
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical and chemical routes to hydride loss from an iridium dihydride†
A. G.
Walden
a,
A.
Kumar
bc,
N.
Lease
b,
A. S.
Goldman
b and
A. J. M.
Miller
*a
aDepartment of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-3290, USA. E-mail: ajmm@email.unc.edu
bDepartment of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, New Brunswick, New Jersey 08903, USA
cDepartment of Chemistry, Indian Institute of Technology Guwahati, Guwahati - 781039, Assam, India
First published on 16th March 2016
Abstract
With a view towards replacing sacrificial hydrogen acceptors in alkane dehydrogenation catalysis, electrochemical methods for oxidative activation of a pincer-ligated iridium hydride intermediate were explored. A 1H+/2e− oxidation process was observed in THF solvent, with net hydride loss leading to a reactive cationic intermediate that can be trapped by chloride. Analogous reactivity was observed with the concerted hydride transfer reagent Ph3C+, connecting chemical and electrochemical hydride loss pathways.
Iridium complexes supported by tridentate R4PCP (R4PCP = κ3-C6H3-2,6-(CH2PR2)2) pincer ligands are prolific dehydrogenation catalysts, enabling landmark transformations such as the dehydrogenation,1,2 metathesis,3 coupling4,5 and dehydroaromatization6 of alkanes.7 Efficient dehydrogenation reactions require a sacrificial hydrogen acceptor, typically an olefin. The hydrogen acceptor alters the overall reaction thermodynamics and activates the iridium dihydride species.7–9 In transfer dehydrogenation, catalyst activation occurs by insertion of the sacrificial olefin into one Ir–H bond, followed by C–H bond-forming reductive elimination with the other Ir–H bond, generating a highly reactive 14e− intermediate capable of alkane C–H bond activation (Scheme 1).
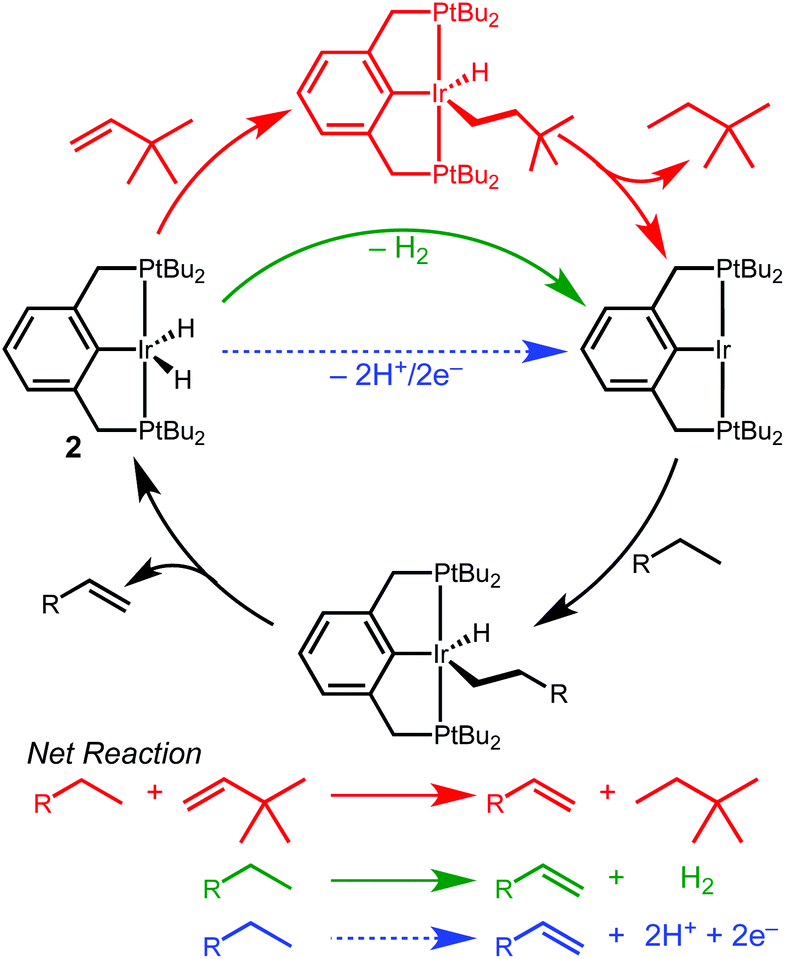 | ||
| Scheme 1 | ||
The requirement of an added stoichiometric reagent represents a significant limitation in dehydrogenation reactions.8,10 In considering new strategies to promote dehydrogenation reactions, we were drawn to electrochemical methods that could decouple the catalyst activating and hydrogen accepting steps.11,12 We envisioned electrochemical oxidation of (R4PCP)Ir(H)2 at an anode, generating a catalytic intermediate while releasing 2H+/2e− (Scheme 1) that could be used to drive any range of reactions at the cathode.
