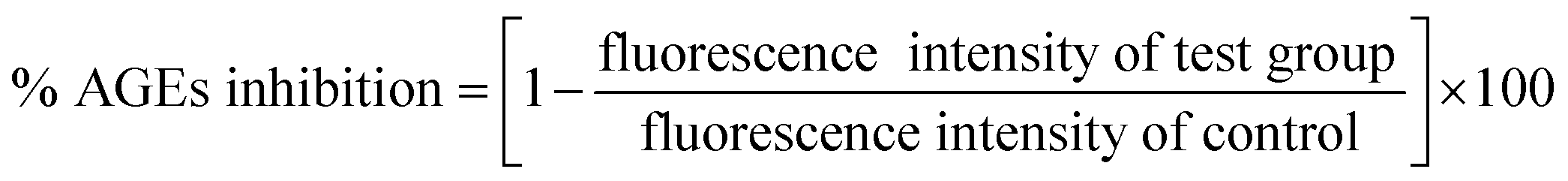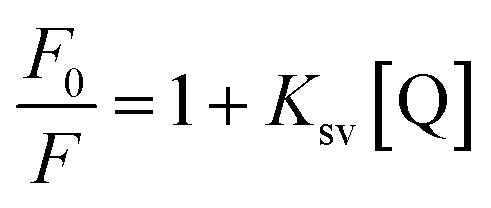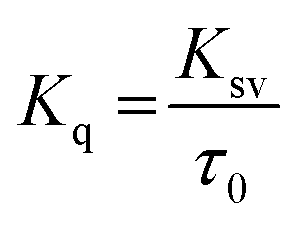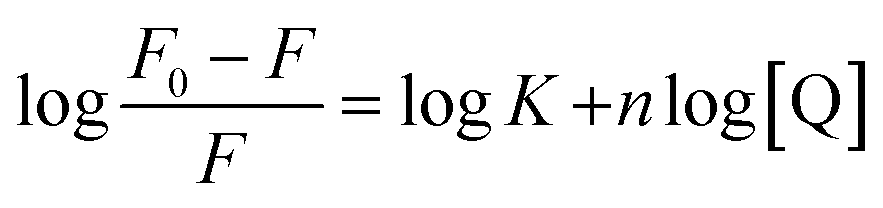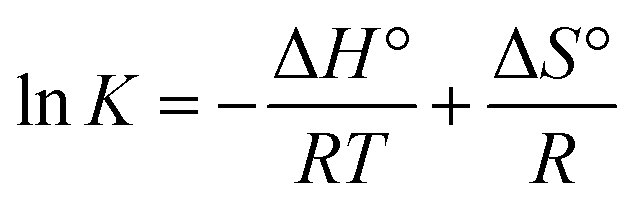DOI:
10.1039/C6RA12321J
(Paper)
RSC Adv., 2016,
6, 65322-65337
Understanding the mechanism of non-enzymatic glycation inhibition by cinnamic acid: an in vitro interaction and molecular modelling study†
Received
12th May 2016
, Accepted 30th June 2016
First published on 11th July 2016
Abstract
Under hyperglycaemic conditions non-enzymatic glycation of proteins gives rise to advanced glycation end products (AGEs). The AGEs thus formed generate free radicals, which foster the development of diabetes and its associated complications. Inhibition of glycation is expected to play a role in controlling diabetes. Plant derived antioxidants like cinnamic acid (CA) are known for limiting AGE formation, however, the mechanism involved is poorly understood. Therefore, we aimed to investigate the possible mechanism of inhibition of AGEs formation by CA through various experimental approaches. Glycation of HSA was achieved by incubating the reaction mixture with glucose for 30 days at 37 °C. The protein samples were tested for levels of free lysine & thiol groups, carbonyl content and reactive oxygen species (ROS). Interaction between CA and HSA was also studied through various biophysical techniques. Thermodynamic studies showed a strong exothermic interaction between CA and HSA. The positive value of TΔS° and negative value of ΔH° indicates that the HSA–CA complex is mainly stabilized by a hydrophobic interaction and hydrogen bond. Further, molecular docking reveals that CA binds to HSA subdomain IIA (Sudlow's site I) with a binding energy of −7.0 kcal mol−1, nearly the same as obtained in isothermal titration calorimetry (ITC) and fluorescence spectroscopy. The results of various spectroscopic techniques along with molecular docking and examination of many biomarkers highlights the role of CA in preventing disease progression.
1. Introduction
The reaction of reducing sugars such as fructose and glucose with proteins, gives rise to Amadori products.1 These further undergo a complex cascade of repeated rearrangements, condensations, oxidative modifications and cause the abnormal cross-linking of proteins to form advanced glycation end products (AGEs).2 The major AGEs in vivo appear to be formed from highly reactive intermediate carbonyl groups, known as oxoaldehydes or -dicarbonyls, including 3-deoxyglucosone, methylglyoxal and glyoxal.3,4 These can accumulate in the body and cause massive damage to the tissues by stimulating reactive oxygen species (ROS) production. AGEs directly stimulated by NADPH oxidase (NOX) after interaction with AGE receptor (RAGE) are present on the cell surface in several tissues or may indirectly increase ROS levels by altering antioxidant proteins. Of note, the expression of both NOX and antioxidant enzymes might also be affected by AGEs and thereby trigger age-related disorders and even diabetic complications such as neuropathy, nephropathy and retinopathy.5
Diabetes is a heterogeneous metabolic disorder characterized by hyperglycaemia and altered carbohydrate, fat and protein metabolism.6 The mechanism of diabetes in humans is subjected to wide scrutiny on a physiological, molecular and genetics basis. Despite advances in understanding the disorder, mortality and the morbidity due to this disease is still high. One mechanism increasingly considered as a fundamental cause of diabetic tissue damage is glycation (non-enzymatic glycosylation).7 Although many drugs and interventions are available to manage diabetes, they are increasingly expensive and have inherent adverse effects. As such, there is a growing interest among researchers to use natural products with combined anti-glycation and antioxidant properties to prevent tissue damage. A primary approach has been the use of plant derived products to control disease progression. Various plant herbal medicine and phytocompounds have shown antidiabetic activities in vitro and in vivo. Due to their safety, and lesser side effects a wide variety of plant products have been explored as therapeutic intervention options.8
It is well established that dietary micronutrients contain bioactive compounds, that inhibit AGEs formation in vitro and in vivo.9,10 One such component is cinnamic acid. It is derived from cinnamon which has been used in several cultures as a traditional medicine. Many studies have shown antioxidant and antidiabetic properties of cinnamic acid and its derivatives.11,12 It has been reported to modulate gluconeogenesis, glycogenesis and accelerate the insulin sensitivity in diabetic rats.13,14 However the mechanism involved in still unclear. In our previous study, our group demonstrated the inhibitory effect of plant flavonoid, quercetin in the formation of AGEs.15 To the best of our knowledge, the mechanism of antiglycation of cinnamic acid is not yet fully explored. Therefore, the aim of this study, was to determine the inhibitory effect of cinnamic acid against glycation using human serum albumin (HSA) as a protein model and to understand its possible mode of interference through in vitro interaction studies and molecular modelling.
2. Material and methods
2.1. Materials
Fatty acid-free human serum albumin (HSA) (A3782), TNBSA, ibuprofen and gluconolactone (G2164) were purchased from Sigma-Aldrich, Chemical Company, USA. Cinnamic acid extrapure (034812), aminoguanidine (AG), DMSO, glucose warfarin and DTNB were obtained from SRL chemicals (India). All other chemicals and reagents used were of analytical grade.
2.2. Human serum albumin (HSA) in vitro glycation assay
To determine the antiglycation activity of test compound, the method by Alam et al. was adopted with minor modifications.15 Human serum albumin (300 μM) was incubated with glucose (165 mM) in 10 mM PBS (pH 7.4) containing 0.02% NaN3 to avoid microbial contamination, at 37 °C for 30 days. Aminoguanidine (10 mM) incubated with HSA (300 μM) in presence of glucose (165 mM) was taken as positive control. HSA was incubated with glucose in the same concentration as mentioned above in the presence of varying concentration of cinnamic acid (25, 50, 100, 200 and 500 μM) for test group. On completion of incubation, all samples were dialyzed overnight against PBS to remove excess amount of glucose. All samples were stored at −20 °C for further examination and protein concentration in each sample were estimated by Lowry method.16
2.2.1. AGEs-fluorescence measurements. Advanced glycation end products (AGEs) show strong fluorescence emission when excited between 300 and 400 nm. The formation of AGEs was detected by exciting all the samples at 322, 335, 365 and 375 nm.17 All samples were diluted to 3 μM in 10 mM PBS (pH 7.4). The emission spectra were recorded in the range of 300–600 nm using fluorescence spectrophotometer (RF-5301PC, Shimadzu, Japan).
2.2.2. ANS fluorescence. Each protein sample was diluted to 3 μM in PBS (10 mM). Briefly, 5 μL ANS (8-anilino-1-naphthalenesulfonic acid) from 5 mM stock solution was mixed with of 2.995 mL diluted samples just before scanning. All the samples were excited at 385 nm and fluorescence emission spectra were recorded in the range 400–600 nm at 298 K.18 The increase in ANS fluorescence intensity indicates the extent of exposure of hydrophobic domains of HSA.
2.2.3. Methylglyoxal–HSA reactivity. Methylglyoxal–HSA reactivity assay was performed according to Lee et al. (1998) with minor modifications.19 HSA (50 mg mL−1) was incubated with 40 mM methyl-glyoxal (MG) in absence and presence of 25, 50, 100 and 200 μM cinnamic acid. Aminoguanidine (10 mM) was taken as positive control. All the sample were prepared in 10 mM sodium phosphate buffer and incubated for 14 days at 37 °C. The percent inhibition in AGEs formation was calculate using formula given below:| |
 | (1) |
2.2.4. Thiol and free lysine group estimation. The quantitative estimation of free thiol (–SH) group was performed by Ellman's method.20 Briefly, 50 μL DTNB (5,5′-dithiobis, 2-nitrobenzoic acid) (3 mM) and 150 μL of EDTA (0.1 M) was added to 50 μL of each sample in 100 mM Tris–HCl (pH 8.0) making a final volume of 1.5 mL. Double distilled water was used in place of sample for control. Absorbance of each sample was recorded at 412 nm after incubation for 20 min at 37 °C and the free thiol group was calculated using the standard curve of L-cysteine.The amount of free amino groups in all samples were estimated by using 2,4,6-trinitrobenzene sulfonic acid (TNBSA).21 Briefly, all protein samples were diluted to 0.2 mg mL−1 in 0.1 M sodium bicarbonate buffer (pH 8.5). To all diluted samples (0.5 mL), 0.25 Ml of the 0.01% (w/v) TNBSA solution was added and then the reaction mixture was incubated for 2 h at 37 °C. After incubation, 0.25 mL SDS (10%) and 0.125 mL of 1 N HCl was added to each sample. For blank, distilled was added instead of protein. The absorbance of each sample was recorded at 335 nm against blank.
2.2.5. Carbonyl content estimation. Total carbonyl content in the HSA was estimated to study the level of protein oxidation. Carbonyl content in all treated and control samples were examined by Levine method.22 The absorbance was noted at 360 nm and the amount of carbonyl content was calculated using molar absorption coefficient of 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1. All the results are expressed in nmol per mg protein.
000 M−1 cm−1. All the results are expressed in nmol per mg protein.
2.2.6. Determination of secondary structure by circular dichroism. Circular dichroism spectra of all the samples were recorded by using JASCO spectropolarimeter (J-815). All the spectral measurements were done at 25 °C by a thermostatically controlled cell holder attached to Neslab's RTE 110 water bath having a temperature accuracy of ±0.1 °C. To study the changes in the secondary structure of HSA, far-UV CD analysis was evaluated by diluting each protein sample to 0.3 mg mL−1 in 10 mM PBS (pH 7.4) and 1 mm path length quartz cuvettes.
2.2.7. Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE). To examine the change or damages caused by AGEs, native HSA and glycated-HSA were analyzed by SDS-PAGE on 8% polyacrylamide gel, as described previously.23 Fifteen microgram of each samples were loaded into the wells. Electrophoresis was performed at 80 volts for 4 h at room temperature. Protein was stained with Coomassie Brilliant Blue (CBBR-250) for 60 min followed by overnight destaining.
2.2.8. Reactive oxygen species (ROS) generation assay (NBT assay). Generation of the superoxide anion was measured by the nitrobluetetrazolium (NBT) assay with slight modification.24 Briefly, 300 μL sodium phosphate buffer (100 mM), 100 μL NBT (1 mM), 300 μL EDTA (1 mM), 300 μL Triton-X-100 (0.06%) and 50 μL of each native HSA, glycated HSA and treated HSA (25–200 μM cinnamic acid) was added and the final volume was made to 3 mL using double distilled water. Aminoguanidine was taken as positive control and 50 μL of double distilled water was used in place of sample for blank. The absorbance of all the samples was recorded at 560 nm at the interval of 30 min for 3 hours.
