
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The Bull–James assembly as a chiral auxiliary and shift reagent in kinetic resolution of alkyne amines by the CuAAC reaction†‡
William D. G.
Brittain
 ab,
Brette M.
Chapin
ab,
Wenlei
Zhai
ab,
Vincent M.
Lynch
b,
Benjamin R.
Buckley
c,
Eric V.
Anslyn
*b and
John S.
Fossey
*a
ab,
Brette M.
Chapin
ab,
Wenlei
Zhai
ab,
Vincent M.
Lynch
b,
Benjamin R.
Buckley
c,
Eric V.
Anslyn
*b and
John S.
Fossey
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, West Midlands B15 2TT, UK. E-mail: j.s.fossey@bham.ac.uk
bDepartment of Chemistry, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: anslyn@austin.utexas.edu
cDepartment of Chemistry, Loughborough University, Loughborough, Leicestershire, LE11 3TU, UK
First published on 6th September 2016
Abstract
The Bull–James boronic acid assembly is used simultaneously as a chiral auxiliary for kinetic resolution and as a chiral shift reagent for in situ enantiomeric excess (ee) determination by 1H NMR spectroscopy. Chiral terminal alkyne-containing amines, and their corresponding chiral triazoles formed via CuAAC, were probed in situ. Selectivity factors of up to s = 4 were imparted and measured, accurate to within ±3% when compared to chiral GC.
One of the bottlenecks in the analysis of asymmetric reaction outcomes is the enantiomeric excess (ee) determination by HPLC analysis using a chiral stationary phase.2 The development of enantiomer separation conditions, followed by often lengthy elution times means that even a relatively fast reaction can require hours to properly analyse the stereochemical efficiency of the transformation. Therefore, the development of high-throughput screening (HTS) techniques to determine ee is important, especially where hundreds to thousands of reactions need to be analysed swiftly in order to avoid holding up industrial processes.2,3
Nuclear magnetic resonance (NMR) spectroscopy is one method that could be utilised in some cases as a HTS ee determination strategy. Proton NMR spectra can usually be obtained in less than five minutes, whereas HPLC runs often require approximately tens of minutes.4 The increased throughput in combination with circumvention of chiral chromatography method development makes 1H NMR spectroscopy an attractive approach for HTS.5 Determination of enantiopurity has been successfully carried out via NMR spectroscopy previously. One popular strategy is the use of chiral shift reagents, with the most common reagents being relatively expensive lanthanide-based complexes e.g. Eu(hfc)3.6 A weakly paramagnetic chiral lanthanide complex forms an in situ mixture of diastereoisomers whose diastereomeric ratio (dr) is measured to determine the ee of the chiral analyte.6c
Chiral derivatisation of compounds to form distinguishable diastereomers is one strategy for indirect determination of ee via NMR spectroscopy.7 This technique was most notably employed by Mosher (Mosher's acid) for the analysis of enantiomer ratios of chiral alcohols and amines.8 However, this method requires one to sacrifice a small amount of sample in order to carry out the analysis and is therefore less than ideal for HTS applications.9
A boronic acid-based supramolecular assembly for the 1H NMR spectroscopic analysis of the enantiopurity of chiral amines and diols were first reported by Bull, James and co-workers.10 This system takes advantage of the diastereomeric complexes formed from a boronic acid, chiral diol and a chiral primary amine. Using a stereo-defined amine component, the ee of a chiral alcohol could be analysed, and vice versa.10a–e,g,k The commercial availability and relatively inexpensive nature of the Bull–James assembly components gives it an economic edge over lanthanide-based chiral shift reagents.
