
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Handheld real-time PCR device†
Christian D.
Ahrberg
a,
Bojan Robert
Ilic
b,
Andreas
Manz
a and
Pavel
Neužil
*acd
aKIST-Europe, Microfluidics Group, Campus E7.1, 66111 Saarbrücken, Germany. E-mail: pavel.neuzil@gmail.com
bNational Institute of Standard and Technology (NIST), Center for Nanoscale Science and Technology, 100 Bureau Drive, MS 6201, Gaithersburg, MD 20899-6201, USA
cBrno University of Technology (BUT), Central European Institute of Technology (CEITEC), Technická 3058/10, CZ-616 00 Brno, Czech Republic
dNorthwestern Polytechnical University (NPU), School of Mechanical Engineering, Department of Microsystem Engineering, 127 West Youyi Road, Xi'an Shaanxi, 710072, PR China
First published on 16th December 2015
Abstract
Here we report one of the smallest real-time polymerase chain reaction (PCR) systems to date with an approximate size of 100 mm × 60 mm × 33 mm. The system is an autonomous unit requiring an external 12 V power supply. Four simultaneous reactions are performed in the form of virtual reaction chambers (VRCs) where a ≈200 nL sample is covered with mineral oil and placed on a glass cover slip. Fast, 40 cycle amplification of an amplicon from the H7N9 gene was used to demonstrate the PCR performance. The standard curve slope was −3.02 ± 0.16 cycles at threshold per decade (mean ± standard deviation) corresponding to an amplification efficiency of 0.91 ± 0.05 per cycle (mean ± standard deviation). The PCR device was capable of detecting a single deoxyribonucleic acid (DNA) copy. These results further suggest that our handheld PCR device may have broad, technologically-relevant applications extending to rapid detection of infectious diseases in small clinics.
The invention of polymerase chain reaction (PCR) 32 years ago is considered to be one of the greatest inventions of the last century.1 Over the years, many variants of the original system have been developed. One of the most important advancements is the real-time PCR analysis system.2 The approach enables real-time PCR amplification, monitoring, and quantification of the number of deoxyribonucleic acid (DNA) copies in the sample under consideration. This method is commonly referred to as quantitative PCR (qPCR).2 The main advantage of real-time PCR is the elimination of any post-processing, such as electrophoresis or hybridization to detect the PCR product.
The PCR reaction is performed by thermal cycling in the presence of specific oligonucleotides, the enzyme polymerase, free nucleic acids and bivalent salts such as MgSO4 or MgCl2. This cocktail is commonly referred to as the PCR master mix. The detection of PCR product amplification is conducted by monitoring the fluorescence amplitude during the PCR. In the presence of an intercalating dye, such as SYBR Green I, the fluorescence amplitude is proportional to the concentration of the DNA amplicon, the product of the PCR. In order to verify amplification specificity, upon PCR completion, employment of an intercalating dye enables performing melting curve analysis (MCA). Another improvement of the PCR is the addition of the reverse transcriptase enzyme to the PCR cocktail, forming reverse transcription PCR (RT-PCR).
PCR has become the method of choice for the detection of DNA and RT-PCR to detect RNA. These two reactions have revolutionized genetics. Furthermore, PCR has many diverse applications in infectious disease diagnostics for detection of viruses or bacteria,3 in forensic science,4,5 paternity tests,6 security applications7 and myriad other commercial applications.8 Commercial systems are typically rather large table-top tools used for high throughput mass screenings and are impractical for use in point-of-care applications (POC), where the most important system parameters are portability and power consumption. The quest for a miniaturized PCR version suitable for POC diagnostics was initiated at Lawrence Livermore Laboratories9,10 more than two decades ago. Agrawal et al. have developed a pocket-sized conventional PCR system11 that requires extensive sample post-processing to identify the presence of an amplicon. In contrast, real-time PCR eliminates the need for sample processing once PCR is completed.
Real-time PCR systems consist of a heater, temperature sensor, and fluorescence excitation and detection unit. Temperature cycling is performed by heating and cooling of samples. Within the PCR process, the cooling rate is one of the primary limiting factors. Bulky commercial systems have large heat capacities, hence heat removal is challenging, and is typically accomplished by using a thermoelectric cooler (TEC), commonly known as the Peltier element. Since these bulky systems consume a considerable amount of power, they are generally unsuitable for field testing POC applications.
Small PCR instruments are often based on microfluidic devices, so-called “lab-on-a-chip” devices.12 These systems comprise two major groups, spatial-domain and time-domain PCRs. On the one hand, time-domain PCRs have a single heater with samples placed in direct contact. Here, temperature cycling is carried out by changing the heater element temperature. On the opposite end of the spectrum, the spatial-domain PCR has several heaters, each held at a different temperature. In this scenario, temperature cycling is accomplished by moving samples between heaters.
