
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmallest molecular chalcogenidometalate anions of the heaviest metals: syntheses, structures, and their interconversion†
G.
Thiele
,
C.
Donsbach
,
R.
Riedel
,
M.
Marsch
,
K.
Harms
and
S.
Dehnen
*
Philipps-Universität Marburg, Fachbereich Chemie and Wissenschaftliches Zentrum für Materialwissenschaften, Hans-Meerwein-Straße 4, D-35043 Marburg, Germany. E-mail: dehnen@chemie.uni-marburg.de; Fax: +49 6421 282 5653; Tel: +49 6421 282 5751
First published on 16th November 2015
Abstract
The syntheses of the first molecular meta-selenidomercurate(II), ortho-telluridothallate(III) and a hydrate of an ortho-selenidoplubate(IV) are presented alongside an improved and facile synthesis of the selenidobismuthate(III) with almost quantitative yields. By means of quantum chemical calculations, the energetics of the interconversions of small metalate anions is discussed and the existence of the heaviest homologues of [NO2]−, [NO3]−, [PO4]2− and [CO3]2− are predicted.
1. Introduction
Chalcogenides of the heavy metals possess intriguing opto-electronic1 and thermo-electric2 properties and have therefore been used in numerous applications.3 Those properties can be fine-tuned and adjusted via dimensional reduction,4 the choice of elemental combination,5 or further incorporation of additional metal species towards multinary compounds.6 Conventional solid state syntheses, solvothermal reaction pathways and solution-based methods have been shown to facilitate the generation of a large variety of such compounds, including chalcogenidometalate salts.7Whereas the class of oxidometalates has extensively been studied – regarding both their fundamental properties and their potential applications – only a few reports have been dealing with the elemental combination of atoms of the heaviest metals with heavier chalcogen ligands. Here, we will concentrate on salts of the smallest of the heaviest chalcogenidometalate anions, hence with the general formulae [MxChy]z− (M = Hg, Tl, Pb, Bi; x = 1 or 2; Ch = Se or Te, y = 2–4), of which only the following are known to date. Besides the mononuclear ortho-chalcogenidomercurate anion [HgCh4]6−, received from solid state reactions,8 only the linear [HgCh2]2− anion, synthesized in solution,9 has been reported thus far. Molecular chalcogenidothallates and chalcogenidoplumbates have only been obtained from solution. Molecular chalcogenidothallates have so far been restricted to Schrobilgen's dinuclear [Tl2Te2]2−.10 The largest number of small anions is currently found among the chalcogenidoplumbates. Here, one knows the ortho-selenidoplumbate anion [PbSe4]4−,11 the dinuclear, trigonal bipyramidal [Pb2Ch3]2− anion,12 and the mononuclear trigonal pyramidal species [PbTe3]4−.13 For bismuthates, only the trigonal pyramidal [BiCh3]3− is known from solid state14 and solution-based approaches.15
Based on a broad experience in chalcogenidogermanate and chalcogenidostannate chemistry,16 we are currently about to extend the investigations on the heaviest tetrel congener as well as related anions of the neighbouring heavy metal elements. For this, we are addressing the synthesis of new small and soluble metalate anions, besides the exploration of their physical properties as well as their reactivity in solution or under solvothermal conditions. During the course of our investigations, we were able to synthesize a number of according compounds comprising new such anions with unprecedented elemental combinations, and were thus interested in general tendencies of the energetics of such species and their hypothetical interconversion. Here we report about our findings for the first time.
2. Experimental and computational details
2.1 General remarks and synthesis details
All manipulations and reactions were performed in an argon atmosphere using standard Schlenk or glovebox techniques under strict exclusion of air and external moisture. Multinary solid mixtures of the nominal parent composition “AmA′nMoM′pChq” (A, K; A′ = Na; M = Hg, Tl, Bi; M′ = Pb; Ch = Se, Te) were prepared by fusion of the elements and K2Ch, unless stated otherwise, with an oxygen/methane burner for at least 15 min until homogeneity was achieved. The m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n:o:p:q ratios, which are specified in the Discussion section, are those that actually produced the named products, usually after testing a variety of different ratios. If necessary, metallic by-products were manually removed. The multinary product was pestled and further worked-up as described below. Solvents ethane-1,2-diamine (en), N,N-dimethylformamide (DMF) and pyridine were freshly distilled from CaH2, H2O was degassed at least three times to a final pressure of p < 10−5 mbar and successively saturated with argon atmosphere. K2Ch was synthesized from the elements in liquid NH3. Metal salts were evacuated to p < 10−3 mbar for at least 8 h unless otherwise noted.
:n:o:p:q ratios, which are specified in the Discussion section, are those that actually produced the named products, usually after testing a variety of different ratios. If necessary, metallic by-products were manually removed. The multinary product was pestled and further worked-up as described below. Solvents ethane-1,2-diamine (en), N,N-dimethylformamide (DMF) and pyridine were freshly distilled from CaH2, H2O was degassed at least three times to a final pressure of p < 10−5 mbar and successively saturated with argon atmosphere. K2Ch was synthesized from the elements in liquid NH3. Metal salts were evacuated to p < 10−3 mbar for at least 8 h unless otherwise noted.
In the case of solvothermal reaction conditions, the reactants and 2 mL of a chosen solvent were placed in a glass vial within a Teflon inlay which was then placed in a stainless steel autoclave. The autoclave has been tightly closed and heated to 150 °C (Ch = Se) or to 130 °C (Ch = Te) for 7 days and was allowed to slowly cool down to room temperature over 1 day. The products were hand-selected under a light microscope unless stated otherwise. The product mixture for the synthesis of thallate and plumbate compounds always contained amorphous (side-)products that were not further investigated, and K2Ch2 and K2Ch3 that were identified via single crystal diffraction. Consequently, the provision of yields is afflicted with high uncertainty. Energy dispersive X-ray spectroscopy was performed on all samples to verify the given heavy atom composition. All yields are given with respect to the heaviest element involved in the respective case.
