, was discovered.32–36 These LPMO enzymes have a single copper active site and also have a histidine brace ligand. Based on these observations, there have been suggestions that the ‘bimetallic’ site present on pMMO may in fact contain only one copper active centre.33,35,37
Overall, though, the binuclear copper center seems to be the most accepted proposal for the active site of pMMO.29,30 However, the oxidation state of the two copper ions in the active catalytic site of pMMO is also a matter of debate in the literature. This is mainly due to the fact that several O2-derived reactive intermediates can be formed upon the reaction with O2.37,38 Possible dicopper-oxo reactive intermediates are cis or trans (μ-1,2-peroxo)Cu(II)2, (μ-oxo)Cu(II)2, (μ-η2:η2-peroxo)Cu(II)2 and (bis-μ-oxo)Cu(III)2. Mixed valence reactive intermediates of the type bis(μ-oxo)Cu(II)Cu(III), (μ-oxo)(μ-hydroxo)Cu(II)Cu(III) can also be formed. A detailed discussion about these intermediates can be found in the comprehensive review of Solomon and co-workers.38
Biochemical and spectroscopic studies with model compounds of pMMO have shed some light on the possible O2-derived copper intermediates involved in the oxidation of alkanes. Symmetrical Cu(III) complexes of the type bis(μ-oxo)Cu(III)2 have been reported to be involved in C–H bond activation.39,40 Some authors have demonstrated that the existence of a given Cu2O2 isomer will depend on the chemical environment generated by the ligands.41,43 Stack et al.42,43 showed that primary amines can stabilize Cu(III) and reported the first example of bis(μ-oxo)Cu(III)2 complexes ligated by primary amines that exhibited oxidative reactivity towards C–H bond activation. This may explain a possible role of the histidine brace (R-NH2) in stabilizing Cu(III) species in the dicopper active site of pMMO. Karlin, Solomon and co-workers41 showed that steric demand around the coordination site can also stabilize a given Cu2O2 isomer. Employing a polypyridyl ligand, they showed that the bis(μ-oxo)Cu(III)2 isomer is preferred over (μ-1,2-peroxo)Cu(II)2, when slightly bulkier substituents are introduced into the polypyridyl ligand.
Studies with the pMMO enzyme, however, revealed that a symmetrical Cu(II)2-peroxo species is involved. Rosenzweig et al. showed, from spectroscopic studies involving the interaction between pMMO and a recombinant protein fragment of the soluble region of pMMO with O2 and H2O2, that a chemical species of type (μ-η2:η2-peroxo)Cu(II)2 can be an intermediate capable of activating the C–H bond of methane.44 However, the structure of this possible peroxide adduct remains elusive and, in addition, the details about how the reaction proceeds are not totally understood: it is conceivable that this Cu(II)2-peroxo intermediate can isomerize to the bis(μ-oxo)Cu(III)2 along the reaction coordinate. Mixed valence complexes of the type (bis-μ-oxo)Cu(II)Cu(III), generated by addition of one electron from an exogenous source,29,31 have also been proposed as catalytic species for the oxidation of methane catalyzed by pMMO. Recently Rosenzweig, Hoffman et al.31 used EPR measurements to propose a valence-localized mixed-valence Cu(I)Cu(II) pair at the dicopper site of pMMO. According to the authors, this mixed-valence dicopper center is different from any other studied previously.
Thus, based on these several possibilities two important questions arise: (i) which species is in fact more reactive? And (ii) by which mechanism does the activation and hydroxylation of methane occur? In this regard, electronic structure calculations can contribute to the analysis of the stability of the possible O2-derived reactive intermediates and also be used to investigate possible mechanistic routes. In fact, computational studies have shed some light on these questions. The equilibrium involving the (μ-η2:η2-peroxo)Cu(II)2 and bis(μ-oxo)Cu(III)2 has been extensively investigated using density functional theory and wave function-based methods,45–55,58 with studies focusing on the Cu2O2 core and different ligand environments. The predicted relative stability of the two isomeric forms varies significantly, depending on the theoretical level employed and also on which ligands are modeled. For instance, Shiota and Yoshizawa59 reported a difference of 8.3 kcal mol−1 in favor of the (μ-η2:η2-peroxo)Cu(II)2 isomer, using the B3LYP* functional and three histidine ligands, as appears in the crystal structure. Flock and Pierloot,46 using the complete active space self-consistent field/complete active space second-order perturbation theory (CASSCF/CASPT2) found a value of 12.7 kcal mol−1 in favor of the bis(μ-oxo)Cu(III)2 isomer with six ammonia ligands. More recently Liakos and Neese55 revisited this problem using the wave function-based local-pair natural orbital coupled cluster (LPNO-CCSD) method and several DFT exchange–correlation functionals, including dispersion corrections. They found the bis(μ-oxo)Cu(III)2 isomer to be 6.8 kcal mol−1 more stable than the (μ-η2:η2-peroxo)Cu(II)2 is in the presence of two ethylene diamine ligands. According to the authors, dispersion interactions, solvent and relativistic effects all contribute substantially to the stabilization of the bis(μ-oxo)Cu(III)2 form. The authors also suggest that inconsistent handling of relativistic effects may explain some of the discrepancies found in previous computational studies.
Computational studies also gave important contributions to the understanding of the reactivity of pMMO active site towards the C–H bond activation of methane.56–59 Chan and co-workers applied DFT to investigate the reaction mechanism of pMMO, considering mono, di and trinuclear copper models and concluded that the trinuclear compound is the most reactive species for methane activation.56 In this computational investigation the authors proposed a concerted mechanism in which the cleavage of the C–H bond occurs via formation of a nonlinear C⋯O⋯H transition state, in which the insertion of oxygen into the C–H bond takes place after the coordination of methane to one of the copper ions present in the trinuclear cluster model. Despite providing good insight into the C–H bond activation mechanism promoted by copper-containing enzymes such as pMMO, some caveats must be placed upon the conclusions given the nature of the models used in these calculations. First, no evidence for a trinuclear copper complex was obtained from the crystal structure of pMMO.11 Second, in the model, NH3 ligands were used to represent the histidine residues. Third, Chen and Chan did not consider the possible isomerization reaction involving the peroxide and bis-oxide species that, according to studies with other copper-containing proteins and binuclear copper synthetic complexes, is an important step to understand the reactivity of this class of metallic compounds.5,20,28–31 Moreover, Chen and Chan calculated the reaction pathway only for the singlet state, although there is a possibility of other electronic states. For the case involving the trinuclear copper cluster, only the doublet state was studied.
Yoshizawa and co-workers also investigated the catalytic mechanism of pMMO using pure QM and QM/MM models.57–59 These studies have yielded important contributions for understanding the pMMO reactivity. For instance, they showed from DFT calculations that the mononuclear copper and zinc sites are considerably less reactive in comparison with the binuclear copper site.58 This result provides one more indication in favor of the binuclear copper complex as the possible catalytic site of pMMO. Regarding the reactivity of the binuclear copper center, Yoshizawa et al. showed that a bis(μ-oxo)Cu(II)Cu(III) intermediate is more reactive than a bis(μ-oxo)Cu(III)2 species towards the C–H bond activation of methane.57,58 However, these calculations were performed using a model in which the Glu 35 residue was present as a ligand of the first coordination sphere, even though its side-chain oxygen atoms are more than 4 Å away from the copper ions in the crystal structure solved by Rosenzweig and co-workers.11 In a very recent study, Yoshizawa and co-workers59 revisited the pMMO mechanism using a new model where the residues Tyr 374 and Glu 35 were included in the second coordination sphere of the binuclear copper center. From DFT calculations they proposed that the Tyr 374 acts by transferring a hydrogen atom to one oxygen of the (μ-η2:η2-peroxo)Cu(II)2 complex in order to form a partially reduced intermediate of the type (μ-oxo)(μ-hydroxo)Cu(II)Cu(III), which is found by computation to be more reactive than the bis(μ-oxo)Cu(III)2 species. This was the first work aimed at understanding the role of residues beyond the first coordination sphere on the reactivity of pMMO. It should however be noted that according to the crystal structure of pMMO the oxygen atom of the OH group of Tyr 374 is ∼5 Å away from the binuclear copper site.