Following previously published work from some of the co-authors of this report (Brittain, Buckley and Fossey), we wished to tackle the difficult task of kinetic resolution (KR) of terminal alkynes using copper catalysed azide alkyne cycloadditions (CuAACs) (Scheme 1). As there is, to the best of our knowledge, only one reported example of this type of resolution,11 we believed this to be a robust test of the Bull–James sensing assembly in the challenging forum of kinetic resolution, where both the ee of starting material and of product are simultaneously probed. Kinetic resolution takes advantage of the difference in rate of reaction of enantiomers11 and allows for the recovery of enantioenriched starting materials and products. Additionally, it is an asymmetric approach that has been successfully used to obtain many enantioenriched species, including scaffolds with proven biological activity.12 Enantioenriched, unreacted alkynes, as well as product triazoles, can be obtained.13
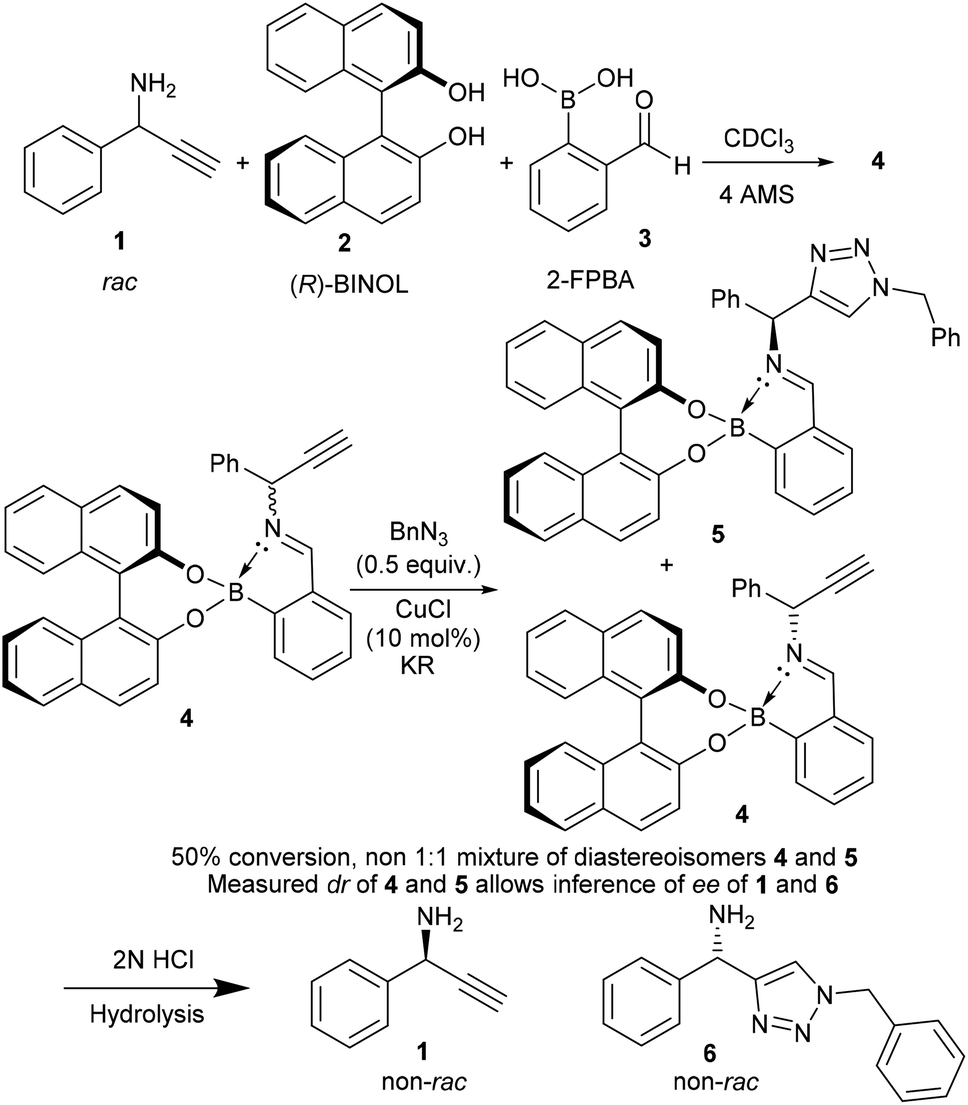 | ||
| Scheme 1 Boronic acid assembly is used as a chiral auxiliary and an in situ ee NMR determination methodology.17 | ||
To test if the assembly was suitable for inference of ee for a primary amine alkyne, 1-phenylprop-2-yn-1-amine 1 was required. The alkyne 1 was selected because it is an important building block for several biologically active compounds such as peptide isosteres,13 analogues of oxotremorine,14 and benzo[b]furan compounds.15 Additionally, the associated triazole has previously been studied for its antimicrobial activity.16 Therefore, kinetic resolution would allow access to compounds and intermediates with previously proven biological activity in an enantioenriched form.