2.2 General computational details
All molecular electronic structure calculations were carried out with the TURBOMOLE program package V6.6.17 COSMO was used for the compensation of negative charges, applying the default parameters with ε = ∞.18 Structure optimizations were undertaken by employment of the RIDFT program, using the BP86 functional19 and def2-TZVP basis sets with respective ECPs.20 The optimized structures were confirmed to be local minima on the energy hypersurface by means of calculations of the 2nd derivative of the energy. Further details are given in the ESI.†3. Results and discussion
Our primary objective was the synthesis and characterization of small and heavy chalcogenidometalate homologues of their lighter congeners, e.g. stannates or indates.21 We therefore systematically investigated solvothermal reactions of phases of the nominal composition “AxMyChz” (A = alkali(ne earth) metal, M = Hg, Tl, Pb, Bi; Ch = Se, Te), in most cases with x:y:z such that the ratio corresponds with a charge balance for ortho-metalates (x: 1:4) or meta-metalates (x: 1:3). We started out with selenium as the chalcogen component, since according phases are generally thermally more stable and less sensitive. Upon identification of suitable reaction conditions for the selenium containing compounds, analogous reactions were attempted with tellurium phases. Due to the lower thermal stability of the latter, the reaction temperatures were then decrease by at least 20 °C, and the amount of starting materials was increased by a factor of two or more due to higher solubility.
The phases were then treated under solvothermal reaction conditions with diverse solvents, such as amines, alcohols, thiols and ethers. Until now, only the application of amines yielded products that were suitable for single crystal diffraction experiments, apart from the formation of a vast variety of single-crystalline polyselenides from other solvents.22,23
We also tested whether water, amines like NH3, 2,6-dimethylmorpholine (dmmp) or 4-(dimethylamino)pyridine (dmap), ammonium or phosphonium salts (NMe4Cl, NBu4Cl, PPh4Cl), or Brønstedt bases (AOH, A2CO3) might serve as auxiliary reactants for enhanced hydrogen bonding, and/or to change the basicity. However, only addition of water, or (in reactions of thallium compounds) addition of Brønstedt bases led to the formation of crystalline products (see below).
Upon solvothermal treatment of phases that contained mercury as KxHgySez (with all combinations of x = 2–6, y = 1–10, z = 1–12), we exclusively observed the formation of known compounds K2Hg3Se4,24 K2Hg2Se3,25 and polyselenides.22 We thus changed the mixture of starting materials to potassium monochalcogenides, K2Ch, and mercury salts. This way, we crystallized K4[HgSe3]·H2O (1) or K4[HgTe3]·H2O (2) upon utilization of K2Ch and HgSO4·nH2O (0.1 − n − 2.7 according to Karl Fischer titrations) in en as a solvent. The reaction towards the molecular meta-mercurate 1 is straight forward, according to a reaction scheme given in eqn (1):
| 3K2Se + HgSO4 + H2O → K4[HgSe3]·H2O (1) + K2SO4 | (1) |
The precipitation of K2SO4 seems to be the driving force, leading to an almost quantitative yield. Furthermore, the reaction can be scaled-up to 25 g per batch, only restricted by the volume of the glass vial within the autoclave. Solvent water in 1 stem from the crystal water in HgSO4·nH2O. The use of dried HgSO4 (n = 0) results in the precipitation of a yellow amorphous powder, which can, however, be recrystallized in the presence of water to yield 1. The amount of crystal water in HgSO4·nH2O varies with the time of storage from newly purchased (n ≈ 0.1) to years of storage under ambient conditions and use (n ≈ 2.7).
The synthesis of the homologous meta-telluridomercurate K4[HgTe3]·H2O 2 can be carried out with the quoted, typical variations for a replacement of Se with Te: treatment of K2Te and HgSO4·nH2O in en under solvothermal reaction conditions at slightly reduced temperature (130 °C). However, the yields are lower, since K2Te2,26 K2Te3,27 and amorphous side products are obtained along with 2. Intending to increase both yield and purity, we changed the reaction conditions to the use of a solid mixture containing sodium besides potassium. This way, we thought to allow for more flexibility in crystallization conditions according to various ionic radii. Further, we used DMF as solvent, as it is known that [Na(dmf)6]+ cations are easily formed and well soluble. However, in the present case, not 2 or its sodium salt was formed, but the meta-telluridomercurate K10Na2[HgTe3]2(HCO2)3 (3). 3 comprises a formiate moiety and Na+ as well as K+ counter ions. The formation of sodium formiate is often observed as a by-product due to decomposition of DMF under the given conditions; for this, the presence of the additional anion was not unexpected.
With a solvothermal reactions starting from a phase of the nominal composition K2PbHgSe2 and pyridine as solvent, we investigated whether we might access a mixture of metalate anions, either in a double salt or a salt of a ternary anion, or not. The latter turned out to be the case, as small amounts of K2[HgSe2]·H2O (4) emerged from the experiment upon addition of 0.5 vol% of water to the reaction mixture. Complete absence of water, or other volume ratios than 995:5 led to no or lower yields.
For us, such hydrate compounds are particularly interesting for their apparent stability in water and thus the potential for subsequent reactions in aqueous media, which are, however, beyond the scope of this report.