Several possible mechanisms (radical, non-radical and concerted) have been proposed for hydroxylation of methane catalyzed by the soluble form of the MMO enzyme.10 Nevertheless, regarding the pMMO, the few studies published so far have considered only the concerted mechanism firstly proposed by Chan et al. A non-synchronous concerted mechanism has been preferred for pMMO based on experimental results obtained by Chan and coworkers, which indicated that the hydroxylation of alkanes by pMMO proceeds with the retention of configuration.60,61 However, these studies were carried out using membrane preparations based on a trinuclear copper center that was not identified in the crystal structure of pMMO. Furthermore, in an investigation involving the mechanism of aliphatic hydroxylation catalyzed by a bis(μ-oxo)Cu(III)2 complex, Spuhler and Holthausen showed from DFT calculations that the aliphatic hydroxylation reaction can also take place through an OH rebound mechanism.62 Hence, we believe that a radical mechanism, similar to that postulated for sMMO, cannot be totally discarded for pMMO. In addition, from a theoretical point of view there are some important questions about the accuracy of DFT in describing the energetic properties involving binuclear copper complexes similar to those proposed for the pMMO model, which needs to be investigated in detail.
In this present work, we investigated the methane hydroxylation reaction catalyzed by the binuclear copper site of pMMO through a radical rebound mechanism. The B3LYP functional with inclusion of dispersion effects was used to calculate the structures and energies of all intermediates involved in the mechanism investigated. As we shall see, the radical rebound mechanism proposed in this work is kinetically equivalent to the concerted mechanism proposed before and provides an alternative pathway for this reaction to take place. In addition, the accuracy of the B3LYP functional was assessed with the results obtained from the CCSD(T)-F12 method for a small model. This article is organized as follows: first, the computational procedure used to study the mechanism is presented followed by discussion of the accuracy of DFT to describe the energetics of binuclear copper complexes and after, the structural and energetic aspects results for the rebound mechanism are presented and discussed.
2. Computational details
The initial coordinates of the dicopper model were taken from the crystallographic structure obtained from the Protein Data Bank server (PDB code 1YEW). Our starting model is composed of two Cu(I) ions together with their first coordination sphere ligands, made up by the sidechains of residues His 33, His 137 and His 139. Based on the crystal structure of pMMO, the amino-terminal nitrogen of His 33 was also included, and modeled as bound to one of the copper ions.11 Restraints were applied to the atomic coordinates of some atoms of the model (see the ESI† for details) in order to take into account the steric effects of the protein. The electrostatic effects of the broader protein environment were modeled using the Polarizable Continuum Model (PCM) with a dielectric constant equal to 4.63The geometry optimizations were carried out using the hybrid GGA B3LYP functional64,65 including the D2 dispersion correction proposed by Grimme and coworkers.66 Recent B3LYP-D calculations on hydroxylation reaction catalyzed by bioinorganic systems have shown the importance of including dispersion effects.67,68 The 6-31G(d) basis set69,70 was used for H, C, N and O atoms. All electrons of copper atoms were treated explicitly using the 6-311G(d) basis set.71 Hereafter this level of calculation will be referred to as B3LYP-D2/BSI. Open-shell species were described using an unrestricted approach. All geometry optimization and frequency calculations were performed using the C.01 version of the Gaussian 09 program.72 Given that the positions of some atoms were maintained frozen during the optimization procedure, standard computation of vibrational frequencies is not meaningful. In order to obtain approximate frequencies for computation, e.g. of entropic contributions, a simple block-Hessian procedure73 was used. The full Hessian matrix was obtained from a standard Gaussian frequency calculation, but then, only the sub-block corresponding to the N atoms whose positions were optimized was mass-weighted and diagonalized. This yields 3N vibrational eigenmodes and frequencies, which, for the chemically most important regions well ‘inside’ the sub-system resemble well the values obtained through full frequency analysis of a much larger system. This analysis was carried out using a homemade program.
Single-point energy calculations using DFT and the explicitly correlated version of the coupled cluster theory74 CCSD(T)-F12 were carried out to refine the energetics, using the MOLPRO program package.75 The coupled-cluster calculations were performed using a reference determinant constructed from Kohn–Sham orbitals obtained using the restricted open-shell ansatz and the B3LYP functional, as described later in the text. In these calculations the relativistic effects were included according to the Douglas–Kroll method.76
3. Results and discussion
3.1 Accuracy of DFT-D calculations
Prior to the study of the structural and energetic aspects of the species involved in the C–H bond activation of methane, we have tested the accuracy of the B3LYP-D2 method employed. Despite several previous computational studies of dioxygen activation by copper-containing enzymes, the accuracy of DFT to describe the energetics of these processes is still a matter of debate. The relative energies involved in the isomerization process between the peroxo and bis-oxo species are usually used as reference for benchmarking proposals. Previous studies using wavefunction- and DFT-based methods have provided different conclusions about the stability of the peroxo and bis-oxo intermediates.54,55 Recently, Liakos and Neese used highly ab initio correlated methods, including relativistic and solvation effects, to show that the bis(μ-oxo)Cu(III)2 is in fact more stable than (μ-η2:η2-peroxo)Cu(II)2, in agreement with the experimental results.55 Furthermore, Liakos and Neese also investigated the performance of different exchange–correlation functionals to describe the relative energies involved in the peroxo–bis-oxo isomerization and found that the B3LYP-D functional presented the best agreement with the LPNO-CCSD results. The mean absolute error computed for the deviation of the B3LYP results in relation to LPNO-CCSD was 4.4 kcal mol−1.In order to evaluate the accuracy of the B3LYP-D2/BSI level of theory to describe the energetics related to the dinuclear copper complexes investigated in this present work, we performed accurate single-point energy calculations employing the new explicitly correlated version of the coupled-cluster theory [CCSD(T)-F12].74 Due to considerable computational demand required by this method, smaller symmetrical models, shown in Fig. 2, were used. Geometry optimizations were performed using the B3LYP-D2 method with the BSI basis set, which employs the 6-31G(d) basis for O, N and H atoms and 6-311G(d) for the Cu atom. Single-point CCSD, CCSD(T) and CCSD(T)-F12 energies were calculated on the B3LYP-D2 optimized geometries using the MOLPRO2010.1 program.75 CCSD(T) calculations were also performed including relativistic effects via a second-order Hamiltonian according to the Douglas–Kroll (DK) method.76 In these calculations, the copper atom was described using the cc-pwCVTZ basis set,77 while the oxygen and nitrogen atoms were treated using the cc-pVTZ and cc-pVDZ basis sets78 for hydrogen (with appropriate modified forms for the calculations involving DK relativistic effects). This basis set is denoted hereafter as BSII. Auxiliary basis was included in the basis set BSII in order to enable density fitting, as described in the ESI.†
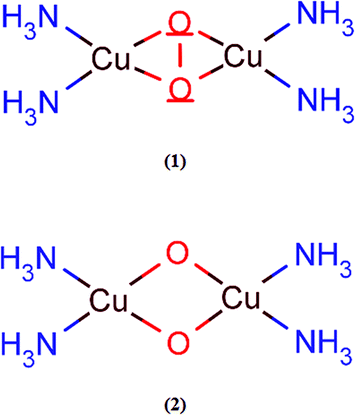 | ||
| Fig. 2 Model compounds used in the coupled-cluster calculations. | ||
The following model reactions were used: (i) isomerization of (μ-η2:η2-peroxo)Cu(II)2, in the triplet state, triplet-(1), to bis(μ-oxo)Cu(III)2 in its singlet state, singlet-(2), (triplet-(1) → singlet-(2)) and (ii) conversion of singlet-(2) to the triplet-(2) (singlet-(2) → triplet-(2)). The results are quoted in Table 1. The first process involves the scission of the O–O bond in (1) to produce (2), in the singlet state. The B3LYP-D2 method predicts the formation of the singlet to be exothermic by 11.0 kcal mol−1, compared to 16.6 kcal mol−1 computed at the CCSD(T)-F12-DK/BS1 level of theory, so the difference is 5.6 kcal mol−1. Discarding the relativistic effects, i.e., comparing the B3LYP-D2 results with CCSD(T)-F12, the difference is negligible. Our coupled-cluster and B3LYP-D2 results show the singlet of (2) as the most stable intermediate, as can be seen from the energies involved in the singlet-(2) → triplet-(2) reaction. Liakos and Neese obtained a similar conclusion from the LPNO-CCSD and DFT calculations involving other binuclear copper complexes.55
| Reaction | ΔE (kcal mol−1) | |||||
|---|---|---|---|---|---|---|
| CCSD | CCSD(T) | CCSD(T)-F12 | CCSD(T)-DK | CCSD(T)-F12-DKb | B3LYP-D2 | |
| a Coupled cluster calculations were performed using the BSII basis set and using a reference wave function constructed from Kohn–Sham orbitals. The B3LYP-D2 results were obtained with the BSI basis set. b Results obtained using CCSD(T)-DK minus CCSD(T) as a correction to CCSD(T)-F12. | ||||||
| Triplet (1) → singlet (2) | 21.78 | −10.04 | −11.29 | −15.38 | −16.63 | −11.05 |
| Singlet (2) → triplet (2) | 33.09 | 21.68 | 22.75 | 23.47 | 24.55 | 20.91 |
The B3LYP-D2 method predicts the singlet–triplet separation of (2) to be 20.9 kcal mol−1, whereas the CCSD(T)-F12-DK level of theory predicts this separation to be 24.5 kcal mol−1. In this case, the B3LYP-D2/BSI level underestimates the explicitly correlated method CCSD(T)-F12-DK/BS1 by only 3.6 kcal mol−1. It is worth noting that the CCSD method was unable to predict the correct stability of the isomers and overestimates the singlet–triplet gap by 10 kcal mol−1. The summary of these results shows that the use of B3LYP functional including dispersion effects provides relative energies in reasonable agreement with correlated ab initio methods. Application of highly correlated ab initio methods for studies involving bioinorganic systems still remains challenging, mainly with respect to the computational demand required.79 Hence, DFT methods remain as the main choice for investigations of biological systems of moderate size. The results obtained in this comparative study, as well as those reported by Liakos and Neese, support the B3LYP functional including dispersion effects as a good computational method for studies involving binuclear copper complexes like the one investigated here.