Originally, the Bull–James three-component assembly was selected for use as only a tool to measure the ee of 1 and 6 following exposure of compound 1 to our previously reported resolution conditions, which had successfully resolved racemic quaternary alkyne oxindoles.11 We had hoped to use the Bull–James assembly to measure the ee of 1 and 6 simultaneously and in situ during resolution using our previously developed KR methodology.11 Upon mixing amine 1 with the assembly components (to form 4), we observed signals corresponding to imine diastereomers in the 1H NMR spectra (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 integration). Thus, the imine signals (8.74 ppm and 8.88 ppm) were selected for dr measurement and subsequent ee determination of amine 1. Carrying out the same process with triazole 6 (to form 5) we also observed clear imine signals (8.56 ppm and 9.05 ppm, 1:1 integration) which did not overlap with resonances of 4. The assembly was deemed suitable for the determination of ee for this resolution. Unfortunately, we found that the free base of amine 1 decomposed and was not suitable for resolution using our previously reported system and that research was suspended.11
:1 integration). Thus, the imine signals (8.74 ppm and 8.88 ppm) were selected for dr measurement and subsequent ee determination of amine 1. Carrying out the same process with triazole 6 (to form 5) we also observed clear imine signals (8.56 ppm and 9.05 ppm, 1:1 integration) which did not overlap with resonances of 4. The assembly was deemed suitable for the determination of ee for this resolution. Unfortunately, we found that the free base of amine 1 decomposed and was not suitable for resolution using our previously reported system and that research was suspended.11
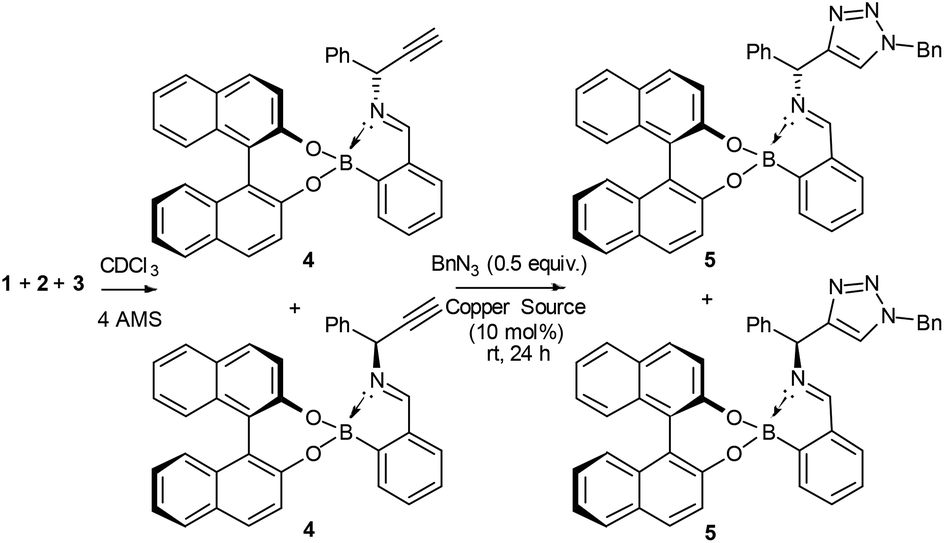
Since the imine in assembly 4 is more stable than the free primary amine 1, we tested if it was possible to form the assembly and then carry out a CuAAC reaction to measure conversion and circumvent the stability issues of 1.
To our surprise, we found that combining 1, 2 and 3 to form the assembly, followed by addition of half an equivalent of benzyl azide and 10 mol% CuCl, led to formation of the triazole assembly 5. Proton NMR spectroscopy revealed that integrations corresponding to diastereomers of both alkyne and triazole imine peaks (in 4 and 5 respectively) were not 1:1 (Fig. 1). Since the reaction was carried out on a 1:1 mixture of diastereomers of the assembly 4, the alkyne of one diastereomeric assembly is consumed more rapidly than the other. Therefore, the Bull–James assembly was acting as a chiral auxiliary for KR, simultaneously with its precedented use as an ee determination tool. In addition, the reversible nature of boron–diol binding means that after hydrolysis all of the remaining alkyne 1 and formed triazole 6 could be recovered, thus giving the approach an advantage over irreversible chiral derivatisations such as Mosher's acid. It should be noted that half an equivalent of benzyl azide was used, in accordance with typical kinetic resolution reactions, in order to avoid full conversion (full conversion would lead to 1:1 diastereomeric assembly 5 and thus racemic triazole 6).