A typical representative of a spatial-domain system is the continuous PCR-on-a-chip.13 Within this system, the sample flow in the microfluidic chip is positioned over the heaters, each kept at a different temperature. The sample flows through tubes, thereby achieving thermal cycling. Here, PCR duration is only limited by the flow rate and the heat transfer between the sample and the side walls, for both heating and cooling. The two major drawbacks of a flow-through PCR are system complexity and a high likelihood of sample-to-sample cross-contamination.
An alternative version was introduced a few years ago where the sample was in the form of a virtual reaction chamber (VRC).14–16 The VRC self-assembly system consists of a water droplet covered with mineral oil, preventing water evaporation from the sample. In this scenario, the water droplet contained the PCR master mix with a pre-determined number of DNA copies. Here, the VRC with DNA was separated from the micromachined silicon heaters by a disposable, hydrophobically-coated microscope cover slip. To eliminate sample-to-sample contamination, the glass cover slip was a disposable part of the system, and therefore each cover slip was a single use component. The sample contained magnetic particles which facilitated sample motion between heaters.17 A pocket-size real-time PCR system capable of processing a single sample was introduced a few years later.18 The system had an integrated miniaturized optical detection unit, LCD display and control electronics. One of the key features was the implementation of lock-in amplification for optical signal processing.17 The lock-in amplification feature allowed for ambient system operation without light protection, thereby rendering the system robust and user-friendly. One of the system drawbacks was processing a single sample at a time and furthermore, the device was bulky.
A practical system to conduct PCR for POC applications requires simultaneous processing of 4 or more samples. Diagnoses of clinical samples should be concurrently conducted with positive and negative control samples, thereby eliminating false negative or positive events.
In our work, we introduce a new portable PCR system (Fig. 1) capable of concurrently analyzing four ≈200 nL volume samples. The system speed is determined by the heating and cooling rates. The heating rate collectively arises from the VRC thermal capacitance (H) and the dissipated Joule heat. The rate of passive cooling applied within our system is given by the thermal time constant (τ) of the system, which is given by H/G, where G is the thermal conductance. Since the specific heat of water is exceptionally large, the thermal properties of our system are strongly dependent on the sample water volume. Consequently, a smaller sample size results in a faster system.19
 | ||
| Fig. 1 (A) CAD design drawing of the handheld PCR. The illustration shows a display with a compartment accommodating 4 samples in the VRC form. (B) Fabricated and assembled complete real-time PCR device packaged within a 3D printed casing. | ||
System samples consist of a negative control, also called no template control (NTC), a positive control, and two samples of interest. The four sample system architecture represents the minimal number of samples required for practical applications. Our system conducts 40 PCR cycles in less than ≈35 min, while simultaneously processing the results. Furthermore, our portable real-time PCR is capable of detecting a single DNA copy.
The PCR performance was evaluated by detecting a complementary DNA from the avian influenza virus (H7N9) as well as two human transcripts, hypoxanthine phosphoribosyltransferase (HPRT) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). To the best of our knowledge, our system platform represents the smallest real-time PCR system.
Our PCR instrument has two key features:
1. The four samples are in the VRC form and are placed on a disposable glass cover slip over micromachined silicon heaters. Upon PCR completion, the single-use, disposable glass element is removed and a new glass cover slip is placed on top of the silicon heater.
2. The fluorescence excitation/detection system is based on a lock-in amplifier, thereby rendering the system immune to ambient light. The PCR instrument is equipped with a graphical 84 × 48 pixel liquid crystal display (LCD) with a diagonal size of ≈38.1 mm to show the reaction progress and final results. The captured data is stored in an internal memory and can be uploaded for external processing via a universal serial bus (USB). The system is powered by an external 12 V battery.