2.3. Interaction studies
2.3.1. Sample preparation. Phosphate Buffer Saline (PBS) 10 mM of pH 7.4 was prepared in double distilled water. The buffer was then filtered through a 0.22 mm syringe filter. HSA was dissolved in 1 mL PBS to make 300 μM stock solution and it was diluted in same buffer for further use. For the 3 mM cinnamic acid stock, 4.44 mg of cinnamic acid was dissolved in double distilled water containing 1% ethanol and volume was adjusted to 10 mL.
2.3.2. UV-visible spectroscopic study. The UV spectra of HSA and HSA–cinnamic acid complex were measured using Shimadzu 1800 UV-vis spectrophotometer and spectra were recorded in the range of 200 to 600 nm. A fixed concentration of HSA (3 μM) was titrated in absence and presence of cinnamic acid (3–30 μM). Dilution from the stock solution to working concentration was made in 10 mM PBS (pH 7.4). The final volume in cuvette was maintained to 3 mL and base line correction was done with the same buffer.
2.3.3. Steady state fluorescence. All the fluorescence studies were performed on Spectrofluorophotometer RF-5301PC, Shimadzu Scientific Instruments, Japan. Briefly, HSA (3 μM) was excited at 280 nm and the fluorescence emission spectra was recorded in the range of 285–600 nm. The change in the fluorescence intensity of the HSA was monitored with increasing concentration of cinnamic acid (3–30 μM) at three different temperatures (298 K, 303 K, and 310 K). The final volume of the reaction mixture was made to 3 mL by adding 10 mM PBS.
2.3.4. Synchronous fluorescence measurement. To measure synchronous fluorescence, spectrofluorometer cell containing 3 mL of 3 μM HSA was titrated with increasing concentration of cinnamic acid (0–30 μM) at 298 K. The excitation wavelength was set at 240 nm and emission spectra was recorded 255–600 nm, thereby, maintaining Δλ = 15 nm for tyrosine residue. Similarly, for tryptophan residue, Δλ = 60 nm was kept fixed with excitation wavelength at 240 nm and emission from 300–600 nm.
2.3.5. Competitive displacement assay. For site specific binding experiment, HSA (3 μM) was first saturated with site specific markers (ibuprofen and warfarin), in which the ratio of HSA to the site marker was 1:2. The HSA-site marker complex was excited at 280 nm and emission spectra was recorded from 285 nm to 600 nm both in absence and presence of cinnamic acid (3–30 μM).
2.3.6. Effects of ionic strength. This experiment was performed to check presence of any ionic interaction between HSA and cinnamic acid. Briefly, HSA (3 μM) was saturated with cinnamic acid (6 μM) and the final volume was made to 3 mL in 10 mM PBS. This HSA–cinnamic acid complex was titrated with increasing concentration of NaCl (10–100 mM). The excitation wavelength was set to 280 nm and emission spectra were record in the range of 285 nm to 600 nm.
2.3.7. Isothermal titration calorimetric measurements (ITC). The thermodynamics of HSA and cinnamic interaction was measured using a VP-ITC titration microcalorimeter (MicroCal Inc., Northampton, MA). HSA, cinnamic acid and the reference buffer were degassed in a thermovac for 15 min prior to their loading. The 20 μM HSA and 10 mM PBS was loaded into the sample and reference cell, respectively. 29 successive injections of cinnamic acid (1.5 mM) of 10 μL each were titrated to sample cell containing 20 μM HSA with an initial delay of 60 s. Time duration for each injection was set to 20 s and spacing between two consecutive injections was kept at 180 s. The stirring speed and the reference power were fixed at 307 rpm and 16 μcal s−1, respectively.
2.3.8. Molecular docking. In order to perform molecular docking, AutoDock-vina program was deployed since it does more accurate calculations than Autodock software and is reported to perform faster.25,26 To perform molecular modelling measurements, the three dimensional crystal structure of HSA was downloaded from RCSB Protein Data Bank [PDB: 1AO6]. To avoid hindrance while docking, all water molecules were removed. Kollman charges were added to HSA after merging all the non-polar hydrogen atoms and the coordinate file was converted into PDBQT format using MGL Tools-1.5.6.27 The size of the grid was set to 52 × 48 × 48 Å with maximum spacing (1 Å) to cover all the active site residues having centre of the grid at x = −22.717, y = −33.725, z = 38.864. The 3D structure of cinnamic acid [CID: 444539] was downloaded from https://pubchem.ncbi.nlm.nih.gov in SDF format which was converted to pdb format using Chimera 1.10.2. All other docking parameters were kept as default and post modelling analysis was done using Accelrys Discovery Studio 4.5.
3. Results and discussion
3.1. Antiglycation activity of cinnamic acid
3.1.1. UV-visible spectral analysis for glycation. All the samples were analysed by UV-visible absorption spectra for the structural change in HSA after incubation. Native HSA exhibited a characteristic peak at 280 nm (ESI Fig. S1†). Glycated sample showed maximum hyperchromicity, up to 3 times the absorbance of native samples. This increase in absorbance is thought to result from unfolding of protein due to glycation. This can affect protein function. Samples incubated in the presence of CA exhibited reduced absorption compared to glycated HSA. The change in native conformation of HSA results in loss of its function which is main carrier protein in human blood. Treatment of varying concentrations of cinnamic acid protected HSA from unfolding and thereby, retaining its function.
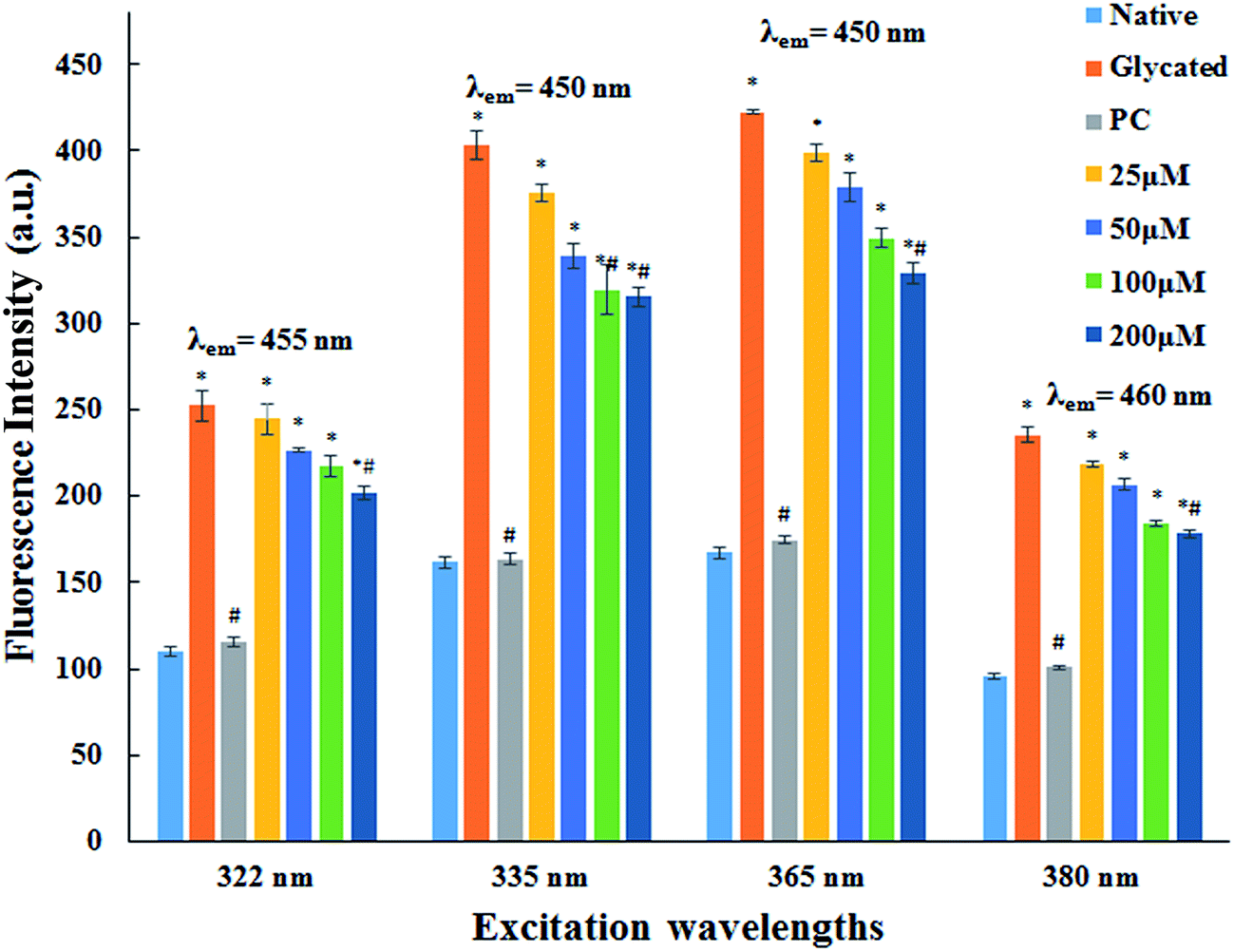
3.1.2. AGE fluorescence examination. AGEs are one of the major factors responsible for pathogenicity of several age related problems cardiovascular complications of diabetes mellitus. AGEs specific fluorescence is commonly employed for their detection.28,29 The fluorescence emission spectra of native HSA and HSA incubated with cinnamic acid is shown in Fig. 1 at specific excitations (322 nm, 335 nm, 365 nm and 380 nm) for the determination of fluorescent AGEs. The intensity of fluorescence decreased with increasing concentration of cinnamic acid indicating that CA prevents protein glycation. This result illustrates a dose dependent inhibitory effect of cinnamic acid in the formation of AGEs.
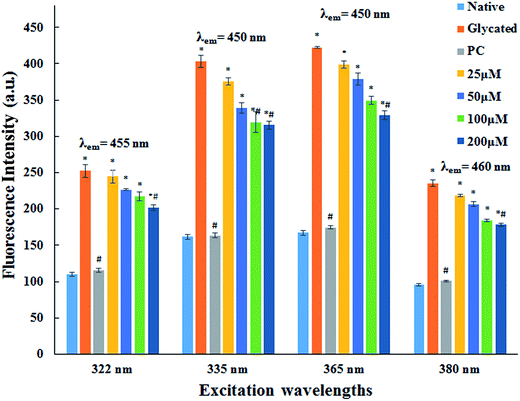 |
| | Fig. 1 Fluorescence emission spectra of native HSA, glycated HSA, HSA with aminoguanidine (PC) and HSA with different concentrations of cinnamic acid. All the data have been expressed in mean ± SEM for three independent experiments. | |
3.1.3. ANS fluorescence. ANS (1-anilinonaphthalene-8-sulfonate) is a fluorescent molecular probe, widely used for the detections of hydrophobic domains of proteins.30 ANS shows very strong fluorescence when bound to protein after excited at 385 nm while it is non-fluorescent when present in free form in the solution.31 Glycated sample showed more than two-fold increase in the ANS fluorescence emission signal as compared to native HSA. This is either due to the exposure of hydrophobic domains of HSA upon glycation or due to the formation of Amadori products.32 Results showed that dose dependant treatment with cinnamic acid decreased ANS fluorescence intensity as shown in Fig. 2. Treatment with 25, 50, 100 and 200 μM of cinnamic acid decreased fluorescence intensity by 3.7%, 6.5%, 12.2% and 17.4% respectively, as compared to glycated sample. This result demonstrates the burial of hydrophobic domains of HSA with the treatment of cinnamic acid that shows the inclination towards native conformation. The reduction in ANS fluorescence intensity with increasing concentration of cinnamic acid also resulted in increase in surface hydrophobicity of HSA.