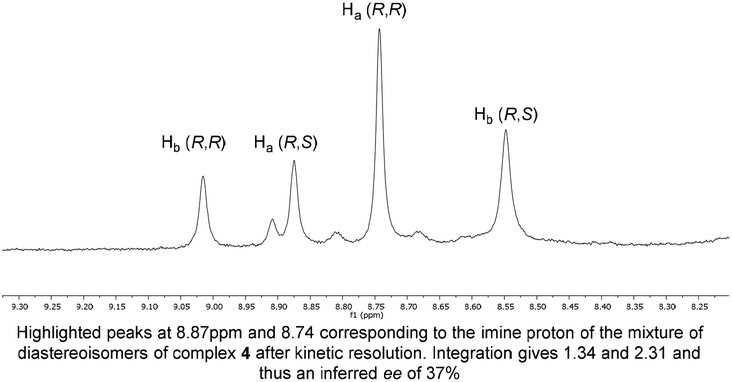 | ||
| Fig. 1 Imine region of NMR following assembly reaction. | ||
In order to confirm that NMR spectroscopic analysis was an accurate measure of assembly dr (and thus ee) during the kinetic resolution process, we first used chiral HPLC as a standard with which to compare the relative NMR peak integrations. Thereby, we would be able to confirm that the measured enantioenrichment of 1 and 6 following kinetic resolution was accurate.
Hydrolysis of the mixture of assemblies 4 and 5 with 2 N HCl and subsequent basic extraction led to the recovery of the amine 1 and triazole 6 with minimal amounts of 2-FPBA and BINOL remaining (Scheme 1). The amine was then protected using benzoyl chloride to give two pairs of HPLC-separable enantiomers (see ESI‡). The HPLC traces showed peaks with the correct retention times for both benzoylated starting material 1 and benzoylated product 6 (see ESI‡ for details). The calculated ee's from HPLC peak integrations were within ±5% ee agreement with the level of enantioenrichment determined by in situ1H NMR spectroscopic integration. As additional evidence of accuracy, it was found that amine 1 was suitable for chiral GC without need of benzoylation. The chiral GC integration values were shown to be within ±3% ee of the NMR analysis method. The agreement of the ee values determined by two methods of chiral chromatography with the 1H NMR integrations of the imine signals of the assemblies allowed us to conclude that the assembly was successfully doubling as a dual-function chiral auxiliary and ee measurement ensemble of both the starting material and product in situ.
After confirming that the three-component assembly was acting as a dual ee determination ensemble and chiral auxiliary, we began to screen reaction conditions in the hope of improving selectivity. First we varied the copper source, and a range of commercially available Cu(II), Cu(I) and Cu(0) sources were tested. Of the copper sources tested, CuCl was confirmed as superior in terms of selectivity (Table 1, entry 1) whilst giving reasonable conversion. CuBr gave slightly increased conversion (34%) but with slightly reduced selectivity (s = 3.7) whilst CuI was very sluggish and required doubling of the reaction time to give a reasonable conversion (39%) with poorer selectivity (s = 2.4) (Table 1, entries 2 and 3). Cu(OTf)·0.5toluene gave decreased selectivity (s = 2.8) (Table 1, entry 4). [Cu(CH3CN)4]PF6 gave increased conversion (50%). However, selectivity was lower (s = 3.0) (Table 1, entry 5). In addition to these a wide range of other copper sources were tested (see ESI‡) however none of them could improve on CuCl.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Copper source | Conv.a (%) | %ee 1b | %ee 6b | s |
| a Conversion was determined by integration of 1H NMR of the assembly comparing the imine proton of the starting material and triazolic product. b ee was determined via comparison of the integration values of the imine region diastereomers in the 1H NMR spectrum of the assembly as de = ee (see ESI). c Reaction was slow, thus reaction time was increased to 48 h. | |||||
| 1 | CuCl | 30 | 23 | 39 | 4.1 |
| 2 | CuBr | 34 | 25 | 48 | 3.7 |
| 3 | CuIc | 39 | 21 | 13 | 2.4 |
| 4 | Cu(OTf)·0.5toluenec | 31 | 18 | 31 | 2.8 |
| 5 | [Cu(CH3CN)4]PF6 | 50 | 36 | 16 | 3.0 |
Next solvent effects were investigated to influence both enantioselectivity and spectra analysis. We found that DMSO-d6 and DMF-d7 led to broad spectra, which were difficult to integrate to give reliable values, even after drying over molecular sieves. To our surprise, we found that acetonitrile-d3 and toluene-d8 both led to precipitation of the assembly. On closer inspection of the acetonitrile case, only one diastereomer precipitated (R,R), which was recrystallised and analysed through XRD to obtain structure 7. We did not include dichloromethane-d2 in our screen due to the possibility of unwanted side reactions (Fig. 2).18 Chloroform-d was thus determined to be the best choice.