System setup
Our current system has two new key features: an integrated optical head and simplified control electronics.1. Integrated optical head
A fluorescence detection system for a single spot requires a light source, three filters (excitation, dichroic mirror and emission) and a detector. We redesigned the original head20,21 with 5 filters for the four units (see Fig. 2). Each measurement spot is illuminated with a light emitting diode (LED) with a principal emission wavelength of 470 nm and a luminous intensity in the range of 7.2 cd to 12 cd. Light passes through an excitation band pass filter with a center wavelength of ≈470 nm and a band pass of ≈40 nm, blocking light from the LED with wavelengths longer than ≈490 nm. Light is then reflected off of a long pass dichroic mirror with a cut-off wavelength of ≈495 nm, and focused by a lens with a focal length of ≈3.1 mm, a numerical aperture of ≈0.68 and an antireflective coating in the range of ≈350 nm to ≈700 nm. The emitted fluorescence (F) is collimated by the same lens, passing through the dichroic mirror. The residual blue light is suppressed by a long pass emission filter with a cut-off wavelength of ≈510 nm, and fluorescence is captured by the conventional silicon photodiode with a radiant sensitive area of ≈7.5 mm.2 The cross section schematic of the handheld PCR system illustrating the optical path is shown in Fig. S1 (ESI†). The resulting photocurrent is converted into voltage using an ultra-low bias current operational amplifier with dielectrically-isolated field effect transistor inputs (diFET) as a transconductance amplifier. In this configuration, the four sample systems share optical filters. Four LEDs are mounted in two pairs. Within each pair, the LEDs were parallel, thus requiring only two excitation and two dichroic filters for all 4 LEDs. Finally, there is only a single emission filter for all photodiodes. A potential expansion to eight systems would require additional LEDs and photodiodes with amplifiers. | ||
| Fig. 2 Schematic illustration of the integrated optical head. Blue arrows show the optical path from one LED with filters to the VRC. The green arrow shows the optical path of excited fluorescence to the photodiode. Light emitted from a blue LED passes through the blue filter in order to remove the green portion of emitted light, then reflects off of the dichroic mirror through a donut-shaped heater and is focused on the sample by an aspherical lens. Excited fluorescence is collimated by the same lens, passing through the dichroic mirror with the blue portion filtered out by a green filter. Residual green light interacts with a photodiode and induces a photocurrent, which is further processed. | ||
2. Control electronics
A previously published system18 had one lock-in amplifier for a single fluorescence detection system and a second one for temperature measurement. This scenario was very inefficient since fluorescence was monitored for ≈2 s during each PCR cycle. The second lock-in amplifier for temperature measurement was used during the entire PCR operation. Our current system employs a single lock-in amplifier to monitor the sample temperature and capture fluorescence from all four spots.The system heaters were connected in a serial–parallel combination, wherein the system controlled the average temperature of all four heaters. We used a similar AC biased Wheatstone bridge to convert the resistance of the resistance temperature detector (RTD) into a DC voltage as previously described.18
The control electronics for the optical system was a simplified version of our previous work.18 Here, each PCR system (spot) had its own LED, collimating lens and a photodiode with a respective transconductance amplifier while the optical filters are shared. We activated one LED at a time, thereby feeding the signal to a single, corresponding transconductance amplifier. Outputs of the four amplifiers were connected together and processed as one signal. The complete schematic of the PCR system is shown in Fig. S2 (ESI†).
The incident photocurrents from the photodiodes were converted into voltage using four dedicated operational amplifiers. The amplifier outputs were connected together and a single composite signal was further processed. The cross talk between four measured spots was minimized since one LED was activated at a time; therefore the resulting total amplitude of the processed photocurrent originated from a single dedicated PCR reaction. All important devices are listed in Table S1 (ESI†).
Experimental
A typical real-time PCR protocol with an intercalating dye such as SYBR-Green I initiates with a hot start to activate the polymerase. PCR cycles consist of denaturation, annealing and extension steps. The fluorescence amplitude is measured at the end of the extension step for a period of ≈2 s. We controlled the heater temperature using the proportional integrative-derivative (PID) closed feedback loop method. The amount of heat delivered through dissipated Joule heating was controlled using a pulse-width modulation (PWM) technique. The last ≈2 s of the amplification cycle were used for fluorescence monitoring (see Fig. 3). In this step, following temperature stabilization, we monitored the duty cycle of the PWM and calculated its average. During the last ≈2 s, the feedback loop was disconnected, the temperature was not monitored and the average value of the PWM was employed. During the system development phase, we monitored the heater temperature and found that the method described above gives us a temperature variation of less than ±0.5 °C over a ≈2 s time interval. The sample temperature follows the heater temperature with a ≈1.5 s delay,22 therefore a ±0.5 °C temperature variation at the heater does not affect the PCR performance. The measured temperature profile from ≈6 PCR steps is shown within the system liquid crystal display (LCD) in Fig. 4A. | ||