Electrochemical dehydrogenation relies on (sometimes coupled) electron transfer and proton transfer steps,13,14 while chemical dehydrogenation often involves concerted hydride transfer.7,15,16 Recent reports have started to draw connections between chemical and electrochemical processes, however. For example, inspired by a report of (R4PCP)Ir-catalyzed hydrogenation of CO2 to formate,17 Brookhart and Meyer developed an analogous electrochemical reduction of CO2 to formate catalyzed by (tBu4POCOP)Ir complexes (tBu4POCOP = κ3-C6H3-2,6-(OPtBu2)2).18–20 A striking oxidative example involves two different catalysts for the same alcohol oxidation reaction that operate by two different mechanisms, either a concerted H2 loss mechanism or an outer-sphere electron transfer mechanism in which a chemical oxidant (not an electrode) and a base facilitate 2H+/2e− loss.21
Studies of electrochemical reactions that parallel well-known organometallic oxidations can help bridge the divide between chemical and electrochemical methods. This report focuses on the oxidation of a pincer-ligated iridium dihydride. Net loss of hydride (H+/2e−) is promoted by either electrochemical or chemical methods to produce an iridium monohydride species.
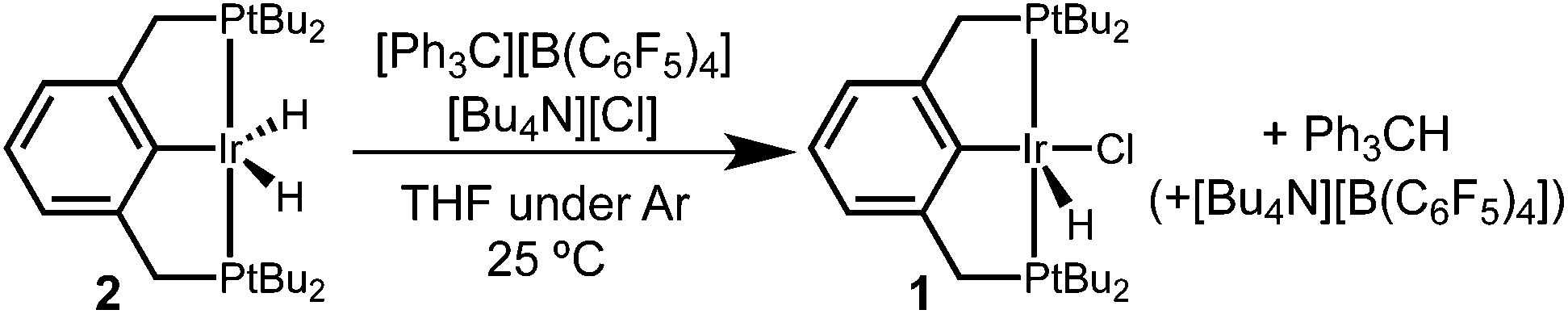
The dihydride complex was prepared according to previously reported procedures by dehydrohalogenation of (tBu4PCP)Ir(H)(Cl) (1) under an H2 atmosphere.1,22 This procedure affords a mixture of the five-coordinate dihydride (tBu4PCP)Ir(H)2 (2) and (tBu4PCP)Ir(H)4 (3).23 Samples could be stirred in pentane, filtered, and dried under vacuum to remove the dihydrogen ligand and provide pure 2.‡
The oxidation of dihydride 2 was initially explored using cyclic voltammetry (CV). When a solution of 2 in argon-saturated THF containing [Bu4N][PF6] supporting electrolyte was assessed by a CV sweep to oxidative potentials, a single irreversible feature was observed at −0.08 V vs. Cp2Fe+/0 (Fig. 1). No return reduction process was apparent, even as the scan rate was increased to 1 V s−1.