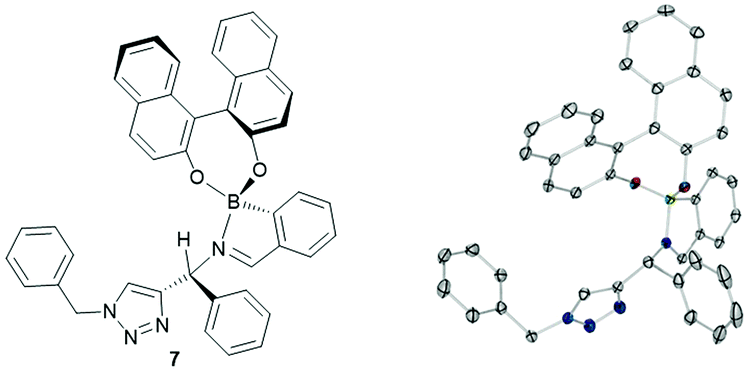 | ||
| Fig. 2 Crystal structure of assembly precipitated from mixture of components in acetonitrile, chirality of the diastereoisomer (R,R) was calculated from the known chirality of the (R)-BINOL. Full details in ESI‡ and CCDC submission. | ||
We next varied the diol component to determine whether this had an effect on the observed enantioselectivity and ee determination capability. However, after testing a range of diols it was confirmed that BINOL was the best choice for this system (see ESI‡). BINOL was found to have the correct solubility and steric bulk to achieve selectivity; bulkier diols tried were found not to be soluble at the concentrations required for NMR analysis and thus were disregarded.
Due to the reversible nature of boronic acid–diol binding, adventitious water led to the appearance of additional peaks in the proton NMR spectrum. We found that rigorous drying of the BINOL and 2-FPBA and the addition of molecular sieves to all solutions led to the minimisation of these side-products.
These preliminary findings show that a chiral boronic acid-based assembly, designed for the analysis of ee of amines via1H NMR spectroscopy, can also be used as a chiral auxiliary. This allows for the catalysis and analysis of the reaction mixture to be carried out by the same auxiliary. It was shown that this assembly can effect, and accurately measure, the enantioenrichment of primary amine alkynes and triazoles in a kinetic resolution reaction. This system allows for rapid and relatively accurate measurement of an on-going asymmetric transformation in situ, thus analysis time is shortened compared with standard chiral chromatography. We anticipate that the concept of using a chiral appendage as both an asymmetric induction unit and as an NMR chiral shift reagent will be applicable to many other asymmetric transformations and we look forward to reporting on these in due course.19
The authors thank Tony D. James and Steven D. Bull for inventing and developing the ee analysis protocol. Without their breakthrough technology none of this work would have been possible. WDGB and WZ are grateful to the University of Birmingham for PhD support. The China Scholarship Council (CSC) are also thanked for providing support (WZ). JSF thanks the University of Birmingham for support, the Royal Society for an Industrial Fellowship (6955) and the EPSRC for funding (EP/J003220/1). WDGB, WZ, BMC, JSF and EVA thank the Royal Society International Joint Project and Research Grant Schemes for underpinning this project. WDGB would like to thank the Royal Society of Chemistry, School of Chemistry at the University of Birmingham and The Society of the Chemical Industry for travel grants. EVA also thanks the Welch Regents Chair (F-0046) and the NIH (GM077437) for support. The Catalysis and Sensing for our Environment (CASE) group is thanked for providing essential networking opportunities.20
Notes and references
- Two co-authors to this manuscript are pleased to be inaugural winners of MSMLG 2016 Czarnik Award (EVA) and MSMLG 2016 Czarnik Emerging Investigator Award (JSF).