3.2 Reaction mechanism – structural remarks
At this point, it is important to highlight the differences between the model used in this work and the models used previously by Yoshizawa and Shiota.57 In their model they assumed a negatively charged Glu 35 residue as part of the first coordination sphere of one copper ion, even though its side-chain oxygen atoms are more than 4 Å away from the copper ions in the crystal structure solved by Rosenzweig and co-workers.11 Additionally, the terminal nitrogen of the His 33 residue was protonated and, therefore, unable to coordinate to the copper ion as a histidine brace, present in the crystal structure. Therefore, this dicopper active site model has an initial Cu(I)/Cu(I) configuration and, after the addition of an oxygen molecule and reduction by one electron the Cu(I)/Cu(II) peroxo species is formed. Recently, Yoshizawa, Shiota and Juhász revisited this model59 proposing a Cu(II)/Cu(II) peroxo intermediate composed of one Tyr, one Glu and three histidine residues, one of them acting as a histidine brace. Although this Tyr 374 residue is almost 5 Å away from the binuclear center in the crystal structure, the authors suggest that a hydrogen atom transfer or proton-coupled electron transfer from this Tyr residue to the Cu(II)/Cu(II) peroxo species may generate the more reactive species (μ-oxo)(μ-hydroxo)Cu(II)Cu(III). In our model we considered only the three histidine residues coordinated to the Cu(II)/Cu(II) peroxo species (see Fig. 3), as is presented in the crystal structure, one of them forming a histidine brace, which have been shown to be able to stabilize the Cu(III) oxidation state.42 This peroxo species generates the bis(μ-oxo)Cu(III)2 reactive intermediate through breaking the O–O bond in the peroxo intermediate. Therefore, the formation of the reactive intermediate does not require one electron reduction from an exogenous source. Experimental evidence for the viability of the model proposed in this work comes from two very recent studies.31,43 Stack and co-workers43 have recently characterized several bis(μ-oxo)Cu(III)2 complexes with three imidazoles and one primary amine, showing that the bis(μ-oxo)Cu(III)2 is a thermodynamically plausible intermediate for the C–H activation in pMMO, giving support to our model. These compounds were able to oxidize the C–H bonds of exogenous substrates such as 9,10-dihydro-methylacridine and 1,4-cyclohexadiene.42 In addition, Rosenzweig, Hoffman and co-workers31 used advanced EPR techniques to identify the valence and the coordination environment of the pMMO active site. According to the authors pMMO has a Cu(I)Cu(I) active site which, upon reduction and reaction with O2 or H2O2, generates an absorption at 345 nm, consistent with the formation of the (μ-η2:η2-peroxo)Cu(II)2 species.44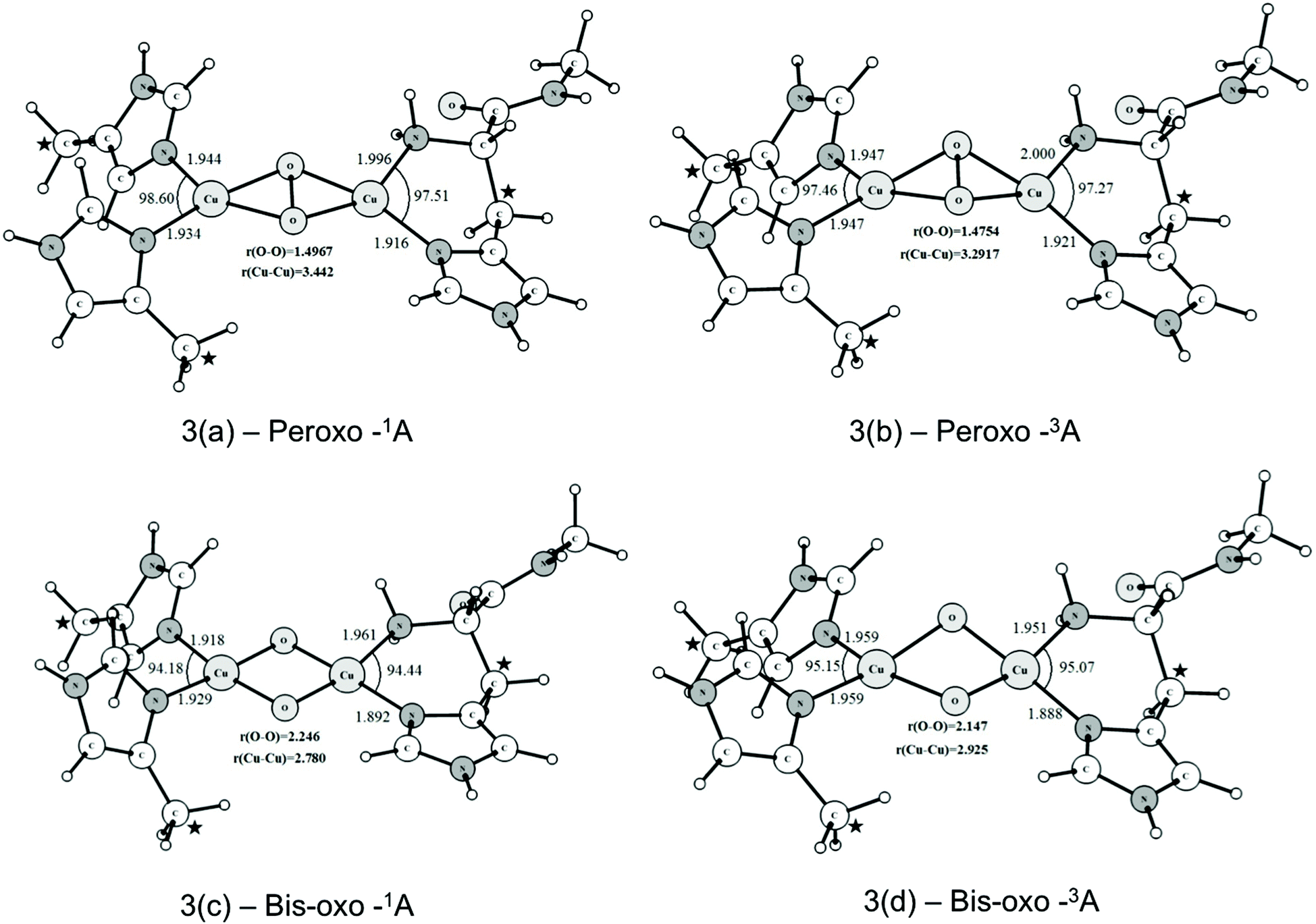 | ||
| Fig. 3 B3LYP-D2/BSI optimized structures of the singlet and triplet states of the peroxo and bis-μ-oxo intermediates generated from the interactions of the O2 molecule with the binuclear copper center of pMMO. Bond length and angles are given in Angstrom and degrees, respectively. The coordinates of the atoms highlighted with a star were kept fixed during the geometry optimization. | ||
According to several computational and experimental investigations involving copper-containing proteins and synthetic models,21,22,37,44 the probable first step of the pMMO cycle consists of the activation of the oxygen molecule by the Cu(I)Cu(I) motif. The interaction of the oxygen molecule with the Cu(I)Cu(I) species (Fig. 1b) generates a (μ-η2:η2-peroxo)Cu(II)2 complex that can isomerize, generating a bis(μ-oxo)Cu(III)2 intermediate which has been suggested as the true species capable of cleaving the C–H bond of alkanes.8,42,43,58 The broken symmetry (BS) approach80,81 was used to describe the d9–d9 antiferromagnetic coupling for the singlet state of the peroxide species. Despite some fundamental problems, the BS method has obtained a considerable success in studies involving bioinorganic systems containing transition metals.82,83 All species located in the triplet PES were described within the unrestricted formalism.