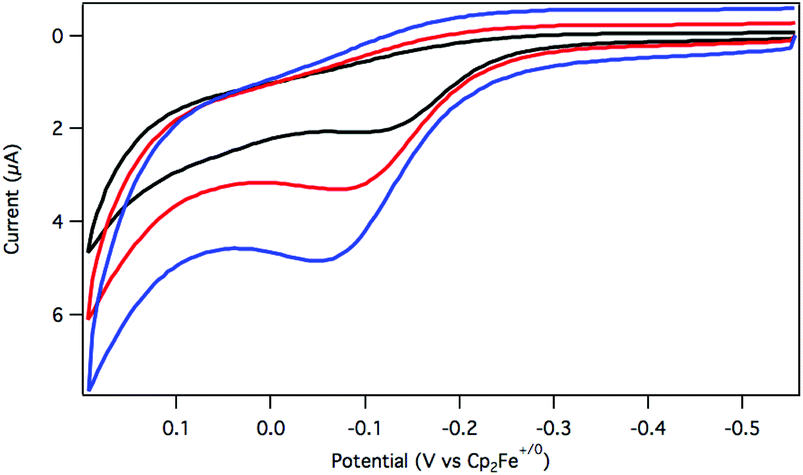 | ||
| Fig. 1 Cyclic voltammetry of 2 at 25 mV s−1 (black), 100 mV s−1 (red), and 250 mV s−1 (blue) in THF solution with 0.1 M [Bu4N][PF6] electrolyte. Glassy carbon working electrode, platinum counter electrode, Ag wire pseudo-reference electrode, 298 K. | ||
An irreversible electrochemical oxidation is consistent with a rapid chemical reaction following electron transfer from 2 to the electrode. The dihydride 2 is more easily oxidized than the hydridochloride complex 1, which exhibited a quasi-reversible oxidation around 0.5 V vs. Cp2Fe+/0 in CH2Cl2 at fast scan rates in a prior study.24
To identify the product formed at positive potentials under argon, a controlled potential electrolysis experiment was carried out. A high-surface-area reticulated vitreous carbon working electrode was submersed in a THF solution of dihydride 2 and polarized to 0.2 V vs. Cp2Fe+/0. The flow of current diminished as a gradual color change from pale orange to pale yellow was observed. The oxidation passed 239 mC of charge, corresponding to 1.9 e− per Ir, but an aliquot analyzed by 31P{1H} NMR spectroscopy revealed a mixture of species.
Considering the possibility that oxidation of 2 would produce a reactive cationic species,25 the oxidative electrochemistry was also carried out in the presence of a chloride ion source as a trapping agent. In the presence of LiCl (and with conditions otherwise similar to those described above), the CV response of 2 was essentially unchanged relative to chloride-free conditions, suggesting that chloride does not influence the initial oxidation process.
Controlled potential electrolysis of a THF solution containing 2 and excess LiCl or [Bu4N][Cl] was conducted at 0.2 V vs. Cp2Fe+/0 (Scheme 2). In the presence of chloride, the solution color changed from pale orange to a much brighter orange, and the 283 mC of charge passed corresponds to a 2e− oxidation (2.3 e− per Ir). Analysis by 31P{1H} NMR spectroscopy now revealed a single phosphorous-containing species (δ 69). The product was isolated from the electrolyte by removal of the THF under vacuum and extraction with pentane. Full NMR spectroscopic analysis in THF-d8 showed a triplet hydride resonance far upfield (δ −42.9) in the 1H NMR spectrum that is diagnostic of (tBu4PCP)Ir(H)(Cl) (1). All of the 31P and 1H NMR signals closely matched the previously reported values.22
 | ||
| Scheme 2 | ||
The electrochemical conversion of dihydride 2 to hydridochloride 1 represents a net hydride abstraction via the loss of 2e− to the anode and loss of H+ (to solution or perhaps to a surface site on the electrode), followed by chloride binding. This two-step electrochemical–chemical (EC) transformation is consistent with the irreversible CV response (prior studies of (pincer)Ir(H)(Cl) also implicated an EC mechanism, but did not identify a product).24 The stability of the product, hydridochloride 1, towards further oxidation at the potentials applied during electrolysis is critical to the success of the reaction.24
Analogous electrochemical hydride loss via a two-electron/one-proton oxidative process has been reported for a series of Group 6 complexes of the type CpM(CO)3H (M = Cr, M, W),26 which may involve a concerted proton-coupled electron transfer event in the tungsten case.27 In contrast, the Rh analogue (tBu4PCP)Rh(H2), which is best described as a Rh(I) dihydrogen complex,28 does not undergo oxidative hydride loss: reversible 1e− oxidation is observed in CH2Cl2, and H2 loss is observed in coordinating solvents.29
To further probe the hydride transfer reactivity, chemical methods that could effect an analogous hydride loss were explored. When dihydride 2 is allowed to react with the hydride abstractor [Ph3C][B(C6F5)4] in THF-d8, the solution changes color from pale orange to pale yellow. NMR spectroscopic monitoring revealed a mixture of products analogous to those observed in the initial electrolysis.