- H. H. Jo, C.-Y. Lin and E. V. Anslyn, Acc. Chem. Res., 2014, 47, 2212–2221 CrossRef CAS PubMed.
- H. H. Jo, X. Gao, L. You, E. V. Anslyn and M. J. Krische, Chem. Sci., 2015, 6, 6747–6753 RSC.
- Y. Okamoto and E. Yashima, Angew. Chem., Int. Ed., 1998, 37, 1020–1043 CrossRef.
- (a) C. Dalvit, M. Flocco, S. Knapp, M. Mostardini, R. Perego, B. J. Stockman, M. Veronesi and M. Varasi, J. Am. Chem. Soc., 2002, 124, 7702–7709 CrossRef CAS PubMed; (b) M. T. Reetz, A. Eipper, P. Tielmann and R. Mynott, Adv. Synth. Catal., 2002, 344, 1008–1016 CrossRef CAS; (c) A. Chen and M. J. Shapiro, J. Am. Chem. Soc., 2000, 122, 414–415 CrossRef CAS.
- (a) H. L. Goering, J. N. Eikenberry and G. S. Koermer, J. Am. Chem. Soc., 1971, 93, 5913–5914 CrossRef CAS; (b) J. Kido, Y. Okamoto and H. G. Brittain, J. Org. Chem., 1991, 56, 1412–1415 CrossRef CAS; (c) M. D. McCreary, D. W. Lewis, D. L. Wernick and G. M. Whitesides, J. Am. Chem. Soc., 1974, 96, 1038–1054 CrossRef CAS.
- (a) J. A. Dale and H. S. Mosher, J. Am. Chem. Soc., 1973, 95, 512–519 CrossRef CAS; (b) M. E. Powell, C. D. Evans, S. D. Bull, T. D. James and P. S. Fordred, in Comprehensive Chirality, Elsevier, Amsterdam, 2012, pp. 571–599, DOI:10.1016/B978-0-08-095167-6.00845-4.
- (a) J. A. Dale, D. L. Dull and H. S. Mosher, J. Org. Chem., 1969, 34, 2543–2549 CrossRef CAS; (b) D. L. Dull and H. S. Mosher, J. Am. Chem. Soc., 1967, 89, 4230–4230 CrossRef CAS.
- D. A. Allen, A. E. Tomaso, O. P. Priest, D. F. Hindson and J. L. Hurlburt, J. Chem. Educ., 2008, 85, 698 CrossRef CAS.
- (a) A. M. Kelly, Y. Perez-Fuertes, J. S. Fossey, S. L. Yeste, S. D. Bull and T. D. James, Nat. Protocols, 2008, 3, 215–219 CrossRef CAS PubMed; (b) Y. Perez-Fuertes, A. M. Kelly, J. S. Fossey, M. E. Powell, S. D. Bull and T. D. James, Nat. Protocols, 2008, 3, 210–214 CrossRef CAS PubMed; (c) A. M. Kelly, S. D. Bull and T. D. James, Tetrahedron: Asymmetry, 2008, 19, 489–494 CrossRef CAS; (d) A. M. Kelly, Y. Pérez-Fuertes, S. Arimori, S. D. Bull and T. D. James, Org. Lett., 2006, 8, 1971–1974 CrossRef CAS PubMed; (e) Y. Pérez-Fuertes, A. M. Kelly, A. L. Johnson, S. Arimori, S. D. Bull and T. D. James, Org. Lett., 2006, 8, 609–612 CrossRef PubMed; (f) M. E. Powell, A. M. Kelly, S. D. Bull and T. D. James, Tetrahedron Lett., 2009, 50, 876–879 CrossRef CAS; (g) D. A. Tickell, M. F. Mahon, S. D. Bull and T. D. James, Org. Lett., 2013, 15, 860–863 CrossRef CAS PubMed; (h) J. S. Fossey, E. V. Anslyn, W. D. G. Brittain, S. D. Bull, B. M. Chapin, C. S. L. Duff, C. V. Manville, T. D. James, G. Lees, S. Lim, J. A. C. Lloyd, D. T. Payne and K. A. Roper, J. Chem. Educ., 2016 DOI:10.1021/acs.jchemed.6b00355; (i) P. Metola, E. V. Anslyn, T. D. James and S. D. Bull, Chem. Sci., 2012, 3, 156–161 RSC; (j) G. Mirri, S. D. Bull, P. N. Horton, T. D. James, L. Male and J. H. R. Tucker, J. Am. Chem. Soc., 2010, 132, 8903–8905 CrossRef CAS PubMed; (k) S. L. Yeste, M. E. Powell, S. D. Bull and T. D. James, J. Org. Chem., 2009, 74, 427–430 CrossRef CAS PubMed.