The crystallization of the hydrate 4 inspired us to revisit the formation of heavy main-group ortho-chalcogenidometalates, which have either not been identified at all (Tl, Bi) or at extremely low yields (Pb)11 to date. Upon solvothermal treatment of a phase of the nominal composition K5TlSe4 and en/H20 as solvent, K6[Tl2Se6]·2H2O (5) has been obtained. The formation of such dinuclear species was reported previously for some indates to be dependent on various reaction parameters, including the pH value.6,21,28 We thus explored the utilization of different amines, addition of various amounts of NH3 or H2O, and Brønstedt bases. However, for the heavier congener of indium, none of these variations yielded the mononuclear ortho-selenidothallate. Additional change of the nominal composition of the used phases afforded further compounds instead, K6[Tl2Se6]·2K2CO3 (6) and K0.94Tl0.06[TlSe2] (7). Compound 6 again comprises dinuclear aggregates of the ortho-selenidothallate anion, while 7 incorporates infinite 1D strands {[TlSe2]−}n of corner sharing [TlSe4] tetrahedra. So far, such chalcogenidothallate strands were only obtained as KxTl1−x[TlSe2] with x < 0.44,29,30 but they are also known for the lighter homologues, for instance from KInTe2.31
Only when changing the chalcogen from selenium to tellurium, and increasing the water content within the en/H2O mixture to 3 vol%, the first known ortho-chalcogenidothallate(III) was obtained as {K5[TlTe4]·2KOH}3·10H2O (8). The high water content is apparently necessary for the formation and/or crystallization of 8; replacement with NH3, for instance, did not lead to crystalline products. However, at the same time the presence of water triggers the formation of polytellurides and large amounts of amorphous side products, hence, the product had to be hand-separated from the reaction mixture.
Application of similar reaction conditions, including a 95:5 en/H2O solvent mixture, to the generation of respective lead compounds resulted in the formation of a hydrate salt of the rare ortho-selenidoplumbate anion, K4[PbSe4]·en·H2O (9). The structure is isostructural to the ammoniate K4[PbSe4]·en·NH3, which was published only recently.11 The crystallographic discrimination from the ammoniate was unambiguous upon collection of a data set of very high quality (see below). Further analytical methods fail here, as both ammonia N–H and water O–H vibrations are overlaid by the en N–H vibrations, and as elemental analyses tend to produce erroneous results for these heavy metal salts. The existence of a hydrate is not less amazing than the discovery of the [PbSe4]4− anion itself – the latter is anticipated to be highly redox active, due to the relatively poor stabilization of formal Pb(IV) by selenide and furthermore supposed to be highly oxophilic. Its relative stability in the presence of H2O, instead of spontaneous formation of [PbO4]4− or even spontaneous reduction, was thus unexpected. Counterintuitively again, the yield was dramatically increased from a few crystals of the ammoniate, to approx. 20% of the hydrate 9, although separation of the crystals by hand from the reaction mixture remained necessary.
Finally, we attempted solvothermal treatment of phases of the nominal composition KxBiySez. Due to its thermodynamic preference, K3[BiSe3] (10) was obtained from all batches. The structure was reported in the literature, however upon a rather cumbersome experimental procedure: Bi2O3 was reduced with K2[CO3] in a selenium-charged hydrogen stream at 850 °C.1510 can alternatively be obtained from simple fusion of the elements and successive work up via excessive washing with en until the filtrate turns colorless. 10 is then obtained as micro-crystalline powder in almost quantitative yields. Single crystals suitable for X-ray analysis were obtained via recrystallization by solvothermal treatment in en.
3.1 Crystal structure description
 | ||
| Fig. 1 Fragment of the crystal structure of 1. Ellipsoids are drawn at 50% probability. Selected structural parameters [Å, °]: Hg–Se: 2.552(2)–2.621(2), K⋯Se 3.233(5)–3.620(5), K⋯O 2.78(2)–3.19(2), Se–Hg–Se 114.02(6)–129.76(7). | ||
Each K+ ion is situated in a distorted octahedral environment of five Se atoms and one O atom, with the exception of K4 with coordination number (c.n.) 7 (see ESI†).
 | ||
| Fig. 2 Fragment of the crystal structure of 2. Ellipsoids are drawn at 50% probability. Selected structural parameters [Å, °]: Hg–Te: 2.7256(6)–2.7775(6), K⋯Te 3.468(2)–3.774(2), K⋯O 2.800(7)–3.258(7), Te–Hg–Te 114.427(18)–126.824(18). | ||
K+ and [K(μ-H2O)K]2+ ions separate the anions from each other, again with distorted octahedral coordination environments of K+ ions (see ESI†).
 | ||
| Fig. 3 Fragment of the crystal structure of 3. Ellipsoids are drawn at 50% probability. Selected structural parameters [Å, °]: Hg–Te: 2.7397(4)–2.7795(5), K–Te 3.5028(10)–3.7766(10), K⋯O 2.701(4)–2.854(4), Na⋯Te 3.5342(13), Na⋯O 2.898(4), Te–Hg–Te 119.246(8)–121.482(16). | ||
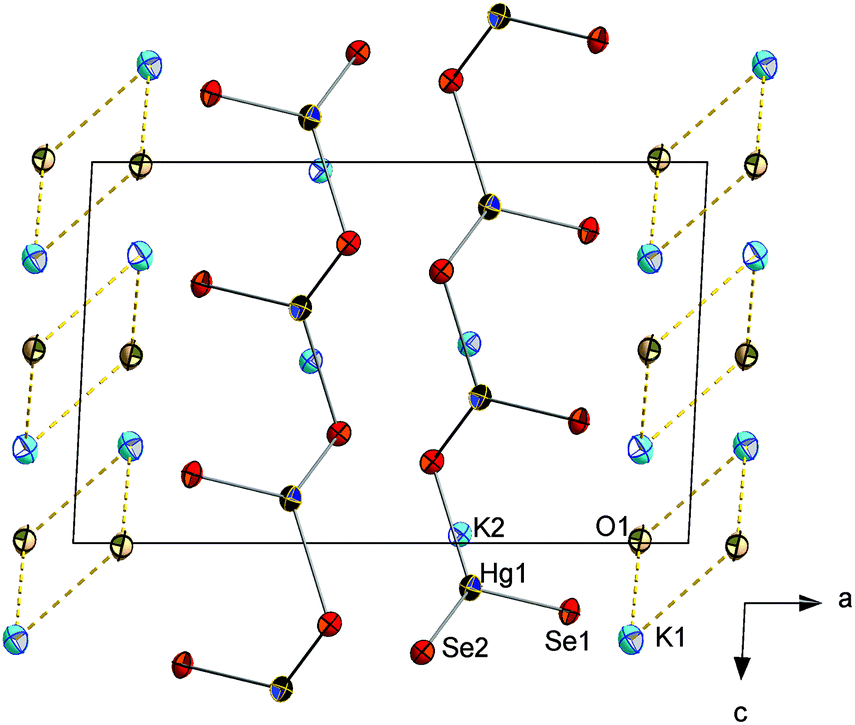 | ||
| Fig. 4 Fragment of the crystal structure of 4. Ellipsoids are drawn at 50% probability. Selected structural parameters [Å, °]: Hg–Se1 2.5097(10), Hg–Se2 2.5908(10)–2.6090(11), K⋯Se 3.193(2)–3.497(2), K⋯O 2.789(9)–3.426(9), Se–Hg–Se 107.19(3)–132.38(3). | ||
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one formula unit in the unit cell. Each (formal) Tl3+ ion is coordinated by two terminal Se atoms and two μ-bridging Se atoms, thus forming a molecular, dinuclear [Tl2Se6]6− anion. The anions are separated by K+ ions and [K2(μ-H2O)]2+ units (see Fig. 5). K+ ions are each coordinated by five Se atoms and one O atom in a distorted octahedral fashion (see ESI†).