Hydride abstraction was next attempted in the presence of a chloride source. Treatment of dihydride 2 with 1 equiv. [Ph3C][B(C6F5)4] and 5 equiv. [Bu4N][Cl] led to a color change from pale orange to a much brighter orange, coinciding with the appearance of the characteristic signals of hydridochloride complex 1 by 31P{1H} and 1H NMR spectroscopy (Scheme 3). Triphenylmethane is also observed by 1H NMR spectroscopy, clearly identifying the fate of the hydride.
 | ||
| Scheme 3 | ||
We suggest that the electrochemical and chemical hydride abstractions proceed via a shared intermediate, given the similar product distributions under various reaction conditions. As shown in Scheme 4, we hypothesize that oxidation of dihydride 2 occurs as a net 1H+/2e− process (via one of the pathways described above) to generate a reactive monohydride cation, [(tBu4PCP)Ir(H)]+ (4). Chemical hydride transfer from 2 to [Ph3C][B(C6F5)4] would also afford 4. We are not aware of any prior reported isolation of cation 4. An analogous [(tBu4POCOP)Ir(H)]+ species, isolated as an acetone or dichloromethane adduct, is an active hydrosilylation catalyst.30,31
 | ||
| Scheme 4 | ||
From this shared intermediate cation 4, trapping with chloride ion can generate the hydridochloride 1. In the absence of chloride, we suspect that cation 4 decomposes through reactions with itself and/or the solvent, the details of which are currently under investigation. The observation of identical products under electrochemical and chemical reaction conditions suggests that future electrochemical oxidations (even in non-polar solvents)32–34 can be modeled after existing hydride abstraction reactions.
By implicating a key monohydride cation intermediate and building an analogy between well-defined organometallic hydride abstraction reactions and electrochemical oxidation processes, these joint chemical/electrochemical studies provide a foundation for future development of electrochemical dehydrogenation processes.
Acknowledgements
The authors acknowledge funding from NSF Center for Enabling New Technologies through Catalysis (CENTC), CHE-1205189. The Templeton group generously provided access to an argon-filled glovebox.Notes and references
- M. Gupta, C. Hagen, R. J. Flesher, W. C. Kaska and C. M. Jensen, Chem. Commun., 1996, 2083–2084 RSC.
- A. Kumar, T. Zhou, T. J. Emge, O. Mironov, R. J. Saxton, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2015, 137, 9894–9911 CrossRef CAS PubMed.
- A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Science, 2006, 312, 257–261 CrossRef CAS PubMed.
- D. C. Leitch, Y. C. Lam, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2013, 135, 10302–10305 CrossRef CAS PubMed.
- J. A. Labinger, D. C. Leitch, J. E. Bercaw, M. A. Deimund and M. E. Davis, Top. Catal., 2015, 58, 494–501 CrossRef CAS.
- R. Ahuja, B. Punji, M. Findlater, C. Supplee, W. Schinski, M. Brookhart and A. S. Goldman, Nat. Chem., 2011, 3, 167–171 CrossRef CAS PubMed.
- J. Choi, A. H. Roy MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed.
- K. Krogh-Jespersen, M. Czerw, N. Summa, K. B. Renkema, P. D. Achord and A. S. Goldman, J. Am. Chem. Soc., 2002, 124, 11404–11416 CrossRef CAS PubMed.