- W. D. Brittain, B. R. Buckley and J. S. Fossey, Chem. Commun., 2015, 51, 17217–17220 RSC.
- (a) G. C. Fu, Acc. Chem. Res., 2004, 37, 542–547 CrossRef CAS PubMed; (b) V. S. Martin, S. S. Woodard, T. Katsuki, Y. Yamada, M. Ikeda and K. B. Sharpless, J. Am. Chem. Soc., 1981, 103, 6237–6240 CrossRef CAS; (c) B. Hu, M. Meng, Z. Wang, W. Du, J. S. Fossey, X. Hu and W.-P. Deng, J. Am. Chem. Soc., 2010, 132, 17041–17044 CrossRef CAS PubMed.
- (a) F. Amblard, J. H. Cho and R. F. Schinazi, Chem. Rev., 2009, 109, 4207–4220 CrossRef CAS PubMed; (b) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed; (c) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed; (d) M. Whiting, J. Muldoon, Y.-C. Lin, S. M. Silverman, W. Lindstrom, A. J. Olson, H. C. Kolb, M. G. Finn, K. B. Sharpless, J. H. Elder and V. V. Fokin, Angew. Chem., Int. Ed., 2006, 45, 1435–1439 CrossRef CAS PubMed; (e) D. K. Scrafton, J. E. Taylor, M. F. Mahon, J. S. Fossey and T. D. James, J. Org. Chem., 2008, 73, 2871–2874 CrossRef CAS PubMed; (f) N. V. Sokolova and V. G. Nenajdenko, RSC Adv., 2013, 3, 16212–16242 RSC; (g) W. D. G. Brittain, B. R. Buckley and J. S. Fossey, ACS Catal., 2016, 6, 3629–3636 CrossRef CAS.
- B. M. Nilsson, H. M. Vargas, B. Ringdahl and U. Hacksell, J. Med. Chem., 1992, 35, 285–294 CrossRef CAS PubMed.
- F. Messina, M. Botta, F. Corelli, M. P. Schneider and F. Fazio, J. Org. Chem., 1999, 64, 3767–3769 CrossRef CAS PubMed.
- D. Castagnolo, F. Dessì, M. Radi and M. Botta, Tetrahedron: Asymmetry, 2007, 18, 1345–1350 CrossRef CAS.
- In this report wedged and dashed bonds are used as follows: (i) wedges and dashes with the same width as either end indicates relative stereochemistry; (ii) wedges and dashes with a thin and thick end indicate absolute stereochemsitry the thin end if the bond is the furthest away.
- Organic azides in pure forms especially with low molecular weights or C:N ratios <2 can spontaneously explode.
- Author contributions: JSF and EVA are the primary investigators and corresponding authors, who led the research across their laboratories jointly. JSF initiated the use of the Bull–James assembly for monitoring ee of starting material and product whilst at the same time using it as a chiral auxiliary. WDGB, BMC and WZ carried out experimental work at both the University of Birmingham (UK) and the University of Texas at Austin (USA). VML solved the crystal structure and helped with preparation of ESI.‡ BRB gave intellectual input and contributed to preparation of the manuscript.
- (a) J. S. Fossey and W. D. G. Brittain, Org. Chem. Front., 2015, 2, 101–105 RSC; (b) D. T. Payne, J. S. Fossey and R. B. P. Elmes, Supramol. Chem., 2016 DOI:10.1080/10610278.2016.1150595.
Footnotes |
| † Dedicated to the 60th Birthday of Tony Czarnik.1 |
| ‡ Electronic supplementary information (ESI) available. CCDC 1477891. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob01623e |
| This journal is © The Royal Society of Chemistry 2016 |