The optimized structures of the peroxo and bis-oxo intermediates are shown in Fig. 3. From Fig. 3(a) and (b) it is possible to see that the main geometrical parameters of singlet and triplet structures are slightly different. The peroxide species have a nonplanar bridging μ-η2:η2-peroxo butterfly-like structure. This type of structure has been reported in a variety of other binuclear copper complexes also involved in O2 activation.5,21,38 Masuda and coworkers reported the synthesis of a (μ-η2:η2-peroxo)Cu(II)2 complex containing bidentate nitrogen ligands in the coordination sphere,84 showing this butterfly geometry. The Cu–Cu distance determined by Masuda et al. was 3.265 Å, which is in good agreement with the mean value (for the singlet and triplet states) of 3.362 Å calculated at the B3LYP-D2/BSI level. The O–O distance determined from X-ray diffraction was 1.498 Å, which is also in good agreement with the values of 1.497 and 1.475 Å calculated for the singlet and triplet structures, respectively. The computed values also agree very well with the O–O bond length of 1.480 Å found in the tyrosinase enzyme (PDB code: 1WX2). In summary, these results indicate that our binuclear model including restraints in some atom coordinates is a reasonable choice to represent the possible molecular structure of peroxide species generated in the pMMO catalytic cycle. Further, this brief analysis of the geometrical results also shows that the B3LYP-D2/BSI level of theory describes adequately the main structural aspects of the pMMO active site.
As can be seen from Fig. 3(c) and (d), the μ-η2:η2-peroxo-Cu(II)2 (Fig. 3a and b) species can evolve further to generate the bis-μ-oxoCu(III)2 species where the O–O bond is totally broken. Such bis(μ-oxo)copper species are already well known, and are known to have singlet ground states, corresponding to low spin d8 Cu(III) centers. However, as will be seen below, in line with the previous work57–79 we found the singlet state to be relatively unreactive towards methane. Hence we also considered the possible role of a triplet spin-state of the bis(μ-oxo) form. For singlet and triplet optimized structures the O–O distances are 2.246 and 2.147 Å, respectively, as expected for a system in which O–O bonding no longer occurs.
Unlike what is observed with the peroxide species, the core of the bis(μ-oxo)Cu(III)2 complex has an almost planar structure. Fig. 4(a) and (b) show the atomic charges and spin densities calculated for the triplet states of the peroxo-Cu(II)2 and bis(μ-oxo)Cu(III)2 complexes. The peroxo species has significant spin on both coppers, as expected, given the Cu(II) oxidation state. Spin polarization of the peroxo ligand can account for its non-negligible spin density. Cleavage of the O–O bond leads to an increase in the negative charge and also the spin density on the oxygen atoms belonging to the core of bis(μ-oxoCu)(III)2. The electronic structure can be described based on the closed shell singlet bis(μ-oxoCu)(III)2 as involving formal removal of a spin-down electron from an oxygen-based orbital, and transfer to mainly a copper-based orbital. This yields two oxygen groups with a mixed oxo- and oxyl character, and two copper ions with a mixed Cu(II) and Cu(III) character. The spin densities suggest that the copper atom involved in the histidine brace is the one that has undergone most of this formal reduction to Cu(II). The presence of a significant oxyl/unpaired electron character on the oxygen atoms has been described as an essential factor for the reactivity of metal-oxo complexes and clusters towards the C–H bond activation of alkanes and other inert substrates.85–91 As will be seen later, this oxyl radical character in the triplet state of the bis(μ-oxo)Cu(III)2 complex plays an important role in determining the preferred pathway for the oxidation of methane.
 | ||
| Fig. 4 Calculated Mulliken spin densities and atomic charges for the triplet state of peroxo-Cu(II)2 and bis(μ-oxo)Cu(III)2 complexes. Spin densities are shown in red and atomic charges in black. | ||
Starting from the singlet and triplet optimized structures of the bis-oxo form, (3c) and (3d), we calculated the transition states related to the activation of the C–H bond of methane. The optimized structures of the transition states are presented in Fig. 5. These transition states were characterized by having imaginary frequencies of 1519i and 800i for the triplet (4b) and singlet (4a) states, respectively. The eigenvectors associated with these imaginary frequencies indicate a concerted hydrogen atom transfer mechanism in which at the same time as the C–H bond is broken the O–H bond is formed, generating a CH3 radical and a Cu(μ-O)(μ-OH)Cu μ-hydroxo species, with partially reduced Cu(2.5) centers. As can be seen, the transition state structures for the singlet and triplet reaction pathways are considerably different regarding the relative position of the methane molecule to the Cu2O2 core. The TS for the triplet state of (4b) has a near-linear O–H–CH3 angle of 157.2 degrees, whereas in the singlet state (4a) this angle is 111.2°. As discussed later our results show that the triplet state of the bis-oxo species is considerably more reactive than the singlet state towards the cleavage of the C–H bond of methane. Hence, after the activation of the C–H bond of methane the other steps of the catalytic cycle were investigated only for the triplet pathway.
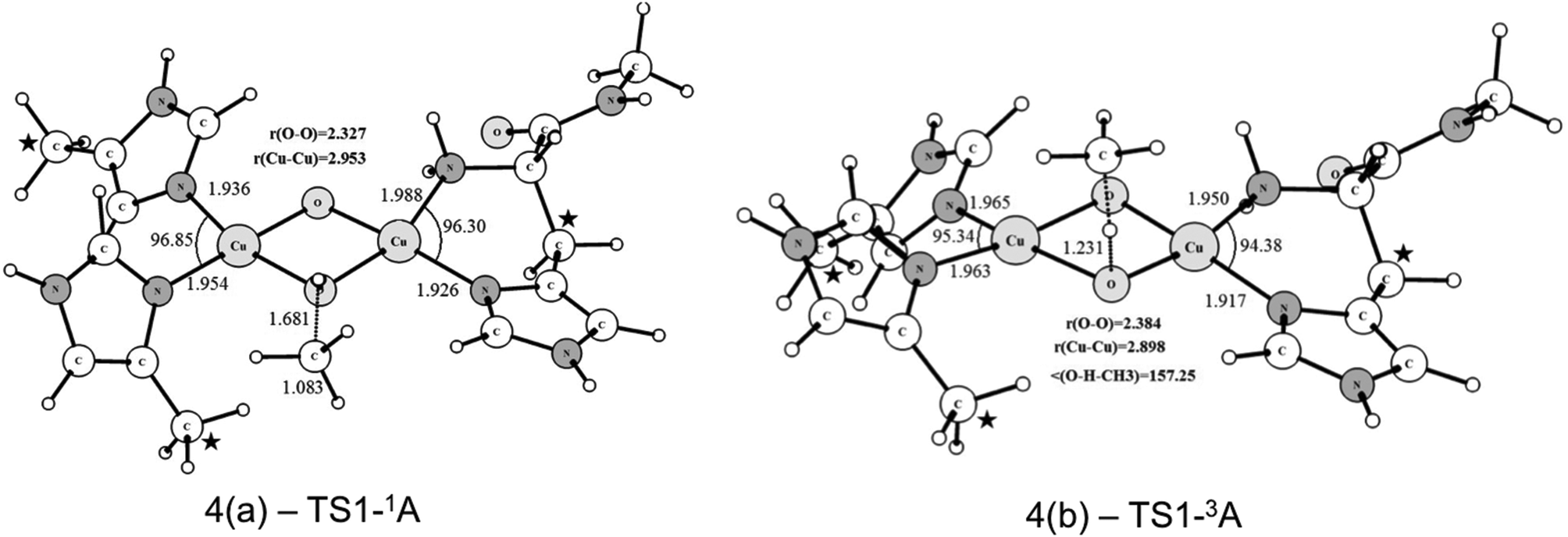 | ||
| Fig. 5 Optimized structures of the transition states (singlet and triplet) involved in the C–H bond activation step. Bond length and angles are given in Angstrom and degrees, respectively. The coordinates of the atoms highlighted with a star were kept fixed during the geometry optimization. | ||
Fig. 6 shows the UB3LYP/BSI optimized structures obtained for the CH3 rebound step. The transition state has an imaginary frequency of 330i cm−1, associated with the formation of the C⋯OH bond through a radical mechanism. As can be seen from Fig. 6, the transition state (TS2) leads to the formation of a methanol-complex, in which the methanol molecule is coordinated only to one of the copper ions.