| Fig. 3 PCR thermal profile of a single amplification step. The heater temperature (red part) is linearly proportional to the built-in lock-in amplification output, which was captured with an oscilloscope. While performing denaturation at ≈93 °C, annealing at ≈56 °C and most of the extension step at ≈72 °C, the built-in lock-in amplifier is utilized to measure the average temperature of all four heaters. In the last two seconds of the extension step, the lock-in amplifier is used to sequentially process the fluorescence signal (green part) from the 4 measurement spots (circled area). These data are stored in their original format in analog-to-digital converter units (ADC units) within the memory of a microcontroller. Once fluorescence is measured, the heater is powered by the average value of pulse width modulation obtained during the extension phase. | ||
 | ||
| Fig. 4 (A) An assembled PCR system with four VRCs, each consisting of a ≈0.5 μL sized sample covered with ≈1.5 μL of M5904 mineral oil, showing the PCR temperature profile (protocol) within the LCD display. The device size is 82 mm × 45 mm × 20 mm (length, width and height). Scale bar is 40 mm. The protocol started by a “hot start” to activate the polymerase enzyme for ≈10 min at ≈95 °C, followed by 40 cycles of PCR amplification. Each amplification cycle consisted of 3 steps. First, denaturation for ≈10 s at ≈95 °C, then annealing for ≈10 s at ≈50 °C and the last step was extension for ≈15 s at ≈68 °C (instead of typical ≈72 °C), during which fluorescence was measured. The LCD display shows 6 cycles. At the completion of each PCR amplification cycle, fluorescence amplitude at each spot was calculated and amplification curves were plotted in (B). We placed NTC at position 1, a low concentration of complementary DNA (cDNA) from H7N9 HA gene at position 3, a medium concentration at position 2 and the highest concentration (positive control) at position 4. The results show that the PCR reaction was successfully accomplished without PCR amplification of the NTC sample. Furthermore, the results prove that the samples were not cross contaminated, thereby eliminating false positive outcomes. Positive control results at position 4 indicate the absence of false negative results, thereby showing a successful PCR amplification process. | ||
During the last two seconds of the extension step, the fluorescence measurement system was activated. Sequentially, each LED was individually powered for ≈0.5 s and the emitted fluorescence was captured by the respective photodiode and lock-in amplifier. At the completion of an amplification cycle, the system was switched back into the temperature measurement mode, initiating the start of a new cycle. The PCR amplification curves were plotted for each spot and the captured amplitude of fluorescence was displayed on the LCD display. The typical PCR amplification curve is shown in Fig. 4B. Directly following the PCR process, melting curve analysis (MCA) was performed. Since temperature measurement during the MCA is time consuming, we performed this step without a feedback loop. In the course of the PCR, we monitored and recorded the PWM duty cycle of three fixed temperature points: denaturation, annealing and extension temperatures. These three points were used to calculate the required duty cycle for temperature scans ranging from ≈68 °C to ≈94 °C without a closed feedback loop. The system was stabilized at ≈68 °C. The temperature of each heater was then gradually increased to ≈95 °C while the fluorescence in each of the four spots was sequentially measured. The measurement of each spot required a duration of ≈0.5 s, hence ≈2 s was required to measure all 4 VRCs. This measurement setup allowed for fluorescence measurement from each spot with an offset of ≈0.25 °C between adjacent VRCs. Once the MCA was completed, we stabilized the sample at the assumed temperature of ≈95 °C and then measured the actual temperature. Directly following this, the system's central processing unit (CPU) performed two corrections. First, the MCA was recalculated based on the actual final temperature. Second, the correction accounts for the temperature offset between individual VRCs. Consequently, the MCAs for each spot were recalculated accordingly. Finally, a negative derivative value of fluorescence with respect to temperature (−dF/dT) was calculated.
The device performance was evaluated using synthetic complementary DNA (cDNA) for the hemagglutinin of the H7N9 avian influenza virus. Forward and reverse primers were chosen as suggested earlier:23 forward primer: TACAGGGAAGAGGCAATGCA, reverse primer: AACATGATGCCCCGAAGCTA, giving a total amplicon length of 104 base pairs with a melting temperature of ≈81.1 °C, as measured using a commercial real time PCR system.
The PCR master mix was prepared by mixing ≈2 μL of FastStart DNA Master SYBR Green I, ≈2 μL of MgCl2 solution, ≈2 μL of sample HA (5 × 10−5 ng μL−1), ≈0.3 μL of ≈400 mg mL−1 BSA solution and primers in a final concentration of ≈1.8 × 10−6 mol L−1. We added deionized (DI) water with a resistivity higher than 18 MΩ cm at 25 °C to create the final volume of ≈20 μL. The NTC sample had ≈2 μL HA gene volume replaced with DI water.