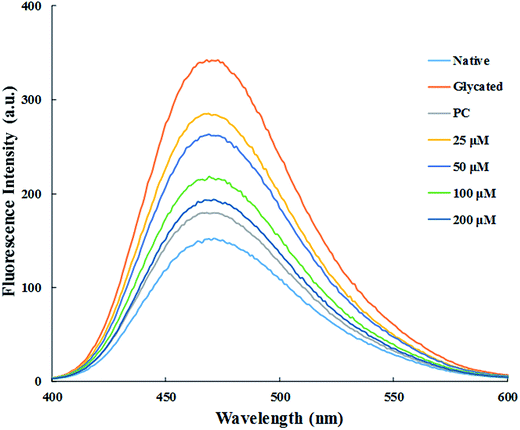 |
| | Fig. 2 Fluorescence spectra of ANS binding to HSA excited at 380 nm (PC is aminoguanidine treated). All the data have been expressed in mean ± SEM for three independent experiments. | |
3.1.4. Methylglyoxal–HSA reactivity assay. Methylglyoxal (MG) is a metabolic intermediate of glucose metabolism which is formed through polyol pathway. In hyperglycaemic conditions, there is increased flux of glucose that leads to the formation and accumulation of AGEs.33 MG is one of the most reactive α-ketoaldehydes formed through glycolytic pathway in vivo. This causes the modification of arginine residues of serum proteins leading to the formation of fluorescent AGEs (mainly argpyrimidine).34 This assay was performed for the quantitative evaluation of inhibition in AGEs formation. The result obtained (Fig. 3) shows that there is dose dependent increase in AGEs inhibition with varying concentration of cinnamic acid. There was 7.6%, 14.1%, 19.4% and 23.7% inhibition in AGEs formation upon treated with 25, 50, 100 and 200 μM cinnamic acid (Fig. 3). Aminoguanidine resulted in 34.3% AGEs inhibition of MG-mediated HSA-glycation. The reduction in AGEs formation by cinnamic acid is due to the antioxidant capacity and ability to scavenge reactive carbonyl species.
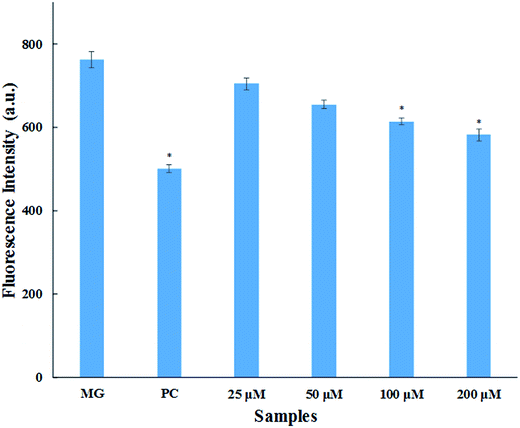 |
| | Fig. 3 MG–HSA assay. Inhibitory effect of cinnamic acid on middle stage of HSA glycation (MG is methylglyoxal as a control; PC is aminoguanidine (10 mM) as a positive control). All the data have been expressed in mean ± SEM for three independent experiments. * indicates significantly different from MG at p ≤ 0.05. | |
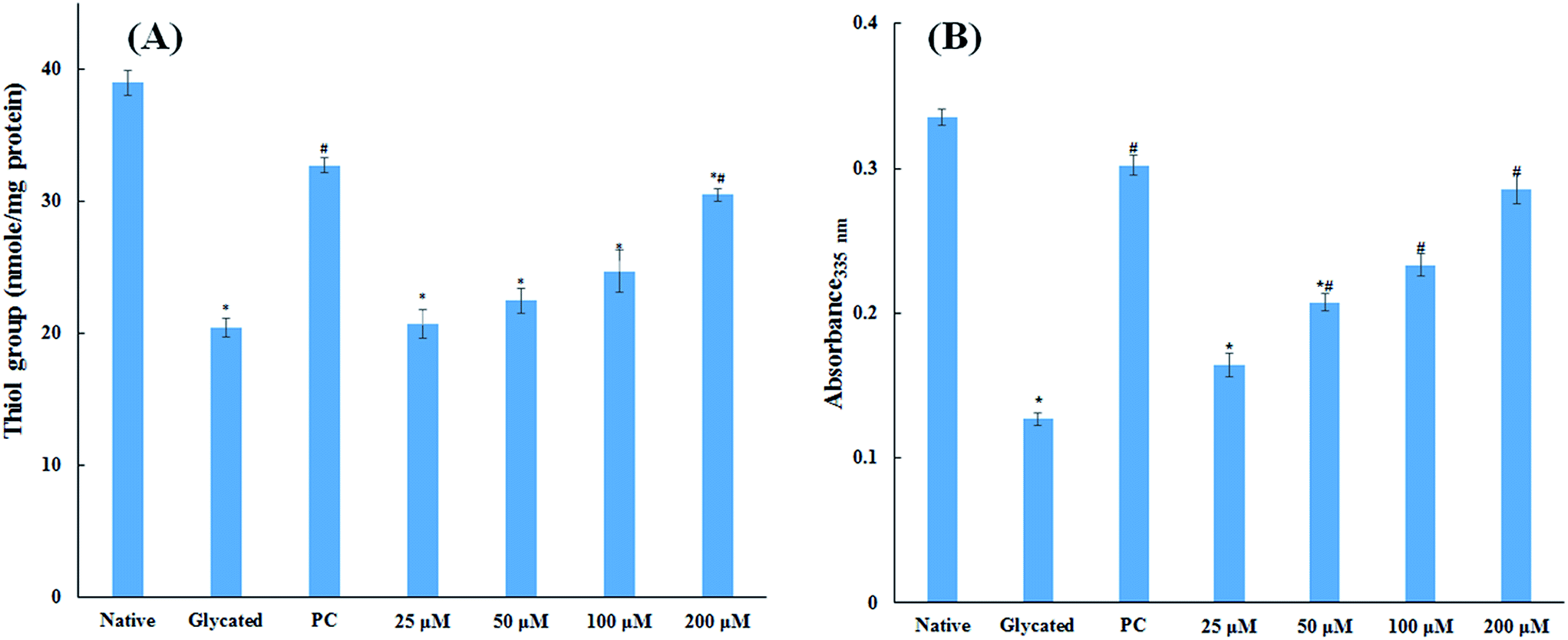
3.1.5. Thiol and free lysine group measurements. The amount of free sulfhydryl group is a direct parameter to analyse the redox status of a system. The oxidative modification in protein samples was studied by estimating the free sulfhydryl group content. It is evident from Fig. 4A that there was a remarkable decrease (47.5%) in free thiol group in glycated protein sample as compared to native HSA. Addition of 25, 50, 100 and 200 μM cinnamic acid to HSA showed 46.9%, 42.4% 36.6% and 21.8% decrease in free thiol group content. The measurement of thiol group in HSA is significant to evaluate the generation of free radical as the degradation of Amadori products may generate free radicals by oxidizing the proteins.20 It is evident from our findings that addition of cinnamic acid to a HSA significantly dampens protein oxidation even in presence of glucose.
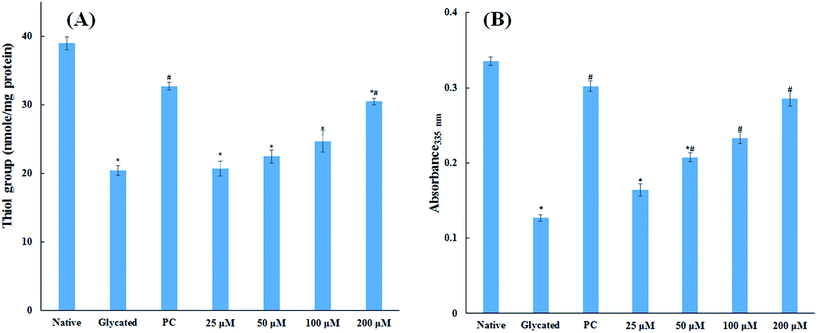 |
| | Fig. 4 Thiol group and free lysine estimation. Concentration free thiol group in native HSA, glycated HSA and HSA treated with various concentration of cinnamic acid (A). Effect of cinnamic acid on free ε-NH2 group of lysine determined by TNBSA assay on native HSA, glycated HSA and HSA treated with various concentration of cinnamic acid (B) (PC is aminoguanidine treated HSA). All the data have been expressed in mean ± SEM for three independent experiments. * indicates significantly different from control at p ≤ 0.05. # indicates significantly different from group B at p ≤ 0.05. | |
The major site for non-enzymatic glycation of proteins is free amino group present in lysine, but arginine, cysteine and histidine have also found to be involved in this phenomenon.23 In this process, free ε-NH2 group of arginine and lysine react with carbonyl group of various sugars to form Amadori product and finally leading to the formation of heterogeneous class of advanced glycated end products such as carboxymethyllysine (CML), vesperlysine (VESP) and carboxyethyllysine (CEL) etc.35 One of the most common mechanism involved in antiglycation process is the masking of lysine and arginine residues by small molecule inhibitors. It is clear from Fig. 4B that glycation of HSA with glucose for 28 days lead to 58.75% decrease in free amino group of lysine. The treatment of 25, 50, 100 and 200 μM of cinnamic acid to HSA showed 29.1%, 63.2%, 83.7% and 124.9% increment in availability of free ε-NH2 group of lysine as compared to glycated HSA. This result reveals that cinnamic acid has potent antiglycation activity which might be due to the masking of free amino groups in HSA.
3.1.6. Carbonyl content estimation. Protein glycation via Schiff base leads to the formation of stable ketoamines called Amadori products. These can undergo enediol reaction, resulting in the formation of carbonylated protein.36 These ketoamines are further converted into superoxide radical causing oxidative and cellular damage.37 Carbonyl content of protein is well known biomarker of cellular oxidative stress. The level of carbonyl content in glycated sample was found to be more than three fold as compared to the native HSA sample as shown in Fig. 5. While the samples treated with 25, 50, 100 and 200 μM cinnamic acid resulted in 26.7%, 46.5%, 55.1% and 58.2% decrease in carbonyl content compared to glycated sample respectively. The result verified the free carbonyl group trapping ability of cinnamic acid that was apparent from the reduced carbonyl content detected in the presence of cinnamic acid.
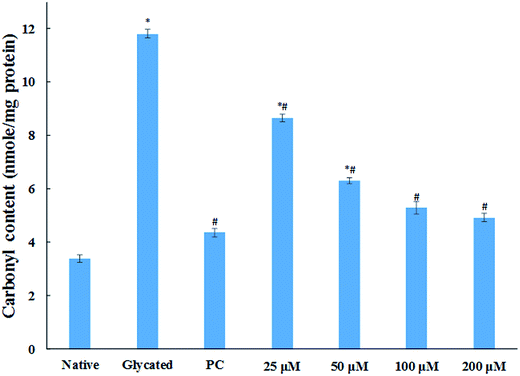 |
| | Fig. 5 Carbonyl content of native, glycated and cinnamic acid treated HSA (PC is aminoguanidine treated). All the data have been expressed in mean ± SEM for three independent experiments. * indicates significantly different from native at p ≤ 0.05. # indicates significantly different from glycated HSA at p ≤ 0.05. | |
3.1.7. Time dependent ROS generation. NBT assay was performed to detect the generation of reactive oxygen species. Fig. 6 demonstrates the generation of ROS which was found to be maximum in glycated sample. While, native HSA did not exhibit any significant ROS production. The generation of superoxide anion decreased remarkably in concentration dependent fashion by treatment with cinnamic acid. It is clear from the Fig. 6 that the ROS quenching ability of cinnamic acid which might be due to its antioxidant property. These results suggest that formation of AGE's which led to the development highly reactive species that might be responsible for the degradation of HSA.