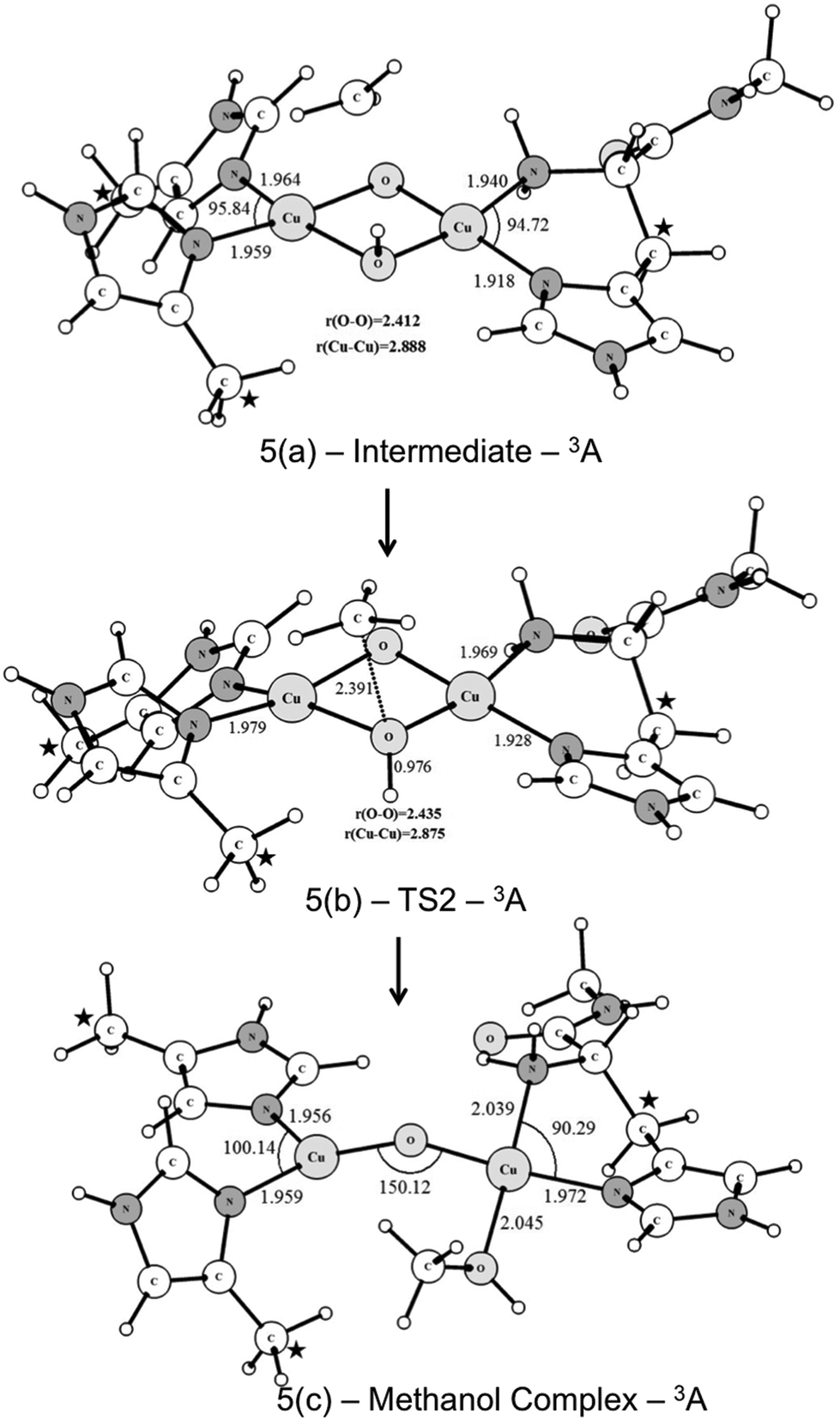 | ||
| Fig. 6 Optimized structures of the intermediate and transition states involved in the CH3 rebound step. Bond length and angles are given in Angstrom and degrees, respectively. The coordinates of the atoms highlighted with a star were kept fixed during the geometry optimization. | ||
3.3 Reaction mechanism – energetics
Fig. 7 shows the energy profile for the hydroxylation of methane promoted by the binuclear copper center of pMMO. The energies in Fig. 7 include the ZPE corrections obtained from diagonalization of the mass-weighted block Hessian matrix obtained by removing the Hessian elements associated with the atoms kept frozen during the geometry optimization. According to the data of Fig. 7, the singlet is the ground state of the peroxide and bis-oxide species. The formation of the bis-oxo complex is thermodynamically unfavorable for both pathways (singlet and triplet). For the reactive intermediate bis(μ-oxo)Cu(III)2, the singlet–triplet splitting is computed as 20.6 kcal mol−1.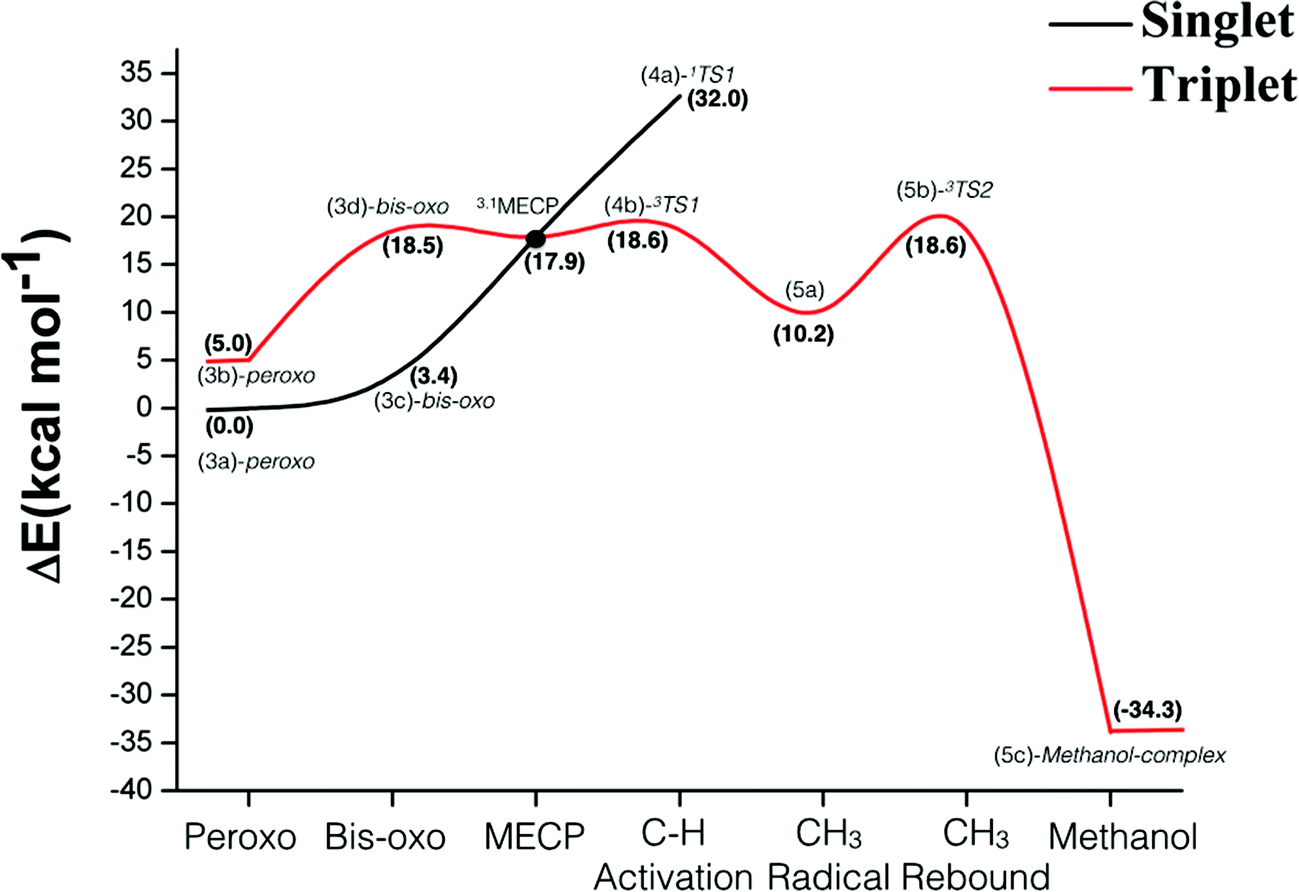 | ||
| Fig. 7 Energetic profile of the hydroxylation reaction of methane catalysed by the binuclear copper site of the pMMO enzyme obtained at the B3LYP-D2/BSI level of theory. The values in parenthesis are given in kcal mol−1 and were obtained relative to the singlet state of the (3a)-peroxo intermediate. | ||
The overall analysis of the entire energy profile of Fig. 7 shows that the singlet peroxo-Cu(II)2 plays an important role as the lowest energy species prior to C–H bond activation. However, for the methane activation step, our results show that the singlet state of bis(μ-oxo)Cu(III)2 is not as reactive as the triplet state, showing a high energy barrier towards the hydrogen abstraction of 32.0 kcal mol−1. The magnitude of this barrier is in agreement with the singlet PES calculated by Yoshizawa in a previous investigation involving a concerted non-synchronous mechanism.58 On the other hand, the energy profile of Fig. 7 shows that the triplet state of bis(μ-oxo)Cu(III)2 is very reactive towards the activation of the strong C–H bond of methane, displaying an activation energy barrier of 18.6 kcal mol−1 for the triplet TS (3b) relative to the ground state of the peroxo-Cu(II)2 species.