First, we performed basic real-time PCR with different contents of cDNA per μL as shown in Table 1 with NTC at position 1 and different contents at positions 2 to 4. The amplification curves are shown in Fig. 5A. Once the PCR process was completed, we also conducted a MCA (not shown here). The MCA shape is not suitable for performing high-resolution analysis;24 nevertheless, it does show that a specific DNA was amplified with the melting temperature of 83.36 ± 0.63 °C (mean ± standard deviation), in close proximity to the measured value of TM ≈ 81.1 °C. The marginal difference in the TM values is due to the uncertainty of calibration precision of the commercial PCR system used as a benchmark tool. The TM resolution is sufficient to verify specific DNA amplification; however, it may not be suitable for performing high-resolution melting curve analysis.25
| Position | Mean CT | Standard deviation | Concentration HA (ng μL−1) |
|---|---|---|---|
| 1 | — | — | — |
| 2 | 28.7 | 1.5 | ≈5.0 × 10−8 |
| 3 | 27.0 | 1.0 | ≈7.5 × 10−8 |
| 4 | 20.0 | 1.0 | ≈5.0 × 10−6 |
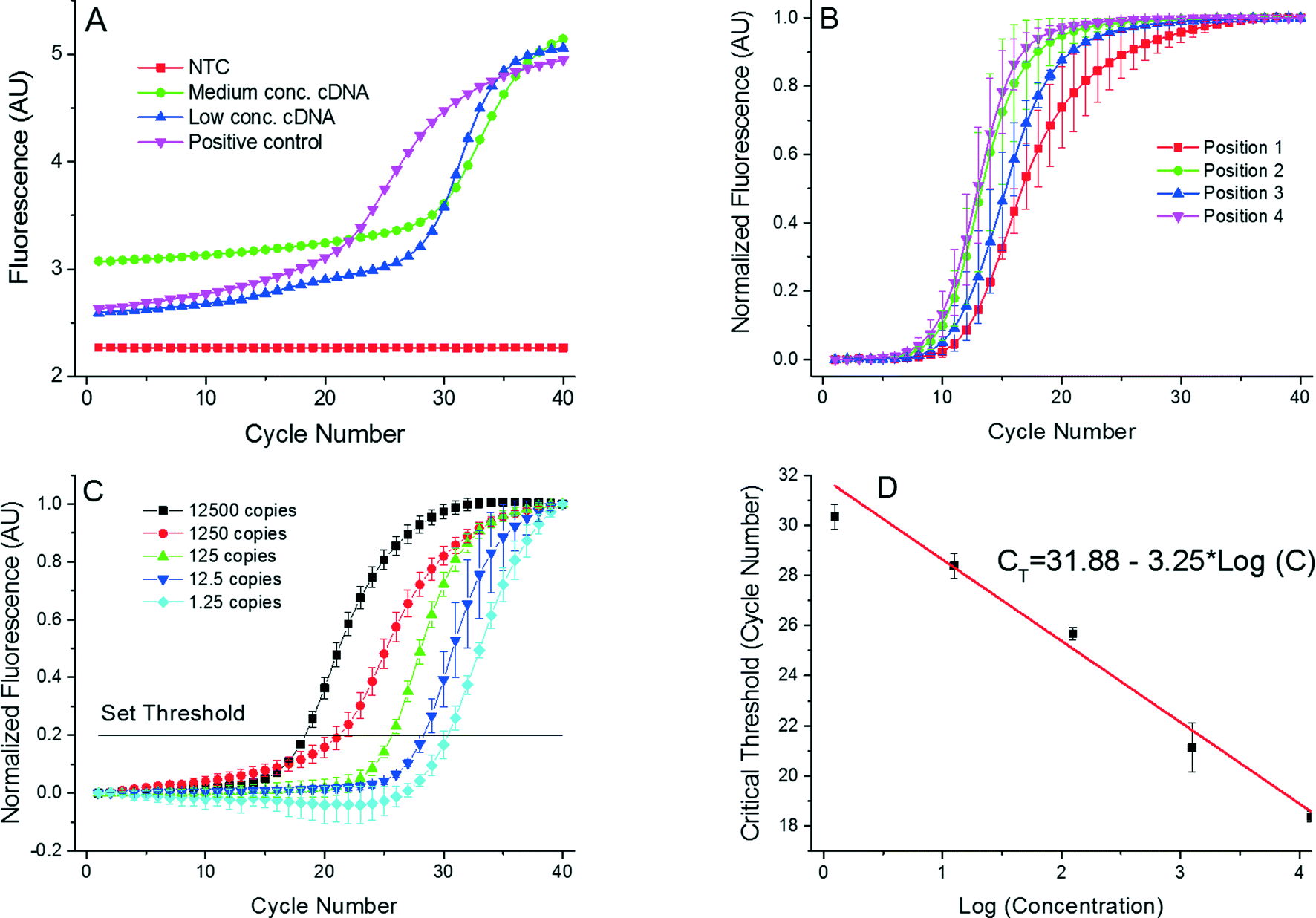 | ||
Fig. 5 (A) Single PCR run as it appears in the PCR display. These PCR amplification curves show results from the 4 positions of the PCR device. We used complementary DNA (cDNA) from the H7N9 HA gene for testing purposes. AU stands for arbitrary units. Once the PCR was completed, the MCAs were performed with a melting temperature value of 83.36 ± 0.63 °C (mean ± standard deviation). Measurement uncertainties emanate from the manual placement of the droplet. Slight droplet misalignments at the heater cause temperature variations between various experimental runs, thereby affecting the overall PCR efficiency. Also, due to these temperature variations, we readily observe a slight shift in the measured melting temperature. (B) Measurements performed at the 4 positions with identical concentration of all samples performed three times to suppress random error. The corresponding extracted critical thresholds (CT) are shown in Table 2. A greater value of CT at position 4 suggests a lower amplification efficiency at that position. This may be caused by a temperature variation due to a non-optimized bonding process of the silicon chip to the PCB. (C) PCR results from position 1 with the calculated number of cDNA copies in the sample from ≈12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 down to ≈1.25. These numbers are calculated by a 10× dilution starting from ≈12500. Since only whole numbers of cDNA copies per sample exist, fractional values imply the statistically most probable value. Each experiment was performed three times. (D) Extracted standard real-time PCR curve from results in Fig. 5C showing the CT value as a function of LOG (cDNA concentration). The slope of −3.02 ± 0.16 cycles at threshold per decade (mean ± standard deviation) corresponds to the PCR efficiency of 0.91 ± 0.05 per cycle (mean ± standard deviation). 