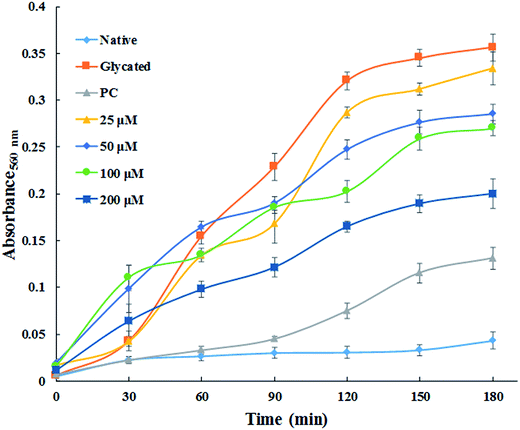 |
| | Fig. 6 Time dependent generation of reactive oxygen species from different samples by NBT assay. All the data have been expressed in mean ± SEM for three independent experiments. | |
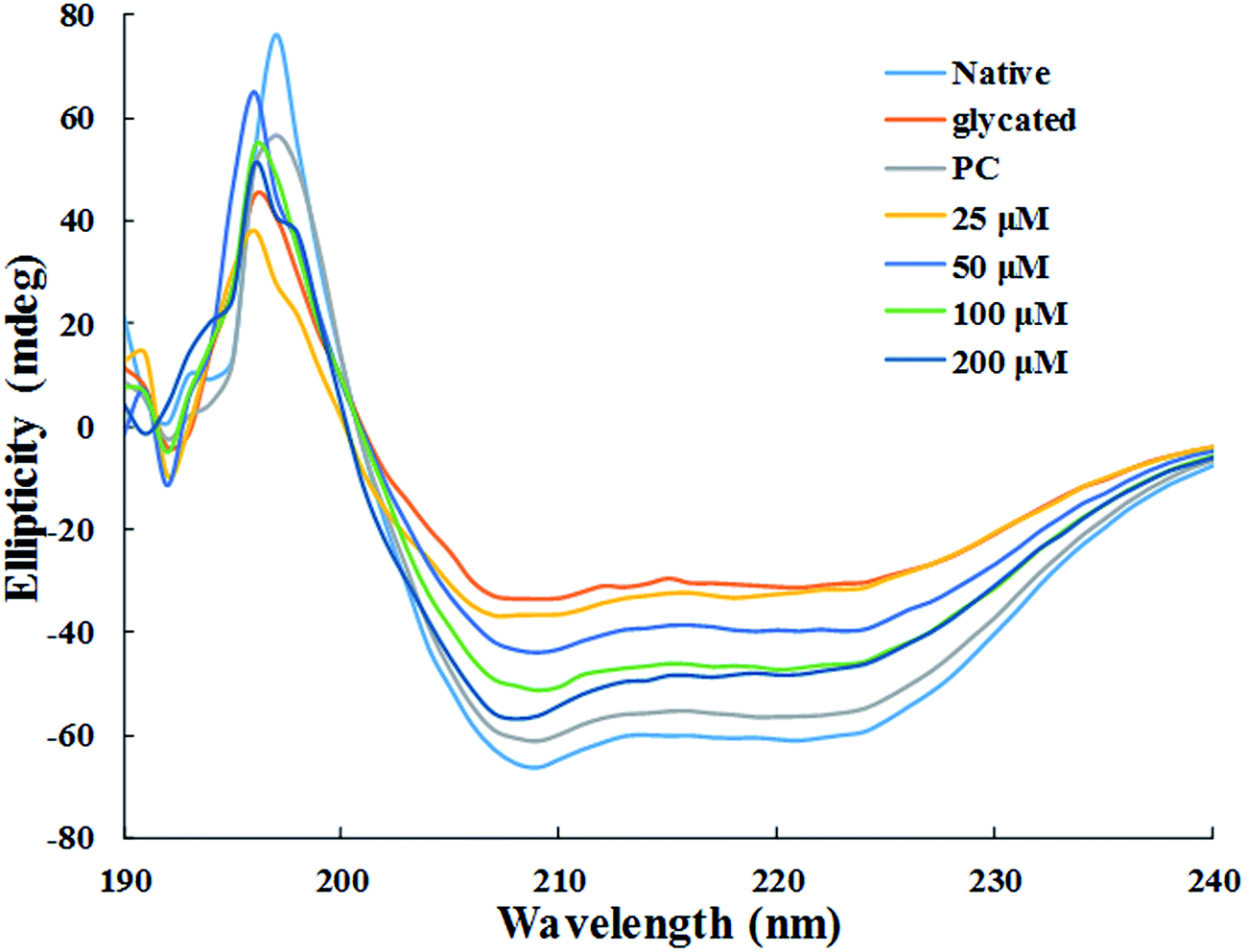
3.1.8. Circular dichroism measurements. The changes secondary structure of HSA was analysed by far-UV CD spectroscopy of each samples. The CD spectra for all the samples showed 2 negative bands, at 208 nm and 222 nm, and a positive band at 190 nm respectively (Fig. 7). DICHROWEB was used for the quantitative estimation of the secondary structural components in all the sample and details is enlisted in Table 1.38 Glycation of HSA resulted in 42.7% loss in regular α-helix as compared to native HSA. It is evident from the data presented in Table 1 that the loss in secondary structures of glycated HSA was accompanied by unordered or random coil conformation. In HSA samples treated with 25 μM, 50 μM, 100 μM and 200 μM of cinnamic acid, the regular α-helix was found to be 16.1%, 22.6%, 25.5% and 26.3% respectively. This indicates that HSA got more inclined to its native or defined secondary structure after treatment with cinnamic acid which evidently prevented glycation to a remarkable extent.
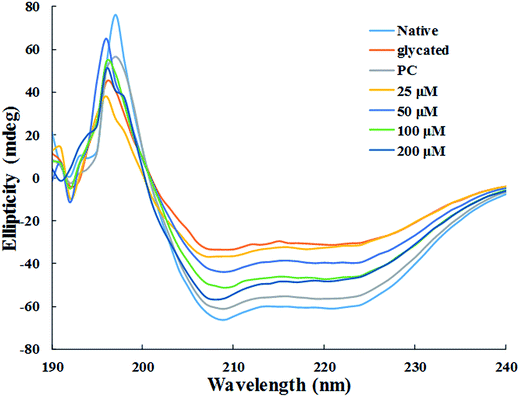 |
| | Fig. 7 Far UV-CD spectra of native, glycated and cinnamic acid treated HSA at various concentrations. All the data have been expressed in mean ± SEM for three independent experiments. | |
Table 1 Secondary structure contents of native HSA, glycated HSA and treated HSA estimated from the CD spectra (Fig. 7) using DICHROWEB (http://dichroweb.cryst.bbk.ac.uk)a
| Samples |
Helix 1 (%) |
Helix 2 (%) |
Strand 1 (%) |
Strand 2 (%) |
Turns (%) |
Unordered (%) |
| Helix 1, Helix 2, Strand 1 and Strand 2 indicate a regular α-helix, distorted α-helix, regular β-strand and distorted β-strand, respectively. |
| Native |
30.2 |
24.3 |
07.1 |
0.90 |
12.9 |
24.5 |
| Glycated |
17.3 |
18.5 |
09.2 |
02.0 |
17.0 |
36.0 |
| Aminoguanidine |
28.8 |
25.4 |
04.9 |
0.40 |
14.0 |
26.4 |
| CA (200 μM) |
26.3 |
27.8 |
00.6 |
0.60 |
17.9 |
26.8 |
| CA (100 μM) |
25.5 |
25.7 |
01.8 |
0.20 |
16.2 |
30.6 |
| CA (50 μM) |
22.6 |
25.4 |
03.8 |
0.9 |
16.9 |
30.4 |
| CA (25 μM) |
16.1 |
18.1 |
08.4 |
02.8 |
19.4 |
35.2 |
3.1.9. Electrophoresis (SDS-PAGE). The migration pattern of native and glycated human serum albumin samples are shown on SDS-PAGE in Fig. 8. Native HSA showed a single band while glycated-HSA showed more than one band. This could be due the protein fragmentation caused by ROS generated by AGE's. All samples treated with cinnamic acid, exhibited a single band showing the protective role of cinnamic acid against the formation of AGE's. There was negligible fragmentation in all the treated samples including the one with aminoguanidine.
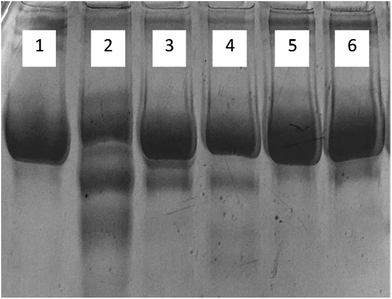 |
| | Fig. 8 SDS-polyacrylamide gel electrophoresis of HSA on 8% SDS-PAGE for 4 h at 80 V. Protein samples (10 μg in each lane) were loaded on well. Lanes: (1) native HSA; (2) glycated HSA; (3) aminoguanidine; (4) 50 μM; (5) 100 μM; (6) 200 μM cinnamic acid respectively. | |
3.2. Interaction studies
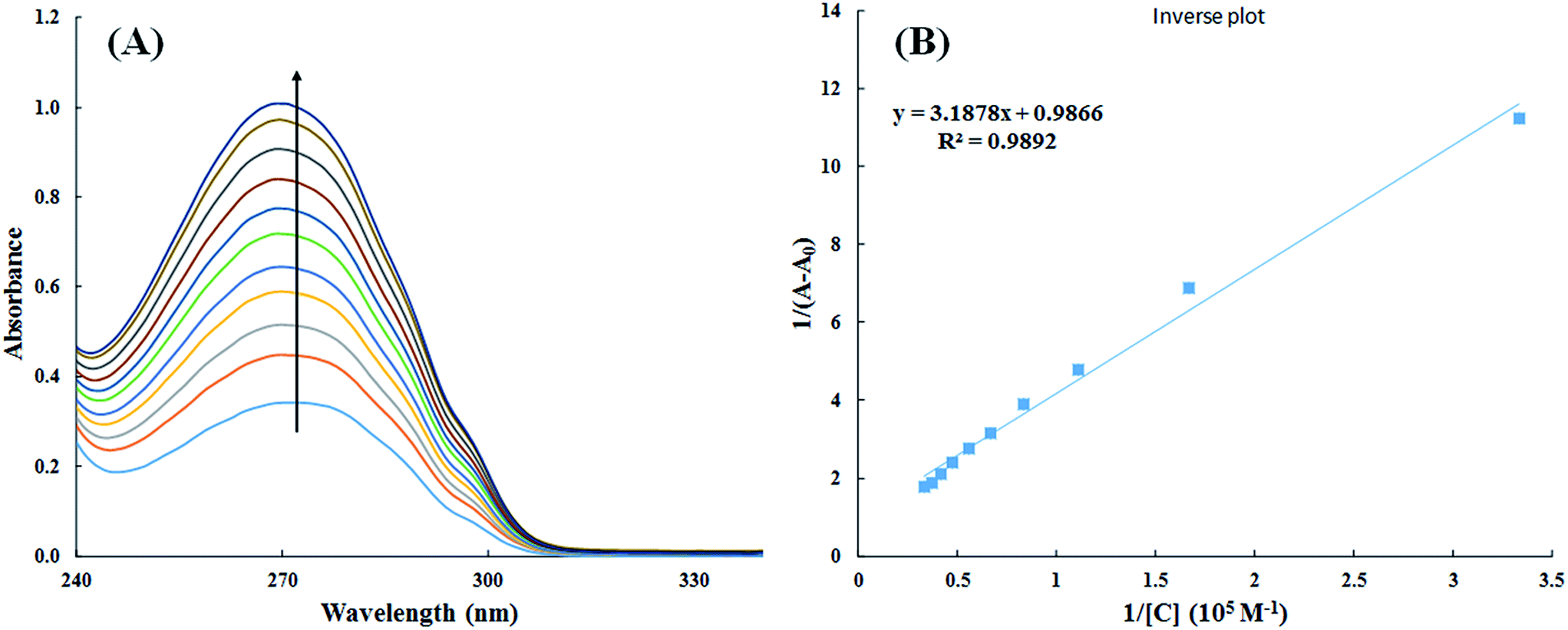
3.2.1. UV-visible spectroscopy. Interaction of small molecules that result in the formation of drug–protein complex are also measured by UV-visible spectroscopy and the extent of interaction is characterized by changes in absorbance or shift in the position of peak (λmax).39,40 Titration of HSA with increasing concentration of cinnamic acid resulted in hyperchromism of UV-visible spectrum of HSA with a negligible change in peak position Fig. 9A. These changes in the UV-visible spectrum of HSA may be due to corresponding changes in the conformation of HSA upon interaction with cinnamic acid.
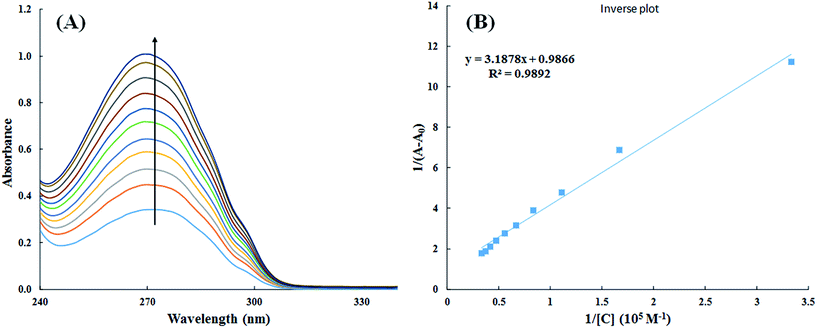 |
| | Fig. 9 UV-visible absorption spectra of HSA in absence and presence of varying concentration of cinnamic acid (A) and plot of 1/(A − A0) vs. 1/[cinnamic acid] (B). | |
The value of binding constant was calculated using eqn (2).41
| |
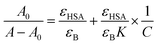 | (2) |
where
A0 and
A are the absorbance of HSA in the absence and presence of cinnamic acid,
εHSA and
εB are the molar extinction coefficient of HSA alone and the bound complex respectively,
K is the binding constant and
C is the concentration of cinnamic acid.
The double reciprocal plot of 1/(A − A0) vs. 1/C (Fig. 9B) is linear. The value of binding constant was estimated from the ratio of the intercept to that of slope of the above mentioned plot. The binding constant for the interaction of cinnamic acid with HSA was found to be 3.09 × 104 M−1 which signifies strong interaction between HSA and cinnamic acid.