with one formula unit in the unit cell. Each (formal) Tl3+ ion is coordinated by two terminal Se atoms and two μ-bridging Se atoms, thus forming a molecular, dinuclear [Tl2Se6]6− anion. The anions are separated by K+ ions and [K2(μ-H2O)]2+ units (see Fig. 5). K+ ions are each coordinated by five Se atoms and one O atom in a distorted octahedral fashion (see ESI†).
 | ||
| Fig. 5 Fragment of the crystal structure of 5. Ellipsoids are drawn at 50% probability. Selected structural parameters [Å, °]: Tl–Se(13) 2.5905(15)–2.5918(15), Tl–Se(2) 2.6876(15)–2.7132(15), K⋯Se 3.223(4)–3.908(4), K⋯O 2.766(11)–2.901(12), Se(1)–Tl–Se(3) 114.53(5), Se(1,3)–Tl–Se(2) 106.85(5)–116.66(5), Se(2)–Tl–Se(2′) 96.17(4), K⋯O⋯K 96.5(3). | ||
with one formula unit in the unit cell. The constitution of [Tl2Se6]6− anions is the same as found in 5. Carbonate ions are coordinating to eight K+ ions, three of which are chelated by the same one carbonate ion. Five further terminal K+ ions, in turn, are situated between two adjacent carbonate ions to form a complicated 2D [K10(CO3)2]6+ sheet extending between layers of the anions parallel to the ab plane by interconnecting strands of [K5(CO3)]3+ along the crystallographic a direction (see Fig. 6 and ESI†).
 | ||
| Fig. 6 Fragment of the crystal structure of 6. Ellipsoids are drawn at 50% probability. Selected structural parameters [Å,°]: Tl–Se(1,3) 2.5824(11)–2.5828(12), Tl–Se(2) 2.6629(10)–2.7273(11), K⋯Se 3.246(2)–3.919(3), K⋯O 2.681(9)–3.366(9), C–O 1.270(13)–1.327(12), Se(1)–Tl–Se(3) 115.80(3), Se(1,3)–Tl–Se(2) 103.64(4)–116.71(4), Se(2)–Tl–Se(2′) 93.17(3). | ||
:6 ratio for K:Tl on the positions.
 | ||
| Fig. 7 Fragment of the crystal structure of 7. Ellipsoids are drawn at 50% probability. Two-colored K1/Tl2 ellipsoids indicate statistical disorder on the respective positions. Selected structural parameters [Å,°]: Tl–Se 2.6653(7), K⋯Se 3.4299(4), Tl–Tl 3.5052(5), Se–Tl–Se 97.77(3), 115.619(14). | ||
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c with six formula units in the unit cell. The (formal) Tl3+ ions are surrounded by Te atoms in a tetrahedral mode, thus forming molecular [TlTe4]3− anions that are embedded in and separated by a complex 3D {K⋯(OH)⋯K⋯(H2O)}n substructure (see Fig. 8). Most of the K+ ions are hexacoordinate in an octahedral manner (K5⋯Te6), a distorted octahedral manner (K3⋯Te4O2), an distorted tetrahedral (K2⋯Te3O) or a trigonal prismatic manner (K1⋯Te3O3). One of the cations is nonacoordinate (K4⋯Te2O7). O atoms in turn are either surrounded by 6 K+ ions in a distorted octahedral fashion (O1, O4) or possess a tetragonal pyramidal environment by 5 K+ ions (O2, O3) (see ESI†). As illustrated in Fig. 8, the crystal structure of this first ortho-chalcogenidothallate salt is extreme complex, with a c axis of 39.226(6) Å and a unit cell volume of 9718(2) Å3, which is very large for an ortho-chalcogenidometalate salt. Maybe the steric requirements or even the electrostatic properties of the [TlCh4]5− anion impede a suitable and efficient packing of alkali(ne earth) metal cations along with the anions; hence, co-crystallization with two formula units of KOH might be essential, however leading to the shown structural complexity.