- K. B. Renkema, Y. V. Kissin and A. S. Goldman, J. Am. Chem. Soc., 2003, 125, 7770–7771 CrossRef CAS PubMed.
- W. Xu, G. P. Rosini, K. Krogh-Jespersen, A. S. Goldman, M. Gupta, C. M. Jensen and W. C. Kaska, Chem. Commun., 1997, 2273–2274 RSC.
- P. Driscoll, E. Deunf, L. Rubin, O. Luca, R. H. Crabtree, C. Chidsey, J. Arnold and J. Kerr, ECS Trans., 2011, 35, 3–17 CAS.
- B. Rausch, M. D. Symes and L. Cronin, J. Am. Chem. Soc., 2013, 135, 13656–13659 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Rev., 2010, 110, PR1–PR40 CrossRef PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS.
- C. R. Waidmann, A. J. M. Miller, C.-W. A. Ng, M. L. Scheuermann, T. R. Porter, T. A. Tronic and J. M. Mayer, Energy Environ. Sci., 2012, 5, 7771–7780 CAS.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed.
- P. Kang, C. Cheng, Z. Chen, C. K. Schauer, T. J. Meyer and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 5500–5503 CrossRef CAS PubMed.
- P. Kang, T. J. Meyer and M. Brookhart, Chem. Sci., 2013, 4, 3497–3502 RSC.
- P. Kang, S. Zhang, T. J. Meyer and M. Brookhart, Angew. Chem., Int. Ed., 2014, 53, 8709–8713 CrossRef CAS PubMed.
- P. J. Bonitatibus, S. Chakraborty, M. D. Doherty, O. Siclovan, W. D. Jones and G. L. Soloveichik, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1687–1692 CrossRef CAS PubMed.
- C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1976, 1020–1024 RSC.
- T. J. Hebden, K. I. Goldberg, D. M. Heinekey, X. Zhang, T. J. Emge, A. S. Goldman and K. Krogh-Jespersen, Inorg. Chem., 2010, 49, 1733–1742 CrossRef CAS PubMed.
- F. Novak, B. Speiser, H. A. Y. Mohammad and H. A. Mayer, Electrochim. Acta, 2004, 49, 3841–3853 CrossRef CAS.
- M. Gupta, W. C. Kaska and C. M. Jensen, Chem. Commun., 1997, 461–462 RSC.
- O. B. Ryan, M. Tilset and V. D. Parker, J. Am. Chem. Soc., 1990, 112, 2618–2626 CrossRef CAS.
- M. Bourrez, R. Steinmetz, S. Ott, F. Gloaguen and L. Hammarström, Nat. Chem., 2015, 7, 140–145 CrossRef CAS PubMed.
- K. Huang, J. H. Han, C. B. Musgrave and E. Fujita, Organometallics, 2007, 26, 508–513 CrossRef CAS.
- M. D. Doherty, S. J. Konezny, V. S. Batista and G. L. Soloveichik, J. Organomet. Chem., 2014, 762, 94–97 CrossRef CAS.
- J. Yang and M. Brookhart, J. Am. Chem. Soc., 2007, 129, 12656–12657 CrossRef CAS PubMed.
- J. Yang and M. Brookhart, Adv. Synth. Catal., 2009, 351, 175–187 CrossRef CAS.
- W. E. Geiger and F. Barrière, Acc. Chem. Res., 2010, 43, 1030–1039 CrossRef CAS PubMed.
- R. J. LeSuer, C. Buttolph and W. E. Geiger, Anal. Chem., 2004, 76, 6395–6401 CrossRef CAS PubMed.
- A. P. Abbott and D. J. Schiffrin, J. Chem. Soc., Faraday Trans., 1990, 86, 1453–1459 RSC.
- R. Ghosh, M. Kanzelberger, T. J. Emge, G. S. Hall and A. S. Goldman, Organometallics, 2006, 25, 5668–5671 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, electrochemical data, and NMR spectra. See DOI: 10.1039/c6dt00522e |
| ‡ Solutions containing hydrides 2 and 3 are stable under Ar or H2, but decompose under N2 or air to a mixture of products with distinct electrochemical responses.35 |
| This journal is © The Royal Society of Chemistry 2016 |