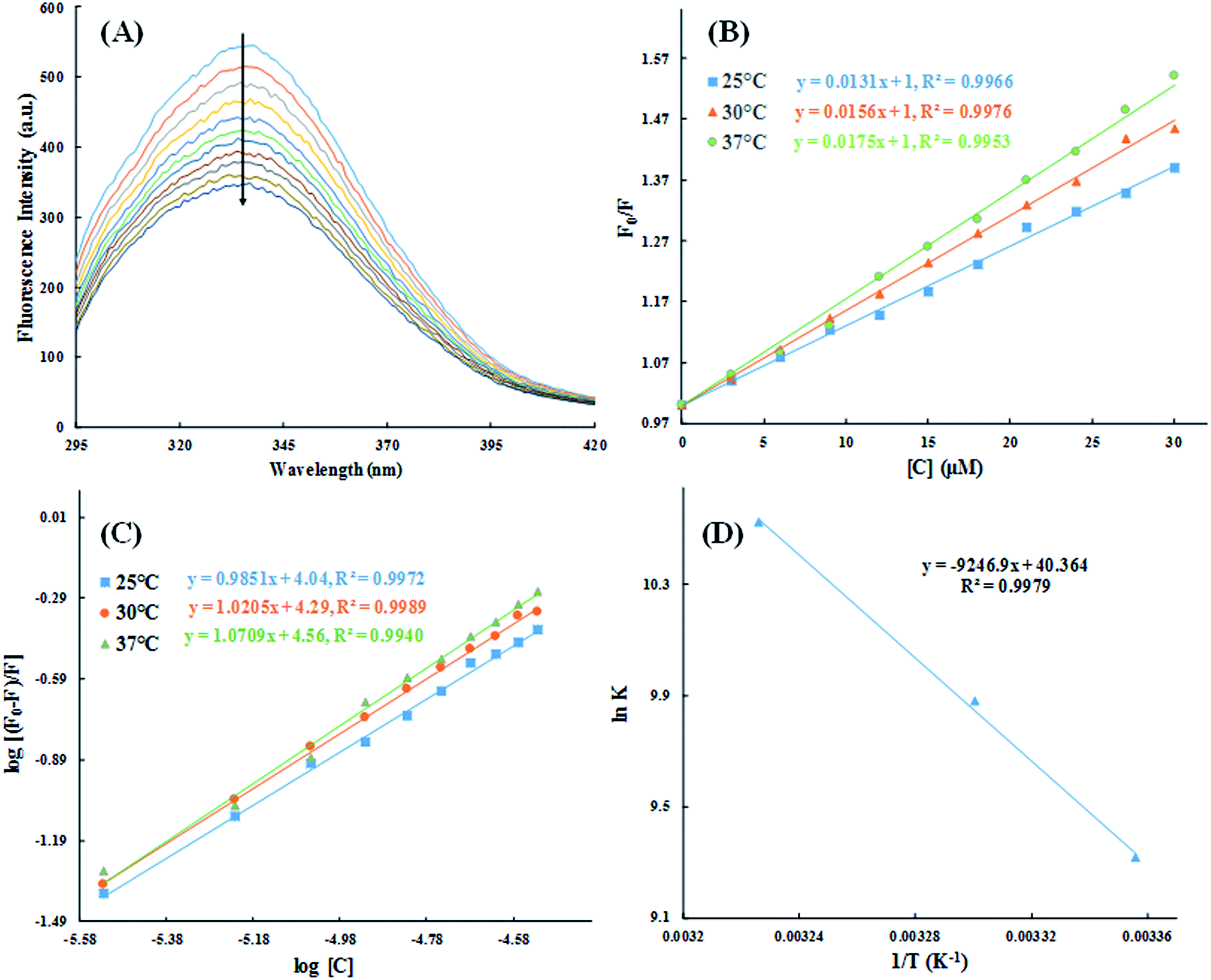
3.2.2. Fluorescence quenching studies. To further understand the interaction of HSA with cinnamic acid, we conducted fluorescence quenching experiment at three different temperatures. Fig. 10A shows the fluorescence emission spectra of HSA in absence and presence of varying concentration of cinnamic acid. HSA exhibited a strong fluorescence spectrum with λmax = 336 nm when excited at 280 nm. In the presence of increasing concentration of cinnamic acid a gradual quenching of the fluorescence intensity of HSA was observed suggesting the formation of a complex between cinnamic acid and HSA.
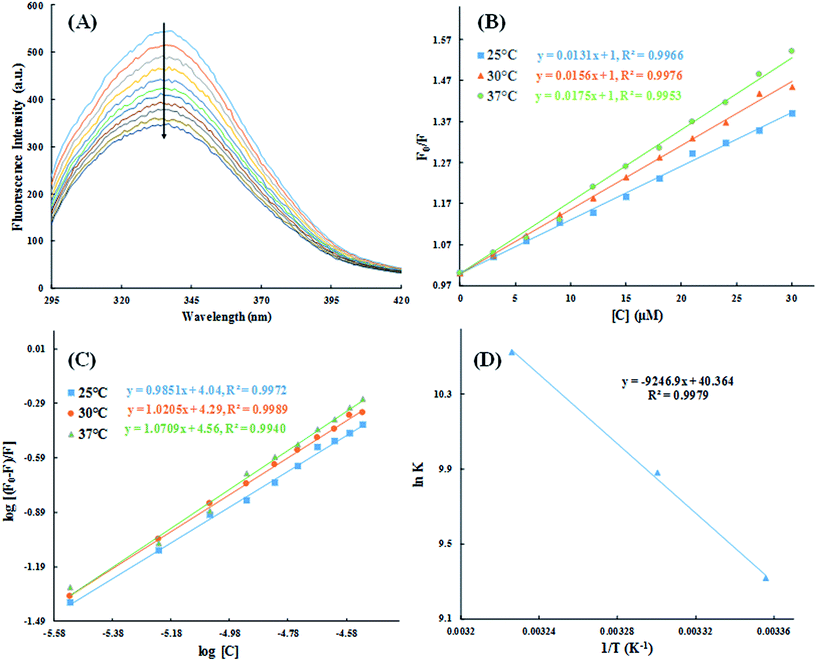 |
| | Fig. 10 Steady-state fluorescence spectra of HSA in absence and presence of varying concentration of cinnamic acid (A) and the Stern–Volmer plots for the HSA–cinnamic acid interaction at different temperatures (B). log[(F0 − F)/F] vs. log[Q] plots for HSA–cinnamic acid interaction (C) and Van't Hoff plot (D). | |
The quantitative analysis of fluorescence quenching data at different temperatures (298, 303 and 310 K) was performed through Stern–Volmer equation.42
| |
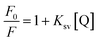 | (3) |
where,
F0 is the peak fluorescence intensity of free HSA,
F is the peak fluorescence intensity of HSA in presence of quencher (cinnamic acid),
Ksv is the Stern–Volmer constant and [Q] is the concentration of quencher (cinnamic acid). The Stern–Volmer plot of
F0/
F vs. [Q] is shown in
Fig. 10B. Slope of the Stern–Volmer plot at fixed intercept after linear regression of this plot, gives the Stern–Volmer constant as illustrated in
Table 2.
Table 2 Stern–Volmer constant and quenching constant of the HSA–cinnamic acid complex at different temperatures
| pH |
Temp (K) |
Ksv (×104 M−1) |
Kq (×1012 M−1 s−1) |
R2 |
| 7.4 |
298 |
1.310 |
2.267 |
0.9966 |
| 303 |
1.560 |
2.699 |
0.9976 |
| 310 |
1.819 |
3.146 |
0.9953 |
Generally, there are two types of fluorescence quenching mechanism i.e. dynamic and static which are governed by the way of interaction between quencher and HSA.43 If any quencher molecules possess sufficient amount of energy to collide with the excited state fluorophore of HSA (Trp-214) and take it back to the ground state, then this is due to the dynamic quenching mechanism. While in case of static quenching, there is formation of non-fluorescent ground state complex between quencher and fluorophore.44
To examine whether interaction of cinnamic acid with HSA is either static or dynamic quenching, eqn (4) was deployed.
| |
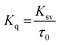 | (4) |
where,
Kq is the apparent bimolecular quenching rate constant and
τ0 is the average integral fluorescence lifetime of tryptophan which is ∼5.78 × 10
−9 s.
45 The quenching constant (
Kq) increased with increase in temperature (
Table 2). The maximum scatter collision quenching constant (
Kq) for various kinds of quenchers with biopolymer is approximately 2 × 10
10 L mol
−1 s
−1.
46 The values of quenching constants by cinnamic acid were found to be greater than the
Kq of the scatter mechanism. These results confirm that the fluorescence quenching mechanism of formation of HSA–cinnamic acid complex involved a static quenching procedure rather than dynamic.
47
Fluorescence quenching of HSA in presence of quencher are also used to quantitate the binding constant (K) and the number of binding sites (n) by modified Stern–Volmer eqn (5):
| |
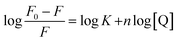 | (5) |
The values of n and K are obtained from the slope and Y-axis intercept from a linear regression of the plot of log[(F0 − F)/F)] vs. log[Q] (Fig. 10C). These values, as a function of temperature, are mentioned in Table 3. The values of K increases with increase in temperature, while the value of n was found to be approximately one at different temperatures. These results suggest the formation of a stable HSA–CA complex which might be more stable at 310 K (physiological temperature) as compared to lower temperature.