c with six formula units in the unit cell. The (formal) Tl3+ ions are surrounded by Te atoms in a tetrahedral mode, thus forming molecular [TlTe4]3− anions that are embedded in and separated by a complex 3D {K⋯(OH)⋯K⋯(H2O)}n substructure (see Fig. 8). Most of the K+ ions are hexacoordinate in an octahedral manner (K5⋯Te6), a distorted octahedral manner (K3⋯Te4O2), an distorted tetrahedral (K2⋯Te3O) or a trigonal prismatic manner (K1⋯Te3O3). One of the cations is nonacoordinate (K4⋯Te2O7). O atoms in turn are either surrounded by 6 K+ ions in a distorted octahedral fashion (O1, O4) or possess a tetragonal pyramidal environment by 5 K+ ions (O2, O3) (see ESI†). As illustrated in Fig. 8, the crystal structure of this first ortho-chalcogenidothallate salt is extreme complex, with a c axis of 39.226(6) Å and a unit cell volume of 9718(2) Å3, which is very large for an ortho-chalcogenidometalate salt. Maybe the steric requirements or even the electrostatic properties of the [TlCh4]5− anion impede a suitable and efficient packing of alkali(ne earth) metal cations along with the anions; hence, co-crystallization with two formula units of KOH might be essential, however leading to the shown structural complexity.
 | ||
| Fig. 8 Fragment of the crystal structure of 8. Ellipsoids are drawn at 50% probability. K and O atoms are drawn in opaque mode, and some of the tetrahedral anions are shown as polyhedra for clarity. Selected structural parameters [Å, °]: Tl–Te 2.8386(12)–2.8458(13), K⋯Te 3.537(4)–3.916(5), K⋯O 2.753(15)–3.053(6), Te–Tl–Te 107.26(3)–122.43(3). | ||
with two formula units in the unit cell. Compound 9 is isostructural with K4[PbSe4]·en·NH3.11 The presence of water instead of ammonia was unambiguously confirmed by structure solution and refinement using a very precise data set: the refinement of nitrogen atoms on the respective positions led to not positive-defined displacement parameters; furthermore, the two hydrogen atoms per water molecule were localized on the difference Fourier map and refined freely. Thus, 9 indeed represents the second known ortho-chalcogenidoplumbate(IV) salt (see Fig. 9). Bond lengths and angles are in good agreement with the reported structure parameters of the ammoniate.
 | ||
| Fig. 9 Fragment of the crystal structure of 9. Ellipsoids are drawn at 50% probability. H atoms are omitted for clarity. Selected structural parameters [Å,°]: Pb–Se 2.5985(4)–2.6157(4), K⋯Se 3.2861(9)–3.4920(9), K⋯O 2.776(3)–3.038(3), K⋯N 2.801(3)–3.384(4), Se–Pb–Se 107.02(2)–110.94(2). | ||
3.2 Quantum chemical calculations
The compounds presented above served to fill some of the existing gaps in the spectrum of known mononuclear or dinuclear chalcogenidometalate anions of the heaviest metals (see Table 1 below). However, many combinations have still not been observed experimentally. We were thus interested, whether according, yet missing species are (a) local minima on the respective energy hypersurfaces, and whether they are (b) accessible via chemical reactions in principle.|
|
|
|
|
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Higher formal oxidation state of M | Lower formal oxidation state of M | Higher formal oxidation state of M | Lower formal oxidation state of M | Higher formal oxidation state of M | ||||||
| Se | Te | Se | Te | Se | Te | Se | Te | Se | Te | |
| Hg | ⊗ | × | — |

|
✓ | — | ⊗ | × | ||
| Tl | ○ | ○ | ○ | ○ | ○ | |||||
| Pb | — | — | ○ | ○ | × |

|
||||
| Bi | ○ | ○ | ○ |

|
× | ○ | ||||





This might then allow for recommendations for future synthetic approaches: in the case of endoenergetic reactions, classical solid state reaction would more likely be successful, while in the case of an exoenergetic reaction, a reaction in solution should in principle be also possible or even preferred.
In a first step, we simultaneously optimized electronic and geometric structures of all mononuclear anions (meta and ortho type) and the pseudo-dimeric ones by means of DFT calculations, followed by the determination of second derivatives of the energy to verify the local minimum character of the shown anions on the energy hypersurface (non-minimum structures to be found in the ESI†). In a second step, we compared the energies of the respective anions and potential elimination products to calculate hypothetic reaction energies.
The accuracy of the calculated structures are within the expected range, i.e., bond lengths by a maximum of 0.067 Å larger than experimentally found ones. Hence, the chosen method is reliable enough for investigation of reaction energies.
It should be noted that the energy values that result upon application of COSMO are afflicted with systematic errors. This is due to a mismatch of gradient and absolute energy convergence in all cases, and even more pronounced if the numbers and/or charges of educts and products differ. Still, the errors are comparably small compared to the errors that are made during calculations of anions without the use of COSMO. In the present case, the errors are identical or at least very similar for all calculations as all species are highly related, such that a discussion of relative energies is reasonable. Furthermore, for the calculations, we chose appropriate reactants to account for the named problems with the quantum chemical treatment of anions: by using H2Se and (HSe)− for balancing the number of selenium ions (instead of Se2−), and by the choice of synthesis steps that do not require the consideration of naked metal cations Hg2+, Tl1/3+, Pb2/4+, or Bi3/5+ we cared for a minimum of charges and charge differences to be dealt with. The results are presented in the following, on the example of the respective selenides.