500 down to ≈1.25. These numbers are calculated by a 10× dilution starting from ≈12500. Since only whole numbers of cDNA copies per sample exist, fractional values imply the statistically most probable value. Each experiment was performed three times. (D) Extracted standard real-time PCR curve from results in Fig. 5C showing the CT value as a function of LOG (cDNA concentration). The slope of −3.02 ± 0.16 cycles at threshold per decade (mean ± standard deviation) corresponds to the PCR efficiency of 0.91 ± 0.05 per cycle (mean ± standard deviation). | ||
We then performed a series of four identical measurements using an HA content of ≈5 × 10−5 ng in a μL of cDNA (see data in Table 2 and a graphical representation in Fig. 5B). Different PCR locations resulted in performance variation, consequently producing mean CT values in the range of ≈8 to ≈9.6 with a standard deviation ranging between ≈0.8 and ≈1.5. The difference in CT values at positions 2 to 4 might be caused by imperfections due to manual VRC placement. The VRC at position 1 appears to have a lower heat transfer rate than VRCs at other positions. We attribute the variation to a slower transition from amplification to saturation of the DNA amplification curve (Fig. 5B). We further presume that heater defects at position 1 could give rise to a temperature that is different in comparison with the other three positions, consequently leading to a differing PCR efficiency. Additionally, this discrepancy could be attributed to the stress induced during chip-to-PCB soldering, giving rise to bending of the chip. The silicon chip deformations could cause both variations in the intermediate oil layer thickness and a differing heat rate.
| Position | Mean CT | Standard deviation | Concentration HA (ng μL−1) |
|---|---|---|---|
| 1 | 9.6 | 1.5 | ≈5.0 × 10−5 |
| 2 | 9.0 | 1.2 | ≈5.0 × 10−5 |
| 3 | 9.5 | 0.6 | ≈5.0 × 10−5 |
| 4 | 8.0 | 0.8 | ≈5.0 × 10−5 |
Finally, we obtained standard PCR curves. The samples were prepared by the following procedure. We mixed a sample with cDNA corresponding to 12500 copies per 200 nL volume. This sample was diluted 10×, yielding 1250 copies per 200 nL volume; the next dilution yielded 125 copies per 200 nL volume. The last two dilutions had a statistical number of 12.5 copies per 200 nL and 1.25 copy per 200 nL volume, respectively. The PCR results as well as the normalized data are shown in Fig. 5C and D, demonstrating that our portable real-time PCR is able to detect a single DNA copy with an excellent efficiency of 0.91 ± 0.05 per cycle (mean ± standard deviation), which is well within the required range of PCR efficiency between 0.8 and 1.0.