Table 3 Thermodynamic parameters, binding constant and number of binding sites for HSA–cinnamic acid complex formation at various temperatures
| pH |
Temp (K) |
K (×104 M−1) |
n |
R2 |
ΔG° (kcal mol−1) |
ΔH° (kcal mol−1) |
TΔS° (kcal mol−1) |
| 7.4 |
298 |
1.111 |
0.985 |
0.9972 |
−5.527 |
18.373 |
23.901 |
| 303 |
1.949 |
1.033 |
0.9989 |
−5.928 |
24.301 |
| 310 |
3.711 |
1.046 |
0.9940 |
−6.489 |
24.863 |
3.2.3. Thermodynamics of HSA–cinnamic acid interactions. The change in entropy, enthalpy and free energy at different temperatures were analysed from the van't Hoff's equations mentioned below.| |
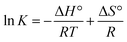 | (6) |
andwhere, K is the binding constant, ΔH° is the enthalpy change, ΔG° is the free Gibbs energy change, ΔS° is the entropy change, and R is the universal gas constant (1.987 cal mol−1 K−1).The nature of forces involved in the formation of HSA–cinnamic acid complex can be depicted from the magnitude and sign of various thermodynamic parameters.48 Therefore, we plotted the van't Hoff's plot to know the involvement of molecular forces in the HSA–cinnamic acid complex from the binding constants earlier obtained at their respective temperatures (Fig. 10D). The values of ΔH° and TΔS° mentioned in Table 3 were determined from the slope and Y-axis intercept of linear regression of van't Hoff's plot using eqn (6). The negative value of ΔG obtained from the eqn (7) suggest that the formation of HSA–cinnamic acid complex in spontaneous. Non-covalent interactions, mainly van der Waal's forces and hydrogen bonds played major role in cinnamic acid–HSA interactions.48 The positive TΔS° is also a strong manifestation that water molecules have been excluded from the interface of binding site of HSA, because the presence of water at binding site mimic nature of forces responsible for protein–ligand interaction. The positive value of TΔS° is routinely is regarded as quintessential affirmation of hydrophobic interactions.49
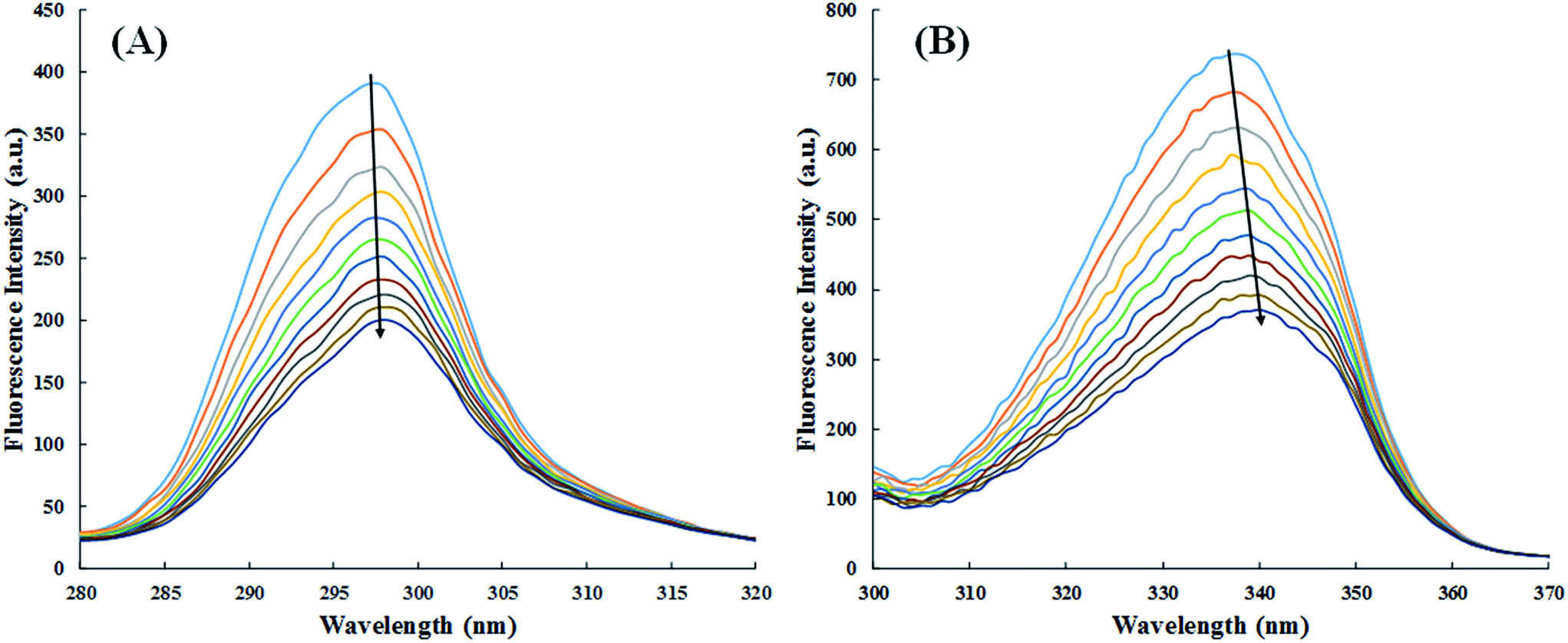
3.2.4. Synchronous fluorescence study. Fluorescence quenching study only tells about the binding constants and various thermodynamic parameters. Synchronous fluorescence spectroscopy was performed to examine whether the binding of cinnamic acid to HSA affects the conformation and/or the molecular environment of HSA's fluorophores.50 Any shift in maximum fluorescence emission wavelength of HSA is governed by the changes of polarity around the fluorophore molecules. When Δλ(λem − λex) is kept at 60 nm or 15 nm, the synchronous fluorescence spectra reveals the characteristic information of the microenvironment of tryptophan or tyrosine residues respectively.51The synchronous fluorescence spectra of HSA in absence and presence of cinnamic acid is shown in Fig. 11. It is evident from the Fig. 11A that there is negligible shift (∼1 nm) on maximum emission wavelength of tyrosine residues, stating that the local environment around tyrosine residue did not had any significant change. While the synchronous fluorescence spectra of tryptophan (Fig. 11B) changed remarkably (3–4 nm) indicating the increase in polarity around the tryptophan residue thereby lowering the hydrophobicity of the residue.52
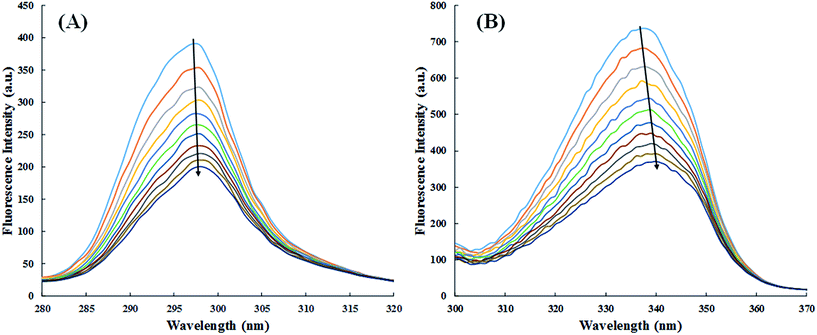 |
| | Fig. 11 Synchronous fluorescence spectra at Δλ = 15 (A) and Δλ = 60 (B) of HSA in absence and in presence of cinnamic acid at different concentrations. | |
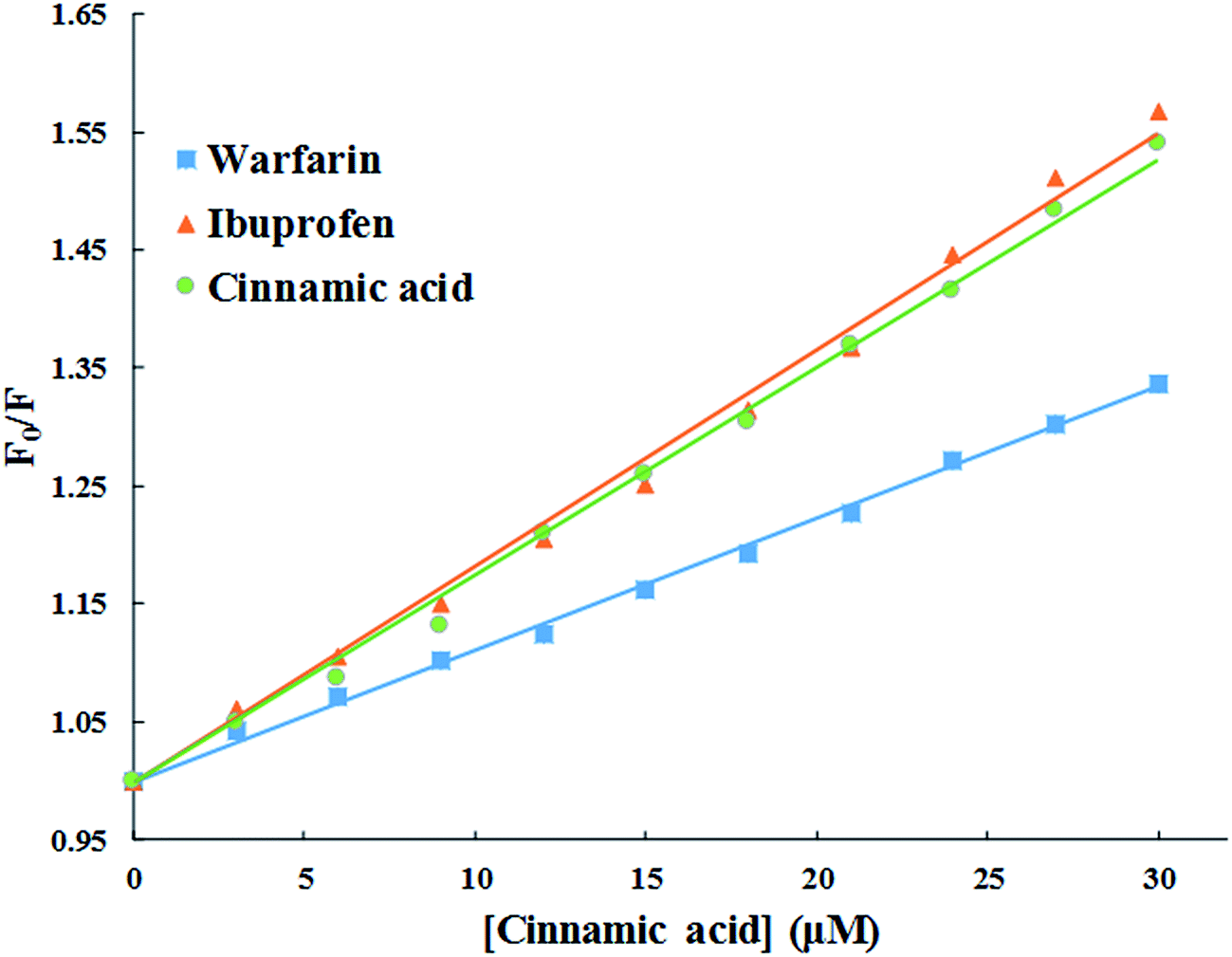
3.2.5. Evaluation of binding site of cinnamic to HSA in the presence of site markers (ibuprofen and warfarin). The crystal structure of HSA unveils that it has three homologous domains named I, II, and III, and each domain contain two sub-domains i.e. A and B.53 To find out the precise binding site, some site specific markers are often used, whose binding sites to HSA are already known. From the X-ray crystallographic studies, it is well known that warfarin is a probe for sub-domain IIA or the Sudlow site I. Similarly, ibuprofen is a probe for sub-domain IIIA or Sudlow site II.54 To facilitate the comparison of the influence of site markers on HSA–cinnamic acid system, a graph was plotted using the Stern–Volmer equation (Fig. 12). It is evident from the figure that the fluorescence property of cinnamic acid–HSA system did not had any significant change in presence of ibuprofen. This result indicates that ibuprofen did not had any effect on the binding of cinnamic acid to its usual location in HSA. In contrast, the fluorescence quenching property by cinnamic acid was remarkably decreased in the presence of warfarin. This illustrates that binding of cinnamic acid was greatly affected due to the presence of warfarin in the solution. The site specific experiment confirms that the binding site of cinnamic acid to HSA is mainly located in Sudlow's site I (sub-domain IIA) of HSA.
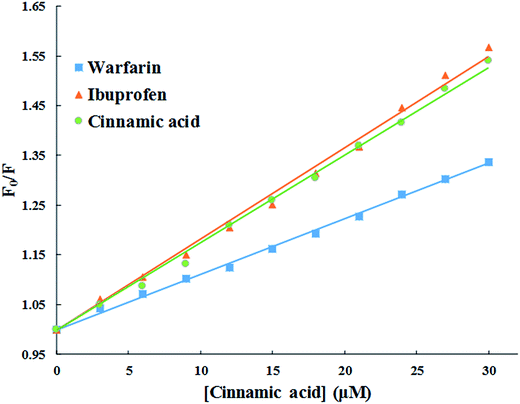 |
| | Fig. 12 Competitive binding of cinnamic to HSA in the presence of site markers at 37 °C [HSA at 3 μM and site markers at 6 μM]. | |
3.2.6. Effects of ionic strength. In this study, we further evaluated the involvement of electrostatic interaction in the stability of HSA–cinnamic acid complex. ESI Fig. S2† shows the titration of different concentration (10–100 mM) of NaCl to a fixed concentration of HSA–cinnamic acid complex which resulted in a very small increase in the fluorescence emission signal. The result suggests a negligible involvement of electrostatic interactions in the formation of HSA–cinnamic acid complex.