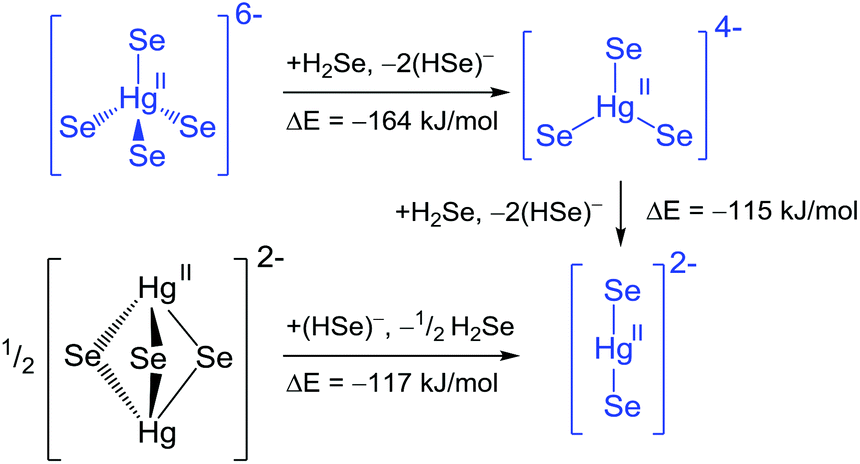 | ||
| Scheme 1 Reaction pathways and energies for the transformation of chalcogenidomercurates. Blue colour indicates experimentally observed anions. | ||
Starting out from the ortho-mercurate anion in a hypothetical transformation pathway through the mononuclear meta-mercurate towards the linear [HgSe2]2− anion under release of an Se2− ligand in each step (considered as (HSe)− for the reasons given above), both reaction steps are exoenergetic. This is in line with the synthesis of [HgCh2]2− from solution and [HgCh4]6− from solid state reactions, and does not contradict an access of the molecular meta-mercurate under solvothermal conditions. Starting from a hypothetical [Hg2Se3]2− anion in a reverse reaction scheme produces two equivalents of the linear [HgSe2]2−, again in an exoenergetic reaction. Thus, although [Hg2Se3]2− is a minimum on the energy hypersurface, it becomes obvious, why no such species has been reported so far: in solution the transformation towards [HgSe2]2− is expected, while a solid state reaction employing the respective 2:2:3 stoichiometric ratio rather produces an extended mercurate framework structure, such as K2Hg2Se3.25 Obviously, this structure type is not available for the 6:1:4 ratio, hence solid state reactions allow for the isolation of the tetrahedral [HgSe4]6− subunit. A comparison of the Hg–Se bond lengths for the planar [HgSe3]4− (2.64 Å) versus [HgSe3]5− (3.01 Å) is in agreement with the formal +II oxidation state at the Hg atoms in 1–3.
To the best of our knowledge, no chalcogenidomercurate(I) anion has been characterized so far. Nevertheless, we calculated the corresponding anions [Hg2Se3]4− (trigonal bipyramidal) and [HgSe3]5− (trigonal planar or pyramidal). The trigonal bipyramidal anion does not converge into a reasonable structure. The meta-mercurate(I) is a minimum on the energy hypersurface in the planar and the pyramidal conformation, with the latter being favoured, but both compounds are open shell systems, which is expected to hamper their isolation.
 | ||
| Scheme 2 Reaction pathways and energies for the transformation of selenido-thallates. Blue colour indicates experimentally observed anions. | ||
Hypothetic reaction cascades starting out from the ortho-thallate anion through a molecular meta-thallate anion [TlSe3]3− and further on to a linear [TlSe2]− anion are exoenergetic, but especially the second step is only slightly exoenergetic. Principally, the existence of the respective species under the simulated conditions should hence be possible. However, this pathway is obviously excelled by the shown and observed dimerization, which is even more exoenergetic. A possible redox reaction yielding a pyramidal [TlSe3]5− anion is energetically highly disfavoured, such that the exoenergetic step to the bent [TlSe2]3− species is not achievable this way. Thus, it becomes perspicuous why an interconversion between thallate(III) and thallate(I) anions have not yet been observed. Regarding the possible thallate(I) species, both [TlSe2]3− and the butterfly-like [Tl2Se2]2− anion should be accessible from an energetic point of view starting out from a (hypothetical) [Tl2Se3]4− bipyramid, but only the second was observed so far. Neither Schrobilgen's comprehensive NMR studies on such systems,10 nor our own attempts16 have so far yielded any indication of further thallate(I) anions. However, a related compound, [TlTe3Pb]3−,12b was published, hence further extension and variations of the synthetic approached should eventually allow for the synthesis of the other species, maybe even by the use of the quoted ternary anion.
 | ||
| Scheme 3 Reaction pathways and energies for the transformation of selenidoplumbates. Blue colour indicates experimentally observed anions, however, in the case of the [PbCh3]2−anion, only the tellurium analogue has been isolated so far. | ||
The degradation of ortho-selenidoplumbate(IV) anion towards a carbonate homologue under release of a selenide ligand is not associated with a significant energy change, hence differences either considering a solvent shell or a counterion environment may easily overcompensate it to an overall preference of the tetrahedral anion. An oxidative elimination of selenium to form the pyramidal [PbSe3]4− anion is considerably endoenergetic, apparently contradicting the experimental observation of the meta-telluridoplumbate(II), but in agreement with a synthesis starting out from a Pb(II) precursor and thus avoiding the disfavoured redox reaction step. Moreover, the distinct energetic preference of the ortho-selenidoplumbate(IV) over its dissociation or dechalcogenation is in line with its formation in and crystallization from solution, even as a hydrate like 9.