Discussion
The PCR protocol consisted of a ≈10 min hot start at ≈95 °C, followed by 40 cycles of ≈10 s at ≈95 °C, ≈10 s at ≈50 °C and ≈15 s at ≈68 °C. Once the PCR amplification was completed, we conducted an MCA with a scan rate of ≈0.2 °C s−1. This protocol required a total time for amplification of less than 35 minutes with an additional ≈150 s for the MCA. The ultimate speed of the PCR was not the primary target of this work. Nevertheless, the time required for DNA amplification can be shortened by using different types of hot starts, Taqman chemistry (or both)19 or even not using the hot start at all.26Here we used the same heater as in our previous work. The dissipated Joule heat P depends on the square of voltage V: P = V2/R, where R is the heater resistance. The Joule heat dissipation and the consequent heating rate were enhanced with either an increased voltage bias or by lowering the heater resistance. In a previous work,22 we increased the bias voltage up to 20 V using an external power supply. Here, the heater is powered using a 12 V external power supply in either an AC/DC converter or a battery type configuration. A change in this voltage would require additional space for a step up voltage converter. The additional feature would further require either a redesign of the heater on a micromachine, requiring a new mask for the metal lithography level, or the use of a thicker metal layer with a lower sheet resistance. Both cases would require the fabrication of new PCR chip architectures.14 The current PCR chip layout is shown in Fig. S3 (ESI†) and the chip fabrication process in section 5 (ESI†).
In principle, the fundamental limitation in the speed of the device is determined by the heat transfer between the heater and the sample, which is ≈1.5 s for each temperature step. Nevertheless, the device can still run as fast as its predecessor achieving ≈9.5 s per PCR cycle, still being considered as one of the fastest real-time PCRs demonstrated at that point in time.19
Furthermore, we enhance the system robustness by not incorporating moving parts. The system light source consists of 4 LEDs with an estimated lifetime of more than 50000 hours. A single 40 cycle PCR run requires each LED to operate for less than 1 min. The most vulnerable part is the micromachined silicon chip mounted directly on the main PCB. We envision a new version of our system, currently under development, with the brittle silicon chip mounted on a dedicated PCB. Therefore, if the fragile part is damaged, a replacement silicon chip can be easily exchanged. In order to limit interference between PWM pulses and temperature sensing signals, the layout of the micromachined silicon chip will incorporate electrical shielding between integrated heaters and sensors. In this scenario, the chip size will increase to ≈18 mm × 18 mm. Also, this configuration provides additional space in order to further modify the optical housing and facilitate the removal of the PCB from the optical path. The current version of the system exhibited a large self-induced fluorescence due to the PCB being illuminated by the blue LEDs. Finally, we plan to reduce the complexity of the optical housing by reducing the number of parts. This would also allow us to replace the silicon chip once variations in PCR efficiency are discovered.
Furthermore, in future experiments, we plan to use either microscope glass cover slips with upfront lyophilized PCR or a single step RT-PCR master mix. In this configuration, the pipetted ≈200 nL volume sample will be entirely composed of DNA (RNA).
Conclusions
We designed and tested one of the smallest real-time PCR devices. It has a length of ≈100 mm, a width of ≈60 mm and a height of ≈33 mm and weighs only ≈90 g. The device measured 4 PCRs simultaneously in less than ≈35 min, including MCA. The sample was processed in the form of a virtual reaction chamber (VRC) where 200 nL of a sample was placed on a disposable glass cover slip covered with mineral oil to prevent water evaporation from the sample. The sample only interacts with the glass cover slip to eliminate possibilities of sample-to-sample cross-contamination; the glass element was a single-use, disposable system component. Our negative control tests further demonstrate the lack of cross contamination between samples. The system depicted in Fig. 4 was successfully utilized for at least 100 distinct PCR runs. We have demonstrated its performance by amplifying the cDNA of an HA gene of the H7N9 avian influenza virus and displayed the results on an integrated LCD display. We demonstrated the capability of simultaneously running 4 samples at a time with good reproducibility. The PCR efficiency was demonstrated by obtaining a PCR standard curve in the range of 12500 to 1.25 copies with an achieved slope of −3.02 ± 0.16 cycles at threshold per decade (mean ± standard deviation). The value corresponds to a PCR efficiency of 0.91 ± 0.05 per cycle (mean ± standard deviation). The system was also capable of detecting a single DNA copy within the sample.The captured data was subsequently transferred to a personal computer (PC) via a USB interface for further processing. This tiny real-time PCR device is a promising diagnostic system for remote clinics as well as a tool for educational institutions demonstrating the power of a real-time PCR as “seeing is believing”. The system throughput can be doubled using a single channel multiplexing method as demonstrated earlier.27
Acknowledgements
P. Neužil acknowledges partial financial support from the Central European Institute of Technology (CEITEC), grant number CZ.1.05/1.1.00/02.0068. The authors gratefully acknowledge the NIST CNST NanoFab staff for helpful discussions and assistance with device fabrication. This article identifies certain commercial equipment, instruments, and materials to specify the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the equipment, instruments, and materials identified are necessarily the best available for the purpose.References
- K. Mullis, F. Faloona, S. Scharf, R. Saiki, G. Horn and H. Erlich, Cold Spring Harbor Symp. Quant. Biol., 1986, 51(Pt 1), 263–273 CrossRef CAS PubMed.