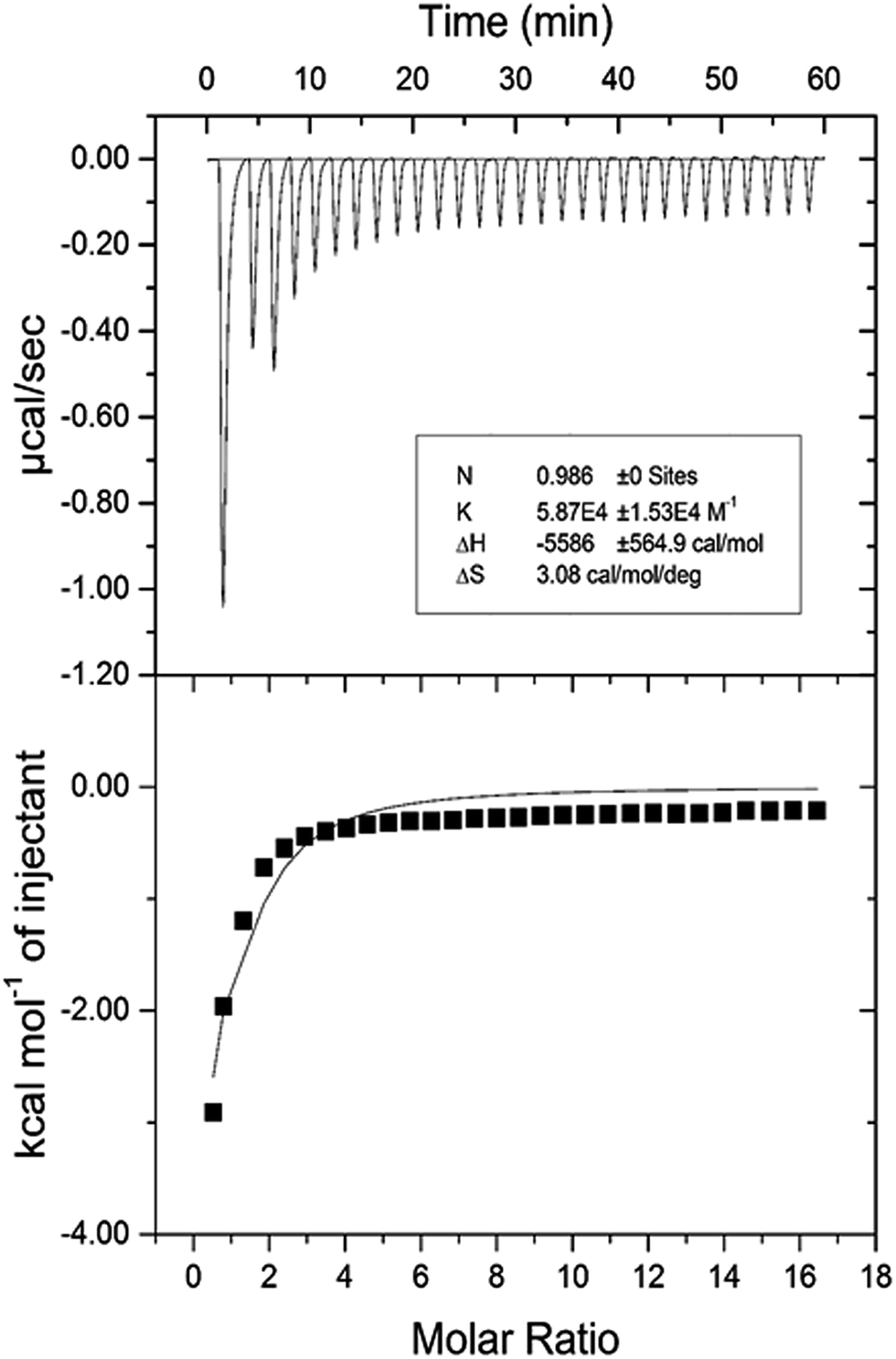
3.2.7. Isothermal titration calorimetric (ITC) characterization of HSA–cinnamic acid interaction. The validation of various thermodynamic parameters of HSA–cinnamic acid was further investigated from ITC studies. ITC experiments not only provides information about thermodynamic quantities such as enthalpy change (ΔH°), entropy change during binding (ΔS°), Gibb's free energy change (ΔG°) but also determines the binding affinity (K) and the number of binding sites (n).55 ITC profiles of the binding of cinnamic acid with HSA is depicted in Fig. 13. Measurements of various thermodynamic quantities are described in Table 4.
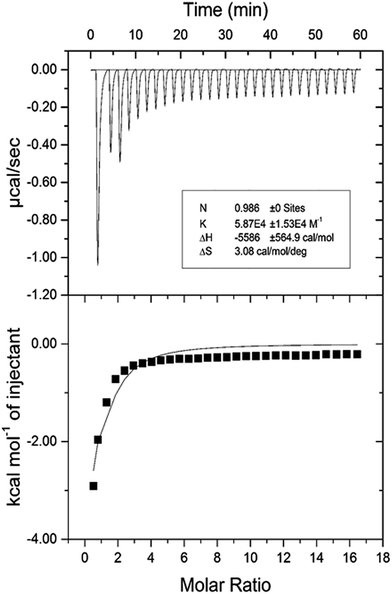 |
| | Fig. 13 Isothermal titration calorimetry profile of HSA and cinnamic acid interaction. | |
Table 4 Isothermal titration calorimetry of HSA and cinnamic acid interaction
| pH |
Temp. (K) |
Kb (×104 M−1) |
n |
ΔG° (kcal mol−1) |
ΔH° (kcal mol−1) |
TΔS° (kcal mol−1) |
| 7.4 |
298 |
5.87 |
0.986 |
−6.503 |
−5.586 |
0.917 |
The negative value of ΔH° confirms that the formation of HSA–cinnamic acid complex is exothermic. The value on n close to unity deciphers that HSA has single binding site for cinnamic acid. The positive value of TΔS° and negative value of ΔH° also indicates that HSA–cinnamic acid complex is mainly stabilized by hydrophobic interaction and hydrogen bond.54 The values of binding constant and Gibb's energy change (ΔG°) obtained from spectroscopy and calorimetry (Tables 3 and 4) are comparable and found to in same order. In contrast, the values of TΔS° and ΔH° differ significantly from that of obtained by the fluorescence spectroscopy. This is due the fact that the value of ΔH° is temperature dependent while fluorescent spectroscopic technique calculates it as temperature-independent parameter.45,56–58 Furthermore, the differences in the values of thermodynamic parameters obtained by fluorescence spectroscopy and ITC is due to the fact that ITC measures a global change in the thermodynamic property, whereas the fluorescence spectroscopy only measures the local changes around the fluorophore (Trp-214).59,60
3.2.8. Molecular docking studies. Molecular docking has been employed for further validation of spectroscopic and calorimetric experimental studies and to get a closer look of the site specific binding of cinnamic acid. HSA is 585 amino acids polypeptide chain having three domains as domain I (residues 1–195), domain II (196–383) and domain III (384–585) with further subdivision of each domain as subdomain A and B. Most common binding sites for drugs are located in the hydrophobic cavity of subdomain IIA of HSA.54 AutoDock-vina resulted in 9 best conformations with increasing order of Gibb's free energy and the conformation having lowest energy is depicted in Fig. 14. Molecular docking result reveals that cinnamic acid binds to subdomain IIA (Sudlow's site I) with a binding energy of −7.0 kcal mol−1, nearly the same value obtained using ITC and fluorescence spectroscopy. Cinnamic acid forms two hydrogen bonds with Ser287 and Arg257 of HSA and hydrophobic interactions with Leu219, Leu234, Leu238, Ile 264 and Leu260. The complex is also being stabilized by van der Walls forces that are formed between Arg222, Ile290, Ala291, Phe223 and Tyr150 of HSA and cinnamic acid. The cinnamic acid was found to be in the binding pocket (Sudlow's site I) of HSA surrounded by many hydrophobic residues such as Ala254, Val241, Leu233, Ile219, Val216, Ser287 etc. is in accordance with our site specific displacement experiments.49
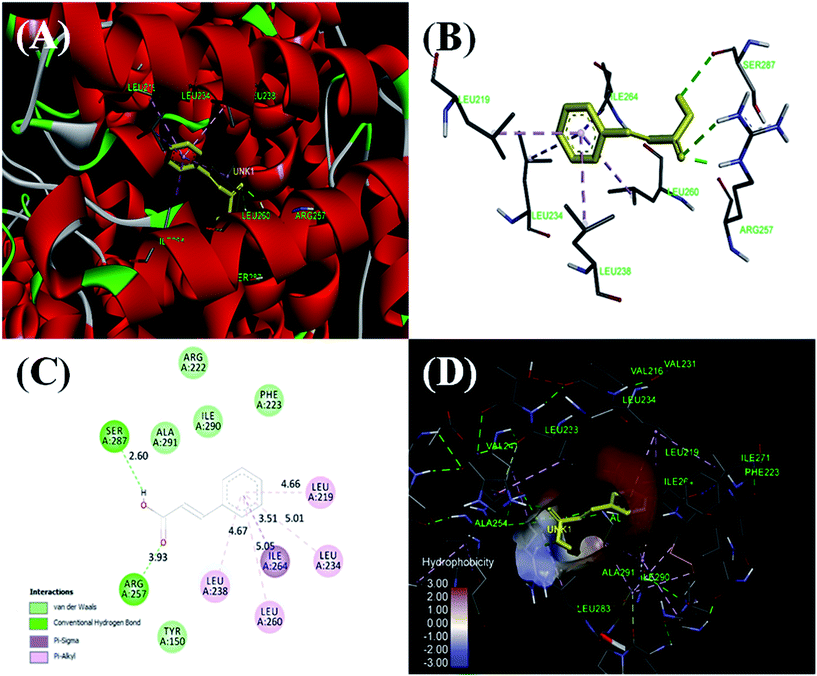 |
| | Fig. 14 Molecular models of HSA complexed with cinnamic acid. (A) Detailed view of the docking poses of the HSA–cinnamic acid complex, selected protein side-chains are shown as ribbons. (B) Cinnamic acid is shown in the binding pocket of HSA with interacting amino acids. (C) Cinnamic acid in hydrophobic pocket of HSA surrounded by hydrophobic amino acids. (D) A 2-dimensional view by Discovery Studio 4.5. | |
4. Conclusion
It is concluded that cinnamic acid strongly inhibits the formation of AGEs and acts as a potent antiglycating agent. The values of binding constants obtained both from UV-visible and fluorescence spectroscopy show a strong interaction between HSA and CA. While The negative value of Gibb's free energy change demonstrated that the process of HSA–CA complex formation is spontaneous, ITC results confirm that the reaction is exothermic in nature. Further, molecular docking studies showed that the interaction between HSA and CA are stabilised mainly through hydrophobic and hydrogen bond. The binding site of CA was at one of the major glycating sites in HSA. In the stable HSA–CA complex, reduced numbers of glycating sites are available to react further with free glucose, thereby inhibiting glycation. Evaluation of secondary structure by far-UV CD shows that treatment with CA, allows HSA to retained its native conformation required for proper function even in the presence of high concentration of glucose. Furthermore, the estimation of various biomarkers such as free lysine group, carbonyl content, free sulfhydryl group, reactive oxygen species etc. depicts that cinnamic acid is a quencher of free radical and could prove beneficial for diabetes and associated complications. Reckoning with these facts, the study may be valuable to compare the effect of different concentrations of CA on HSA–glucose structure and conformation upon glycation. However, most of the investigations in this area are limited to in vitro experimental studies and therefore, further investigations should examine the in vivo efficacy of cinnamic acid in suitable animal model.
Conflict of interest
The authors declare there are no conflicts of interest.
Acknowledgements
We are grateful to Prof. Mohd. Shakir, Chairman, Department of Chemistry, and Prof. Javed Musarrat, Department of Agricultural Microbiology Aligarh Muslim University, Aligarh for providing Instrumentation facilities and encouragement.
References
- F. Akhter, M. S. Khan, U. Shahab and S. Ahmad, Bio-physical characterization of ribose induced glycation: a mechanistic study on DNA perturbations, Int. J. Biol. Macromol., 2013, 58, 206–210 CrossRef CAS PubMed.
- T. J. Lyons, S. R. Thorpe and J. W. Baynes, Glycation and autoxidation of proteins in aging and diabetes, in Hyperglycemia, Diabetes, and Vascular Disease, Springer, New York, 1992, pp. 197–217 Search PubMed.
- M. Brownlee, Biochemistry and molecular cell biology of diabetic complications, Nature, 2001, 414(6865), 813–820 CrossRef CAS PubMed.
- P. J. Thornalley, Advanced glycation and the development of diabetic complications. Unifying the involvement of glucose, methylglyoxal and oxidative stress, Endocrinol. Metab., 1996, 3, 149–166 CAS.
- S. Adisakwattana, W. Sompong, A. Meeprom, S. Ngamukote and S. Yibchok-anun, Cinnamic acid and its derivatives inhibit fructose-mediated protein glycation, Int. J. Mol. Sci., 2012, 13(2), 1778–1789 CrossRef CAS PubMed.
- L. Kardeşler, N. Buduneli, B. Bıyıkoğlu, Ş. Çetinkalp and N. Kütükçüler, Gingival crevicular fluid PGE 2, IL-1β, t-PA, PAI-2 levels in type 2 diabetes and relationship with periodontal disease, Clin. Biochem., 2008, 41(10), 863–868 CrossRef PubMed.
- V. P. Singh, A. Bali, N. Singh and A. S. Jaggi, Advanced glycation end products and diabetic complications, Korean J. Physiol. Pharmacol., 2014, 18(1), 1–4 CrossRef CAS PubMed.
- A. A. Chinchansure, A. M. Korwar, M. J. Kulkarni and S. P. Joshi, Recent development of plant products with anti-glycation activity: a review, RSC Adv., 2015, 5(39), 31113–31138 RSC.
- D. Cervantes-Laurean, D. D. Schramm, E. L. Jacobson, I. Halaweish, G. G. Bruckner and G. A. Boissonneault, Inhibition of advanced glycation end product formation on collagen by rutin and its metabolites, J. Nutr. Biochem., 2006, 17(8), 531–540 CrossRef CAS PubMed.
- S. Sang, X. Shao, N. Bai, C. Y. Lo, C. S. Yang and C. T. Ho, Tea polyphenol (−)-epigallocatechin-3-gallate: a new trapping agent of reactive dicarbonyl species, Chem. Res. Toxicol., 2007, 20(12), 1862–1870 CrossRef CAS PubMed.
- F. Natella, M. Nardini, M. Di Felice and C. Scaccini, Benzoic and cinnamic acid derivatives as antioxidants: structure–activity relation, J. Agric. Food Chem., 1999, 47(4), 1453–1459 CrossRef CAS PubMed.