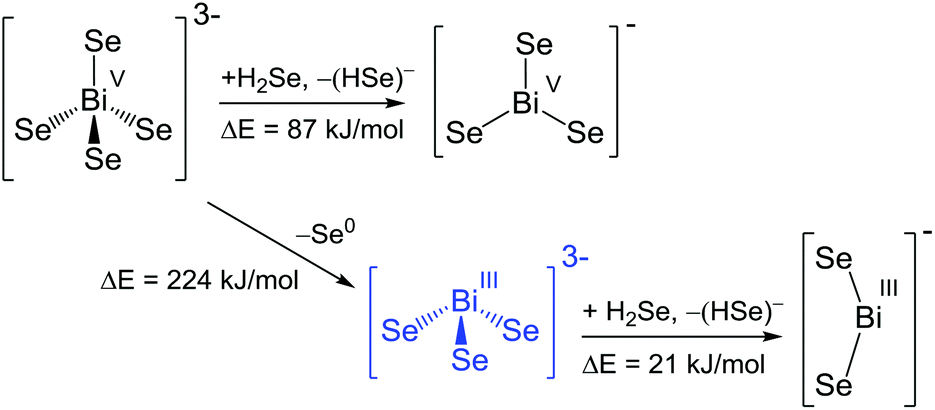 | ||
| Scheme 4 Reaction pathways and energies for the transformation of selenidobismuthates. Blue colour indicates experimentally observed anions. | ||
All reactions starting from a hypothetic ortho-bismuthate(V) anion including stepwise elimination and/or dissociation of Se0/Se2− are endoenergetic. Thus the existence of [BiSe4]3− does not seem to be generally unrealistic. However, very obviously, so far only chalcogenidobismuthates of Bi(III) have been produced that seem to be kinetically stable enough not to undergo the hypothetic, exoenergetic oxidation towards the tetrahedral species. [BiSe3]− would be the second heaviest possible homologue of the nitrate [NO3]− anion, yet its formation is not favourable according to energetic considerations. Also a bent [BiSe2]− species represents a minimum on the energy hypersurface, yet its formation from the observed [BiSe3]3− anion would require another 21 kJ mol−1. For this, although not isolated yet, the list of potentially observable Bi/Se anions definitely include the homologues of [NO3]−, [PO4]3− and [NO2]− in addition to the observed [BiCh3]3− anions. The remaining challenge is finding an appropriate synthetic approach, such as described recently and once more herein for salts of [PbSe4]4−.
4 Conclusions
We presented the synthesis and structure of novel ortho- and meta-chalcogenidometalates of mercury, thallium and lead, including the first molecular meta-chalcogenidomercurate(II), the first ortho-chalcogenidothallate(III) and the second ortho-selenidoplumbate(IV). The latter emerged for the first time and unexpectedly as its hydrate salt. In addition, we reported a new and facile synthetic pathway for the generation of the meta-selenidobismuthate in almost quantitative yield. The existence of all those anions inspired quantum chemical investigations of structures and energetics of the diverse species with different elemental combinations. We discussed hypothetic reaction energies, which helped to rationalize the observations and to predict further species to be isolated in future work. In the case of the chalcogenidoplumbates, the heaviest [CO3]2− homologues are generally stable, however most likely not accessible via solution based techniques. The same is valid for the chalcogenidobismuthate homologues of [NO3]−. Tables 1 and 2 summarize the experimental and theoretical findings for all smallest molecular chalcogenidometalate anions of the heaviest metals, including the new ones.|
|
|
|
||||
|---|---|---|---|---|---|---|
| Lower formal oxidation state of M | Lower formal oxidation state of M | Higher formal oxidation state of M | ||||
| Se | Te | Se | Te | Se | Te | |
| Hg | n/a | ○ | ○ | |||
| Tl | ○ | × | ○ |

|
||
| Pb | n/a | ⊗ | × | ○ | ||
| Bi | n/a | n/a | ○ | |||



Acknowledgements
We are indebted to Jörn Münzer and Marcus Müller for their help with synthetic work. We gratefully acknowledge fruitful discussion with Prof. Dr Werner Massa on the structure refinement of compound 9. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) within the framework of SPP 1415. GT thanks for financial support by the Friedrich–Ebert–Stiftung (FES).References
- C. D. Morris, H. Li, H. Jin, C. D. Malliakas, J. A. Peters, P. N. Trikalitis, A. J. Freeman, B. W. Wessels and M. G. Kanatzidis, Chem. Mater., 2013, 25, 3344 CrossRef CAS.
- M. A. McGuire, T. J. Scheidemantel, J. V. Badding and F. J. DiSalvo, Chem. Mater., 2005, 17, 6186 CrossRef CAS.
- (a) S. Wagner, J. L. Shay, P. Migliorato and H. M. Kasper, Appl. Phys. Lett., 1974, 25, 434 CrossRef CAS; (b) M. G. Panthani, V. Akhavan, B. Goodfellow, J. P. Schmidtke, L. Dunn, A. Dobabalapur, P. F. Barbara and B. A. Korgel, J. Am. Chem. Soc., 1998, 120, 16770 Search PubMed.
- J. Androulakis, S. C. Peter, H. Li, C. D. Malliakas, J. A. Peters, Z. Liu, B. W. Wessels, J.-H. Song, H. Jin, A. J. Freeman and M. G. Kanatzidis, Adv. Mater., 2011, 23, 4163 CrossRef CAS PubMed.
- (a) F. Lips and S. Dehnen, Inorg. Chem., 2008, 47, 5561 CrossRef CAS PubMed; (b) E. Ruzin, E. Zent, E. Matern, W. Massa and S. Dehnen, Chem. – Eur. J., 2009, 15, 5230 CrossRef CAS PubMed.
- (a) J. Heine and S. Dehnen, Z. Anorg. Allg. Chem., 2012, 638, 2425 CrossRef CAS; (b) S. Dehnen and M. Melullis, Coord. Chem. Rev., 2007, 251, 1259 CrossRef CAS; (c) E. Ruzin, A. Fuchs and S. Dehnen, Chem. Commun., 2006, 4796 RSC; (d) E. Ruzin and S. Dehnen, Z. Anorg. Allg. Chem., 2006, 632, 749 CrossRef CAS; (e) S. Dehnen and M. K. Brandmayer, J. Am. Chem. Soc., 2003, 125, 6618 CrossRef CAS PubMed.
- (a) W. S. Sheldrick, in Handbook of Chalcogen Chemistry, ed. F. Devillanova, The Royal Society of Chemistry, London, 2007, ch 9.2 Search PubMed; (b) W. S. Sheldrick and M. Wachhold, Angew. Chem., Int. Ed. Engl., 1997, 36, 206 CrossRef CAS; (c) M. G. Kanatzidis and S.-P. Huang, Coord. Chem. Rev., 1994, 130, 509 CrossRef CAS; (d) M. A. Ansari and J. A. Ibers, Coord. Chem. Rev., 1990, 100, 223 CrossRef CAS.