- R. K. Saiki, D. H. Gelfand, S. Stoffel, S. J. Scharf, R. Higuchi, G. T. Horn, K. B. Mullis and H. A. Erlich, Science, 1988, 239, 487–491 CAS.
- S. Yang and R. E. Rothman, Lancet Infect. Dis., 2004, 4, 337–348 CrossRef CAS PubMed.
- R. Decorte and J. J. Cassiman, J. Med. Genet., 1993, 30, 625–633 CrossRef CAS PubMed.
- M. A. Jobling and P. Gill, Nat. Rev. Genet., 2004, 5, 739–751 CrossRef CAS PubMed.
- M. A. Jobling, A. Pandya and C. TylerSmith, Z. Rechtsmed., 1997, 110, 118–124 CAS.
- S. S. Iqbal, M. W. Mayo, J. G. Bruno, B. V. Bronk, C. A. Batt and J. P. Chambers, Biosens. Bioelectron., 2000, 15, 549–578 CrossRef CAS PubMed.
- M. A. Valasek and J. J. Repa, Adv. Physiol. Educ., 2005, 29, 151–159 CrossRef PubMed.
- P. Belgrader, J. K. Smith, V. W. Weedn and M. A. Northrup, J. Forensic Sci., 1998, 43, 315–319 CAS.
- P. Belgrader, S. Young, B. Yuan, M. Primeau, L. A. Christel, F. Pourahmadi and M. A. Northrup, Anal. Chem., 2001, 73, 391–391 CrossRef CAS.
- N. Agrawal, Y. A. Hassan and V. M. Ugaz, Angew. Chem., Int. Ed., 2007, 46, 4316–4319 CrossRef CAS PubMed.
- P. Neuzil, S. Giselbrecht, K. Laenge, T. J. Huang and A. Manz, Nat. Rev. Drug Discovery, 2012, 11, 620–632 CrossRef PubMed.
- M. U. Kopp, A. J. Mello and A. Manz, Science, 1998, 280, 1046–1048 CrossRef CAS PubMed.
- P. Neuzil, J. Pipper and T. M. Hsieh, Mol. BioSyst., 2006, 2, 292–298 RSC.
- P. Neuzil, T. M. Hsieh and J. Pipper, US Pat., 8216855, 2012 Search PubMed.
- J. Pipper, T. M. Hsieh and P. Neuzil, US Pat., 8124033, 2012 Search PubMed.
- J. Pipper, M. Inoue, L. F. Ng, P. Neuzil, Y. Zhang and L. Novak, Nat. Med., 2007, 13, 1259–1263 CrossRef CAS PubMed.
- P. Neuzil, L. Novak, J. Pipper, S. Lee, L. F. Ng and C. Zhang, Lab Chip, 2010, 10, 2632–2634 RSC.
- P. Neuzil, C. Zhang, J. Pipper, S. Oh and L. Zhuo, Nucleic Acids Res., 2006, 34, e77 CrossRef PubMed.
- L. Novak, P. Neuzil, J. Pipper, Y. Zhang and S. Lee, Lab Chip, 2007, 7, 27–29 RSC.
- P. Neuzil, L. Novak and J. Pipper, Pat. Appl., WO2008024080A, 2008 Search PubMed.
- P. Neuzil, W. Sun, T. Karasek and A. Manz, Appl. Phys. Lett., 2015, 106 Search PubMed.
- V. M. Corman, M. Eickmann, O. Landt, T. Bleicker, S. Brünink, M. Eschbach-Bludau, M. Matrosovich, S. Becker and C. Drosten, Euro Surveill., 2013, 18(16), 20461 CAS . Available online: http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20461.
- G. H. Reed and C. T. Wittwer, Clin. Chem., 2004, 50, 1748–1754 CAS.
- L. Zhou, J. Vandersteen, L. Wang, T. Fuller, M. Taylor, B. Palais and C. T. Wittwer, Tissue Antigens, 2004, 64, 156–164 CrossRef CAS PubMed.
- J. S. Farrar and C. T. Wittwer, Clin. Chem., 2015, 61, 145–153 Search PubMed.
- C. D. Ahrberg and P. Neuzil, Sci. Rep., 2015, 5, 12595 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5lc01415h |
| This journal is © The Royal Society of Chemistry 2016 |