The results related to the C–H bond activation step also show that a crossing between the singlet and triplet PES takes place prior to the H-abstraction by the bis(μ-oxo)Cu(III)2 species. The geometry and energy of this crossing point between singlet and triplet potential energy surfaces were determined from the optimization of the corresponding minimum energy crossing point (MECP). The calculation of this MECP was performed using the method described in ref. 92, where the Gaussian program was used to calculate the energy and gradient on the two potential energy surfaces. A FORTRAN code was used to calculate the effective gradient pointing toward the MECP and update the geometry. This calculation was carried out with constraints at the atomic coordinates of the atom marked with stars in Fig. 3. As can be seen from Fig. 7, the 1,3MECP is approximately 0.7 and 14.0 kcal mol−1 more stable than the transition states 3TS1 and 1TS1, respectively. Based on our experience with previous spin-forbidden reactions of transition metal compounds, while passage from the singlet to the triplet state at the MECP will be somewhat hindered, this should not have a major impact on the rate. Hence the bottle-neck formed by the MECP may be less of an impediment to the reaction than 3TS1 (due to its lower potential energy), or comparable to it. These results suggest that the two-state reactivity with the reaction starting on the singlet surface and then moving to the triplet surface is a credible possibility.
The CH3 radical rebound step takes place through a barrier with an energy of 8.4 kcal mol−1 relative to the 5(a) intermediate, so this step does not constitute the bottle-neck to the oxidation. According to our B3LYP-D2 results, the formation of the methanol-complex, via a radical mechanism, is a quite thermodynamically favorable process. Similar stability has been reported in previous studies.56–59,62 The inspection of the full energy profile of Fig. 7 shows that the hydrogen atom transfer step is the rate-determining step, which is in agreement with experimental and also with previous theoretical results.56,57,59,60
Previous theoretical investigations reported the values in the range of 18–21 kcal mol−1,56–59 for the C–H bond activation step. Chan et al.56 suggested that the trinuclear copper(II, II, III) complex activates the C–H bond through a direct insertion of singlet oxene into the C–H bond in a concerted manner. Yoshizawa et al.57–59 have proposed that the initial Cu(II)–Cu(II) peroxo complex is converted to a Cu(II)–Cu(III) oxo complex through a one electron reduction. This is then suggested to carry out the hydrogen abstraction and thereby generates a methyl intermediate with radical character. The C–O bond is then formed through a rebound of the OH species to CH3. Our proposal instead involves a H-atom abstraction by the triplet state of the bis(μ-oxo)Cu(III)2 complex generating a methyl radical intermediate followed by a CH3 rebound step leading to the methanol-complex. The triplet state of the bis(μ-oxo)Cu(III)2 complex has an oxyl character which facilitates the Hydrogen Atom Transfer (HAT) reaction, leading to a preferred pathway. Since the product formed from C–H abstraction from the singlet and triplet dioxo states is essentially the same, the net final energy is the same in both cases, though the singlet reaction is much more endothermic taking the corresponding dioxo species as reference in each case. Thus, according to the conceptual framework developed by Mayer,93 the intrinsic barrier for the reaction with the triplet state must be much lower.
It is worth noting that the proposed mechanism does not require a redox partner as in the Yoshizawa's proposal. Comparing the previous energetic values obtained for the C–H bond activation step with the ones obtained in this present work, both proposals are a priori kinetically feasible. Hence the results obtained here are not sufficient to prove the radical rebound mechanism as the true reaction pathway. In fact, this mechanism may be difficult to prove experimentally. It is worth mentioning that the mechanism proposed here could in principle occur with any oxidase containing a μ-dioxo-copper dimer in the active site. However, why the present system can oxidize the very strong C–H bond of methane, in contrast with other oxidases, is not immediately clear and further work is needed in order to understand this behavior, including the effects of the protein environment.
It is also important to mention that recently a class of enzymes involved in the oxidative breaking of polysaccharides chains has been identified.32–35 This reaction is of great economic and environmental interest since it can exploit the potential of cellulose as a biofuel. These enzymes termed lytic polysaccharide monooxygenases (LPMOs) possess a single copper atom in the active site. The mechanism of the reaction was recently investigated by Kim et al.94 using an active site model and it was obtained, also for this reaction, that a copper-oxyl intermediate abstracts a hydrogen atom and subsequently hydroxylates the substrate via an oxygen rebound step. These results show that copper–oxygen compounds are involved in the oxidative C–H bond activation of important substrates such as methane and cellulose and may have an impact on the future of fuel and feedstock generation as was recently discussed by Yoon and Karlin.37 We note also that these enzymes contain mononuclear copper centers capable of activating strong C–H bonds. There is also structural and spectroscopic data suggesting that the pMMO ‘dinuclear’ site may also contain only one copper.35,37 If this is the case, then the mechanism modeled here would obviously need to be modified – but would remain a potential mechanism for other dicopper enzymes.
4. Final remarks
In the present study, DFT calculations at the B3LYP-D2 level of theory were carried out to evaluate a proposed radical rebound mechanism for the methane hydroxylation reaction catalysed by the binuclear copper site of the pMMO enzyme. Two potential energy surfaces were evaluated: (i) a singlet and (ii) a triplet. The relative energies calculated for the isomerization involving peroxo and dioxo species revealed that the singlet peroxo-Cu(II)2 plays an important role as the lowest energy species prior to C–H bond activation. However, the singlet state of bis(μ-oxo)Cu(III)2 formed from isomerization of peroxo-Cu(II)2 species is not able to activate methane with a low barrier based on our calculations. We showed from the MECP calculation that, before the cleavage of the C–H bond, a crossing between the singlet and triplet PES can occur. Our B3LYP-D2/BSI results suggest that the triplet state of bis(μ-oxo)Cu(III)2 is very reactive towards the activation of the strong C–H bond of methane. The overall energetic profile indicated C–H bond activation as the rate-determining step of the reaction. The activation energy of 18.6 kcal mol−1 is in agreement with values in the range of 18.0 to 21.0 kcal mol−1 calculated in the previous work with different models and different mechanisms. Comparison between these results indicates that the non-synchronous concerted (previously proposed) as well as radical rebound mechanisms are both kinetically possible based on computational insights. Nevertheless, the results reported in this present contribution are not enough to prove the radical rebound mechanism. Anyhow, this present work quantified the possibility of an alternative mechanism.In summary, our mechanistic proposal is that one possible mechanism for the oxidation of methane by the binuclear copper site of pMMO proceeds through three fundamental steps: (1) activation of molecular oxygen by the Cu(I)Cu(I) compound (Fig. 1b), (2) C–H bond cleavage (step 2) by the triplet state of the bis(μ-oxo)Cu(III)2 complex and (3) the CH3 rebound step, which leads to the formation of the methanol-complex. We assume that immediately following the hydrogen atom abstraction, the CH3 radical interacts with the bridged hydroxo species, forming a “bound radical” species, and further interaction leads to the final product. That is, the hydrogen atom transfer and methyl radical rebound must be almost synchronized.