- H. Sommer and R. Hoppe, Z. Anorg. Allg. Chem., 1978, 443, 201 CrossRef CAS.
- C.-W. Park, D. M. Smith, M. A. Pell and J. A. Ibers, Inorg. Chem., 1997, 36, 942 CrossRef CAS.
- (a) R. C. Burns and J. D. Corbett, J. Am. Chem. Soc., 1981, 103, 2627 CrossRef CAS; (b) H. Borrmann, J. Campbell, D. A. Dixon, H. P. A. Mercier, A. M. Pirani and G. J. Schrobilgen, Inorg. Chem., 1998, 37, 1929 CrossRef CAS.
- G. Thiele, T. Krüger and S. Dehnen, Angew. Chem., Int. Ed., 2014, 53, 4699 CrossRef CAS PubMed.
- (a) M. Björgvinsson, J. F. Sawyer and G. J. Schrobilgen, Inorg. Chem., 1987, 26, 741 CrossRef; (b) H. Borrmann, J. Campbell, D. A. Dixon, H. P. A. Mercier, A. M. Pirani and G. J. Schrobilgen, Inorg. Chem., 1998, 37, 6656 CrossRef CAS PubMed; (c) M. Björgvinsson, J. F. Sawyer and G. J. Schrobilgen, Inorg. Chem., 1991, 30, 2231 CrossRef; (d) C.-W. Park, R. J. Salm and J. A. Ibers, Can. J. Chem., 1995, 73, 1148 CrossRef CAS.
- C. D. W. Jones, F. J. DiSalvo and R. C. Haushalter, Inorg. Chem., 1998, 37, 821 CrossRef CAS.
- (a) W. Bronger, A. Donike and D. Schmitz, Z. Anorg. Allg. Chem., 1996, 622, 1003 CrossRef CAS; (b) B. Eisenmann and R. Zagler, Z. Kristallogr., 1991, 197, 257 CAS.
- G. Thiele, L. Vondung and S. Dehnen, Z. Anorg. Allg. Chem., 2015, 641, 247 CrossRef CAS.
- (a) L. Protesescu, M. Nachtegaal, O. Voznyy, O. Borovinskaya, A. J. Rossini, L. Emsley, C. Copéret, D. Günther, E. H. Sargent and M. V. Kovalenko, J. Am. Chem. Soc., 2015, 137, 1862 CrossRef CAS PubMed; (b) E. Ruzin, A. Fuchs and S. Dehnen, Chem. Commun., 2006, 4796 RSC; (c) M. W. DeGroot, H. Rösner and J. F. Corrigan, Chem. – Eur. J., 2006, 12, 1547 CrossRef CAS PubMed; (d) M. Melullis, R. Clérac and S. Dehnen, Chem. Commun., 2005, 6008 RSC; (e) C. Zimmermann, C. E. Anson, F. Weigend, R. Clérac and S. Dehnen, Inorg. Chem., 2005, 44, 5686 CrossRef CAS PubMed; (f) M. K. Brandmayer, R. Clérac, F. Weigend and S. Dehnen, Chem. – Eur. J., 2004, 10, 5147 CrossRef CAS PubMed; (g) C. Zimmermann, M. Melullis and S. Dehnen, Angew. Chem., Int. Ed., 2002, 41, 4269 CrossRef CAS.
- Turbomole Version 6.6, Turbomole GmbH 2014, Turbomole is a development of University of Karlsruhe and Forschungszentrum Karlsruhe 1989–2007, Turbomole GmbH since 2007; (a) F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, WIREs Comput. Mol. Sci., 2014, 4, 91 CrossRef CAS; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC.
- (a) D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef PubMed; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822 CrossRef.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC; (c) M. Dolg, H. Stoll, A. Savin and H. Preuss, Theor. Chim. Acta, 1989, 75, 173 CrossRef CAS; (d) H. Stoll, B. Metz and M. Dolg, J. Comput. Chem., 2002, 23, 767 CrossRef CAS PubMed.
- (a) J. Heine and S. Dehnen, Z. Anorg. Allg. Chem., 2008, 634, 2303 CrossRef CAS; (b) E. Ruzin, S. Jakobi and S. Dehnen, Z. Anorg. Allg. Chem., 2008, 634, 995 CrossRef CAS.
- G. Thiele, L. Vondung, C. Donsbach, S. Pulz and S. Dehnen, Z. Anorg. Allg. Chem., 2014, 640, 2684 CrossRef CAS.
- G. Thiele, L. Vondung and S. Dehnen, unpublished results.
- M. G. Kanatzidis and Y. Park, Chem. Mater., 1990, 2, 99 CrossRef CAS.
- G. Thiele, S. Lippert, F. Fahrnbauer, P. Bron, O. Oeckler, A. Rahimi-Iman, M. Koch, B. Roling and S. Dehnen, Chem. Mater., 2015, 27, 4114 CrossRef CAS.
- P. Boettcher, J. Getzschmann and R. Keller, Z. Anorg. Allg. Chem., 1993, 619, 476 CrossRef CAS.
- B. Eisenmann and H. Schaefer, Angew. Chem., Int. Ed. Engl., 1978, 90, 731 CAS.
- P. Feng, X. Bu and N. Zheng, Acc. Chem. Res., 2005, 38, 293 CrossRef CAS PubMed.
- J. Weis, R. Schäfer and G. Schön, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1976, 31, 1336 Search PubMed.
- J. A. A. Ketelaar, W. H. t'Hart and D. Polder, Z. Kristallogr., 1939, 101, 396 CAS.
- E. R. Franke and H. Schäfer, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1972, 27, 1308 CAS.
- E. Dönges, Z. Anorg. Allg. Chem., 1951, 265, 56 CrossRef.
- P. W. Lange, Naturwissenschaften, 1939, 27, 133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1430781–1430790. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03996g |
| This journal is © The Royal Society of Chemistry 2016 |