- I. M. Liu, F. L. Hsu, C. F. Chen and J. T. Cheng, Antihyperglycemic action of isoferulic acid in streptozotocin-induced diabetic rats, Br. J. Pharmacol., 2000, 129(4), 631–636 CrossRef CAS PubMed.
- D. W. Huang and S. C. Shen, Caffeic acid and cinnamic acid ameliorate glucose metabolism via modulating glycogenesis and gluconeogenesis in insulin-resistant mouse hepatocytes, J. Funct. Foods, 2012, 4(1), 358–366 CrossRef CAS.
- W. Arlt, P. Neogi, C. Gross and W. L. Miller, Cinnamic acid based thiazolidinediones inhibit human P450c17 and 3beta-hydroxysteroid dehydrogenase and improve insulin sensitivity independent of PPARgamma agonist activity, J. Mol. Endocrinol., 2004, 32(2), 425–436 CrossRef CAS PubMed.
- M. M. Alam, I. Ahmad and I. Naseem, Inhibitory effect of quercetin in the formation of advance glycation end products of human serum albumin: an in vitro and molecular interaction study, Int. J. Biol. Macromol., 2015, 79, 336–343 CrossRef CAS PubMed.
- O. Classics Lowry, N. Rosebrough, A. Farr and R. Randall, Protein measurement with the Folin phenol reagent, J. Biol. Chem., 1951, 193, 265–275 Search PubMed.
- M. Bohlooli, M. Ghaffari-Moghaddam, M. Khajeh, Z. Aghashiri, N. Sheibani and A. A. Moosavi-Movahedi, Acetoacetate promotes the formation of fluorescent advanced glycation end products (AGEs), J. Biomol. Struct. Dyn., 2016, 1–9 Search PubMed.
- N. Sattarahmady, A. A. Moosavi-Movahedi, F. Ahmad, G. H. Hakimelahi, M. Habibi-Rezaei, A. A. Saboury and N. Sheibani, Formation of the molten globule-like state during prolonged glycation of human serum albumin, Biochim. Biophys. Acta, Gen. Subj., 2007, 1770(6), 933–942 CrossRef CAS PubMed.
- C. Lee, M. B. Yim, P. B. Chock, H. S. Yim and S. O. Kang, Oxidation-reduction properties of methylglyoxal-modified protein in relation to free radical generation, J. Biol. Chem., 1998, 273(39), 25272–25278 CrossRef CAS PubMed.
- B. Bouma, L. M. Kroon-Batenburg, Y. P. Wu, B. Brünjes, G. Posthuma, O. Kranenburg, P. G. de Groot, E. E. Voest and M. F. Gebbink, Glycation induces formation of amyloid cross-β structure in albumin, J. Biol. Chem., 2003, 278(43), 41810–41819 CrossRef CAS PubMed.
- M. L. Kakade and I. E. Liener, Determination of available lysine in proteins, Anal. Biochem., 1969, 27(2), 273–280 CrossRef CAS PubMed.
- R. L. Levine, J. A. Williams, E. P. Stadtman and E. Shacter, [37] Carbonyl assays for determination of oxidatively modified proteins, Methods Enzymol., 1994, 233, 346–357 CAS.
- Neelofar, J. Ahmad and K. Alam, Impact of in vitro non-enzymatic glycation on biophysical and biochemical regimes of human serum albumin: relevance in diabetes associated complications, RSC Adv., 2015, 5(78), 63605–63614 RSC.
- M. A. Husain, T. Sarwar, S. U. Rehman, H. M. Ishqi and M. Tabish, Ibuprofen causes photocleavage through ROS generation and intercalates with DNA: a combined biophysical and molecular docking approach, Phys. Chem. Chem. Phys., 2015, 17, 13837–13850 RSC.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31(2), 455–461 CAS.
- P. Pandya, L. K. Agarwal, N. Gupta and S. Pal, Molecular recognition pattern of cytotoxic alkaloid vinblastine with multiple targets, J. Mol. Graphics Modell., 2014, 54, 1–9 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function, J. Comput. Chem., 1998, 19(14), 1639–1662 CrossRef CAS.
- J. Leclère and I. Birlouez-Aragon, The fluorescence of advanced Maillard products is a good indicator of lysine damage during the Maillard reaction, J. Agric. Food Chem., 2001, 49(10), 4682–4687 CrossRef.
- M. E. Westwood and P. J. Thornalley, Molecular characteristics of methylglyoxal-modified bovine and human serum albumins. Comparison with glucose-derived advanced glycation endproduct-modified serum albumins, J. Protein Chem., 1995, 14(5), 359–372 CrossRef CAS PubMed.
- D. Matulis and R. Lovrien, 1-Anilino-8-naphthalene sulfonate anion-protein binding depends primarily on ion pair formation, Biophys. J., 1998, 74(1), 422–429 CrossRef CAS PubMed.
- S. A. Bhat, A. Sohail, A. A. Siddiqui and B. Bano, Effect of non-enzymatic glycation on cystatin: a spectroscopic study, J. Fluoresc., 2014, 24(4), 1107–1117 CrossRef CAS PubMed.
- T. A. Khan, M. Saleemuddin and A. Naeem, Partially folded glycated state of human serum albumin tends to aggregate, Int. J. Pept. Res. Ther., 2011, 17(4), 271–279 CrossRef CAS.
- R. Nagai, K. Matsumoto, X. Ling, H. Suzuki, T. Araki and S. Horiuchi, Glycolaldehyde, a reactive intermediate for advanced glycation end products, plays an important role in the generation of an active ligand for the macrophage scavenger receptor, Diabetes, 2000, 49(10), 1714–1723 CrossRef CAS PubMed.
- R. Gomes, M. S. Silva, A. Quintas, C. Cordeiro, A. Freire, P. Pereira, A. Martins, E. Monteiro, E. Barroso and A. P. Freire, Argpyrimidine, a methylglyoxal-derived advanced glycation end-product in familial amyloidotic polyneuropathy, Biochem. J., 2005, 385(2), 339–345 CrossRef CAS PubMed.
- S. Awasthi and N. T. Saraswathi, Vanillin restrains non-enzymatic glycation and aggregation of albumin by chemical chaperone like function, Int. J. Biol. Macromol., 2016, 87, 1–6 CrossRef CAS PubMed.
- M. Khajehpour, J. L. Dashnau and J. M. Vanderkooi, Infrared spectroscopy used to evaluate glycosylation of proteins, Anal. Biochem., 2006, 348(1), 40–48 CrossRef CAS PubMed.
- M. F. Beal, Oxidatively modified proteins in aging and disease 1, 2, Free Radicals Biol. Med., 2002, 32(9), 797–803 CrossRef CAS PubMed.
- N. Sreerama and R. W. Woody, A self-consistent method for the analysis of protein secondary structure from circular dichroism, Anal. Biochem., 1993, 209(1), 32–44 CrossRef CAS PubMed.
- J. Jaumot and R. Gargallo, Experimental methods for studying the interactions between G-quadruplex structures and ligands, Curr. Pharm. Des., 2012, 18(14), 1900–1916 CrossRef CAS PubMed.
- H. Sun, J. Xiang, Y. Liu, L. Li, Q. Li, G. Xu and Y. Tang, A stabilizing and denaturing dual-effect for natural polyamines interacting with G-quadruplexes depending on concentration, Biochimie, 2011, 93(8), 1351–1356 CrossRef CAS PubMed.
- S. Tabassum, W. M. Al-Asbahy, M. Afzal, F. Arjmand and R. H. Khan, Interaction and photo-induced cleavage studies of a copper based chemotherapeutic drug with human serum albumin: spectroscopic and molecular docking study, Mol. BioSyst., 2012, 8(9), 2424–2433 RSC.
- M. R. Eftink, Fluorescence quenching reactions, in Biophysical and biochemical aspects of fluorescence spectroscopy, Springer, US, 1991, pp. 1–41 Search PubMed.
- J. Mariam, P. M. Dongre and D. C. Kothari, Study of interaction of silver nanoparticles with bovine serum albumin using fluorescence spectroscopy, J. Fluoresc., 2011, 21(6), 2193–2199 CrossRef CAS PubMed.
- A. Bhogale, N. Patel, P. Sarpotdar, J. Mariam, P. M. Dongre, A. Miotello and D. C. Kothari, Systematic investigation on the interaction of bovine serum albumin with ZnO nanoparticles using fluorescence spectroscopy, Colloids Surf., B, 2013, 102, 257–264 CrossRef CAS PubMed.
- M. Ishtikhar, S. Khan, G. Badr, A. O. Mohamed and R. H. Khan, Interaction of the 5-fluorouracil analog 5-fluoro-2′-deoxyuridine with ‘N’and ‘B’isoforms of human serum albumin: a spectroscopic and calorimetric study, Mol. BioSyst., 2014, 10(11), 2954–2964 RSC.
- Q. Saquib, A. A. Al-Khedhairy, S. A. Alarifi, S. Dwivedi, J. Mustafa and J. Musarrat, Fungicide methyl thiophanate binding at sub-domain IIA of human serum albumin triggers conformational change and protein damage, Int. J. Biol. Macromol., 2010, 47(1), 60–67 CrossRef CAS PubMed.
- S. Monti, S. Ottani, F. Manoli, I. Manet, F. Scagnolari, B. Zambelli and G. Marconi, Chiral recognition of 2-(3-benzoylphenyl) propionic acid (ketoprofen) by serum albumin: an investigation with microcalorimetry, circular dichroism and molecular modelling, Phys. Chem. Chem. Phys., 2009, 11(40), 9104–9113 RSC.
- P. D. Ross and S. Subramanian, Thermodynamics of protein association reactions: forces contributing to stability, Biochemistry, 1981, 20(11), 3096–3102 CrossRef CAS PubMed.
- M. Ishtikhar, G. Rabbani and R. H. Khan, Interaction of 5-fluoro-5′-deoxyuridine with human serum albumin under physiological and non-physiological condition: a biophysical investigation, Colloids Surf., B, 2014, 123, 469–477 CrossRef CAS PubMed.
- N. Ibrahim, H. Ibrahim, S. Kim, J. P. Nallet and F. Nepveu, Interactions between antimalarial indolone-N-oxide derivatives and human serum albumin, Biomacromolecules, 2010, 11(12), 3341–3351 CrossRef CAS PubMed.
- J. N. Miller, Recent advances in molecular luminescence analysis, in Proc Anal Div Chem Soc, 1979, vol. 16, no. 7, pp. 203–208 Search PubMed.
- U. Kragh-Hansen, Molecular aspects of ligand binding to serum albumin, Pharmacol. Rev., 1981, 33(1), 17–53 CAS.
- T. Chakraborty, I. Chakraborty, S. P. Moulik and S. Ghosh, Physicochemical and conformational studies on BSA–surfactant interaction in aqueous medium, Langmuir, 2009, 25(5), 3062–3074 CrossRef CAS PubMed.
- G. D. Sudlow, D. J. Birkett and D. N. Wade, The characterization of two specific drug binding sites on human serum albumin, Mol. Pharmacol., 1975, 11(6), 824–832 CAS.
- M. W. Freyer and E. A. Lewis, Isothermal titration calorimetry: experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions, Methods Cell Biol., 2008, 84, 79–113 CAS.
- M. Ishtikhar, G. Rabbani and R. H. Khan, Interaction of 5-fluoro-5′-deoxyuridine with human serum albumin under physiological and non-physiological condition: a biophysical investigation, Colloids Surf., B, 2014, 123, 469–477 CrossRef CAS PubMed.
- A. A. Thoppil, R. Sharma and N. Kishore, Complexation of β-lactam antibiotic drug carbenicillin to bovine serum albumin: energetics and conformational studies, Biopolymers, 2008, 89(10), 831–840 CrossRef CAS PubMed.
- E. Ahmad, G. Rabbani, N. Zaidi, S. Singh, M. Rehan, M. M. Khan, S. K. Rahman, Z. Quadri, M. Shadab, M. T. Ashraf and N. Subbarao, Stereo-selectivity of human serum albumin to enantiomeric and isoelectronic pollutants dissected by spectroscopy, calorimetry and bioinformatics, PLoS One, 2011, 6(11), e26186 CAS.
- H. Watanabe, S. Tanase, K. Nakajou, T. Maruyama, U. Kragh-Hansen and M. Otagiri, Role of Arg-410 and Tyr-411 in human serum albumin for ligand binding and esterase-like activity, Biochem. J., 2000, 349(3), 813–819 CrossRef CAS PubMed.
- M. T. Rehman, H. Shamsi and A. U. Khan, Insight into the binding mechanism of imipenem to human serum albumin by spectroscopic and computational approaches, Mol. Pharmaceutics, 2014, 11(6), 1785–1797 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra12321j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.