In addition, the accuracy of the B3LYP-D2 method was evaluated by comparison with the results obtained using the newly developed version of the coupled-cluster method with explicit correlation [CCSD(T)-F12-DK]. We estimate that the B3LYP-D2 relative energies involving binuclear copper peroxo and dioxo species are accurate to within 4.5 kcal mol−1. These results suggest that the B3LYP-D2 method provides a reasonably accurate level of theory to describe the energetics of the pMMO mechanism.
Continued experimental and theoretical investigation is necessary in order to refine our understanding of the methane hydroxylation mechanism catalyzed by pMMO. Experimentally, this means that new structural, biochemical, spectroscopic and kinetic studies are needed to provide a more accurate description with respect to the coordination environment of the metal centers, the precise location, as well as the electronic properties of these binding sites. In terms of computational work, besides the necessity of accurate methods to describe the electronic structure of the reactive region, the use of methods that allow evaluation of the structural and dynamic effects related to protein environment is also required. In this context, computational simulations using QM/MM schemes are the main candidates.
Acknowledgements
The authors would like to thank the CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, INCT-Catálise) and FAPEMIG (Fundação de Amparo à Pesquisa do Estado de Minas Gerais) for financial support and research grants. CENAPAD-SP (Centro Nacional de Processamento de Alto Desempenho em São Paulo). J. C. S. S. also thanks the CAPES-PNPD for a scholarship grant.Notes and references
- J. E. Bercaw and J. A. Labinger, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6899 CrossRef CAS PubMed.
- C. Hall and R. N. Perutz, Chem. Rev., 1996, 96, 3125 CrossRef CAS PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS PubMed.
- J. M. Bollinger Jr. and J. B. Broderick, Curr. Opin. Chem. Biol., 2009, 13, 51 CrossRef PubMed.
- L. Que and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef CAS PubMed.
- D. E. T. Pazmiño, M. Winkler, A. Glieder and M. W. Fraaije, J. Biotechnol., 2010, 146, 9 CrossRef PubMed.
- R. S. Hanson and T. E. Hanson, Microbiol. Rev., 1996, 60, 439 CAS.
- A. S. Hakemian and A. C. Rosenzweig, Annu. Rev. Biochem., 2007, 76, 223 CrossRef CAS PubMed.
- M. H. Sazinsky and S. J. Lippard, Acc. Chem. Res., 2006, 39, 558 CrossRef CAS PubMed.
- M.-H. Baik, M. Newcomb, R. A. Friesner and S. J. Lippard, Chem. Rev., 2003, 103, 2385 CrossRef CAS PubMed.
- R. L. Lieberman and A. C. Rosenzweig, Nature, 2005, 434, 177 CrossRef CAS PubMed.
- G. Villar-Acevedo, E. Nam, S. Fitch, J. Benedict, J. Freudenthal, W. Kaminsky and J. A. Kovacs, J. Am. Chem. Soc., 2011, 133, 1419 CrossRef CAS PubMed.
- M. Suzuki, Acc. Chem. Res., 2007, 40, 609 CrossRef CAS PubMed.
- F. Zhong, G. P. Lisi, D. P. Collins, J. H. Dawson and E. V. Pletneva, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E306 CrossRef CAS PubMed.
- R. L. Lieberman, D. B. Shrestha, P. E. Doan, B. M. Hoffman, T. L. Stemmler and A. C. Rosenzweig, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3820–3825 CrossRef CAS PubMed.
- P. Basu, B. Katterle, K. K. Andersson and H. Dalton, Biochem. J., 2003, 369, 417 CrossRef CAS PubMed.
- R. L. Lieberman and A. C. Rosenzweig, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 147 CrossRef CAS PubMed.
- K. R. Vinothkumar and R. Henderson, Q. Rev. Biophys., 2010, 43, 65 CrossRef CAS PubMed.
- S. J. Opella, Annu. Rev. Anal. Chem., 2013, 6, 305 CrossRef CAS PubMed.
- A. C. Rosenzweig and M. H. Sazinsky, Curr. Opin. Struct. Biol., 2006, 16, 729 CrossRef CAS PubMed.
- C. Gerdemann, C. Eicken and B. Krebs, Acc. Chem. Res., 2002, 35, 183 CrossRef CAS PubMed.
- H. Decker, T. Schweikardt and F. Tuczek, Angew. Chem., Int. Ed., 2006, 45, 4546 CrossRef CAS PubMed.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563 CrossRef CAS PubMed.
- M. Torrent, D. G. Musaev, H. Basch and K. Morokuma, J. Comput. Chem., 2002, 23, 59 CrossRef CAS PubMed.
- B. F. Gherman, M.-H. Baik, S. J. Lippard and R. A. Friesner, J. Am. Chem. Soc., 2004, 126, 2978 CrossRef CAS PubMed.
- S. I. Chan, Y.-J. Lu, P. Nagababu, S. Maji, M.-C. Hung, M. M. Lee, I.-J. Hsu, P. D. Minh, J. C.-H. Lai, K. Y. Ng, S. Ramalingam, S. S.-F. Yu and M. K. Chan, Angew. Chem., Int. Ed., 2013, 52, 3731 CrossRef CAS PubMed.
- M. Martinho, D. W. Choi, A. A. DiSpirito, W. E. Antholine, J. D. Semrau and E. Münck, J. Am. Chem. Soc., 2007, 129, 15783 CrossRef CAS PubMed.
- R. Balasubramanian, S. M. Smith, S. Rawait, L. A. Yatsunyk, T. L. Stemmler and A. C. Rosenzweig, Nature, 2010, 465, 115 CrossRef CAS PubMed.
- R. A. Himes, K. Barnese and K. D. Karlin, Angew. Chem., Int. Ed., 2010, 49, 6714 CrossRef CAS PubMed.
- M. A. Culpepper and A. C. Rosenzweig, Crit. Rev. Biochem. Mol. Biol., 2012, 47, 483 CrossRef CAS PubMed.
- M. A. Culpepper, G. E. Cusail III, W. A. Gunderson, B. M. Hoffman and A. C. Rosenzweig, J. Am. Chem. Soc., 2014, 136, 11767 CrossRef CAS PubMed.
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. Liu, H. Zhai, M. Sørlie and V. G. H. Eijsink, Science, 2010, 330, 219 CrossRef CAS PubMed.
- R. J. Quinlan, M. D. Sweeney, L. L. Leggio, H. Otten, J.-C. N. Poulsen, K. S. Johansen, K. B. R. M. Krogh, C. I. Jørgensen, M. Tovborg, A. Anthonsen, T. Tryfona, C. P. Walter, P. Dupree, F. Xu, G. J. Davies and P. H. Walton, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15079 CrossRef CAS PubMed.
- G. R. Hemsworth, B. Henrissat, G. J. Davies and P. H. Walton, Nat. Chem. Biol., 2014, 10, 122 CrossRef CAS PubMed.
- C. H. Kjaergaard, M. F. Qayyum, S. D. Wong, S. D. Wong, F. Xu, G. R. Hemsworth, D. J. Walton, N. A. Young, G. J. Davies, P. H. Walton, K. S. Johansen, K. O. Hodgson, B. Hedman and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8797 CrossRef CAS PubMed.
- L. L. Leggio, K. S. Johansen, T. J. Simmons, J.-C. N. Poulsen, K. E. H. Frandsen, G. R. Hemsworth, M. A. Stringer, P. von Freiesleben, M. Tovborg, K. S. Johansen, L. De Maria, P. V. Harris, C.-L. Soong, P. Dupree, T. Tryfona, N. Lenfant, B. Henrissat, G. J. Davies and P. H. Walton, Nat. Commun., 2015, 6, 5961 CrossRef PubMed.
- J. Yoon and K. D. Karlin, Curr. Opin. Chem. Biol., 2015, 25, 184 CrossRef PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659 CrossRef CAS PubMed.
- S. Itoh, H. Nakao, L. M. Berreau, T. Kondo, M. Komatsu and D. Fukuzumi, J. Am. Chem. Soc., 1998, 120, 2890 CrossRef CAS.
- J. Shearer, C. X. Zhang, L. N. Zakharov, A. L. Rheingold and K. D. Karlin, J. Am. Chem. Soc., 2005, 127, 5469 CrossRef CAS PubMed.
- D. Maiti, J. S. Woertink, A. A. N. Sarjeant, E. I. Solomon and K. D. Karlin, Inorg. Chem., 2008, 47, 3787 CrossRef CAS PubMed.
- C. Citek, B.-L. Lin, T. E. Phelps, E. C. Wasinger and T. D. P. Stack, J. Am. Chem. Soc., 2014, 136, 14405 CrossRef CAS PubMed.
- C. Citek, J. B. Gary, E. C. Wasinger and T. D. P. Stack, J. Am. Chem. Soc., 2015, 137, 6991 CrossRef CAS PubMed.
- M. A. Culpepper, G. E. Cutsail, B. M. Hoffman and A. C. Rosenzweig, J. Am. Chem. Soc., 2012, 134, 7640 CrossRef CAS PubMed.
- C. J. Cramer, B. A. Smith and W. B. Tolman, J. Am. Chem. Soc., 1996, 118, 11283 CrossRef CAS.
- M. Flock and K. Pierloot, J. Phys. Chem. A, 1998, 103, 95 CrossRef.
- P. E. M. Siegbahn and M. Wirstam, J. Am. Chem. Soc., 2001, 123, 11819 CrossRef CAS PubMed.
- C. J. Cramer, A. Kinal, M. Wloch, P. Piecuch and L. Gagliardi, J. Phys. Chem. A, 2006, 110, 11557 CrossRef CAS PubMed.
- C. J. Cramer, M. Wloch, O. Piecuch, C. Puzzarini and L. Gagliardi, J. Phys. Chem. A, 2006, 110, 1991 CrossRef CAS PubMed.
- P. A. Malmqvist, K. Pierloot, A. R. M. Shahi, C. J. Cramer and L. Gagliardi, J. Chem. Phys., 2008, 128, 204109 CrossRef PubMed.
- L. Kong and M. Nooijen, Int. J. Quantum Chem., 2008, 108, 2097 CrossRef CAS.
- T. Saito, Y. Kataoka, Y. Nakanishi, T. Matsui, Y. Kitagawa, T. Kawakami, M. Okumura and K. Yamagychi, Int. J. Quantum Chem., 2009, 109, 3649 CrossRef CAS.
- B. T. Op't Holt, M. A. Vance, L. M. Mirica, D. E. Heppner, T. D. P. Stack and E. I. Solomon, J. Am. Chem. Soc., 2009, 131, 6421 CrossRef PubMed.
- B. F. Gherman and C. J. Cramer, Coord. Chem. Rev., 2009, 253, 723 CrossRef CAS.
- D. G. Liakos and F. Neese, J. Chem. Theor. Comput., 2011, 7, 1511 CrossRef CAS PubMed.
- P. P.-Y. Chen and S. I. Chan, J. Inorg. Biochem., 2006, 100, 801 CrossRef CAS PubMed.
- K. Yoshizawa and Y. Shiota, J. Am. Chem. Soc., 2006, 128, 9873 CrossRef CAS PubMed.
- Y. Shiota and K. Yoshizawa, Inorg. Chem., 2009, 48, 838 CrossRef CAS PubMed.
- Y. Shiota, G. Juhász and K. Yoshizawa, Inorg. Chem., 2013, 52, 7907 CrossRef CAS PubMed.
- B. Wilkinson, M. Zhu, N. D. Priestley, H.-H. T. Nguyen, H. Morimoto, P. G. Williams, S. I. Chan and H. G. Floss, J. Am. Chem. Soc., 1996, 118, 921 CrossRef CAS.
- S. S.-F. Yu, L.-Y. Wu, K. H.-C. Chen, W.-I. Luo, D.-S. Huang and S. I. Chan, J. Biol. Chem., 2003, 278, 40658 CrossRef CAS PubMed.
- P. Spuhler and M. C. Holthausen, Angew. Chem., Int. Ed., 2003, 42, 5961 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- R. Lonsdale, J. N. Harvey and A. J. Mulholland, J. Chem. Theory Comput., 2012, 8, 4637 CrossRef CAS PubMed.
- R. Lonsdale, J. N. Harvey and A. J. Mulholland, J. Phys. Chem. Lett., 2010, 1, 3232 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS.
- M. M. Francl, W. J. Petro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zhe, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. Ghysels, H. L. Woodcock III, J. D. Larkin, B. T. Miller, Y. Shao, J. Kong, D. Van Neck, V. Van Speybroeck, M. Waroquier and B. R. Brooks, J. Chem. Theory Comput., 2011, 7, 496 CrossRef CAS PubMed.
- T. B. Adler, G. Knizia and H.-J. Werner, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, K. R. S. Manby, M. Schutz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, A. J. D. T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Koppl, Y. Liu, D. P. O. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nickla, K. P. P. Palmieri, D. Peng, R. T. Uger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, and M. W. T. Thorsteinsson and M. Wang, MOLPRO, version 2010.1, a package of ab initio programs, see http://www.molpro.net Search PubMed.
- W. A. de Jong, R. J. Harrison and D. A. Dixon, J. Chem. Phys., 2001, 114, 48 CrossRef CAS.
- N. B. Balabanov and K. A. Peterson, J. Chem. Phys., 2005, 123, 64107 CrossRef PubMed.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef.
- J. N. Harvey, J. Biol. Inorg. Chem., 2011, 16, 831 CrossRef CAS PubMed.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737 CrossRef CAS.
- L. Noodleman, J. G. Norman, J. H. Osborne, A. Aizman and D. A. Case, J. Am. Chem. Soc., 1985, 107, 3418 CrossRef CAS.
- D. A. Pantazis, V. Krewald, M. Orio and F. Neese, Dalton Trans., 2010, 39, 4959 RSC.
- F. Neese, Coord. Chem. Rev., 2009, 253, 526 CrossRef CAS.
- Y. Funahashi, T. Nishikawa, Y. Wasada-Tsutsui, Y. Kajita, S. Yamaguchi, H. Arii, T. Ozawa, K. Jitsukawa, T. Tosha, S. Hirota, T. Kitagawa and H. Masuda, J. Am. Chem. Soc., 2008, 130, 16444 CrossRef CAS PubMed.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908 CrossRef CAS PubMed.
- J. F. Hull, D. Balcells, E. L. O. Sauer, C. Raynaud, G. W. Brudvig, R. H. Crabtree and O. Eisenstein, J. Am. Chem. Soc., 2010, 132, 7605 CrossRef CAS PubMed.
- S. Ye and F. Neese, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1228 CrossRef CAS PubMed.
- N. Dietl, M. Schlangen and H. Schwarz, Angew. Chem., Int. Ed., 2012, 51, 5544 CrossRef CAS PubMed.
- K.-B. Cho, S. Shaik and W. Nam, J. Phys. Chem. Lett., 2012, 3, 2851 CrossRef CAS.
- S. Ye, C.-Y. Geng, S. Shaik and F. Neese, Phys. Chem. Chem. Phys., 2013, 15, 8017 RSC.
- E. M. C. Alayon, M. Nachtegaal, A. Bodi and J. A. van Bokhoven, ACS Catal., 2014, 4, 16 CrossRef CAS.
- J. N. Harvey, M. Aschi, H. Schwarz and W. Koch, Theor. Chem. Acc., 1998, 99, 95 CrossRef CAS.
- J. M. Mayer, Acc. Chem. Res., 2011, 44, 36 CrossRef CAS PubMed.
- S. Kim, J. Ståhlberg, M. Sandgren, R. S. Paton and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 149 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The total energy (a.u.) and Cartesian coordinates for all structures described in this work. See DOI: 10.1039/c5dt02638e |
| This journal is © The Royal Society of Chemistry 2016 |